

Abstract
Recent studies showed loss of CD36 or scavenger receptor-AI/II (SR-A) does not ameliorate atherosclerosis in hyperlipidemic mouse model, suggesting receptors other than CD36 and SR-A may also contribute to atherosclerosis. In this report, we show that apoE-CD16 double knockout mice (apoE-CD16 DKO) have reduced atherosclerotic lesions compared with apoE KO mice. In vivo and in vitro foam cells analyses showed apoE-CD16 DKO macrophages accumulated less neutral lipids. Reduced foam cell formation in apoE-CD16 DKO mice is not due to change in expression of CD36, SR-A and LOX-1. This led to a hypothesis that CD16 may have scavenger receptor activity. We presented evidence that a soluble form of recombinant mouse CD16 (sCD16) bound to malondialdehyde-modified low-density lipoprotein (MDALDL), and this binding is blocked by molar excess of MDA-BSA and anti-MDA mAbs, suggesting CD16 specifically recognizes MDA epitopes. Interestingly, sCD16 inhibited MDALDL binding to macrophage cell line as well as sCD36, sSR-A and sLOX-1, indicating CD16 can cross-block MDALDL binding to other scavenger receptors. Anti-CD16 mAb inhibited IC binding to sCD16, while partially inhibited MDALDL binding to sCD16, suggesting MDALDL binding site may be in close proximity to the IC binding site in CD16. Loss of CD16 expression resulted in reduced levels of MDALDL induced pro-inflammatory cytokine expression. Finally, CD16 deficient macrophages showed reduced MDALDL-induced Syk phosphorylation. Collectively our findings suggest scavenger receptor activity of CD16 may in part contribute to the progression of atherosclerosis.
Introduction
Oxidative modification of native low-density lipoprotein (nLDL) 3 resulting in the generation of oxidized-LDL (oxLDL) enhances lipid accumulation and chronic inflammation in the arterial intima leading to development of atherosclerotic lesions (1, 2). Under hyperlipidemic conditions, reactive aldehydes including malondialdehyde (MDA) formed during LDL lipid oxidation form adducts with ε-amino group of lysine in apolipoprotein B, a protein backbone of nLDL, to generate MDA-modified LDL (MDALDL) (3, 4). Recent studies have revealed that MDALDL level is increased in number of diseases including coronary artery disease (5–9), diabetes and hypertriglyceridemia (10). Moreover elevated levels of MDALDL were reported in patients with acute coronary syndrome (5, 6). These findings suggest a causal link between MDALDL and cardiovascular disease.
Oxidatively modified LDL binds to a group of transmembrane receptors called scavenger receptors. Currently scavenger receptors can be broadly classified into eight classes, A-H (11, 12), and based on a recent report on scavenger receptor nomenclature it may be increased to 10 classes (A-J) (13). Of these scavenger receptors, functions of SR-A and CD36 class A, and B scavenger receptors, respectively, have been implicated to contribute to the progression of atherosclerosis (12, 14). Earlier studies have reported that SR-A or CD36 deficiency in hyperlipidemic apolipoprotein E knockout (apoE KO) mice had reduced atherosclerotic lesions (15, 16). However in a recent study by the Freeman group showed loss of CD36 or SR-A in apoE KO mice did not ameliorate atherosclerotic lesions (17). Moreover, overexpression of human SR-A in bone marrow-derived cells did not result in the anticipated increase in atherosclerotic lesions in hyperlipidemic mouse model (18, 19). Similarly LDL receptor (LDLR)-CD36 double knockout mice did not show difference in lesions compared to LDLR KO mice (20). Collectively, these findings suggest a role for other cell surface receptors expressed on macrophages with scavenger receptor functions contributing to foam cell formation and atherosclerosis.
In mice, four different classes of FcγRs have been recognized: FcγRI, FcγRII, FcγRIII, and FcγRIV (21). FcγRII and FcγRIII, FcγRIV (CD32b, CD16 and CD16.2, respectively) are low affinity receptors for monomeric IgG (21), while FcγRI (CD64) is the only high affinity FcγR. Functionally, FcγRs can be classified into the activating (CD64, CD16 and FcγRIV) and inhibitory (CD32b) receptors. Fcγ-chain is the signaling subunit with ITAM for all the activating FcγRs (22, 23). Assembly and cell-surface expression of the activating FcγRs (CD16, CD64 and FcγRIV) require the co-expression of Fcγ-chain. FcγR plays an important role in inflammatory cell activation, clearance, presentation of Ag and maintaining IgG homeostasis (21). In addition to its binding to IC, FcγR have been shown to bind to non-IgG ligands. Stein et al (24) have presented evidence that human CD64 and CD32 binds to human CRP. Murine CD32b expression has been shown to bind to a non-IgG ligand expressed on mouse thymic stromal cells (25). Recent studies have presented evidence that murine CD16 binding to a E. coli component (26) negatively regulates the function of MARCO, a class A scavenger receptor implicated in host defense by promoting pathogen clearance (27, 28). Notably, mouse CD32b has been shown to bind oxLDL (29), however functional implications of oxLDL binding function of mouse CD32b has not been explored. Findings from these reports indicate that FcγRs can bind to non-IgG ligands. In search of receptors other than CD36, SR-A and LOX-1 to bind to oxLDL, we investigated whether murine CD16 specifically binds to oxLDL. We hypothesize that murine CD16 expressed constitutively on monocytes and macrophages binds oxLDL and subsequently leads to inflammatory responses. In the present study, we showed deficiency of CD16 in hyperlipidemic apoE KO mouse model showed attenuated atherosclerotic lesions, reduced foam cell formation, without affecting the expression of other scavenger receptors. We also presented evidence that CD16 recognized MDA epitopes in MDALDL and CD16-MDALDL interaction resulted in induction of pro-inflammatory cytokine and chemokines.
Materials and Methods
Recombinant proteins
A soluble form of recombinant mouse CD16 (sCD16) prepared by fusing cDNA encoding extracellular domain of CD16ala21-thr211 with 10-histdine (His) tag at the C terminal end and purified from supernatant of myeloma cells was purchased from RND Systems. Similarly soluble form of recombinant mouse SR-A and LOX-1 (sSR-A, and sLOX-1, respectively) were prepared by fusing cDNA encoding extracellular domains of these receptors (SR-Atrp83-Ser458, and Lox-1arg60-ile363) with 9–10-His tag at the N-terminal end (RND systems). sCD36 was constructed by fusing cDNA encoding extracellular domain of CD36gly30-lys439 with human IgG1pro100-lys330 with a six amino acid linker in between CD36 and hIgG1 (RND systems). GASP-1 (growth and differentiation factor-associated serum protein-1), a His-tag protein (RND Systems) was used as a negative control.
Antibodies and chemicals
Anti-CD16/CD32 mAbs (also known as FcBlock) clone 2.4G2 (rat IgG1), and clone 93 (rat IgG2a) and corresponding isotype controls (rat IgG1 and IgG2a) were purchased from BD Biosciences, and Biolegend, respectively. Other antibodies used in this study include: PE-conjugated anti-CD36 (JC63.1, mIgA, Cayman Chemical), anti-SR-A (clone 268318, rat IgG2b, RND systems), anti-LOX-1 (clone 214012, rat IgG2a, RND systems) and corresponding isotype control antibodies. Biotinylated anti-poly His mAb (clone AD1.1.1.10, mIgG1) was purchased from RND systems. Affinity purified anti-MDA IgG and anti-OVA IgG were purchased from Academy Biomedicals and MyBioSource, respectively. Streptavidin-alkaline phosphatase, streptavidin-PE, streptavidin-HRP, peroxidase/anti-peroxidase immune complex (PAP-IC) and peroxidase-conjugated donkey anti-rabbit IgG, F(ab′)2-goat anti-rat IgG were purchased from Jackson Immunoresearch (West Grove, PA). In some experiments PAP-IC was biotinylated using EZ-link NHS biotin labeling kit (Pierce, Rockford, IL) and used as a soluble IC ligand. Total Syk (#2712) and phosphoSykTyr525/526 (#2711) antibodies were purchased from Cell signaling Technologies (Danvers, MA). Syk inhibitor III (3,4-methylenedioxy-β-nitrostyrene) was purchased from Calbiochem. All other chemicals were purchased from Sigma.
Animals
C57BL/6 (wild type), apoE KO (stock#002052), CD16 KO (stock#003171), and transgenic mice expressing enhanced green fluorescent protein (C57BL/6-GFP; stock#004353) mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed in microisolator cages with filter tops and maintained on a 12-h light/dark cycle in a temperature-controlled room. To generate apoE-CD16 double knockout (DKO) mice, apoE KO and CD16 KO mice on a C57BL/6 background were mated, and F1 progeny (apoE-CD16 heterozygous) were mated to generate apoE-CD16 DKO mice. Genotype analyses of ApoE was done as described (30), and CD16 were done using following primers (common, gtggctgaaaagttgctgctg; mutant, gcacgagactagtgagacgtg; WT, ctacatcctccatctctctag) and separated PCR method as described in the Jax website. The wild type and mutant CD16 will yield 238 bp, and 500 bp, respectively. ApoE KO and apoE-CD16 DKO mice (5 wk. of age) were fed a high-fat Western diet (TD 05576; Harlan Laboratories, Madison, WI) for 10 wk. Macrophages were isolated from apoE KO and apoE-CD16 DKO mice fed normal chow or high fat diets. Thioglycollate-elicited mouse peritoneal macrophages were obtained 3 days after injection of 4% thioglycollate (1 ml/mouse). The peritoneal cells were plated in RPMI-1640 supplemented with 10% (v/v) FBS, 2 mM L-glutamine, penicillin, streptomycin, and sodium pyruvate. Non-adherent cells were removed after 2 h and macrophages were used after 48 h. This study was reviewed and approved by the Institutional Animal Care and Use Committee at University of Pittsburgh. Part of the animal studies carried out at University of Arkansas for Medical Sciences, and was approved by the IACUC committee at University of Arkansas for Medical Sciences.
Determination of atherosclerotic lesions
Mice fed high fat diet were anesthetized with isoflurane and euthanized at 15 weeks of age, and blood was collected by the cardiac puncture into heparin-coated tubes. Plasma was separated and stored at −80°C until further analysis. The heart was excised and fixed in amphibian PBS/4% formalin/30% sucrose overnight before mounted in optimal cutting medium and frozen at −70°C. Aortic sinus cryosections (10 μm) were stained with Oil Red O (31). For quantitative analysis of atherosclerosis, the percent lesion area in each of five sections from each mouse was obtained.
Plasma lipid analyses
Concentrations of plasma total-cholesterol and high-density lipoprotein (HDL) cholesterol were determined by enzymatic methods using kits from BioVision (Mountain View, CA) as described earlier (31).
Lipoprotein modifications
nLDL isolated from human plasma was purchased from Kalen Biomedical (Montgomery Village, MD). nLDL was biotinylated using Chromalink biotinylation kit (Solulink, San Diego, CA). MDALDLbiotin and MDALDL were prepared by incubating nLDLbiotin or nLDL with freshly prepared MDA (32). Briefly, MDA was generated by incubating 1,1,3,3-tetramethoxypropane malonaldehyde-bis-dimethyl acetal (704 μl) with 4N HCl (96 μl) and water 3.2 ml for 10 min at 37°C. MDA solution was adjusted to pH 7.4 with 1 M NaOH. Then nLDLbiotin or nLDL (1 mg) was incubated with freshly prepared MDA (100 μl, 0.5 M) at 37°C for 3 h. Free MDA was removed by extensive dialysis in PBS. To confirm CD16 binding to MDALDL prepared in the lab, MDALDL was also purchased from two different commercial sources (Academy Biomedical, Houston, TX and Kalen Biomedical). MDALDL (low and high MDA modification) was purchased from Kalen Biomedical. The extent of MDA modification was assessed by the electrophoretic mobility of MDALDL and nLDL using TITAN (Helena Labs, Houston, TX) agarose gel electrophoresis (33). Differentially oxidized-LDL (oxLDL) was prepared by incubating nLDL with 5 μM CuSO4 at different time period as indicated. nLDL treated without CuSO4 was used as control. Oxidation of LDL was confirmed by TBARS (Cayman Chemical), rabbit anti-MDA IgG (Academy Biomedical) binding, and gel electrophoresis as described (33). Lipoproteins were used for experiments within 3 weeks after preparation. All the reagents used to prepare MDALDL were endotoxin-free and MDALDL was tested for the presence of endotoxin using endotoxin assay kit (Genscript). MDALDL preparation with endotoxin levels >50 pg/mg were not used.
ELISA to determine MDA epitope in MDALDL and oxLDL
MDA epitopes in MDALDL, oxLDL and differentially oxLDL were determined by ELISA using affinity purified rabbit anti-MDA IgG (1 μg/ml, Academy Biomedicals) and saturating concentrations of goat anti-rabbit IgGbiotin followed by streptavidin-HRP.
Cytokine analysis
Cytokine secretion (TNF-α, MCP-1 and RANTES) in supernatants from MDALDL- or IC-stimulated macrophages was determined using cytokine bead array in a BD-LSR Fortessa and analyzed using FCAP array CBA software (BD Biosciences) or MCP-1 Duoset ELISA kit (RND systems).
Quantitative real-time RT-PCR
Total RNA was isolated from macrophages and macrophage cell lines using RNAEasy kit (Qiagen) according to the manufacturer’s instructions and treated with RNase-free DNase. RNA was quantified using Nanodrop spectrophotometer (Nanodrop Products, Wilmington, DE). Gene expression was measured by quantitative RT-PCR after reverse transcription of total RNA (0.5 μg) as described earlier (31). PCR primer pairs were purchased from SA-Biosciences (Frederick, MD). Two-step PCR with denaturation at 95°C for 15 s and annealing and extension at 60°C for 1 min for 40 cycles was conducted in a CFX96 (Bio-Rad). Expression of target genes was calculated by the ΔΔCt method using threshold cycles for β-actin as a normalization reference. All real-time PCR reactions were carried out at least twice from independent cDNA preparations. RNA without reverse transcriptase served as a negative control. Real-time PCR primers were purchased from Qiagen RT2 assays.
Cell culture and siRNA transfection
RAW264.7 and J774, murine macrophage cell lines was maintained in RPMI medium supplemented with 10% heat-inactivated FBS, and antibiotics (penicillin at 100 IU/ml streptomycin at 100 μg/ml). Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. CD16 silencing was done using SMARTpool ON-TARGETplus CD16 siRNA, and non-target control siRNA (Dharmacon) was used as control. Transfections were performed using INTERFERin (Polyplus, New York, NY) with siRNA (1 nM), following manufacture’s protocol. To knockdown CD16 protein expression siRNA transfection was done twice, the second transfection was done 48-h after the first transfection. Knockdown efficiency for mRNA and protein expression was confirmed by qRT-PCR and FACS analyses respectively. CD16 primers (sense, cgaaggaaccgccaagtg; anti-sense, ctagagcaagcgatgacagg) were used to determine CD16 knockdown. CD36, SR-A, LOX-1, β-actin and GAPDH primer assays were purchased from SA-Biosciences.
Immune Complexes
MDALDL or OVA (10 μg/ml) was incubated at 4°C for 18–24 h with affinity purified rabbit anti-MDA IgG or rabbit anti-OVA IgG at 30 μg/ml to prepare soluble MDALDL-IC and OVA-IC. Soluble IC was centrifuged at 10000 rpm for 20 min to remove any particulate IC. To prepare insoluble BSA-IC, IgG-free BSA (10 μg/ml in PBS, Sigma A0281) was incubated with rabbit anti-BSA antiserum (300 μl of 2 mg/ml anti-BSA IgG, Sigma B1520) for 20 min at 37°C. Insoluble BSA-IC was centrifuged at 5000 rpm for 2 min and pellet containing insoluble BSA-IC was washed with PBS three times and resuspended in 310 μl serum free RPMI medium. BSA-IC (15 μl/1 ×106 cells/well) was used to activate macrophages. In all the assays both soluble and insoluble ICs were prepared fresh and used within 48 h.
MDALDL binding to sCD16
sCD16 (50 μl of 2.5 μg/ml in PBS) was coated on 96-well ELISA white plate (corning 2992) overnight at 4°C. Plates were washed twice with PBS/0.01% Tween-20 (PBST) buffer and blocked with TBS-protein free blocking buffer (Pierce, cat#37570) for 1 h at ambient temperature. Biotinylated ligands (MDALDL or nLDL) were added to the plate at indicated concentration and incubated for 1 h at RT. Plates were washed twice with PBST and a saturating concentration of streptavidin-alkaline phosphatase (1:20000 dilution) and incubated further for 1 h at RT. Plates were washed thrice with PBST and Lumiphos 530 (Lumigen, Southfield, MI), a luminescent phosphatase substrate, was added to the plate and luminescence was measured in a Bio-Tek or Tecan microplate reader. Controls used in this assay include: wells coated with CD16 alone without ligands, wells blocked with blocking buffer that received ligands. All the control wells showed very low luminescence values indicating minimal non-specific binding of MDALDL to BSA coated wells. CD16 alone did not bind to streptavidin alkaline phosphatase, detecting reagent. To simulate a polyvalent ligands, in the indicated experiments reverse binding assay was performed in which ligands (such as MDALDL) were coated on the plate followed by the addition of sCD16. CD16 binding was detected using anti-poly His IgGbiotin (RND systems) and streptavidin-alkaline phosphatase.
Surface plasmon resonance (SPR) studies of binding kinetics
SPR experiments were performed using a Biacore 3000 on CM5 biosensor chips (Biacore Life Sciences, GE Healthcare, Piscataway, NJ). Anti-His IgG was captured to the experimental and reference flow cells on CM5 chip surfaces utilizing a Biacore/GE Healthcare His capture kit, by standard amine coupling chemistry, according to manufacturer instructions. The surface density of the sCD16 was tested empirically and optimized for each binding interaction based on standard SPR kinetic considerations. All experiments were carried out in duplicate at 25°C. Two-fold dilutions starting at 100 nM of rat anti-mouse CD16 mAb (2.4G2, BD-Biosciences) were injected at a flow rate of 30 μl/min. Association was measured for three minutes, followed by ten minutes of dissociation in HBS-EP running buffer (0.01M HEPES pH 7.4, 0.15M NaCl, 3mM EDTA, 0.005% surfactant P20; GE Healthcare). Association of PAP-IC with sCD16 was measured using two-fold serial dilutions (starting at 50 nM) at a flow rate of 40 μl/min for three minutes. Dissociation was allowed to occur for ten minutes in HBS-EP buffer alone. sCD16 binding with MDALDL was examined utilizing 1.5-fold serial dilutions of the MDALDL starting at 230 nM at a flow rate of 60 μl/min. Three minutes of association was followed by five minutes of dissociation in HBS-EP buffer. In all three binding interaction studies, surfaces were regenerated upon completion of dissociation with 10 mM glycine-HCl, pH 1.5. All measurements were double referenced by subtracting out the response to the reference flow cell surface as well as a buffer only injection. Binding isotherms were analyzed using BIAevaluation 4.1.1 software (Biacore/GE Healthcare). sCD16 binding to anti-CD16 mAb was fit to a 1:1 Langmuir binding model. The heterogeneous nature of the IC and MDALDL did not allow for curves that fit a 1:1 Langmuir kinetic model analysis with sCD16. Binding affinity was determined for IC and MDALDL interactions with sCD16 using the general fit model of steady state kinetics and Req vs. concentration plots of binding curves.
Cell binding
RAW264 cells (5×105) were incubated with MDALDLbiotin (at indicated concentration) at 4°C for 1 h. MDALDL binding was detected using saturating concentrations of streptavidin-PE. MDALDLbiotin (10 μg/ml) was pre-incubated with sCD16 (5 μg/ml) for 2 h at 22°C to determine if sCD16 cross-block MDALDL binding to macrophage cell line. MDALDL treated with irrelevant His-tag protein (GASP-1) was used as negative control. Cells were washed in fixed in PBS/1% formalin and acquired in a FACSCalibur flow cytometer equipped with CellQuest Pro software (BD-Biosciences, San Jose, CA) and analyzed using FlowJo (TreeStar, Ashland, OR). Alternatively, J774 cells were labeled with calcein-AM and fluorescent-labeled macrophage cell adhesion to MDALDL coated plates were performed as described (33, 34). Briefly, nLDL or MDALDL (50 μl of 10 μg/ml in borate buffer/10 mM EDTA pH 8.5) was coated onto an ELISA plate for 16 h at 4°C. After blocking the plates with binding buffer A (HBSS Ca/Mg free/5 mM EDTA/0.5% BSA) for 2 h. J774 cells (5 × 106/ml) were labeled with 2 μM calcein-AM (Molecular Probes), and the adhesion of labeled cells (1 × 105/well) at 4°C was performed as described earlier (33, 34). The fluorescent intensity of the cells adhered to the plates was measured in a Synergy microplate reader (BioTek) at Ex480/Em530 nm. BSA-coated wells were used as a blank. Adhesion assay was performed in the absence (buffer) or presence of sCD16 or sCD36. GASP-1 and human IgG1 added wells were used as controls. Cells adhered to nLDL and BSA-coated wells were used as negative controls.
MDALDL-induced Syk activation
Thioglycollate-elicited peritoneal macrophages from apoE KO and apoE-CD16 DKO mice were obtained 3 days after injection of thioglycollate. Macrophages (3 × 106 cells/well) in RPMI-1640 medium supplemented with 10% (v/v) FBS, 2 mM L-glutamine, penicillin, streptomycin, and sodium pyruvate was plated in 24-well plates. Non-adherent cells were removed after 2 h and macrophages were used after 48 h. Macrophages were incubated with MDALDL (40 μg/ml) for indicated time. For CD16 cross-linking, macrophages were incubated on ice for 30 min with anti-CD16 mAb (2.4G2, 10 μg/ml). After washing, cells were treated with pre-warmed cross-linking F(ab′)2-goat anti-rat IgG (10 μg/ml) for 30 min. Unstimulated cells incubated on ice followed by 37°C in culture media was used as a control. Macrophages incubated with LPS (100 ng/ml) for 5 min was used as an additional control. Cells were washed with cold PBS containing 1 mM sodium vanadate and lysed ice-cold lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% (v/v) Triton X-100, 1 mM Na3VO4, 1 mM β-glycerophosphate, 1 mM PMSF, protease inhibitor, and phosphatase inhibitors) for 10 min. Supernatants were separated by centrifugation at 13,200 rpm for 10 min at 4°C and protein was estimated using DC protein assay kit (Bio-Rad). Aliquots of total lysates (10 μg) were separated on 8% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). The blots were subjected to immunoblotting with phosphoSykTyr525/526 (1: 2000) and total Syk (1:2000) followed by peroxidase-conjugated donkey anti rabbit IgG (1:5000, Jackson ImmunoResearch) and detected using ECL prime Western blot Detection Reagent (GE Healthcare) and pixel density was determined using QuantityOne software (Bio-Rad). To determine the effect of Syk on MDALDL-induced MCP-1 secretion, macrophages were pre-incubated with Syk inhibitor III (5 μM) for 30 min followed by the addition of ligands (NLDL or MDALDL at 40 μg/ml) or soluble MDALDL-IC (20 μg/ml) or insoluble BSA-IC (15 μl/well) for 24 h. MCP-1 levels in the supernatant was determined using DuoSet MCP-1 ELISA kit (RND systems).
Foam cell assay
Thioglycollate elicited macrophages from apoE KO and apoE-CD16 DKO mice fed high fat diet for 6 weeks were plated on glass cover slip for 2 h. Non-adherent cells were removed after 2 h and cells were stained with Oil Red O next day to determine diet-induced foam cells. To determine MDALDL-induced in vitro foam cell formation, thioglycollate-elicited macrophages from WT and CD16 KO mice (fed chow diet) were plated as described above. Macrophages were exposed to MDALDL (10 μg/ml) for 24 h, stained with oil Red O and DAPI to determine MDALDL-induced in vitro foam cell formation. To determine the effect of CD16 deficiency on in vivo foam cell formation, apoE KO expressing GFP (apoE KO-GFP) mice was generated by crossing apoE KO with C57BL/6-GFP and colonies were screened for apoE deficiency by genomic PCR, and GFP expression in leukocytes was confirmed by flow cytometric analysis. ApoE KO-GFP mice were fed high fat diet for 6 weeks and used as recipient mice. Thioglycollate-elicited macrophages (20 × 106) from WT or CD16 KO mice were transferred to apoE KO-GFP+ mice (total n=6, 3/strain) as recipients. Peritoneal cells were collected from apoE KO-GFP+ recipient mice and analyzed for foam cells after staining macrophages with Oil Red O and counter stained with DAPI. An aliquot of peritoneal cells from apoE KO-GFP+ recipient mice were used to determine GFP positive cells by flow cytometry. Macrophages from WT (GFP negative) and apoE KO-GFP+ mice were used as negative and positive controls, respectively for FACS analysis.
Statistical analyses
Values are expressed as mean ± SD. Differences between the groups were considered significant at p italic> 0.05 using the two-tailed Student t test. All data were analyzed using InStat version 3.1a for Macintosh (GraphPad, San Diego, CA).
Results
Mouse sCD16 blocks MDALDL binding to macrophages
A soluble form of CD36 (sCD36) was evaluated for its ability to block MDALDL binding to J774, a mouse macrophage cell line. To address the specificity of sCD36-mediated blocking human IgG1 and sCD16 was used as negative controls. J774 cells did not adhere to nLDL-coated wells, while it adhered to MDALDL-coated wells (Fig. 1A). As expected, sCD36 blocked mouse macrophage binding to MDALDL (Fig. 1A), while human IgG1 did not block the binding. Surprisingly, sCD16 also inhibited MDALDL binding to macrophage cell line (Fig. 1A). As sCD16 has 10-His tag at the C-terminal end, another irrelevant recombinant protein GASP-1 with His tag was used as a control. GASP-1 did not inhibit MDALDL binding to J774 cells (Fig. 1A). As this finding was unexpected, to confirm sCD16-mediated inhibition of MDALDL binding to J774 cells, MDALDL binding to RAW264 cells, another macrophage cell line was determined. RAW264 macrophages bound to MDALDL dose dependently (Fig. 1B), and pre-incubation of MDALDL with sCD16 inhibited MDALDL binding to cells (Fig. 1C). Similar findings were observed using macrophages from WT mice (data not shown). We then determined if sCD16 inhibits MDALDL binding to membrane expressed CD16. J774, RAW264 (mouse macrophage cell lines) and macrophages constitutively express CD16, and other scavenger receptors: CD36, SR-A and low levels of LOX-1 (data not shown). Hence, we investigated whether sCD16 cross-block MDALDL binding to other scavenger receptors. MDALDL binding to individual scavenger receptors was determined using ELISA plates coated with sCD36, sSR-A, or sLOX-1. Dose dependent binding assay showed the saturation of MDALDL binding to scavenger receptors is about 1–3 μg/ml (data not shown). Pre-incubation of MDADLbiotin with sCD16 blocked about 50% MDALDL binding to sCD16-coated plates (Fig. 1D). Interestingly, pre-incubation of MDALDL with sCD16 also inhibited MDALDL binding to sCD36 (Fig. 1E), sLOX-1 (Fig. 1F), and sSR-A (Fig. 1G). These findings suggest that mouse CD16 binds to MDALDL and cross-blocks MDALDL binding to scavenger receptors including CD36, SR-A, and LOX-1.
Figure 1. Mouse CD16 blocks MDALDL binding to macrophages.

A, CD16 inhibits J774 adhesion. MDALDL-coated ELISA plates were treated with indicated reagents (at 2 μg/ml) or buffer for 30 min. Calein-labeled J774 cells were added to the plates and incubated at 4°C for 30 min. After removing non-adherent cells by inverting the plates in PBS for 5 min, cell adhesion (%) was calculated by dividing fluorescence before and after washing the plate to remove non-adherent cells. B, Dose dependent MDALDL binding to RAW264 cells. RAW264 cells were incubated with MDALDLbiotin at indicated concentration for 1 h at 4°C and binding followed by streptavidin-PE. Cells incubated without MDALDLbiotin was used as a negative control. MDALDL binding was determined by FACS analysis. C, CD16 inhibits MDALDL binding to RAW264 macrophages. MDALDLbiotin (10 μg/ml) was preincubated without (buffer) or with sCD16 (5 μg/ml) for 2 h followed by its binding to RAW264 cells. D–G, Recombinant sCD16 blocks MDALDL binding to CD16. sCD16 or indicated soluble scavenger receptors were coated onto ELISA white plate. MDALDLbiotin was preincubated with 2.5 μg/ml sCD16 or buffer for 2 h and added to sCD16- (D) or sCD36 (E), sLOX-1 (F) or sSR-A- (G) coated plates. Binding was detected using streptavidin-alkaline phosphatase and Lumiphos 530 luminescence substrate as described in methods. In all these experiments values are means ± SD triplicate wells. Shown is a representative of three independent experiments. P, <0.01** or <0.001*** compared to buffer treated wells (no competitor).
Mouse sCD16 directly binds to MDALDL
Based on its ability to block MDALDL binding to macrophages, we characterized the MDALDL, a non-IgG alternative ligand, binding function of CD16 by two approaches. In the first approach, mouse sCD16 was added to MDALDL-coated plates (plate bound) and the binding was detected using anti-poly histidine IgG. Mouse sCD16 bound to MDALDL with no detectable binding to nLDL or BSA (Fig 2A). GASP-1, an irrelevant His-tag protein, did not bind to MDALDL. As plate-bound MDALDL represent a polyvalent ligand, we investigated if sCD16 could bind to MDALDL in solution. In this assay, sCD16 was coated onto the ELISA plate and MDALDLbiotin was used a ligand. MDALDL bound to sCD16 in dose-dependent manner and the binding was detected as low as MDALDL concentration at 1 μg/ml (Fig 2B). Moreover, addition of unlabeled MDALDL inhibited MDALDLbiotin binding to sCD16. The relative affinity of MDALDL binding to plate-bound sCD16 is estimated to be about 20 μM compared to anti-CD16 mAb at 300 pM (Supplemental Figure 1A) and OVA-IC at 90 nM (Supplemental Figure 1B). To confirm the non-Ig ligand binding to sCD16, binding affinity of the sCD16 to MDALDL (Fig 2C), IC and anti-CD16 mAb (Supplemental Figure 1C and D) was determined using surface plasmon resonance spectroscopy. Interaction analysis was designed to achieve 1:1 binding interaction where possible. Anti-CD16 mAb bound to sCD16 at a high affinity of 248 pM. IC and MDALDL binding interactions were not capable of 1:1 Langmuir analyses and steady state kinetic analyses indicated IC and MDALDL bound to sCD16 a low affinities of 78 nM and 25 μM, respectively.
Figure 2. Mouse CD16 specifically binds to MDALDL.

A, CD16 binds to plate-bound MDALDL. MDALDL (10 μg/ml) was coated on ELISA plate overnight. CD16-his (2 μg/ml) was added to the plates and incubated for 1 h at 22°C. CD16 binding was detected using HRP-conjugated anti-poly His IgG followed by the addition of HRP substrate. Wells coated with nLDL or BSA was used as controls. Human GASP-1, a His-tag protein, was used as negative control. P, <0.001*** compared to MDALDL binding to GASP1 coated wells. B, Dose dependent and specificity of MDALDL binding of sCD16. CD16 was coated on ELISA white plate (2.5 μg/mL). MDALDLbiotin was added at different concentration and binding was detected by luminescence assay (relative light units, RLU). Specific MDALDL binding to sCD16 was calculated by subtracting RLU values in the absence (buffer) from RLU values in the presence of molar excess of unlabeled MDALDL (+MDALDL). C, Steady state affinity analysis of MDALDL and sCD16. SPR was used to evaluate sCD16 binding kinetics and affinity for MDA-LDL. Binding surfaces were constructed on CM5 chips as detailed in Materials and Methods. Duplicate response equilibriums (Req) of serial dilutions (grey dots) are plotted as a function of their respective, increasing, log10 concentrations and fit (Chi2 = 4.1) to the general fit model of steady state affinity (black line). Inset isotherm is a representative of the doubled-referenced binding data (grey traces) of RU over time. D, MDABSA competes for MDALDL binding to CD16. MDALDLbiotin (3 μg/ml) binding to sCD16 was inhibited by molar excess of unlabeled MDALDL and MDABSA (30 μg/ml). Fucoidan (40 μg/ml) added to sCD16-coated wells was also used as a competing reagent to block MDALDLbiotin binding to sCD16. nLDL and no competitor (buffer) treated wells were used as controls. P, <0.001*** compared to buffer treated wells (no competitor). E, Anti-MDA IgM mAb inhibits MDALDL binding to CD16. MDALDLbiotin (3 μg/ml) was preincubated with mouse anti-MDA IgM mAb (E012) or control mouse IgM (G8G7) at 1:25 or 1:50 dilution for 2 h followed by the addition to CD16-coated plates. P, <0.01** compared to control mIgM. F, Anti-MDA IgG pAb inhibits MDALDL binding to CD16. MDALDL (3 μg/ml) was preincubated with affinity-purified rabbit IgG (whole molecule) or F(ab′)2 fragment or buffer followed by detecting binding to CD16. MDALDL treated with anti-OVA IgG (whole molecule or F(ab′)2 fragment) were used as controls. In all these experiments values are mean ± SD of triplicate wells. Representation of 2–3 independent experiments is presented. P, <0.001*** compared to buffer treated wells (no competitor).
CD16 recognize MDA epitope in MDALDL
Next we investigated the specificity of sCD16 recognizing MDA epitopes in MDALDL in a competition immunoassay. Unlabeled MDALDL and MDA-BSA inhibited MDALDLbiotin binding to sCD16, while nLDL and BSA did not inhibit the binding (Fig. 2D). Interestingly, fucoidan also inhibited MDALDL binding to sCD16 (Fig. 2D). The specificity of mouse CD16 binding to MDA epitopes was further confirmed using anti-MDA pAb and mAb that specifically recognizes MDA epitope. Anti-MDA IgM mAb and anti-MDA IgG specifically bound to MDALDL, while binding to nLDL was very minimal (Supplemental Figure 2) indicating the specificity of anti-MDA antibodies. Anti-MDA specific IgM mAb (E012) inhibited mouse sCD16 binding to MDALDL (Fig. 2E), while isotype specific mIgM (G8G7) did not inhibit MDALDL binding to sCD16. Similarly rabbit anti-MDA IgG (whole molecule) inhibited MDALDL binding to sCD16 (Fig. 2F), while rabbit anti-OVA IgG did not. As the natural ligand for CD16 is IgG, F(ab′) fragment of anti-MDA IgG was prepared and used in the binding assay. F(ab′)2 fragment of anti-MDA IgG also inhibited MDALDL binding to sCD16 (Fig. 2F). However, under similar conditions rabbit anti-OVA IgG (whole molecule and F(ab′)2 fragment) did not inhibit. These findings indicate that CD16 specifically recognize MDA epitopes present in MDALDL.
Mouse sCD16 does not bind to oxLDL
As MDALDL is one of the modification occurs during the oxidation of LDL (3, 4, 8), we then determined whether sCD16 can bind to oxLDL. Interestingly, sCD16 bound poorly to oxLDL (Fig 3A). To confirm whether oxLDL used in this immunoassay is functionally active, we repeated the binding experiment using other scavenger receptors. OxLDL bound to sCD36, sSR-A, and sLOX-1 (Fig 3B–D). These findings suggest that mouse CD16 does not recognize epitopes in oxLDL. The selective binding of sCD16 to MDALDL is surprising since oxLDL has been reported to contain MDA epitopes (3, 4, 8). One possibility is that MDA epitopes either may be masked or reduced in the extensively oxLDL. To address this possibility, CD16 binding assay was repeated using differentially oxLDL. nLDL was incubated with Cu2+ for indicated time to prepare differentially oxLDL. To confirm differentially oxLDL is indeed oxidized, nLDL and differentially oxLDL electrophoretic mobility was determined. The electrophoretic mobility of differentially oxLDL increased over time of LDL oxidation (Fig. 3E). Kinetics of differentially oxLDL and subsequent binding of differentially oxLDL in solution to sCD16 coated plates showed oxLDL binding to sCD16 was higher at early oxidation of LDL, and this binding to sCD16 decreased considerably with increasing time of LDL oxidation (Fig 3F). We used differentially oxLDL as a soluble ligand, hence it is possible that sCD16 binding may be increased if differentially oxLDL is presented as poly-valent ligand in a plate-bound form. To address this possibility, we developed a reverse binding assay in which differentially oxLDL was coated on the plate followed by the addition of sCD16. Reverse binding assay also showed sCD16 bound to the early oxidation product of LDL better than the products of extensive oxidation of LDL (Fig 3G). These findings collectively suggest that MDA epitopes in the extensively oxLDL may either not accessible for binding to CD16 or MDA epitopes are lost during the extensive oxidation of LDL. To address this possibility, we determined rabbit anti-MDA IgG (specific to MDA epitopes) binding to differentially oxLDL. Anti-MDA IgG binding was higher at early time (1–4 h) of LDL oxidation, while anti-MDA IgG binding decreased in the extensive oxidation (24 h) of LDL (Fig 3H). To confirm this finding we also used oxLDL (24 h oxidation) from a commercial source (Kalen), which also showed reduced anti-MDA IgG binding (Fig 3H). These findings collectively suggest that early oxidation of LDL may have either more MDA epitope and/or MDA epitope is more accessible to anti-MDA IgG binding. Based on these findings we investigated whether the extent of MDA modification influences sCD16 binding. MDALDL was purchased from Kalen with different levels of MDA. Electrophoretic mobility analyses showed more MDA modification resulting in faster mobility of MDALDL, while LDL modified with low amount of MDA showed slower mobility (Supplemental Figure 3A). These findings indicate that electrophoretic mobility of differentially MDA-modified LDL is dependent on the extent of MDA modification. CD16 binding to differentially modified MDALDL showed sCD16 binding was dependent on the extent of MDA modification (Supplemental Figure 3B). These findings suggest that CD16 binding to MDALDL is dependent on extent of MDA modification.
Figure 3. Mouse CD16 does not bind to oxLDL.

MDALDLbiotin or oxLDLbiotin (3 μg/ml) was added at sCD16 (A), sCD36 (B) or sSR-A (C) or sLOX-1 (D) coated plates. Binding was detected by luminescence assay. P, <0.001*** compared to nLDL binding. E, Electrophoretic mobility of differential oxidation of LDL. nLDLbiotin (200 μg/ml) was incubated with CuSO4 for indicated time and reaction was stopped by the addition of 0.1 mM EDTA. An aliquot (1 μg) was loaded on agarose gel and stained with Fat Red. nLDL (Kalen) and nLDLbiotin without the addition of copper (0 h) were used as controls. F, Decreased binding of differential oxLDLbiotin binding to sCD16 coated plates were determined by luminescence based assay using streptavidin-alkaline phosphatase. G, Decreased sCD16 binding to plate bound differential oxLDL. Differential oxLDL was prepared as described in 3F, using unlabeled nLDL. Differential oxLDL (5 μg/ml) was coated onto white ELISA plates for 16 h at 4°C. After blocking the plate with blocking buffer, sCD16 (3 μg/ml) was added sCD16 binding was detected using anti-poly His IgGbiotin followed by the addition of streptavidin-alkaline phosphatase in a luminescence assay. H, Loss of MDA epitopes in extensively oxLDL. Differential oxLDL (5 μg/ml) in PBS/1 mM EDTA was coated on ELISA plates. Affinity purified rabbit anti-MDA IgG binding was detected by ELISA using goat anti-rabbit IgG-HRP and peroxidase substrate. OxLDL (copper oxidized LDL for 24 h) from a commercial source (Kalen) was used as an additional control. In all these assays values are mean ± SD of triplicate wells. Representation of 2–3 independent experiments is presented. P, <0.05* compared to 2 h oxLDL.
MDALDL may bind to IC binding domain in CD16
Thus far we have presented evidence that CD16 binds to MDALDL, a non-Ig ligand. As IC is the natural ligand for CD16 (35), we investigated whether MDALDL and IC binds to similar or overlapping domains in mouse CD16. Pretreatment of sCD16 with anti-CD16 mAb (two different mAbs), which has been reported to block IC binding, completely inhibited IC binding to sCD16 (Fig 4A). Interestingly, pretreatment of sCD16 with blocking anti-CD16 mAb partially inhibited sCD16 binding to MDALDL (Fig. 4B). As IC is the natural IgG containing ligand for CD16, we then determined whether soluble IC could block MDALDL binding to sCD16. Soluble IC addition partially inhibited the MDALDL binding to sCD16 (Fig 4B), while completely inhibited soluble PAP-IC binding to CD16 (Fig 4A). Moreover, MDALDL as well as MDA-BSA competitively inhibited IC binding to sCD16 (Fig 4C). Collectively, these findings suggest that MDALDL may either share IC binding domain or the MDALDL binding domain could be at close proximity to the IC binding domain in mouse CD16.
Figure 4. MDALDL binds to IC binding domain in CD16.
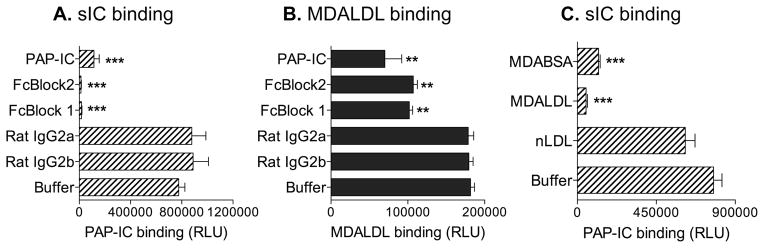
aCD16 was coated on white ELISA plate. Soluble PAP-ICbiotin at 2 μg/ml (A) or MDALDLbiotin at 3 μg/ml (B) binding to sCD16-coated plates were performed by luminescence-based assay. Anti-CD16 blocking mAbs (2 μg/ml, FcBlock 1 or 2) were added to sCD16-coated wells before the addition of ligands. CD16-coated wells incubated with unlabeled PAP-IC or isotype control rat IgG were used as positive and negative controls, respectively. sCD16 coated well without blocking reagent (buffer) shows the total ligand (PAP-ICbiotin or MDALDLbiotin ) binding. C, MDA-modified proteins inhibit soluble IC binding to CD16. Soluble PAP-ICbiotin (2 μg/ml) binding to sCD16-coated wells was performed in the absence (buffer) or presence of unlabeled MDABSA or MDALDL (at 20 μg/ml) by luminescence based assay. nLDL (20 μg/ml) was used a negative control. Values are mean ± SD of triplicate wells. Representation of two independent experiments is presented. P, <0.001*** or <0.01** compared buffer treated (no competitor) wells.
ApoE-CD16 DKO mice show reduced atherosclerotic lesions
If mouse CD16 has MDALDL binding function, we hypothesize that deficiency of CD16 may result in attenuated atherosclerotic lesions. We tested this hypothesis using apoE-CD16 DKO mice. To determine the role of CD16 in the progression of atherosclerosis, CD16 deficiency in apoE KO background was generated. Genotype analyses showed complete knockout for CD16 in the apoE KO background (Fig 5A). Atherosclerotic lesions in apoE KO and apoE-CD16 DKO female mice fed a high fat diet showed 40% reduction in lesions (p bold> 0.001, Student t test) in apoE-CD16 DKO mice compared with apoE KO mice (Fig 5B and C). Male apoE-CD16 DKO mice also showed a similar reduction in lesions (Fig 5D and E), suggesting there is no effect of gender on the extent of lesions. These findings suggest that CD16, one of the activating FcγRs, contribute to the progression of atherosclerosis.
Figure 5. ApoE-CD16 DKO mice show reduced atherosclerotic lesions.

A, Genotype analyses for apoE (left) and CD16 (middle and right) was determined by PCR using DNA isolated from apoE-CD16 DKO mice colony. CD16 genotype analyses were performed using separated PCR with a common and WT primer (middle) or common and mutant primers (right). Wild type (WT), CD16 KO, and apoE KO (single knockout) were used as controls. Number indicates mouse ID. Representative aortic sinus sections from apoE KO and apoE-CD16 DKO mice female (B) and male (D) fed high fat diet were stained with Oil Red O to determine fatty streak lesions. Representative of a 6–8 aortic sinus from apoE KO and apoE-CD16 DKO mice were presented. Quantitative analysis of atherosclerotic lesions in aortic sinus of female (C) and male (E) were determined as described under ‘methods’. Each group contained 6–8 mice. P, <0.01** compared to apoE KO mice.
Mechanisms contributing to attenuated lesions in apoE-CD16 DKO mice
To determine the molecular mechanisms contributing to the decreased atherosclerotic lesions in apoE-CD16 DKO mice, plasma total cholesterol levels were determined. There were no significant differences in plasma total cholesterol levels in apoE-CD16 DKO mice (1113 ± 172 mg/dL vs. 1073 ± 213 mg/dL) compared with apoE KO mice fed a high-fat diet. Similarly HDL-cholesterol levels were also similar in apoE KO and apoE-CD16 DKO mice (data not shown). These data suggest that the reduced lesions in apoE-CD16 DKO mice were not due to change in plasma lipid levels.
Reduced foam cell formation in apoE-CD16 DKO macrophages
Because foam cell formation is an early event in atherosclerosis (36, 37), we subsequently investigated if reduced foam cell formation by CD16 deficient macrophages contributed to the attenuated lesions in apoE-CD16 DKO mice. ApoE KO and apoE-CD16 DKO mice fed high fat diet for six weeks to determine diet-induced foam cell formation in vivo by staining elicited-macrophages with Oil Red O. Macrophages from apoE-CD16 DKO mice showed attenuated diet induced foam cell formation compared to macrophages from apoE KO mice (Fig 6A and B). To confirm the attenuated diet-induced foam cell formation in apoE-CD16 DKO mice, elicited macrophages were treated with MDALDL to determine foam cell formation in vitro. WT macrophages treated with MDALDL showed more than 50% foam cell formation (Fig 6C and D). CD16 single knockout (CD16 KO) macrophages showed reduced (<20%) foam cell formation (Fig 6C and D). Glass group (38) suggested that in vivo foam cell formation reflects lipid-laden macrophage at the lesion site in vivo. Hence, we investigated if CD16 deficiency could result in reduced in vivo foam cell formation using high fat fed-apoE KO-GFP+ mice as recipient, and macrophages from WT and CD16 KO as donors. WT macrophages transferred to recipient apoE KO-GFP+ mice fed with high fat diet showed >50% foam cell formation (Fig 6E and F). However, transfer of CD16 deficient macrophages to hyperlipidemic apoE KO-GFP+ mice showed reduced in vivo foam cell formation (Fig 6E and F). FACS analysis showed the recovered peritoneal macrophages from apoE KO-GFP+ recipient mice were negative (>95%) for GFP expression (Fig. 6G), indicating that contribution by resident macrophages from the apoE KO-GFP+ recipient mice was minimal. As other scavenger receptor expression in CD16 KO macrophages could influence MDALDL-induced foam cell formation, expression of CD36 and SR-A was determined. FACS analysis of macrophages showed CD16 KO macrophages have no expression of CD16 (Fig 6H), and low levels expression observed is due to the anti sCD16/CD32 mAb recognizing CD32 expression. CD36 and SR-A expression in CD16 KO macrophage were similar to WT macrophages (Fig 6H), suggesting that decreased foam cell formation in CD16 KO is independent of CD36, SR-A and LOX-1 expression. These findings also suggest that CD16 contributes to foam cell formation.
Figure 6. Reduced foam cell formation in apoE-CD16 DKO macrophages.

A, High fat diet induced foam cell formation is reduced in apoE-CD16 DKO mice. ApoE KO and apoE-CD16 DKO mice were fed high fat diet for 6 wk. Resident peritoneal macrophages were collected, plated on a cover glass and stained with oil Red O and DAPI (nuclear stain) and foam cells (%) were determined (B). n=7–8, P, <0.01** compared to apoE KO mice. C, Attenuated MDALDL-induced foam cell formation in vitro in CD16 KO macrophages. Thioglycollate-elicited macrophages from wild type or CD16 KO mice were treated with MDALDL (10 μg/ml) for 24 h and percent foam cells (D) were determined after staining cells with Oil Red O and DAPI. n=4, P, <0.01** compared to apoE KO mice. E, CD16 KO macrophages show reduced foam cell formation in vivo. Thioglycollate-elicited peritoneal macrophages (3 × 106) from WT or CD16 KO (donor, GFP negative) mice (n=3/strain) were injected to apoE KO-GFP+ recipient mice (n=6) fed high fat diet for 6 wk. Three days after injecting macrophages, peritoneal cells were plated and stained with Oil Red O to determine in vivo foam cell formation (F). FACS analyses showed three days after injecting donor macrophages (WT, 1–3) or CD16 KO (4–6) into apoE KO-GFP+ recipient mice >95% recovered peritoneal macrophages were GFP negative (G). WT and apoE KO-GFP+ (shown in top panel) were used as controls for FACS analysis. Values are means ± SD. P, <0.01** compared to apoE KO (A and B) or <0.001*** compared to WT (C–F) mice. H, Scavenger expression was not altered in CD16 KO macrophages. Thioglycollate-elicited peritoneal macrophages from WT or CD16 KO mice were stained with biotin-conjugated anti-CD16, anti-CD36 or anti-SR-A mAb (closed histogram) followed by streptavidin-PE. Cells stained with corresponding isotype control IgG (open histogram) was used as a control. Stained cells were analyzed in FACSCalibur flow cytometer. Number indicates mean fluorescence.
CD16 deficiency results in reduced MDALDL induced chemokine secretion
We investigated functional implications of scavenger receptor like-function of CD16. For this purpose, initially CD16 expression in J774 macrophages was silenced using a Smartpool siRNA specific for CD16. Smart-pool CD16 siRNA efficiently reduced CD16 mRNA expression by 90%, while control non-target siRNA did not have any effect (Supplemental Figure 4A). CD16 protein expression also reduced by >60% in CD16 siRNA transfected cells (data not shown). CD16 silencing did not affect CD36, SR-A and LOX-1 mRNA (Supplemental Figure 4A) and protein expression (data not shown). CD16 silencing reduced MDALDL and soluble IC binding to J774 cells (Supplemental Figure 4B). Then, we determined the effect of CD16 silencing on MDALDL-induced pro-inflammatory cytokine/chemokine expression. Non-target siRNA transfected J774 cells treated with MDALDL showed increased secretion of TNF-α (Fig 7A), MCP-1 (Fig 7B), and RANTES (Fig 7C). However, MDALDL-induced pro-inflammatory cytokine and chemokine secretion in CD16 silenced J774 macrophages were significantly decreased (Fig. 7A–C). To confirm that the attenuated cytokine expression is due to CD16 silencing, J774 (non-target and CD16 siRNA transfected) cells were treated with BSA-IC or MDALDL-IC to determine IC-mediated chemokine expression. Non-target siRNA transfected J774 cells treated with BSA-IC or MDALDL-IC, natural ligand for CD16, also induced TNF-α (Fig 7D), MCP-1 (Fig 7E), and RANTES (Fig 7F). However, in CD16 silenced cells BSA-IC and MDALDL-IC induced cytokine chemokine expression was reduced (Fig. 7D–F). To confirm the findings from macrophage cell line, experiments were repeated using primary macrophages from CD16 KO mice. MDALDL and soluble IC binding was lower in CD16 KO macrophages compared to WT macrophages (Fig. 7G) and there was no difference in oxLDL binding between WT and CD16 KO macrophages (Fig. 7G). WT macrophages exposed to MDALDL-IC induced MCP-1 secretion (Fig 7H), while CD16 KO macrophages showed reduced response. Interestingly, CD16 KO macrophages exposed to MDALDL showed significantly reduced MCP-1 secretion compared with WT macrophages (Fig 7H). These findings collectively suggest that scavenger receptor like function of sCD16 contributes to MDALDL-induced chemokine secretion.
Figure 7. CD16 deficiency reduces MDALDL induced chemokine secretion.

CD16 silenced and non-target siRNA treated J774 cells were incubated with MDALDL at 40 μg/ml (A–C) or soluble MDALDL-IC (20 μg/ml) or insoluble BSA-IC, 15 μl/well (D–F) for 24h. Secretion of TNF-α (A, D), MCP-1 (B, E), and RANTES (C, F) were determined by cytokine bead array kit. Samples were analyzed in BD-Fortessa flow cytometer. P, <0.01** or <0.001*** compared to media treated cells. G, CD16 deficiency reduces MDALDL and sIC binding. Thioglycollate-elicited macrophages from WT and CD16 KO mice were plated on 96-well plate for 24 h. Indicated ligands (all biotinylated) were added to macrophages and incubated for 2 h at RT, followed by the addition of streptavidin-HRP. Binding of ligands was detected after the addition of HRP substrate and OD at 450 nm was measured. P, <0.01** compared to WT mice. H, MDALDL-induced MCP-1 secretion in CD16 KO macrophages. Thioglycollate-elicited macrophages from WT and CD16 KO mice were plated on 96-well plate for 24 h. Macrophages were treated with MDALDL (10 μg/ml) or MDALDL-IC (20 μg/ml) and supernatant collected 24h after treatment. MCP-1 levels in the supernatant were determined by DuoSet ELISA kit. Values are mean ± SD of triplicate wells. Representation of two independent experiments is presented. P, <0.01** or <0.001*** compared to WT mice.
MDALDL-induced Syk phosphorylation
Activation of Syk is essential for CD16-induced macrophage activation (22, 23, 39). We were therefore keen to understand whether the MDALDL binding to CD16 is able to transduce signals similar to those described for IC binding to CD16 (22, 23, 39). CD16-mediated Syk phosphorylation was examined using macrophages derived from CD16 KO mice. CD16 cross-linking with anti-CD16 mAb induced Syk phosphorylation in wild type macrophages (Fig 8A and B). However anti-CD16 mAb-induced Syk phosphorylation is significantly reduced in CD16 KO macrophages (Fig 8A and B). MDALDL binding to WT macrophages also induced Syk phosphorylation (Fig 8A and B). Interestingly MDALDL-induced Syk phosphorylation is reduced in CD16 KO macrophages (Fig 8A and B) without affecting total Syk expression (Fig 8A and C). We also determined whether blocking Syk activation would affect MDALDL-induced MCP-1 secretion. Pre-incubation of macrophages with selective inhibitors of Syk activity (40, 41) inhibited MDALDL- and IC-induced MCP-1 secretion (Fig. 8D). This confirms that CD16 binding to MDALDL induced Syk phosphorylation and subsequent MCP-1 secretion.
Figure 8. Decreased MDALDL-induced Syk activation in CD16 deficient macrophages.

A, Thioglycollate-elicited macrophages from apoE KO and apoE-CD16 DKO mice were plated and incubated with MDALDL (40 μg/ml) at indicated time. Macrophages incubated with anti-CD16 mAb (2.4G2, 5 μg/ml) followed by cross-linking with F(ab′)2-goat anti-rat IgG (10 μg/ml) was used as a positive control. LPS (100 ng/ml) treated cells were used as an additional control. Phospho-Syk (B) and total Syk (C) levels were determined by Western blot using specific antibodies and band intensities (pixel density) were determined in BioRad Quantity One software. C, Syk inhibitor blocked MDALDL-induced MCP-1 secretion. Macrophages were pre-incubated with Syk inhibitor III (5 μg/ml) for 30 min followed by the addition of MDALDL (40 μg/ml) or MDALDL-IC (20 μg/ml) for 18 h. Cells incubated with BSA-IC (15 μl/well) and nLDL (40 μg/ml) was used as positive and negative controls, respectively. Cells without the additional of Syk inhibitor or indicated reagents (media) was used to determine basal level of MCP-1 secretion. MCP-1 levels were determined by DuoSet ELISA kit from RND systems. Representation of two independent experiments is presented.
Discussion
In the current study, we tested the hypothesis that non-IgG scavenger receptor function of the CD16, one of the activating FcγRs, contributes to the progression of atherosclerosis. We presented evidence that mouse CD16 blocks MDALDL binding to macrophages and cross-blocks MDALDL binding to CD36, SR-A and LOX-1. We also showed sCD16 binds selectively to MDALDL and MDALDL binding site may be either shared and/or in close proximity to IC binding domain of CD16. We showed a significant reduction in arterial lesions in apoE-CD16 DKO mice after a high-fat diet. Remarkably, the reduction in atherosclerotic lesion progression in CD16 deficiency in hyperlipidemic apoE KO mouse model was associated with reduction in foam cell formation and MDALDL-induced inflammatory cytokine and chemokine expression and Syk activation.
Role of SR-A and CD36 in the progression of atherosclerosis is not resolved because earlier studies have reported that apoE-SR-A DKO or apoE-CD36 DKO mice (mixed genetic background 129sv/B6) had reduced atherosclerotic lesions (15, 16). However, recent study showed loss of CD36 or SR-A in apoE KO mice (B6 congenic) did not ameliorate atherosclerotic lesions (17). The difference in genetic background was attributed to the differences in the findings from both studies. Similar findings were also observed in LDLR-CD36 DKO mice fed high fat diet (20). Moreover bone marrow transplantation of human SR-A overexpressing bone marrow-derived cells to apoE KO or LDLR KO did not show increase in atherosclerotic lesions (18, 19). These reports raise a possibility that other cell surface receptors expressed on macrophages with scavenger receptor functions may also contribute to foam cell formation and atherosclerosis. Interestingly during the course of our study to determine the efficacy of sCD36 to block MDALDL-induced macrophage function, we made a surprising finding that sCD16 (used as a negative control) also blocked MDALDL binding to macrophages. This led to the present study to investigate the scavenger receptor like function of mouse CD16. We demonstrated that mouse CD16 bound to MDALDL, one of the molecular species generated during LDL lipid peroxidation (3, 4, 8). Specificity analyses showed MDALDL binding to mouse sCD16 was competitively inhibited by MDA-BSA. Moreover, anti-MDA IgM mAb and IgG (mAb and pAb, respectively) recognizing MDA epitopes inhibited the binding of sCD16 to MDALDL, indicating CD16 specifically recognize MDA epitope. Notably, sCD16 blocked MDALDL binding to macrophages as well as cross-blocked MDALDL binding to other scavenger receptors CD36, SR-A and LOX-1. These findings revealed a novel non-IgG ligand binding function of CD16.
CD16 has two Ig-domains in the extra cellular regions and competition studies using mAb that block IC binding to CD16 have mapped IC binding domain resides in the membrane proximal Ig-domain (42, 43). In the present study we showed anti-CD16 mAb and IC partially inhibited MDALDL binding to CD16. Conversely, cold MDALDL also inhibited IC binding to sCD16. These findings suggest that mouse CD16 binding to MDALDL may occur via domains that have been reported to bind to IC (natural ligand). However, amino acids present in the IC-binding domain that specifically recognizing MDA epitope need to be established. Notably, CD16 has been shown to recognize non-IgG ligands, which includes thymic stromal cell antigen (44), a uncharacterized ligand expressed on NK target cells (45), E. coli K12 (26). All these non-IgG ligands have been shown to bind to IC binding domain of mouse CD16.
Several studies have presented evidence that oxidation of nLDL results in complex mixture of products including MDALDL, minimally modified-LDL (46), oxidized-phospholipids (47, 48) and oxidized polyunsaturated fatty acids such as HETE and HODE (48). This raises the question that what is the clinical significance of MDALDL and it’s binding to CD16. Using mAb that specifically recognize oxLDL or MDALDL clinical studies has shown that plasma oxLDL and MDALDL levels were elevated in patients with coronary artery disease (6–9). Interestingly, elevated plasma MDALDL has been associated with myocardial infarction (5, 6), and elevated plasma MDALDL levels have been suggested to be a biomarker for myocardial infarction. Moreover, the significance of MDALDL contributing to the progression of atherosclerosis is supported by the presence of MDA epitopes in atherosclerotic lesions of rabbits and human (1). Collectively, these reports suggest that MDA-modified LDL binding to cell surface receptors such as CD16 expressed on monocytes, neutrophil, dendritic cells and macrophages could lead to inflammatory responses.
Clinical studies have suggested a strong positive correlation between CD16 polymorphisms and a higher incidence of inflammatory diseases in human including coronary artery disease (49). Earlier studies using CD16 deficiency in LDLR, another hyperlipidemic mouse model, have shown attenuated lesions and the reduced lesions was attributed to increased IL-10 secretion by T cells (50). However, in the same studies the authors have also reported that T cells from CD16-LDLR DKO mice have higher IFN-γ levels (50). Though the relative contribution by anti-inflammatory IL-10 (Th2) and pro-inflammatory IFN-γ (Th2) was not addressed, it was suggested macrophages and innate cells could also be contributing to attenuated lesions. Hence, it is possible that the attenuated lesions in CD16 deficient mice could also be mediated by mechanisms other than or in addition to altered T cell immune response. In this study, we showed that apoE-CD16 DKO mice have reduced lesions and diet induced foam cell formation. Additionally, CD16 single knockout macrophages also showed reduced MDALDL-induced foam cell formation. CD16 binding to E. coli has been shown to bacterial clearance functions of MARCO (26) a class-A scavenger receptor implicated in bacterial clearance and host defense (27, 28). As MARCO can also binds to modified-LDL it is conceivable that the attenuated lesions and foam cell formation in apoE-CD16 DKO mice may be mediated by MARCO function in CD16 KO mice. This possibility is not likely because CD16 has been shown to negatively regulate bacterial clearance function of MARCO without affecting the expression of (26). Based on these reports one should expect deficiency of CD16 would result in increased MARCO-dependent modified-LDL binding and subsequent inflammatory response. However our data show that MDALDL binding is reduced in CD16 KO macrophages, and decreased MDALDL binding to CD16 KO macrophages is also associated with blunted inflammatory cytokine response. As expression of CD36, SR-A and LOX-1 was not altered in CD16 KO and apoE-CD16 DKO macrophages, the reduction in lesions and foam cell formation is independent of previously characterized scavenger receptor expression.
In macrophages FcγRs including CD16 associates with the ITAM-containing common Fcγ-chain, and this association is necessary for downstream signaling (22, 23). IC binding to activating FcγRs including CD16 initiates signaling events via activation of Src family tyrosine kinases resulting in rapid and transient phosphorylation of the ITAMs on the associated Fcγ-chain of CD16 (39, 51–53). Phosphorylated ITAM creates docking sites for Syk and subsequent phosphorylation of Syk (54, 55). Number of studies has shown that Syk activation play a critical role in promoting FcγR-mediated inflammatory responses and phagocytosis (39, 56–58). To examine the functional significance of MDALDL binding to CD16, we investigated whether CD16 binding to MDALDL increased Syk tyrosine phosphorylation and MCP-1 secretion. The results reported here demonstrate that MDALDL induced Syk activation resulting in Syk phosphorylation in CD16+ WT macrophages. However, MDALDL-induced phosphorylation of Syk is significantly reduced in CD16 deficient macrophages. We also showed MDALDL induced MCP-1 secretion is reduced in macrophages from CD16 KO mice. Moreover, Syk inhibitor significantly inhibited MDALDL-induced MCP-1 secretion. Collectively, the findings from this study suggest CD16 expressed in monocytes/macrophages has an alternative function to bind MDALDL and the scavenger receptor like function of CD16 expressed in monocytes/macrophages may have functional significance in inflammatory processes associated with atherosclerosis.
In summary, our investigation demonstrated that murine CD16 binds to MDALDL and scavenger receptor activity of CD16 in part contributed to the abridged lesions in CD16 deficient mice in hyperlipidemic conditions. Our results strongly suggest that scavenger receptor function of CD16 resulting in binding to MDALDL induced chemokine secretion, which may promote monocyte migration and progression of atherosclerosis. In addition to hypercholesterolemic condition, increased formation of MDA-protein adducts have been suggested to contribute to acceleration of lupus in lupus-prone MRL/lpr mice (59) and systemic lupus erythematous patients (60). Similarly, MDA adducts have also been implicated in the progression of experimental autoimmune encephalitis (61) and alcohol induced liver disease (62). These clinical and pre-clinical observations suggest a possibility that MDA-adducts formed in autoimmune and alcohol liver diseases may bind to constitutively expressed CD16 on inflammatory cells resulting in pro-inflammatory responses and subsequent progression of autoimmune diseases. These studies further suggest broader implication of scavenger receptor function of CD16. It will also be important to determine human CD16 also has similar scavenger receptor like function.
Supplementary Material
Acknowledgments
The authors acknowledge technical support from Ramona Burris and Jessica Jordan. The authors also acknowledge Dr. Linda Curtiss, Scripps Research Institute for providing anti-MDA IgM and control IgM mAbs.
Footnotes
Supported by NIH grant R01HL086674 (SN). Also supported by intramural funds from the Experimental Pathology, Department of Pathology and Vascular Medicine Institute, University of Pittsburgh, PA.
Abbreviations used in this paper: ApoE, apolipoprotein E, BSA-IC, BSA immune complex, DKO, double knockout, nLDL, native low-density lipoprotein, MDA, malondialdehyde, oxLDL, oxidized LDL, scavenger receptor-AI/II (SR-A), sIC, soluble immune complex, TBA, thiobarbituric acid, TBARS, thiobarbituric acid reactive substances,
References
- 1.Yla-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, Witztum JL, Steinberg D. Evidence for the presence of oxidatively modified LDL in atherosclerotic lesions of rabbits and man. J Clin Invest. 1989;84:1086–1095. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2311–2316. doi: 10.1161/ATVBAHA.108.179697. [DOI] [PubMed] [Google Scholar]
- 3.Esterbauer H, Gebicki J, Puhl H, Jurgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radical Biol Med. 1992;13:341–390. doi: 10.1016/0891-5849(92)90181-f. [DOI] [PubMed] [Google Scholar]
- 4.Levitan I, Volkov S, Subbaiah PV. Oxidized LDL: diversity, patterns of recognition, and pathophysiology. Antioxid Redox Signal. 2010;13:39–75. doi: 10.1089/ars.2009.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holvoet P, Perez G, Zhao Z, Brouwers E, Bernar H, Collen D. Malondialdehyde-modified low density lipoproteins in patients with atherosclerotic disease. J Clin Invest. 1995;95:2611–2619. doi: 10.1172/JCI117963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holvoet P, Vanhaecke J, Janssens S, Van de Werf F, Collen D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98:1487–1494. doi: 10.1161/01.cir.98.15.1487. [DOI] [PubMed] [Google Scholar]
- 7.Boaz M, Matas Z, Biro A, Katzir Z, Green M, Fainaru M, Smetana S. Serum malondialdehyde and prevalent cardiovascular disease in hemodialysis. Kidney Int. 1999;56:1078–1083. doi: 10.1046/j.1523-1755.1999.00613.x. [DOI] [PubMed] [Google Scholar]
- 8.Tanaga K, Bujo H, Inoue M, Mikami K, Kotani K, Takahashi K, Kanno T, Saito Y. Increased circulating malondialdehyde-modified LDL levels in patients with coronary artery diseases and their association with peak sizes of LDL particles. Arterioscler Thromb Vasc Biol. 2002;22:662–666. doi: 10.1161/01.atv.0000012351.63938.84. [DOI] [PubMed] [Google Scholar]
- 9.Tamer L, Sucu N, Polat G, Ercan B, Aytacoglu B, Yucebilgic G, Unlu A, Dikmengil M, Atik U. Decreased serum total antioxidant status and erythrocyte-reduced glutathione levels are associated with increased serum malondialdehyde in atherosclerotic patients. Arch Med Res. 2002;33:257–260. doi: 10.1016/s0188-4409(01)00381-2. [DOI] [PubMed] [Google Scholar]
- 10.Kondo A, Muranaka Y, Ohta I, Notsu K, Manabe M, Kotani K, Saito K, Maekawa M, Kanno T. Relationship between triglyceride concentrations and LDL size evaluated by malondialdehyde-modified LDL. Clin Chem. 2001;47:893–900. [PubMed] [Google Scholar]
- 11.Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182:1–15. doi: 10.1016/j.atherosclerosis.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 12.van Berkel TJ, Out R, Hoekstra M, Kuiper J, Biessen E, van Eck M. Scavenger receptors: friend or foe in atherosclerosis? Curr Opin Lipidol. 2005;16:525–535. doi: 10.1097/01.mol.0000183943.20277.26. [DOI] [PubMed] [Google Scholar]
- 13.Prabhudas M, Bowdish D, Drickamer K, Febbraio M, Herz J, Kobzik L, Krieger M, Loike J, Means TK, Moestrup SK, Post S, Sawamura T, Silverstein S, Wang XY, El Khoury J. Standardizing scavenger receptor nomenclature. J Immunol. 2014;192:1997–2006. doi: 10.4049/jimmunol.1490003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kzhyshkowska J, Neyen C, Gordon S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology. 2012;217:492–502. doi: 10.1016/j.imbio.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, Takashima Y, Kawabe Y, Cynshi O, Wada Y, Honda M, Kurihara H, Aburatani H, Doi T, Matsumoto A, Azuma S, Noda T, Toyoda Y, Itakura H, Yazaki Y, Kodama T, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 16.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore KJ, V, Kunjathoor V, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Eck M, De Winther MP, Herijgers N, Havekes LM, Hofker MH, Groot PH, Van Berkel TJ. Effect of human scavenger receptor class A overexpression in bone marrow-derived cells on cholesterol levels and atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:2600–2606. doi: 10.1161/01.atv.20.12.2600. [DOI] [PubMed] [Google Scholar]
- 19.Herijgers N, de Winther MP, Van Eck M, Havekes LM, Hofker MH, Hoogerbrugge PM, Van Berkel TJ. Effect of human scavenger receptor class A overexpression in bone marrow-derived cells on lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knockout mice. J Lipid Res. 2000;41:1402–1409. [PubMed] [Google Scholar]
- 20.Kennedy DJ, Kuchibhotla SD, Guy E, Park YM, Nimako G, Vanegas D, Morton RE, Febbraio M. Dietary cholesterol plays a role in CD36-mediated atherogenesis in LDLR-knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:1481–1487. doi: 10.1161/ATVBAHA.109.191940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 22.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 23.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen IA, Schmidt RE, Sandor M, Capel PJ, Daeron M, Van de Winkel JGJ, Verbeek JS. Impaired IgG-dependent anaphylaxis and Arthus reaction in FcgRIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 24.Stein MP, Mold C, Du Clos TW. C-reactive protein binding to murine leukocytes requires Fc gamma receptors. J Immunol. 2000;164:1514–1520. doi: 10.4049/jimmunol.164.3.1514. [DOI] [PubMed] [Google Scholar]
- 25.Sandor M, Galon J, Takacs L, Tatsumi Y, Mueller AL, Sautes C, Lynch RG. An alternative Fc gamma-receptor ligand: potential role in T-cell development. Proc Natl Acad Sci USA. 1994;91:12857–12861. doi: 10.1073/pnas.91.26.12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinheiro da Silva F, Aloulou M, Skurnik D, Benhamou M, Andremont A, Velasco IT, Chiamolera M, Verbeek JS, Launay P, Monteiro RC. CD16 promotes Escherichia coli sepsis through an FcR gamma inhibitory pathway that prevents phagocytosis and facilitates inflammation. Nat Med. 2007;13:1368–1374. doi: 10.1038/nm1665. [DOI] [PubMed] [Google Scholar]
- 27.Dorrington MG, Roche AM, Chauvin SE, Tu Z, Mossman KL, Weiser JN, Bowdish DM. MARCO is required for TLR2- and Nod2-mediated responses to Streptococcus pneumoniae and clearance of pneumococcal colonization in the murine nasopharynx. J Immunol. 2013;190:250–258. doi: 10.4049/jimmunol.1202113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Laan LJ, Dopp EA, Haworth R, Pikkarainen T, Kangas M, Elomaa O, Dijkstra CD, Gordon S, Tryggvason K, Kraal G. Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J Immunol. 1999;162:939–947. [PubMed] [Google Scholar]
- 29.Stanton LW, White RT, Bryant CM, Protter AA, Endemann G. A macrophage Fc receptor for IgG is also a receptor for oxidized low-density lipoprotein. J Biol Chem. 1992;267:22446–22451. [PubMed] [Google Scholar]
- 30.Ng HP, Burris RL, Nagarajan S. Attenuated atherosclerotic lesions in apoE-Fcgamma-chain-deficient hyperlipidemic mouse model is associated with inhibition of Th17 cells and promotion of regulatory T cells. J Immunol. 2011;187:6082–6093. doi: 10.4049/jimmunol.1004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thampi P, Stewart BW, Joseph L, Melnyk SB, Hennings LJ, Nagarajan S. Dietary homocysteine promotes atherosclerosis in apoE-deficient mice by inducing scavenger receptors expression. Atherosclerosis. 2008;197:620–629. doi: 10.1016/j.atherosclerosis.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 32.Freigang S, Horkko S, Miller E, Witztum JL, Palinski W. Immunization of LDL receptor-deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arterioscler Thromb Vasc Biol. 1998;18:1972–1982. doi: 10.1161/01.atv.18.12.1972. [DOI] [PubMed] [Google Scholar]
- 33.Stewart BW, Nagarajan S. Recombinant CD36 inhibits oxLDL-induced ICAM-1-dependent monocyte adhesion. Mol Immunol. 2006;43:255–267. doi: 10.1016/j.molimm.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Nagarajan S. Anti-OxLDL IgG blocks OxLDL interaction with CD36, but promotes FcgammaR, CD32A-dependent inflammatory cell adhesion. Immunol Lett. 2007;108:52–61. doi: 10.1016/j.imlet.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Nagarajan S, Chesla SE, Cobern L, Anderson P, Zhu C, Selvaraj P. Ligand binding and phagocytosis by CD16 (Fc gamma receptor III) isoforms. J Biol Chem. 1995;270:25762–25770. doi: 10.1074/jbc.270.43.25762. [DOI] [PubMed] [Google Scholar]
- 36.Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43. [DOI] [PubMed] [Google Scholar]
- 37.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, Raetz CR, Wang EW, Kelly SL, Sullards MC, Murphy RC, Merrill AH, Jr, Brown HA, Dennis EA, Li AC, Ley K, Tsimikas S, Fahy E, Subramaniam S, Quehenberger O, Russell DW, Glass CK. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151:138–152. doi: 10.1016/j.cell.2012.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VL, DeFranco AL. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raeder EM, Mansfield PJ, Hinkovska-Galcheva V, Shayman JA, Boxer LA. Syk activation initiates downstream signaling events during human polymorphonuclear leukocyte phagocytosis. J Immunol. 1999;163:6785–6793. [PubMed] [Google Scholar]
- 41.Desaulniers P, Fernandes M, Gilbert C, Bourgoin SG, Naccache PH. Crystal-induced neutrophil activation. VII. Involvement of Syk in the responses to monosodium urate crystals. J Leukoc Biol. 2001;70:659–668. [PubMed] [Google Scholar]
- 42.Tamm A, Schmidt RE. The binding epitopes of human CD16 (Fc gamma RIII) monoclonal antibodies. Implications for ligand binding. J Immunol. 1996;157:1576–1581. [PubMed] [Google Scholar]
- 43.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406:267–273. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 44.Lynch RG, Hagen M, Mueller A, Sandor M. Potential role of Fc gamma R in early development of murine lymphoid cells: evidence for functional interaction between Fc gamma R on pre-thymocytes and an alternative, non-Ig ligand on thymic stromal cells. Immunol Lett. 1995;44:105–109. doi: 10.1016/0165-2478(95)00200-o. [DOI] [PubMed] [Google Scholar]
- 45.Mandelboim O, Malik P, Davis DM, Jo CH, Boyson JE, Strominger JL. Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc Natl Acad Sci USA. 1999;96:5640–5644. doi: 10.1073/pnas.96.10.5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25:1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- 47.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 48.Nagy L, Tontonoz P, Alvarez JGA, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 49.Gavasso S, Nygard O, Pedersen ER, Aarseth JH, Bleie O, Myhr KM, Vedeler CA. Fcgamma receptor IIIA polymorphism as a risk-factor for coronary artery disease. Atherosclerosis. 2005;180:277–282. doi: 10.1016/j.atherosclerosis.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 50.Kelly JA, Griffin ME, Fava RA, Wood SG, Bessette KA, Miller ER, Huber SA, Binder CJ, Witztum JL, Morganelli PM. Inhibition of arterial lesion progression in CD16-deficient mice: evidence for altered immunity and the role of IL-10. Cardiovasc Res. 2010;85:224–231. doi: 10.1093/cvr/cvp300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park JG, Murray RK, Chien P, Darby C, Schreiber AD. Conserved cytoplasmic tyrosine residues of the gamma subunit are required for a phagocytic signal mediated by Fc gamma RIIIA. J Clin Invest. 1993;92:2073–2079. doi: 10.1172/JCI116804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson SA, Pleiman CM, Pao L, Schneringer J, Hippen K, Cambier JC. Phosphorylated immunoreceptor signaling motifs (ITAMs) exhibit unique abilities to bind and activate Lyn and Syk tyrosine kinases. J Immunol. 1995;155:4596–4603. [PubMed] [Google Scholar]
- 53.Bewarder N, Weinrich V, Budde P, Hartmann D, Flaswinkel H, Reth M, Frey J. In vivo and in vitro specificity of protein tyrosine kinases for immunoglobulin G receptor (FcgammaRII) phosphorylation. Mol Cell Biol. 1996;16:4735–4743. doi: 10.1128/mcb.16.9.4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsuda M, Park JG, Wang DC, Hunter S, Chien P, Schreiber AD. Abrogation of the Fc gamma receptor IIA-mediated phagocytic signal by stem-loop Syk antisense oligonucleotides. Mol Biol Cell. 1996;7:1095–1106. doi: 10.1091/mbc.7.7.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonnerot C, Briken V, Brachet V, Lankar D, Cassard S, Jabri B, Amigorena S. syk protein tyrosine kinase regulates Fc receptor gamma-chain-mediated transport to lysosomes. EMBO J. 1998;17:4606–4616. doi: 10.1093/emboj/17.16.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greenberg S, Chang P, Silverstein SC. Tyrosine phosphorylation of the gamma subunit of Fcg-receptors, p72sky, and paxillin during Fc-mediated phagocytosis in macrophages. J Biol Chem. 1994;269:3897–3902. [PubMed] [Google Scholar]
- 57.Cox D, Chang P, Kurosaki T, Greenberg S. Syk tyrosine kinase is required for immunoreceptor tyrosine activation motif-dependent actin assembly. J Biol Chem. 1996;271:16597–16602. doi: 10.1074/jbc.271.28.16597. [DOI] [PubMed] [Google Scholar]
- 58.Kiefer F, Brumell J, Al-Alawi N, Latour S, Cheng A, Veillette A, Grinstein S, Pawson T. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol Cell Biol. 1998;18:4209–4220. doi: 10.1128/mcb.18.7.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang G, Li H, Firoze Khan M. Differential oxidative modification of proteins in MRL+/+ and MRL/lpr mice: Increased formation of lipid peroxidation-derived aldehyde-protein adducts may contribute to accelerated onset of autoimmune response. Free Radic Res. 2012;46:1472–1481. doi: 10.3109/10715762.2012.727209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurien BT, Scofield RH. Autoimmunity and oxidatively modified autoantigens. Autoimmun Rev. 2008;7:567–573. doi: 10.1016/j.autrev.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wallberg M, Bergquist J, Achour A, Breij E, Harris RA. Malondialdehyde modification of myelin oligodendrocyte glycoprotein leads to increased immunogenicity and encephalitogenicity. Eur J Immunol. 2007;37:1986–1995. doi: 10.1002/eji.200636912. [DOI] [PubMed] [Google Scholar]
- 62.Xu D, Thiele GM, Beckenhauer JL, Klassen LW, Sorrell MF, Tuma DJ. Detection of circulating antibodies to malondialdehyde-acetaldehyde adducts in ethanol-fed rats. Gastroenterology. 1998;115:686–692. doi: 10.1016/s0016-5085(98)70148-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.