

Abstract
Glucocorticoid-induced TNFR family-related protein (GITR)-mediated activation of c-Jun N-terminal kinase (JNK) was shown to regulate the suppressive activity of naturally occurring CD4+CD25+ T regulatory cells (nTregs) in wild-type (WT) hosts. Here, CD4+CD25+ T cells were shown capable of becoming pathogenic effector cells in sensitized and challenged CD8−/− recipient mice. Only GITR-expressing CD4+CD25+ T cells but neither GITR knocked-in CD4+CD25− T cells or GITR-silenced CD4+CD25+ T cells enhanced development of lung allergic responses. Inhibition of JNK in WT nTregs or nTregs from GITR−/− and JNK2−/− mice failed to enhance lung allergic responses in sensitized and challenged CD8−/− recipient mice. The failure to enhance responses was associated with increased bronchoalveolar lavage (BAL) fluid levels of IL-10 and TGF-β and decreased levels of IL-5, IL-6, and IL-13. In contrast, nTregs from JNK1−/− mice, similar to WT nTregs, were fully effective in enhancing responses. Thus, GITR stimulation of nTregs and signaling through JNK2 but not JNK1 triggered the loss of regulatory function while concomitantly gaining pathogenic CD4+ T effector cell function responsible for exacerbating asthma-like immunopathology.
Keywords: CD4+CD25+ T regulatory cells, plasticity, c-Jun amino-terminal kinase 1 and 2 (JNK1 and JNK2), asthma
INTRODUCTION
Glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) is involved in a number of activities including cell proliferation, differentiation, survival, and function in both the innate and adaptive immune systems (1). GITR was first described in the mouse as a dexamethasone-inducible molecule in a T cell hybridoma (2) and subsequently, a human equivalent was defined (3). GITR, similar to CD25 and CD152, is constitutively expressed by naturally occurring Foxp3+CD4+CD25+ T regulatory cells (nTregs), a small population (5–10%) of circulating lymphocytes. These cells function in the maintenance of immunological tolerance, prevention of autoimmunity, and immunity against foreign pathogens (4). Ligation of GITR by its natural ligand (GITRL) or an agonistic GITR-specific antibody (DTA-1) is generally considered stimulatory, and has been shown to abrogate the suppressive activity of Foxp3+CD4+CD25+ T regulatory cells (5–9). Expression of GITRL in the lungs of recipient mice was shown to directly modulate the activity of nTregs (5). In vitro stimulation of isolated nTregs with GITRL prior to adoptive transfer abrogated suppression of allergen-induced lung allergic responses in sensitized and challenged wild-type (WT) recipients (9), in contrast to rendering effector cells resistant to the suppressive activities of Tregs (10, 11).
In both humans and animals, allergic asthma is an inflammatory disease of the airways characterized by increases in airway hyperresponsiveness (AHR) and inflammation, Th2 cytokine skewing, goblet cell metaplasia, excessive mucus production, elevated antigen-specific IgE, and structural remodeling of the airways. Increasingly, nTregs have been shown to be important and effective regulators of the development and outcomes of lung responses to allergen sensitization and challenge (5, 9). These activities are mediated by the immunocytokines IL-10 and TGF-β released from regulatory T cells (12, 13) in both an antigen-specific (14) and antigen-nonspecific manner (15, 16).
The phenotypic and functional stability of nTregs has been shown to depend on a number of factors including expression levels of the key transcription factor, Foxp3 (17, 18). Spontaneous mutations of Foxp3 have been associated with multiorgan autoimmune disease in Scurfy mice (19) and X-linked immune dysregulation, polyendocrinopathy, and enteropathy (IPEX) syndrome in humans (20). Cytokines such as IL-6 (21–23) and surface proteins such as CD8 (5, 24) have also been shown to impact nTreg activity. In the absence of or interference with MHC I-CD8 interactions, the regulatory activities of nTregs were altered not only resulting in the loss of suppression but in their conversion to pathogenic IL-13-producing CD4+ T effector cells, enhancing lung allergic responses in recipient mice (5). Pathogenic conversion of Tregs has also been described in other experimental models (25, 26). In addition, both the maintenance of suppressive activities in peripheral tissues and the regulation of endogenous production of IL-6 by nTregs were shown to be dependent on the presence of CD8+ T cells (21). Full restoration of suppressive activities and inhibition of IL-6 production in nTregs from CD8−/− mice could be achieved by reconstituting CD8+ T cells in deficient hosts, suggesting that functional plasticity was still possible after thymic development, differentiation, and emigration.
Previously, the critical role of GITR in the conversion of naturally occurring CD4+CD25+ T regulatory cells to pathogenic CD4+ T effector cells was implicated by the abrogation of enhancement of lung allergic response following administration of anti-GITRL antibody (5). Activation of c-Jun N-terminal kinase (JNK) following GITRL ligation of GITR was associated with the loss of suppressive activity (9). Although signaling cascades through GITR in immune cells have been described (4), there has been little to no evidence describing involvement of these pathways in the functional plasticity of nTregs. Given that the same cells are capable of exhibiting different responses, suppression or enhancement, depending on the CD8 expression status of the host (5, 24), we hypothesized that the plasticity of nTregs may also be determined by GITR-mediated activation through JNK.
MATERIALS AND METHODS
Animals
Pathogen-free, 6–8 week old female C57BL/6, WT, CD8−/−, JNK1−/− and JNK2−/− mice were obtained from Jackson Laboratories (Bar Harbor, ME). GITR−/− mice were provided by Dr. Carlo Riccardi (Perugia, Italy). All mice were maintained on an ovalbumin (OVA)-free diet. All protocols were approved by the Institutional Animal Care and Use Committee at National Jewish Health.
Sensitization and Challenge
Sensitization was carried out by intraperitoneal injection of 20 μg OVA (Sigma Aldrich, St. Louis, MO) emulsified in 2.25 mg alum hydroxide (AlumImject; Pierce, Rockford, IL) in a total volume of 100 μl on days 1 and 14. Sensitized and challenged mice, denoted OVA/OVA, and non-sensitized but challenged littermates (PBS/OVA) received aerosol challenges for 20 minutes each day on three consecutive days (days 26, 27, and 28) with 1% OVA in PBS using an ultrasonic nebulizer (Omron, Vernon Hills, IL) (5).
Measurement of Airway Responsiveness
Airway responsiveness was assessed as previously described (5). Changes in airway function to increasing concentrations of aerosolized methacholine (MCh) administered for 10 seconds (60 breaths/min, 500 μl tidal volume) were monitored. Lung resistance (RL) was continuously computed (Labview, National Instruments, Austin, TX) by fitting flow, volume, and pressure to an equation of motion. Maximum values of RL were taken and expressed as a percentage change from baseline following PBS aerosol.
Bronchoalveolar Lavage (BAL)
Immediately following measurement of AHR lungs were lavaged. Total leukocyte numbers were counted (Countess Automated Cell Counter, Invitrogen Corporation, Carlsbad, CA) and differential cell counts were performed in a blinded manner under light microscopy by counting at least 200 cells on cytocentrifuged preparations (Cytospin 2; Cytospin, Shandon Ltd., Runcorn, Cheshire, UK), stained with Leukostat (Fisher Diagnostics, Middletown, VA) and differentiated by standard hematological procedures.
Cell Preparation and Culture
CD4+CD25+ and CD4+CD25− T cells from lungs of naive C57BL/6, GITR−/−, JNK1−/−, and JNK2−/− donors were isolated by collagenase digestion and enriched using nylon wool columns as described previously (12). Lymphocytes were further purified by CD4+CD25+ Regulatory T cell MACS beads (Miltenyi Biotec, Bergisch-Gladbach, Germany), resulting in a purity of >95% CD4+CD25+ cells and by sorting on Synergy (Sony Biotechnology, Champaign, IL) giving a purity of 99% CD4+CD25+ T cells. For reconstitution experiments, CD8+ T cells were purified from spleens by MACS beads giving a purity of >96%. Following positive selection, CD4+CD25− T cells were isolated to >97% purity.
Cells were washed, counted, and resuspended to a final concentration of 4×106 cells per ml in complete RPMI 1640 (Mediatech Celgro, Manassas, VA) tissue culture medium, containing heat-inactivated fetal calf serum (FCS 10%; Sigma-Aldrich), L-glutamine (5 mM), β-mercaptoethanol (2 mM), hepes buffer (15 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml) (all from Gibco, Grand Island, NY).
In Vitro Treatment and Adoptive Transfer
In some experiments, isolated CD4+CD25− and CD4+CD25+ T cells were either pretreated with an active (SP600125, JNKi) or inactive JNK (JNK II inhibitor, negative control) inhibitor (50 μM, Calbiochem), labeled with 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE, eBioscience, San Diego, CA), cotransfected with GFP expression vector, and control or GITR shRNA lentivirus (Santa Cruz Biotechnology, Santa Cruz, CA), and control or GITR expression vector pORF9-mTNFRSF18 (InvivoGen, San Diego, CA) using the Amaxa Mouse T Cell Nucleofector Kit (Lonza, Lonza Cologne, Germany). Cells were sorted on Synergy, giving 100% CFSE+ and GFP+ cells showing respectively, a >30% reduction and >90% increase in GITR expression. Sorted cells were washed extensively prior to intratracheal adoptive transfer of 5×105 nTregs in 50 μL of PBS into sensitized recipients prior to allergen challenge. In reconstitution experiments, 10×106 naive CD8+ T cells were transferred intravenously into CD8−/− mice prior to sensitization.
Measurement of Cytokine Levels
Cytokine levels in the BAL fluid and supernatants of in vitro cultured lung cells were measured by ELISA, IL-4, IL-5, IL-6, IL-10, IL-13, and IFN-γ from eBioscience and TGF-β from BD Biosciences Pharmingen, San Jose, CA. ELISAs were performed according to the manufacturers’ directions. The limits of detection were 4 pg/ml for IL-4, IL-5, and IL-6, 10 pg/ml for IL-10 and IFN-γ, 8 pg/ml for IL-13, and 6 pg/ml for TGF-β.
FACS Analysis
Isolated cells, following preincubation with naive mouse serum in staining buffer (PBS, 2% FCS, 0.2% sodium azide), were labeled with the following conjugated antibodies purchased from BD Biosciences Pharmingen: anti-CD3 FITC, PE, PerCP, APC (17A2); anti-CD4 FITC, PE, PerCP, APC (L3T4); anti-CD25 FITC (7D4), PE (PC61); anti-GITR FITC, PE, APC; CD152 FITC, PE APC. For intracellular staining, cells were stimulated with PMA (100 ng/ml) and ionomycin (2 μg/ml, Sigma-Aldrich) in complete medium overnight and for 6 hrs in the presence of brefeldin A (10 μg/ml, Sigma-Aldrich). Cells were fixed with 4% formaldehyde in PBS, permeabilized in 0.5% saponin, and stained with anti-IL-10 PE, APC (JES5-16E3): IFN-γ PE, APC (XMG1.2); Foxp3 PE and TGF-β (A75-3.1) (eBioscience). Fluorochrome (FITC, PE, PerCPAPC)-labeled isotype-matched control antibodies were used for background fluorescence staining. Staining was analyzed by FACScalibur flow cytometry (BD Pharmingen,) using CellQuest Pro software. Fluorescence intensity was compared to cells stained with corresponding labeled isotype-matched control antibodies.
Real-Time PCR (RT-PCR)
Real-time cDNA primers and probes for mouse IL-6, IL-10, IL-13, Foxp3, TGF-β and GAPDH were obtained from Applied Biosystems (Grand Island, NY). Real-time PCR was performed using the ABI Prism 7000 Sequence Detection System. Analysis of relative gene expression was carried out using the mathematical calculation model based on delta delta Ct value as described (27).
Statistical Analysis
Analysis of variance (ANOVA) was used to determine statistical significance. Comparisons for all pairs were performed by Tukey-Kramer highest significant difference test. The p values for significance were set to 0.05. Values for all measurements were expressed as the mean±standard error of the mean (SEM).
RESULTS
CD4+CD25+ but not CD4+CD25− T Cells Enhance Development of Lung Allergic Responses in Sensitized and Challenged CD8−/− Mice
To initially demonstrate their functional plasticity, isolated CD4+CD25+ nTregs were administered intratracheally to CD8−/− recipients. To avoid possible confounding effects inherent to the positive selection of nTregs employing anti-CD25, CD4+CD25− T cells in the negative fraction were positively selected with anti-CD4 prior to intratracheal transfer. Consistent with previous reports (5, 24), sensitized and challenged CD8−/− mice, unlike WT mice, failed to develop significant AHR to increasing concentrations of aerosolized MCh as measured by changes in lung resistance (RL) (Figure 1A). Transfer of CD4+CD25+ but not CD4+CD25− T cells into CD8−/− mice resulted in significantly increased AHR, beyond levels induced following sensitization and challenge alone. Increases in numbers of total cells, lymphocytes, and eosinophils in BAL fluid were detected in sensitized and challenged CD8−/− mice compared to those challenged alone and eosinophil numbers were further increased in recipients of CD4+CD25+ but not CD4+CD25− T cells (Figure 1B). Increased levels of IL-5, IL-6, and TGF-β and decreases in levels of IL-10 and IFN-γ were detected in BAL fluid of sensitized and challenged mice compared to those challenged alone (Figure 1C). In recipients of CD4+CD25+ but not CD4+CD25− T cells, increases in the levels of IL-5, IL-6, and IL-13 and decreases in IL-10 and IFN-γ were detected; levels of TGF-β were essentially unaltered in BAL fluid (Figure 1C).
Figure 1.
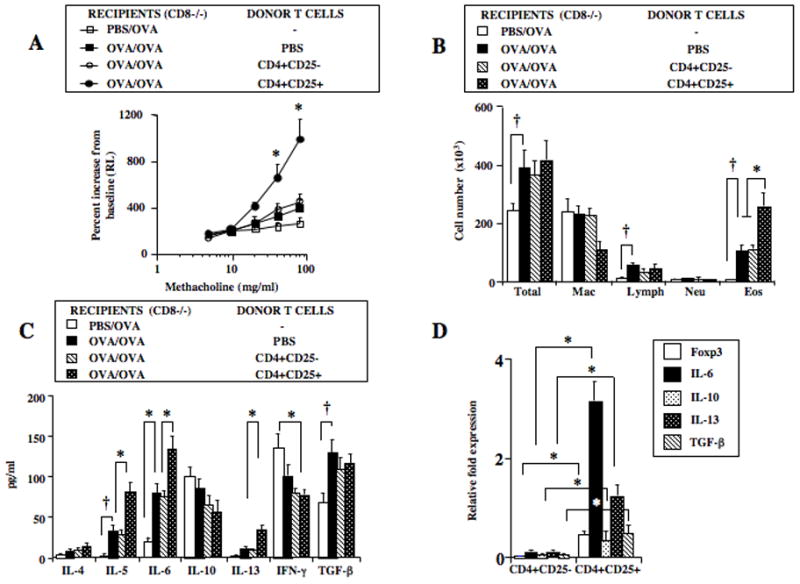
Effect of CD4+CD25+ and CD4+CD25− T cell transfer from naive C57BL/6 donors into sensitized and challenged CD8−/− recipients. Mononuclear cells from lung digests were isolated by density gradient centrifugation. Lymphocytes were enriched over nylon wool columns prior to subset isolation by positive selection. Purified cells (0.5×106) were injected intratracheally prior to allergen challenge. Assays of airway responsiveness to increasing concentrations of aerosolized methacholine and bronchial lavage were assayed 48 hrs following the last allergen challenge. A: AHR, B: BAL fluid inflammatory cell composition (Macrophage:Mac, Lymphocyte:Lymph, Neutrophil:Neu, Eosinophil:Eos), C: BAL cytokine levels, D: RT-PCR of CFSE-labeled cells recovered from CD8−/− recipient lungs. Shown are the means±SEM from 3 independent experiments (4 mice/group, n=12). *p<0.05 comparing sensitized and challenged recipients of CD4+CD25+ T cells to recipients of PBS or CD4+CD25− T cells. †p<0.05 comparing sensitized and challenged mice to mice challenged alone.
Given the instability and fate of nTregs associated with changes in the expression of Foxp3 protein (5), changes in gene expression of Foxp3, IL-6, IL-10, IL-13 and TGF-β in transferred and recovered CD4+CD25+ and CD4+CD25− T cells from lung were analyzed following sensitization and challenge by real-time PCR; values were normalized to those in isolated, non-transferred CD4+CD25+ T cells (assigned a value of 1). Purified CD4+CD25+ and CD4+CD25− T cells were labeled with CFSE prior to transfer, and then recovered from the lungs of recipient mice and enriched by cell sorting. In parallel to the changes in lung physiology and inflammation, there were significant increases in the levels of gene expression of IL-6 and IL-13 in recovered CD4+CD25+ but not in recovered CD4+CD25− T cells (Figure 1D). Levels of expression of Foxp3, IL-10, and TGF-β were higher in the recovered CFSE+CD4+CD25+ compared to CFSE+CD4+CD25− T cells, albeit at lower levels compared to values seen in the isolated, non-transferred CD4+CD25+ T cells. Taken together, these data identified that CD4+CD25+ nTregs but not CD4+CD25− T cells resulted in enhancement of lung allergic responses in association with increased levels of protein and gene expression of the proinflammatory cytokines IL-5, IL-6, and IL-13.
GITRL Alters the Phenotype of CD4+CD25+ and CD4+CD25− T Cells
Addition of GITRL to CD4+CD25+ T cells was shown to trigger the activation of JNK in association with the loss of suppressive activity, changes in cell phenotype, and cytokine production (9). The effects of in vitro addition of GITRL on cytokine production were investigated. Compared to CD4+CD25− T cells, the basal levels of IL-10 and TGF-β in CD4+CD25+ T cells were significantly higher and increased further following stimulation with PMA/ionomycin (Figure 2A). Significant decreases in the levels of IL-10 and TGF-β and increases in IL-6 and IL-13 in the supernatants of CD4+CD25+ but not CD4+CD25− T cells were detected in the presence of GITRL. The basal levels of IL-4, IL-5, IL-6, IL-13, and IFN-γ in both cell types were low and did not change following PMA/ionomycin stimulation.
Figure 2.
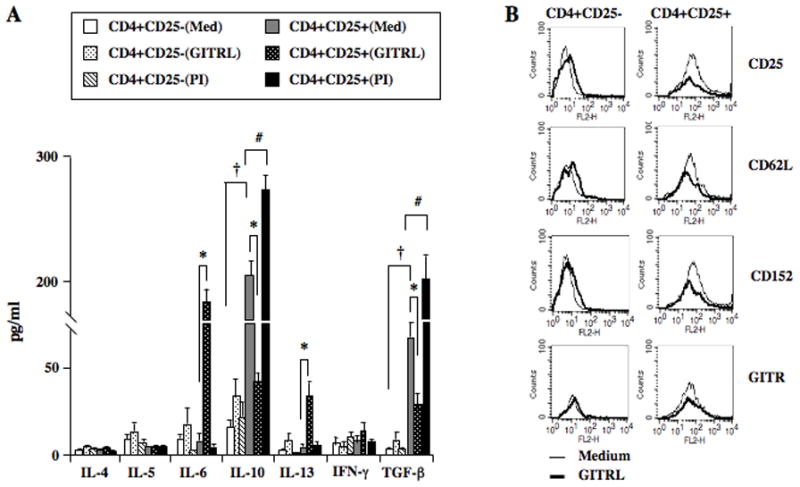
Effect of GITR ligation on in vitro cytokine production and phenotype of CD4+CD25+ and CD4+CD25− T cells. Enriched CD4+CD25+ and CD4+CD25− T cells were cultured in medium alone, in the presence of GITRL, or phorbol/ionomycin. Supernates were recovered for cytokine production and changes in surface marker phenotype were analyzed by FACS. A: Levels of cytokines following 72 hr culture. †p<0.05 comparing basal levels in medium (Med). *p<0.05 comparing levels following GITRL stimulation. #p<0.05 comparing levels in the presence of phorbol/ionomycin (PI). B: FACS analysis following GITRL stimulation.
Changes in cell phenotype with forced (17) and reduced (23) expression of the forkhead transcription factor Foxp3 have been associated with gain and loss of Treg activity, respectively. Lower expression levels of Foxp3, IL-10, and TGF-β associated with changes in phenotype have been demonstrated both in in vivo and in vitro studies (5, 9). When compared to CD4+CD25− T cells, constitutively higher expression levels of CD25, CD62L, CD152, and GITR were detected on CD4+CD25+ T cells (Figure 2B). Addition of GITRL decreased the expression of CD25, CD62L, GITR, and to a lesser degree CD152 in CD4+CD25+ T cells. In contrast, with the exception of GITR expression, a modest increase but low mean fluorescence intensity of these surface markers was detected in CD4+CD25− T cells following GITRL stimulation. Thus, in vitro GITRL stimulation of CD4+CD25+ but not CD4+CD25− T cells increased the production of proinflammatory cytokines, paralleling the in vivo results following adoptive transfer, and induced the changes from a regulatory to an effector phenotype.
Effects of GITR Knock-Down in CD4+CD25+ and GITR Knock-In in CD4+CD25− T Cells
The enhancement of lung allergic responses following transfer of CD4+CD25+ but not CD4+CD25− T cells may have resulted from different levels of GITR expressed on these cell subsets. To determine the role of GITR expression, the levels of GITR were genetically manipulated. In CD4+CD25+ T cells, GITR expression was knocked down by gene silencing using shRNA lentivirus; in CD4+CD25− T cells, expression of GITR was increased using a knock-in strategy. Following knockdown of GITR in CD4+CD25+ T cells, sorted GFP+ cells exhibited a lower mean fluorescence intensity for GITR (Figure 3A). In contrast to recipients of control shRNA-treated cells, sensitized and challenged CD8−/− recipients of sorted GITR shRNA-infected CD4+CD25+ T cells failed to develop significant increases in AHR (Figure 3C) or inflammatory cell accumulation (Figure 3D). Airway responsiveness in these recipients remained higher than in mice that were challenged alone. The diminished enhancement of lung allergic responses was associated with lower levels of IL-5, IL-6, and IL-13, and higher IL-10, IFN-γ, and TGF-β levels in BAL fluid (Figure 3E).
Figure 3.
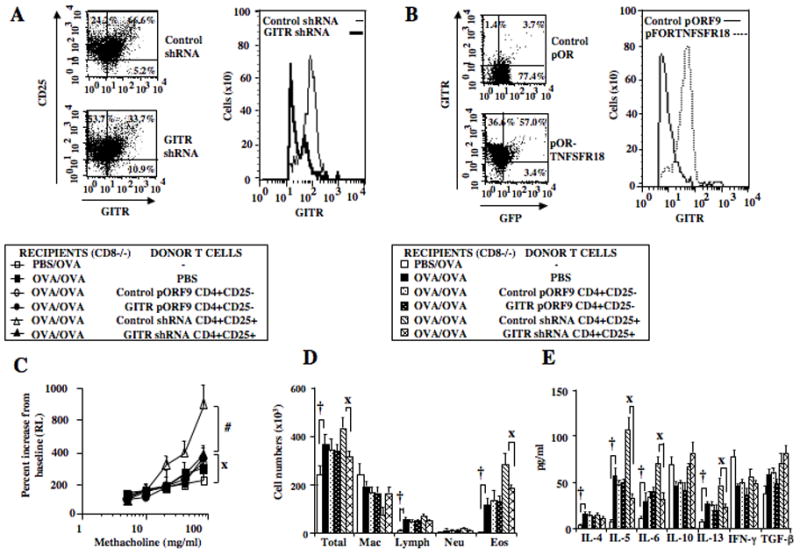
Effect of GITR silencing in CD4+CD25+ and GITR knock-in in CD4+CD25− T cells on enhancement of responses in sensitized and challenged CD8−/− recipients. Purified and activated CD4+CD25+ T cells were cotransfected with GFP/GITRshRNA or GFP/control shRNA lentivirus; CD4+CD25− T cells were nucleofected with GFP/mTNFRSF18 expression plasmids using Amaxa electroporation. GFP+ cells were sorted prior to adoptive transfer. A: FACS analysis of sorted GFP and GITR shRNA-coinfected CD4+CD25+ T cells, B: FACS analysis of sorted GFP and GITR expression vector (pORF9-mTNFSR18)-cotransfected CD4+CD25− T cells, C: AHR, D: BAL fluid inflammatory cell composition, E: BAL cytokine levels. Shown are the means±SEM from 3 independent experiments (4 mice/group, n=12). #p<0.05 comparing recipients given control shRNA to recipients given GITR shRNA transferred cells. Xp<0.05 comparing recipients of GITR shRNA transferred cells CD4+CD25+ T cells to mice challenged alone. †p<0.05 comparing sensitized and challenged mice to mice challenged alone. *p<0.05 comparing recipients given control pORF9-mcs transfected cells to recipients given pORF9-mTNFSR18 transferred cells.
To examine the impact of enhanced expression of GITR in CD4+CD25− T cells, isolated CD4+CD25− T cells were transduced with expression vector pORF9-mTNFRSF18. GFP+ sorted cells exhibited significant increases in mean fluorescence intensity for GITR (Figure 3B). Sensitized and challenged CD8−/− recipients of GITR-overexpressing CD4+CD25− T cells, similar to control-transduced cells or PBS, failed to enhance AHR (Figure 3C) or inflammation (Figure 3D). In parallel, there was little change in levels of IL-5, IL-6, IL-13, IL-10, IFN-γ, and TGF-β in BAL fluid beyond the levels induced following sensitization and challenge alone (Figure 3E). These data demonstrated that the levels of expression of GITR on CD4+CD25+ nTregs, but not CD4+CD25− T cells, were associated with enhancement, suggesting the involvement of GITR in determination of lineage-specific plasticity.
Inhibition of JNK Prevents GITR-Mediated Enhancement of Lung Allergic Responses
SP600125, an anthrapyrazolone inhibitor, prevents activation of JNK by blocking dual phosphorylation of Thr-183 and Tyr-184 of the tripeptide motif (Thr-Xaa-Tyr) (28). We demonstrated that pretreatment of nTregs with the active (SP600125) but not inactive JNK inhibitor decreased GITRL-induced phosphorylation of JNK in vitro. In parallel, the suppressive activity of nTregs in sensitized and challenged WT mice was maintained (9). Since loss of suppression following stimulation with GITRL may be a necessary step in the process of converting nTregs into pathogenic effector T cells, we investigated the involvement of JNK in the GITR-mediated enhancement of lung allergic responses in sensitized and challenged CD8−/− mice. Isolated nTregs were pretreated in vitro with active (JNKi) or inactive inhibitor and washed extensively prior to transfer; this had minimal effect on cell viability. In contrast to untreated nTregs and those treated with the inactive inhibitor, treatment of nTregs with JNKi prevented the enhancement of AHR (Figure 4A) and airway inflammation (Figure 4B). In parallel, lower levels of IL-5, IL-6, and IL-13 and higher IL-10 and TGF-β levels in BAL fluid were detected in recipients of JNKi-treated compared to non-treated and inactive inhibitor-treated nTregs (Figure 4C). Thus, the inhibition of JNK1/2 activation prevented enhancement of lung allergic responses, suggesting involvement of JNK in GITR-mediated plasticity.
Figure 4.
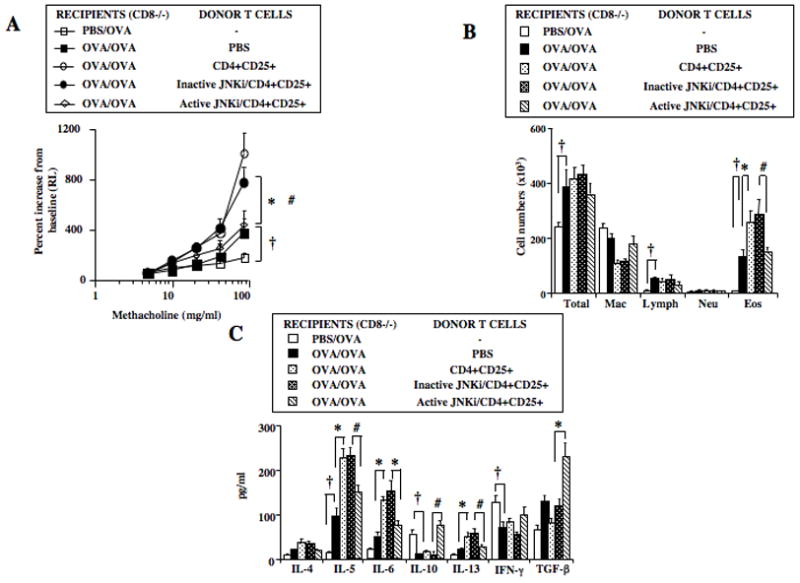
Effect of JNK inhibitor (JNKi) treatment on CD4+CD25+ T cell-enhancement of lung allergic responses. Isolated CD4+CD25+ T cells were pretreated with active or inactive inhibitor in vitro and washed extensively prior to adoptive transfer into sensitized and challenged CD8−/− recipients. A: AHR, B: BAL cell composition, C: BAL cytokine levels. Results shown are means±SEM from 3 independent experiments, n=12. †p<0.05 comparing sensitized and challenged mice to mice challenged alone. *p<0.05 comparing recipients of CD4+CD25+ T cells to those which received PBS. #p<0.05 comparing recipients of inactive inhibitor-treated cells to active inhibitor-treated CD4+CD25+ T cells.
GITR-Mediated Enhancement of Lung Allergic Responses Involves JNK2 but not JNK1
In mammals, JNK1 and JNK2 are ubiquitously and concurrently expressed in all cells and tissues while JNK3 is more tissue-specific and found mainly in the brain, heart, and testes (29). Based on the observation of embryonic lethality in double knock-out of JNK1 and JNK2 (27) mice lacking either JNK1 or JNK2 were viable, suggesting at least partially overlapping roles of these mitogen-activated protein kinases (MAPK) in embryonic development. To address JNK specificity, JNK1−/− and JNK2−/− mice were used to investigate their role in the nTreg-mediated enhancement of responses in sensitized and challenged CD8−/− recipient mice. Results were compared to the activity of nTregs from WT or GITR−/− mice. In contrast to recipients of PBS or nTregs from GITR−/− and JNK2−/− mice, increases in AHR and airway inflammation (Figures 5A, 5B) were observed following transfer of nTregs from WT and JNK1−/− donors, suggesting a critical involvement of GITR and JNK2 in the enhancement response. In association with changes in lung physiologic responses, significantly higher levels of IL-5, IL-6, and IL-13 and lower IL-10 and TGF-β levels in BAL fluid were observed in recipients of nTregs from WT and JNK1−/− but not GITR−/− and JNK2−/− mice (Figure 5C).
Figure 5.
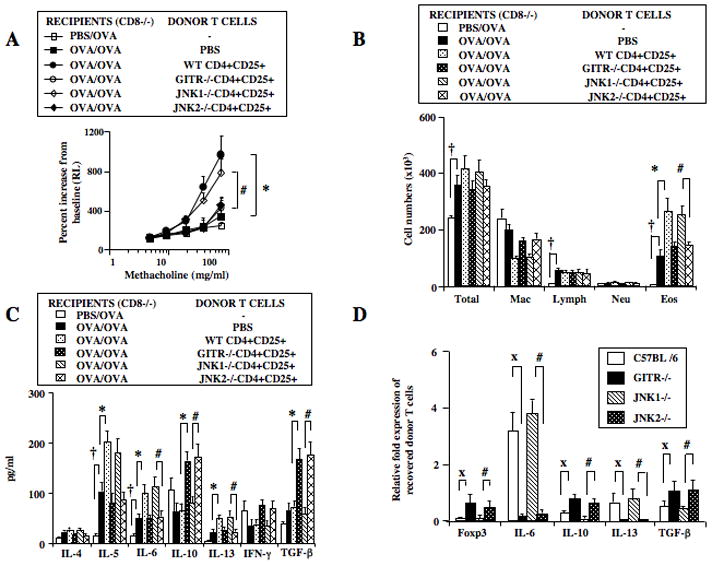
Effect of CD4+CD25+ T cell transfer from naive C57BL/6, GITR−/−, JNK1−/−, and JNK2−/− mice on enhancement of lung allergic responses. Purified CD4+CD25+ T cells from donors were labeled with CFSE prior to adoptive transfer into sensitized and challenged CD8−/− recipients. Following in vivo assays, CFSE+ cells were recovered from recipient lungs. A: AHR, B: BAL cell composition, C: BAL cytokine levels. D: Relative gene expression levels in CFSE-labeled cells recovered from recipient lungs. Results shown are means±SEM from 3 independent experiments, n=12. *p<0.05 comparing recipients of CD4+CD25+ T cells to mice which received PBS. #p<0.05 comparing recipients of CD4+CD25+ T cells from JNK1−/− mice to those from JNK2−/− mice. †p<0.05 comparing sensitized and challenged mice to mice challenged alone. Xp<0.05 comparing recipients of CD4+CD25+ T cells from C57BL/6 to those from GITR−/− mice.
Expression levels of the genes for Foxp3, IL-6, IL-10, IL-13, and TGF-β in CFSE+ cells recovered from the lung were assessed by RT-PCR and expressed as a relative fold increase or decrease compared to corresponding isolated, non-transferred cells. Significantly lower mRNA levels of Foxp3, IL-10 and TGF-β were detected in recovered nTregs from WT and JNK1−/− compared to GITR−/− and JNK2−/− mice (Figure 5D). In parallel, significant increases in gene expression levels of IL-6 and IL-13 were observed in recovered nTregs from WT and JNK1−/− but not from GITR−/− and JNK2−/− mice. Taken together, nTreg-mediated enhancement of lung allergic responses in CD8−/− recipients appeared critically dependent on GITR and JNK2 and was associated with decreases in Foxp3, IL-10 and TGF-β, and increases in IL-6 and IL-13 protein and mRNA expression.
Reconstitution of CD8+ T Cells in CD8−/− Recipients Prevents the Enhancement of Lung Allergic Responses
CD8+ T cells, known effector cells in the pathogenesis of asthma (24, 30–32), were also demonstrated to be critical for the maintenance of nTreg suppressive activity and phenotype in the periphery (21). Differential outcomes, suppression or enhancement, appeared dependent on the interactions of CD8 with nTregs (5, 15, 21, 24). We investigated the effects of reconstitution of CD8+ T cells in CD8−/− mice on GITR/JNK2-mediated enhancement of lung allergic responses. Reconstitution of CD8+ T cells fully restored lung allergic responses in CD8−/− recipients (Figures 6A–C), comparable to the levels seen in CD8−/− recipients of WT nTregs alone. The enhancement of AHR and inflammation, increases in levels of IL-5, IL-6, and IL-13 and decreases in IL-10 and TGF-β in BAL fluid seen following transfer of nTregs from WT or JNK1−/− donors were not observed in recipients of both nTregs and CD8+ T cells. Further, transfer of nTregs from GITR−/− or JNK2−/− mice had no effect in the CD8+ T cell reconstituted CD8−/− recipients. Thus, in recipients of both nTregs and CD8+ T cells, CD8-mediated signals were dominant over those mediated through ligation of GITR.
Figure 6.
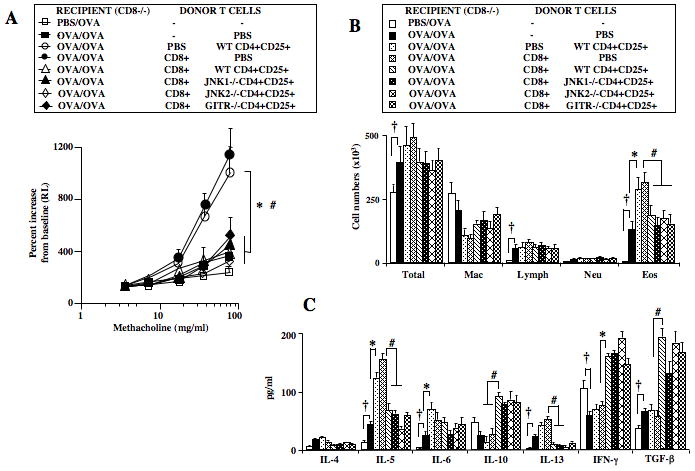
Effect of CD8 reconstitution on enhancement of lung allergic responses in sensitized and challenged CD8−/− recipients. Naive CD8+ T cells were enriched from spleens of WT C57BL/6 donors, and 10×106 cells were injected intravenously prior to sensitization. Isolated CD4+CD25+ T cells from C57BL/6, GITR−/−, JNK1−/−, or JNK2−/− mice were adoptively transferred prior to allergen challenge into sensitized and CD8-reconstituted recipients. A: AHR, B: BAL cell composition, C: BAL cytokine levels. Results represent means±SEM from 3 independent experiments, n=12. †p<0.05 comparing sensitized and challenged mice to those challenged alone. *p<0.05 comparing recipients of CD4+CD25+ or CD8+ T cells to those which received PBS. #p<0.05 comparing recipients given CD4+CD25+ T cells alone to recipients of both CD8+ and CD4+CD25+ T cells from C57BL/6, GITR−/−, JNK1−/−, or JNK2−/− mice.
DISCUSSION
Naturally occurring Foxp3+CD4+CD25+ T regulatory cells (nTregs), a small subset of T cells derived in the thymus (4), are critical to the maintenance of self-tolerance and appropriate immune responses to environmental insults stemming from microbes, allergens, and injury. In the absence of functional Foxp3, the key transcription regulator for Treg development and function (17, 18), severe and life-threatening autoimmune and allergic diseases develop (19, 20). Functional stability and expression of suppressive activities appeared correlated with expression of CD25 and CD152 (3, 5, 6), exposure to factors in the local inflammatory milieu such as IL-6 (21–23), and interactions with other cells including those expressing CD8 (15, 21) and GITRL in the lung (5). Accumulating evidence supports an important role for nTregs in immune homeostasis in the lung to regulate allergen-induced AHR and inflammation. This appears, at least in part, mediated through the actions of IL-10 and TGF-β in mice (12, 13) and in an antigen-nonspecific manner (15).
In light of the importance of CD8 interactions with nTregs in the expression of suppressive activity, the potential for nTreg plasticity and a role in the enhancement of lung allergic responses was investigated. In contrast to CD4+CD25− T cells, CD4+CD25+ nTregs were associated with enhancement of the full spectrum of lung allergic responses following transfer into sensitized and challenged CD8−/− recipients. CFSE-labeled and transferred nTregs recovered from the lungs of these recipients took on the phenotype of pathogenic effector T cells, with reduced gene expression of Foxp3, IL-10, and TGF-β but higher expression levels of IL-6 and IL-13.
Consistent with these results, lower levels of IL-10 and TGF-β and higher IL-6 and IL-13 levels were detected in the supernatants of cultured cells following in vitro stimulation with GITRL, in association with lower expression of CD25, CD62L, GITR, and to a lesser extent CD152. A critical role for GITR was confirmed by the absence of enhancement following both knocking down of GITR expression as well as studies using nTregs from GITR−/− mice. These results were consistent with those targeting GITR using anti-GITRL (5) with all manipulations resulting in the abrogation of enhancement. On the other hand, increasing GITR expression in CD4+CD25− T cells did not alter their activity.
Recently, GITR-mediated loss of suppression had been shown to involve the activation of JNK; inhibition of JNK activity maintained both in vitro and in vivo suppressive activities (9). Conceivably, loss of suppression per se may be one of the necessary steps in the staged process of converting nTregs into pathogenic effector T cells mediating enhancement of lung allergic responses. The involvement of JNK in GITR-mediated enhancement was demonstrated by the prevention of any increases in AHR, airway inflammation, or proinflammatory cytokine levels following in vitro treatment of nTregs with a JNK inhibitor prior to transfer. Pharmacologically, SP600125 indiscriminately inactivates all members of the JNK family. Given the distribution of the JNKs (29), we also determined the activities of nTregs from JNK1−/− and JNK2−/− mice. Interestingly, JNK2 but not JNK1 was identified to be essential in the enhancement activity as demonstrated by the complete absence of any increases in lung allergic responses following transfer of JNK2−/− nTregs. This was surprising as it was thought that JNK1 and JNK2 have overlapping roles, in part since combined JNK1 and JNK2 knock-out is embryonically lethal while mice lacking either JNK1 or JNK2 are viable (27, 29). In contrast to the purported redundancy of function, our data suggested that JNK2 in nTregs has unique and different functions compared to JNK1. Further supporting the critical roles of both GITR and JNK2, nTregs from GITR−/− and JNK2−/− but not WT or JNK1−/− mice recovered from the lungs of sensitized and challenged recipients acquired an effector phenotype highlighted by increased gene expression levels for IL-6 and IL-13 and lower levels of IL-10 and TGF-β. These changes in levels of gene expression were detected only in transferred and recovered nTregs but not in nTregs prior to transfer.
There is support for the notion that rigid lineage commitment and permanent unidirectional function of Tregs may not be valid (33). There is an intriguing degree of plasticity in nTregs despite expression of the key lineage transcriptional regulator Foxp3, which is essential for development and function (4, 18–20). Lineage fidelity can be influenced by the absence and/or presence of extracellular stimuli and/or intracellular signaling molecules that modify the expression of Foxp3 (5, 21, 23, 24). In mice, CD8 appeared to have multiple roles dictating the outcome of lung allergic responses by influencing the expression of nTreg activities (5, 34), inducing the formation of Foxp3+CD4+ T cells in vivo (35) and in vitro (36), and mediating the asthma phenotype as a pathogenic effector cell (30–32). Increasingly, CD8+ T cells have been implicated in human asthma (37, 38) but whether similar influences of CD8+ T cells on nTreg activities occur is unclear at present. Further, optimal expression of nTreg suppressive activity was shown to be critically dependent on the interaction with CD8, and both in vitro and in vivo inhibition or interference with the interaction/engagement of MHC I on naturally occurring CD4+CD25+ T regulatory cells with CD8 effectively abrogated suppression (24). These cell-cell interactions were confirmed microscopically in the lung (15). In addition, CD8 was demonstrated to be critical in the maintenance of suppressive activities in the periphery by controlling endogenous expression of IL-6 (21), a key regulator of nTreg-mediated suppression (22). Together, the results imply a balance between two major pathways governing the functional activity of nTregs. On one hand, the GITR-JNK pathway can lead to abrogation of suppressive activities coupled with phenotypic conversion to a pathogenic effector cell type in the lung in response to allergen sensitization and challenge. In contrast, CD8-MHC I interactions appear necessary for maintenance of nTreg suppressive activity. Since absence (prevention) of enhancement was observed in CD8−/− recipients reconstituted with CD8+ T cells, it appeared that CD8-MHC I interactions supercede GITR-JNK2 signaling in nTregs. At present, it is not clear whether in addition to critical signaling events triggered by CD8, that other chemokines, chemokine receptors, or additional cytokines are involved in maintaining nTreg suppressive activity. The potential integration of signals from both pathways is illustrated in Figure 7.
Figure 7.
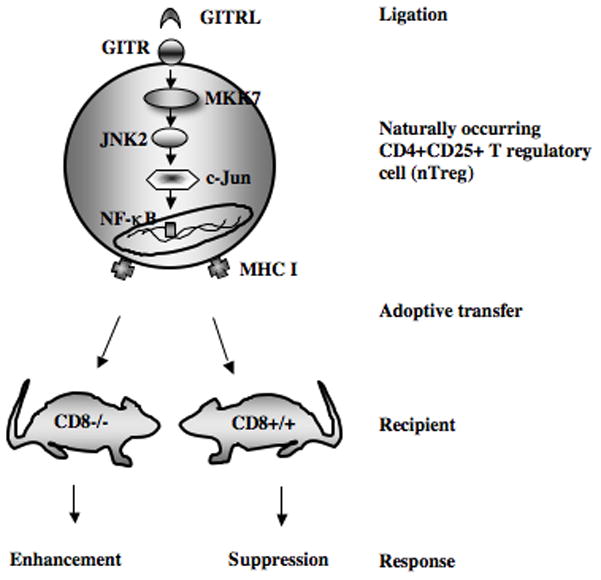
Schematic representation of the integrated signaling pathways through CD8-MHC I and GITRL-GITR-JNK2 in CD4+CD25+ nTregs. In the absence of CD8-MHC I interactions (as in CD8−/− mice), signaling through GITR results in the pathogenic conversion of nTregs and enhancement of lung allergic responses. In CD8+/+ mice signaling through CD8-MHC I dominates and is associated with the maintenance of the regulatory phenotype and suppressive activities of nTregs.
Collectively, the results demonstrated that naturally occurring CD4+CD25+ nTregs but not naive CD4+CD25− or GITR-transduced CD4+CD25− T cells are capable of enhancing the full complement of lung allergic response in sensitized and challenged CD8−/− recipients. The enhancement, associated with the loss of nTreg phenotype and function and gain of effector T cell function, was critically dependent on GITR signaling through JNK2. Signaling through GITR was both necessary and sufficient in the absence of CD8. It appears that signaling by CD8 through MHC I on nTregs predominates over signaling through GITR, abrogating enhancement and maintaining suppressive activity. The data identify a hierarchical and/or temporal integration of signals from interactions between MHC I-CD8 and GITRL-GITR-JNK2 resulting in the functional diversity/plasticity of nTregs.
Acknowledgments
Grant Support: This work was supported by NIH grant AI-77609.
The assistance of Diana Nabighian in the preparation of this manuscript is gratefully acknowledged.
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
References
- 1.Watts HT. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 2.Nocentini G, Giunchi L, Ronchetti S, Krausz LT, Bartoli A, Moraca R, Migliorati G, Riccardi C. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell-receptor-induced apotosis. Proc Natl Acad Sci. 1997;94:6216–21. doi: 10.1073/pnas.94.12.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwon B, Yu KY, Ni J, Yu GL, Jang IK, Kim YJ, Xing L, Liu D, Wang SX, Kwon BS. Identification of a novel activation-inducible protein of the tumor necrosis factor receptor superfamily and its ligand. J Biol Chem. 1999;274:6056–6061. doi: 10.1074/jbc.274.10.6056. [DOI] [PubMed] [Google Scholar]
- 4.Sakaguchi S. Naturally arising CD4+ regulatory T cells form immunologic self-tolerance and negative control of immune response. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 5.Joetham A, Matsubara S, Okamoto M, Takeda K, Miyahara N, Dakhama A, Gelfand EW. Plasticity of regulatory T cells: Subversion of suppressive function and conversion to enhancement of lung allergic responses. J Immunol. 2008;180:7117–7124. doi: 10.4049/jimmunol.180.11.7117. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nature Immunol. 2002;3:135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 7.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 8.Ji H, Liao G, Faubion WA, Abadia-Molina AC, Cozzo C, Laroux FS, Caton A, Terhorst C. Cutting Edge: The natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J Immunol. 2004;172:5823–5827. doi: 10.4049/jimmunol.172.10.5823. [DOI] [PubMed] [Google Scholar]
- 9.Joetham A, Ohnishi H, Okamoto M, Takeda K, Schedel M, Domenico J, Dakhama A, Gelfand EW. Loss of T regulatory cell suppression following signaling through the GITR is dependent on c-Jun N-terminal kinase activation. J Biol Chem. 2012;287:17100–17008. doi: 10.1074/jbc.M111.316943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, Collins Mary, Shevach EM. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. 2004;173:5008–5020. doi: 10.4049/jimmunol.173.8.5008. [DOI] [PubMed] [Google Scholar]
- 11.Cardona ID, Goleva E, Ou LS, Leung DYM. Staphylococcal enterotoxin B inhibits regulatory T cells by inducing glucorticoid-induced TNF receptor-related protein ligand on monocytes. J Allergy Clin Immunol. 2006;117:688–695. doi: 10.1016/j.jaci.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 12.Joetham A, Takeda K, Taube C, Miyahara N, Matsubara S, Koya T, Rha YH, Dakhama A, Gelfand EW. Naturally-occurring lung CD4+CD25+ T-cell regulation of airway allergic responses depends on IL-10 induction of TGF-β. J Immunol. 2006;178:1433–1442. doi: 10.4049/jimmunol.178.3.1433. [DOI] [PubMed] [Google Scholar]
- 13.Presser K, Schwinge D, Wegmann M, Huber S, Schmitt S, Quaas A, Maxeiner JH, Finotto S, Lohse AW, Blessing M, Schramm C. Coexpression of TGF-β1 and IL-10 enables regulatory T cells to completely suppress airway hyperreactivity. J Immunol. 2008;181:7751–7758. doi: 10.4049/jimmunol.181.11.7751. [DOI] [PubMed] [Google Scholar]
- 14.Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, Berry G, Dekruyff RH, Umetsu DT. Antigen-specific regulatory T cells develop via the ICOS-ICOSSL pathway and inhibit allergen-induced airway hyperreactivity. Nature Med. 2002;8:1024–1031. doi: 10.1038/nm745. [DOI] [PubMed] [Google Scholar]
- 15.Joetham A, Takeda K, Okamoto M, Taube C, Matsuda H, Dakhama A, Gelfand EW. Antigen specificity is not required for modulation of lung allergic responses by naturally occurring regulatory T cells. J Immunol. 2009;183:1821–1827. doi: 10.4049/jimmunol.0900303. [DOI] [PubMed] [Google Scholar]
- 16.Szymczak-Workman AL, Workman CJ, Vignali DAA. Cutting Edge: Regulatory T cells do not require stimulation through their TCR to suppress. J Immunol. 2009;182:5188–5192. doi: 10.4049/jimmunol.0803123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcriptional factor FoxP3. Science. 2003;299:1057–1061. [Google Scholar]
- 18.Fontenot JD, Gavin MA, Rudensky AY. FoxP3 programs the development and function of CD4+CD25+ regulatory T cells. Nature Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 19.Clark LB, Appleby MW, Brunkow ME, Willkinson JE, Ziegler SF, Ramshell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. 1999;162:2546–2554. [PubMed] [Google Scholar]
- 20.Bennet CL, Christie J, Ramsdell F, Brunkow ME, Fergurson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome is caused mutations of Foxp3. Nature Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 21.Joetham A, Okamoto M, Takeda K, Schedel M, Ohnishi H, Dakhama A, Gelfand E. CD8 regulates the endogenous production of IL-6 in naturally occurring T regulatory cells and maintains their suppressive phenotype in allergic lung disease. J Immunol. 2011;186:113–120. doi: 10.4049/jimmunol.1001663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dogabci A, Elgenbrod T, Krug N, De Santis GT, Hausding M, Erpenbeck VJ, Haddad E, Schmitt E, Bopp T, Kallen KJ, Herz U, Schmitt S, Luft C, Hecht O, Hohlfeld JM, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Rose-john S, Renz H, Neurath MF, Galle PR, Finotto S. The IL-6Ra chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–325. doi: 10.1172/JCI22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cells regulatory functions. J Immunol. 2007;178:271–279. doi: 10.4049/jimmunol.178.1.271. [DOI] [PubMed] [Google Scholar]
- 24.Joetham A, Takeda K, Miyahara N, Matsubara S, Ohnishi H, Koya T, Dakhama A, Gelfand EW. Functional activation of naturally occurring lung CD4+CD25+ regulatory cells on lung allergic responses requires CD8 and MHC I interaction. Proc Natl Acad Sci. 2007;104:15057–15062. doi: 10.1073/pnas.0706765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan YY, Flavell RA. Regulatory T cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 26.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martines-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to generation of pathogenic memory T cells in vivo. Nature Immunol. 2009;10:1000–1008. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabpathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech Dev. 1999;89:115–119. doi: 10.1016/s0925-4773(99)00213-0. [DOI] [PubMed] [Google Scholar]
- 28.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhapwad SS, Manning AM, Anderson DW. SP600125 an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuan C, Yang DD, Roy DRS, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during brain development. Immunity. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 30.Okamoto N, Takeda K, Joetham A, Ohnishi H, Matsuda H, Swasey CH, Swanson BJ, Yasutomo K, Dakhama A, Gelfand EW. Essential role of Notch signaling in effector memory CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J Exp Med. 2008;2005:1087–1097. doi: 10.1084/jem.20072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyahara N, Takeda K, Kodama T, Joetham A, Taube C, Park JW, Miyahara S, Balhorn A, Dakhama A, Gelfand EW. Contribution of antigen-primed CD8+ T cells to the development of airway hyperresponsiveness and inflammation is associated with IL-13. J Immunol. 2004;172:2549–2558. doi: 10.4049/jimmunol.172.4.2549. [DOI] [PubMed] [Google Scholar]
- 32.Jia Y, Takeda K, Han J, Joetham A, Marcus RA, Lucas JJ, O’Connor BP, Gelfand EW. Stepwise epigenetic alterations poise CD8+T cells to mediate airway hyperresponsiveness and inflammation. J Immunol. 2013;190:4056–4065. doi: 10.4049/jimmunol.1202640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. 2010;20:4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arazi A, Sharabi A, Zinger H, Mozes E, Neumann AU. In vivo dynamical interactions between CD4 Tregs, CD8 Tregs and CD4+CD25− cells in mice. PlosOne. 2009;4:e8447. doi: 10.1371/journal.pone.0008447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colson Y, Christopher K, Glickman J, Taylor KN, Wright R, Perkins DL. Absence of clinical GVHD and the in vivo induction of regulatory T cells after transplantation of facilitating cells. Blood. 2004;104:3829–3835. doi: 10.1182/blood-2004-01-0393. [DOI] [PubMed] [Google Scholar]
- 36.Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, Nussenzweig MC, Steinman RM. CD8+CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol. 2008;181:6923–6933. doi: 10.4049/jimmunol.181.10.6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dakhama A, Collins ML, Ohnishi H, Goleva E, Leung DYM, Alam R, Sutherland R, Martin RJ, Gelfand EW. IL-13-producing BLT1-positive CD8 cells are increased in asthma and are associated with airway obstruction. Allergy. 2013;68:666–673. doi: 10.1111/all.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsitsiou E, Williams AE, Moschos SA, Patel K, Rossios C, Jiang Z, Adams O-D, Macedo P, Booton R, Gibeon D, Chung KF, Lindsay MA. Transcriptome analysis shows activation of circulating CD8+ T cells in patients with severe asthma. J Allergy Clin Immunol. 2012;129:95–103. doi: 10.1016/j.jaci.2011.08.011. [DOI] [PubMed] [Google Scholar]