

Abstract
Mesenchymal stromal cells have emerged as powerful modulators of the immune system. Here we explored how the human macrophage response to tumor necrosis factor (TNF) is regulated by human synovial fibroblasts, the representative stromal cell type in the synovial lining of joints that become activated during inflammatory arthritis. We found that synovial fibroblasts strongly suppressed TNF-mediated induction of an interferon (IFN)-β autocrine loop and downstream expression of IFN-stimulated genes (ISGs), including chemokines CXCL9 and CXCL10 that are characteristic of classical macrophage activation. TNF induced the production of soluble synovial fibroblast factors that suppressed the macrophage production of IFN-β, and cooperated with TNF to limit the responsiveness of macrophages to IFN-β by suppressing activation of Jak-STAT signalling. Genome-wide transcriptome analysis showed that co-cultured synovial fibroblasts modulate the expression of approximately one third of TNF-regulated genes in macrophages, including genes in pathways important for macrophage survival and polarization towards an alternatively activated phenotype. Pathway analysis revealed that gene expression programs regulated by synovial fibroblasts in our co-culture system were also regulated in rheumatoid arthritis (RA) synovial macrophages, suggesting that these fibroblast-mediated changes may contribute to RA pathogenesis. This work furthers our understanding of the interplay between innate immune and stromal cells during an inflammatory response, one that is particularly relevant to inflammatory arthritis. Our findings also identify modulation of macrophage phenotype as a new function for synovial fibroblasts that may prove to be a contributing factor in arthritis pathogenesis.
Introduction
Stromal cells of the mesenchymal lineage have emerged as powerful modulators of the immune system (1). Traditionally viewed as constituents of connective tissue passively providing structural and homeostatic support, stromal cells such as fibroblasts indeed shape innate and adaptive immune responses involved in host defence, tumor immunity and autoimmunity (1, 2). The influential capacity of stromal cells has been widely demonstrated in cancer, for which the stroma impacts tumor growth, metastasis and even drug resistance (3–5). In most settings, the immunomodulatory activity of the mesenchymal lineage has been characterized as immunosuppressive, in part through repressed T cell proliferation and monocyte cytokine production (2, 6, 7). As a result, mesenchymal stem cell transfers are currently being tested as therapies for autoimmune disorders (1, 8). Ultimately within an immune reaction, stromal cells can be viewed as tissue-resident modifiers, functioning in part through cellular crosstalk with cells of hematopoietic origin.
The synovial lining of arthrodial joints is predominantly composed of macrophages and fibroblast-like synoviocytes (hereafter referred to as ‘synovial fibroblasts’). Under homeostatic conditions, these two cell types function coordinately to maintain synovial fluid composition and joint integrity. In pathogenic conditions like rheumatoid arthritis (RA), both macrophages and synovial fibroblasts contribute to chronic inflammation, pannus formation and joint destruction (9–11). This is in part through macrophage secretion of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF), which in turn activates synovial fibroblasts to produce inflammatory cytokines and chemokines like IL-6 and IL-8, tissue-destructive factors such as MMPs, and to assume an invasive and tissue-destructive phenotype (10–15). Although synovial fibroblast contribution to the inflammatory milieu and tissue destruction is well appreciated and their ability to activate T cells and promote B cell survival have been characterized (16, 17), the effects of synovial fibroblasts on macrophage function in an inflammatory setting have not been elucidated (12). It remains to be determined whether under normal physiologic conditions synovial fibroblasts function, like other mesenchymal stromal cells, to limit inflammatory reactions and whether deregulation of this capacity contributes to RA pathogenesis.
Cytokines have emerged as key mediators of autoimmune diseases, of which TNF and type I interferons (IFN) represent two of the most important. TNF promotes classical (also termed M1) inflammatory activation of macrophages, which includes the production of inflammatory cytokines such as IL-1 and chemokines such as IL-8, CXCL9 and CXCL10 (18–20), although TNF can also engage feedback inhibitory pathways (21). The pathogenic capacity of TNF has been established by the efficacy of anti-TNF therapies in rheumatoid arthritis (RA) (9, 22). Alternatively, elevated IFN levels, typically measured as an ‘interferon signature’ gene expression profile, likely contribute to loss of tolerance in systemic lupus erythematosus (SLE) (23–26). It has been proposed that TNF and IFN can directly counteract each other and that deregulation of this interplay contributes to autoimmunity (23, 27–29). In RA, where chronic TNF stimulation is common, an IFN signature has been detected in the synovial tissue and peripheral blood mononuclear cells from a subset of patients (30). In contrast to SLE, TNF contributes to the IFN signature in RA synovial macrophages, including expression of M1 chemokines such as CXCL9 and CXCL10 that are abundant in RA synovium and can lead to recruitment and activation of inflammatory cells within joints (19, 27, 31–34). Although type I IFNs contribute to inflammatory M1 macrophage activation (19, 35), they also have protective effects in autoimmune diseases (36). Thus understanding how their production and function are regulated may lend insight into their role in RA pathogenesis and suggests approaches to modulate IFN responses in a manner beneficial to patients.
Our laboratory previously demonstrated that TNF-stimulated macrophages secrete IFN-β, which acts in an autocrine and paracrine fashion to induce ISGs (31, 37). We also previously reported that the TNF-induced IFN response in macrophages is inhibited by RA synovial fluids (31). As synovial fibroblasts secrete many of the synovial fluid components, we tested whether synovial fibroblasts regulate the TNF-induced IFN response in macrophages. In co-cultures designed to recapitulate cellular cross-regulation that occurs in inflamed synovial tissue, we demonstrate that synovial fibroblasts secrete factors that suppress the IFN response in TNF-stimulated macrophages and affect macrophage activation by modulating Myc- and growth factor-regulated genes important for macrophage survival and polarization towards an alternatively activated (M2) phenotype. We suggest that modulation of TNF-induced macrophage program by synovial fibroblasts results in a mixed M1/M2 macrophage phenotype in inflammatory settings like RA, which may contribute to RA pathogenesis.
MATERIALS AND METHODS
Cell culture
Human CD14+ monocytes were purified from whole blood cell preparations from healthy donors as previously described (37) and differentiated into macrophages for 1–2 days in 10ng/mL MCSF (Peprotech) and RPMI (Gibco) supplemented with 10% defined HBS (Hyclone). Cells were stimulated with 20ng/mL recombinant human TNF and either 20pg/mL or 200pg/mL human IFN-β (Peprotech) for mRNA and protein analyses, respectively. Infliximab (Janssen Biotech), human IgG (Sigma), human IL10/ IL10R (R&D Systems) and human gp130 (R&D Systems) antibodies were added at 5ug/mL.
Human synovial fibroblasts derived from deidentified synovial tissues of RA or osteoarthritis (OA) patients undergoing total joint arthroplasty (protocol approved by Institutional Review Board at the Hospital for Special Surgery) were digested and passaged as previously described (13). The diagnoses of RA was based on the American College of Rheumatology criteria (38).
For co-culture experiments, in vitro differentiated macrophages were replated directly onto adherent synovial fibroblasts, with the fibroblast:macrophage ratio at 1:2.5, respectively. For transwell culture experiments, synovial fibroblasts adhered to polyester chambers with 0.4um pores (Corning) were suspended above the wells containing macrophages. Supernatants collected from synovial fibroblasts cultured with or without TNF for 16 hours were added to macrophage cultures (0.8mL supernatant into 1.2mL standard culture media). Synovial fibroblast supernatants were fractionated by Amicon Ultra Centrifugal Filters (50, 30, 10 and 3kD membranes).
Real-time PCR
RNA obtained using RNAeasy kit (Qiagen) with DNAse treatment was reverse transcribed into cDNA (Fermentas) and analyzed by real-time quantitative PCR (Fast SYBR Green, Applied Biosystems) on a 7500 Fast Real-Time PCR System (Applied Biosystems). Gene-specific primer sequences for CXCL9, CXCL10, IFNβ, Interferon-induced protein with tetratricopeptide repeats 1 gene (IFIT1), Interferon-induced GTP-binding protein Mx1 gene (MX1), GAPDH, CXCL5 and IL1B were as described (37), while the primers for the Natural Killer Cell Granule Protein 7 gene (NKG7) consisted of the following sequences [F:GATCCAGACCTTCTTCTCCT, R:ACAACGCTCAAAACTCATCT]. Expression levels were normalized to GAPDH and either calculated relative to the uninduced macrophage monoculture sample or as a percentage of the TNF- or IFN-induced macrophage monoculture sample.
Western blots
Western blot analyses were performed using standard procedures with the additional step of adding Pefabloc (Sigma) to macrophage cultures before cell lysis to prevent STAT protein degradation. Antibodies used for Western blots include phospho-STAT1 (Y701) (Cell Signalling, #9171), phospo-STAT2 (Y689) (Millipore, #07-224), STAT1 (Santa Cruz, #sc-346), STAT2 (BD, #610188) and p38 (Santa Cruz, sc-535).
RNA-seq
Total RNA was first extracted using RNeasy mini kit (Qiagen). Tru-seq sample preparation kits (Illumina) were then used to purify poly-A transcripts and generate libraries with multiplexed barcode adaptors. All samples passed quality control analysis on a Bioanalyzer 2100 (Agilent). Paired-end reads (50 x 2 cycles, ~75x106 reads per sample) were obtained on an Illumina HiSeq 2500 in the Weill Cornell Medical College Genomics Resources Core Facility. The TopHat program (39) was used to align the reads to the UCSC Hg19 human reference genome, while the Cufflinks program (40) allowed for measurements of transcript abundance (represented by Fragments Per Kilobase of exon model per Million mapped reads (FPKM)). The RNAseq data have been deposited into the GEO Database (http://www.ncbi.nlm.nih.gov/geo/) and assigned accession number GSE57723.
The Ingenuity Pathway Analysis (IPA) program (Ingenuity® Systems, www.ingenuity.com) was used to analyze the genes in TNF-induced macrophages that were differentially regulated by fibroblasts more than 2-fold. The Upstream Regulator analytic was used to predict upstream regulators whose change in expression or function could explain the observed gene expression changes. The p-value relates to the extent of overlap between known target genes for an upstream regulator and genes differentially regulated in the current dataset. The upstream regulators listed in our results were limited to proteins and endogenous small molecules. A z-score of higher absolute value suggests the majority of target genes for that upstream regulator were altered in a direction consistent with either inhibition or activation of the upstream regulator (an absolute value of 2 or higher is considered significant), for which a negative or positive value represents the predicted inhibition or activation of the upstream regulator, respectively. Target gene networks for the predicted Upstream Regulators MYC and epidermal growth factor (EGF) were limited to target genes that were differentially regulated by at least 4-fold and whose expression change was in the direction that is consistent with the prediction of the upstream regulator as either activated or inhibited. For purposes of clarity, the display of networks in Figure 6D was modified from the IPA standard design as follows: 1) target gene color: red indicates increased expression while green indicates decreased expression in the current experimental data set; 2) lines: lines with arrow heads indicate that the upstream regulator (MYC or EGF) is known from prior published findings to induce gene expression of the target whereas lines without arrow heads indicate that the upstream regulator is known to repress gene expression; 3) target gene shapes represent the molecular classification or function of the protein encoded by the gene, as described in the figure legend.
FIGURE 6.

Transcriptome-wide analysis of the synovial fibroblast influence over macrophage TNF responses. (A) Venn diagrams representing the overlap of macrophage genes that TNF induces (left diagram, red circle) or represses (right diagram, green circle), which in the presence of synovial fibroblast are opposingly regulated (left diagram, green circle represents macrophage genes repressed by fibroblasts upon TNF treatment)(right diagram, red circle represents macrophage genes induced by fibroblasts in TNF conditions). Human blood-derived macrophages were treated for 2 days with TNF with or without synovial fibroblasts suspended above the macrophages in transwell chambers and the macrophage RNA was sequenced (RNAseq). TNF-regulated genes included those which became greater than 2-fold induced or repressed in comparison to untreated macrophages. Fibroblast-regulated genes included those with greater than 2-fold differences when comparing macrophages treated with TNF alone versus treated with TNF in the presence of fibroblasts. (B) Heat map depiction of all macrophage genes repressed (left) or upregulated (right) by co-cultured synovial fibroblasts upon TNF treatment (by at least 2-fold) in comparison to macrophages treated with TNF and cultured alone. Colored bars represent the gene expression levels (log2 of FPKM), with red representing higher and green representing lower levels. (C) Proteins and small molecules predicted to mediate the gene expression changes brought on by synovial fibroblasts in TNF-induced macrophages. The dataset of macrophage genes regulated by fibroblasts by at least 2-fold were analyzed by the Ingenuity IPA program. Based on published findings, the Upstream Regulator analytic predicted upstream molecules whose change in expression or function could explain the observed gene expression changes (the p-value relates to the extent of overlap between known targets genes for an upstream regulator and genes altered in the present dataset). A greater absolute z-score value indicates the upstream regulator target genes are mostly altered in a direction consistent with either reduced or increased activity of the upstream regulator, with negative (left panel) or positive (right panel) values indicating predicted inhibition or activation of the upstream regulator, respectively. (D) Target gene networks for the upstream regulators MYC (left) and EGF (right) include genes regulated by fibroblasts more than 4-fold in the presence of TNF and whose direction of expression is consistent with reduced MYC and increased EGF activity. Red versus green shapes indicate increased versus decreased expression of the target genes by fibroblasts, while arrow heads versus inhibitory lines indicate whether the upstream regulator (MYC or EGF) is known to induce or repress the expression of each gene, respectively. The target gene shapes represent the molecular classification or function of the protein encoded by the gene: square, growth factor/cytokine; triangle, transcription regulator; diamond, enzyme; oval, transmembrane receptor; rectangle, nuclear receptor; trapezoid, transporter; circle, other.
Statistical Analyses
A paired two-tailed student’s t-test was used to analyze for statistical significance. P values < 0.05 were considered significant.
RESULTS
Co-cultured synovial fibroblasts suppress the TNF-induced IFN signature in macrophages
Consistent with our previous findings, TNF stimulation of human monocyte-derived macrophages resulted in a type I IFN signature, measured here as increased expression of the well-established ISGs CXCL9, CXCL10, IFIT1 and MX1 (Fig. 1A, first 2 bars). We had previously shown that this response depends on IFN-β production and autocrine stimulation (37). As synovial fluid suppresses the macrophage TNF-induced IFN response (31) and synovial fibroblasts secrete many synovial fluid factors, we wished to test whether co-cultured synovial fibroblasts would suppress the macrophage TNF-induced IFN response. Consistent with published findings (41), TNF also induced ISG expression in RA synovial fibroblasts in monoculture, but to a much lesser extent than in macrophages (Fig. 1A, bars 3 and 4, with the same data plotted with split y-axis in Fig. S1). Notably, consistent with our previous experiments using synovial fluids (31), co-cultured synovial fibroblasts strongly suppressed the TNF-induced IFN response in macrophages (Fig. 1A, bar 6 versus bar 2 and Fig. S1). This included suppression of the IFN-inducible chemokines CXCL9 and CXCL10, as well as the antiviral factors IFIT1 and MX1 (Fig. 1A). Synovial fibroblasts also modestly suppressed basal levels of ISG expression (Fig. S1). Importantly, expression of genes induced by TNF with similar kinetics but not dependent on IFN, including NKG7, IL1B and CXCL5, were not significantly suppressed by co-cultured fibroblasts (Fig. 1B and data not shown). Thus, fibroblast-like cells from RA synovium control specific components of the TNF response in macrophages, including expression of classic ISGs.
FIGURE 1.

Co-cultured synovial fibroblasts suppress the macrophage IFN signature induced by TNF. (A–B) Human macrophages (Mϕ) and RA synovial fibroblasts (Fib) cultured alone or together (Mϕ + Fib) and stimulated with TNF for 16 h were analyzed for mRNA levels by qPCR. Genes analyzed are listed above each graph. Gene expression values represent the mean of 4 experiments; error bars represent standard error (SE). In each experiment, macrophages were derived from a unique blood donor and the synovial fibroblasts were generated from a unique patient, such that n=4 for both cell types. The amount of transcript was normalized to GAPDH levels in the sample and all samples for a given gene are shown relative to the TNF-induced macrophage monoculture sample, which was set to 100. The mean level of mRNA encoded by the indicated genes relative to the internal standard GAPDH mRNA (% of GAPDH) in the TNF-induced samples was: CXCL9, 10%; CXCL10, 600%; IFIT1, 60%; MX1, 50%; NKG7, 1%; CXCL5, 750%; IL1B, 40%. **<p=0.001; NS, no significance.
To gain an understanding of whether the observed impact of synovial fibroblasts on the macrophage TNF response was specific for RA synovial fibroblasts, we tested synovial fibroblasts derived from osteoarthritis (OA) patients and the IMR90 lung fibroblast cell line in similar co-culture experiments. RA, OA, and IMR90 fibroblasts comparably suppressed the macrophage TNF-induced production of CXCL10 (Fig. S2) and other ISGs (data not shown). Thus, the observed inhibitory effect is not specific for RA synovial fibroblasts. It is important to note that the experimental design necessitates that fibroblasts co-cultured with macrophages are also exposed to and activated by TNF, suggesting the results may reflect not a tissue- or disease-specific function of fibroblasts, but rather a function of fibroblasts that becomes apparent under TNF-stimulated inflammatory conditions, such as is often found in RA. These data collectively suggest that while macrophages can produce and respond to type I IFNs during a TNF response, neighboring fibroblasts inhibit the IFN response.
TNF-stimulated synovial fibroblasts produce soluble mediators that inhibit the TNF-induced IFN signature in macrophages
In our co-culture experiments, synovial fibroblast-mediated inhibition of the macrophage IFN response may have resulted from direct cell contact and/or secreted fibroblast factors. To determine if cell contact was required, the fibroblasts were cultured above and separate from the macrophages in porous transwell chambers. This separated arrangement also allowed for isolation and analysis of the cell populations individually. In this transwell system, the TNF-induced IFN response in macrophages was also suppressed by the co-cultured synovial fibroblasts (Fig. 2A). These data suggest that cell contact is not required, but rather soluble fibroblast-derived products inhibit the macrophage TNF-induced IFN response.
FIGURE 2.

Synovial fibroblasts stimulated by TNF produce soluble mediators responsible for restricting the macrophage TNF-induced IFN response. (A) qPCR mRNA analysis of human macrophages stimulated by TNF for 16h in the presence of synovial fibroblasts suspended in porous transwell chambers. Mϕ (Fib), transwell cultures of macrophages with synovial fibroblasts, in which only the macrophage mRNA was analyzed. (B) qPCR mRNA analysis of macrophages stimulated by TNF for 16h in the presence of supernatant media. Supernatants were collected from independent cultures of synovial fibroblast treated with TNF (FibTNF Supe) and without TNF (Fib− Supe) for 16h. (C) qPCR mRNA analysis of macrophages cultured in a similar manner to part B except that the supernatants from TNF-treated fibroblasts were fractionated by molecular weight (left panel) or boiled (right panel) and individually incubated with the macrophages. kDA, kilodalton. (D) qPCR mRNA analysis of macrophages treated with neutralizing antibodies were incubated alone or with synovial fibroblasts in a transwell chamber (Fib). mRNA levels were normalized to GAPDH and made relative to the TNF-induced macrophage monoculture sample, which was set to 100. The mean level of mRNA encoded by the indicated genes relative to GAPDH mRNA (% GAPDH) for the TNF-induced samples in the Transwell cultures in part (A) was: CXCL10, 300%; IFIT1, 20%; and in the supernatant experiments in part (B–D) was: CXCL10, 1400%; IFIT1, 40%. Mean of n=3 in part A, n=4 in part B and n=2 in parts C–D; error bars represent SE; ***<p=0.0001; **<p=0.001; *<p=0.02; NS, no significance.
Mesenchymal stromal cell-derived immunomodulatory factors are commonly secreted in response to inflammatory stimuli (1, 2). In our co-culture and transwell systems, the synovial fibroblasts were stimulated by TNF along with the macrophages. Thus, we next sought to determine whether TNF stimulation of the synovial fibroblasts was required for production of the soluble inhibitory factors. Supernatant media collected from TNF-treated synovial fibroblast cultures inhibited the macrophage TNF-induced IFN response, while supernatants from unstimulated synovial fibroblasts did not significantly alter the response (Fig. 2B). These data indicate that stimulation of synovial fibroblasts with TNF leads to production of factors that cross-regulate the macrophage TNF program, specifically inhibiting the ensuing IFN response.
The transwell and supernatant experiments demonstrated that a soluble factor produced by the fibroblasts confers the suppressive effect. In order to gain a better understanding of the biochemical nature of the factor, we fractionated fibroblast supernatants by molecular weight. Fractions containing molecules greater than 50kD, consistent with the size of macromolecules, significantly inhibited the TNF-induced CXCL10 expression to a similar extent as the complete supernatant (Fig. 2C, left panel). Furthermore, heat denaturation of the fibroblast supernatants partially reduced the suppressive activity, suggesting a protein component to the inhibitory activity (Fig. 2C, right panel). However, smaller molecular weight fractions retained some inhibitory activity and only 50% of the inhibitory activity was abrogated by boiling, suggesting that small molecules resistant to denaturation by boiling can contribute to inhibitory activity. Lastly, we directly tested whether blocking the function of candidate soluble factors that have immunosuppressive activity and are known to be present in synovial fluids could reverse the suppressive activity of fibroblast culture supernatants. However, neither blockade of IL-10 nor the IL-6 family of cytokines that signal via the gp130 receptor subunit had any effect on fibroblast-mediated suppression of macrophage IFN responses (Fig. 2D). We also tested whether TGFβ, a well-characterized anti-inflammatory factor, mediated the suppression by fibroblasts. However, TGFβ was surprisingly required for TNF-induced IFN response (data not shown), thus in fact functioning as a positive regulator of this pathway. Taken together, the results suggest that TNF stimulates synovial fibroblasts to produce soluble factor(s) of greater than 50 kD in size, likely including a protein component, that acts in trans on macrophages to suppress TNF-induced IFN responses.
Fibroblast products suppress both macrophage IFN-β expression and macrophage responsiveness to exogenous IFN-β
Fibroblast-mediated inhibition of TNF-induced expression of ISGs in macrophages could result from suppression of IFN-β production or suppression of macrophage responsiveness to autocrine IFN-β (or both). We found that synovial fibroblasts suppressed TNF-induced IFNB expression in both transwell and co-culture systems (Fig. 3A, left and middle, and Supplementary Fig. S1). IFNB expression was also inhibited by synovial fibroblast supernatants (Fig. 3A, right). Thus, soluble synovial fibroblast factors inhibit TNF-induced IFN-β production, and this suppression likely contributes to the capacity of synovial fibroblasts to inhibit ISG induction downstream of TNF (Figs. 1A and 2) (37).
FIGURE 3.

Synovial fibroblast products suppress IFN-β expression and responsiveness in TNF-stimulated macrophages. (A) Macrophages stimulated by TNF for 16 h with synovial fibroblasts in standard co-cultures, transwell cultures or with synovial fibroblast supernatants were analyzed for mRNA levels by qPCR. The supernatant media (FibTNF Supe) was collected from independent fibroblast cultures stimulated with TNF for 16 h. (B) qPCR mRNA analysis of macrophages cultured with control media (media) or supernatant media (FibTNF Supe) from fibroblasts independently cultured with TNF for 16h. Supernatants obtained from TNF-induced fibroblast cultures were first treated with Infliximab (Ifx) to eliminate TNF activity or isotype control immunoglobulin (IgG). Following the 16h supernatant incubation, the macrophages were stimulated with IFN-β for 3h. mRNA levels were normalized to GAPDH and made relative to the TNF-induced macrophage alone sample (part A) or IFN-induced macrophage with control media and IgG (part B), which were set to 100. The mean level of IFNB mRNA relative to GAPDH mRNA (% GAPDH) in part (A) was: coculture, 0.1%; transwell, 0.1%; supernatant, 0.1%; and the IFN-β-induced responses in part (B): CXCL10, 800%; IFIT1, 140%. Mean of n=4 donor experiments; error bars represent standard error (SE); ***<p=0.0001; **<p=0.001; *<p=0.05.
While suppression of TNF-induced ISG expression likely results at least in part from reduced IFN-β levels (Fig. 3A), we tested whether synovial fibroblast products also suppress the responsiveness of macrophages to type I IFNs. Strikingly, synovial fibroblast supernatants blocked induction of CXCL10 and IFIT1 by exogenously added IFN-β (Fig. 3B, condition 3 versus condition 5). While a portion of this inhibition resulted from residual TNF activity in the supernatants (Fig. 3B, condition 4 versus condition 5), blockade of TNF by the neutralizing antibody infliximab (Ifx) demonstrated that fibroblast factors alone significantly inhibited macrophage responsiveness to IFN-β (Fig. 3B, condition 4 versus condition 2). This suggests that synovial fibroblast products can suppress responses downstream of the receptor for type I IFNs, referred to as the IFNα receptor (IFNAR). Together these data demonstrate that synovial fibroblast products inhibit the TNF-induced IFN signature in macrophages via two distinct but complementary mechanisms, namely by inhibiting IFN-β production and restricting IFNAR function.
Fibroblast products suppress TNF-mediated induction of Jak-STAT signalling and protein effectors of IFN response
Rapid induction of ISGs by type I IFNs is mediated by STAT1 and STAT2 proteins, which become activated by tyrosine phosphorylation upon IFNAR engagement and function as transcriptional activators of ISGs (36). In addition, when cells are exposed to IFNs for longer time periods, such as those in our experiments, STAT1 and STAT2 expression increases and this increased expression contributes to the increased pool of tyrosine phosphorylated STAT1 and 2 and increased transcriptional activity (36, 37, 42). As predicted, overnight stimulation of macrophages alone with TNF induced expression and tyrosine phosphorylation of STAT1, which we showed previously is mediated by autocrine IFN-β (Fig. 4A) (37). TNF also weakly increased STAT2 expression while robustly increasing STAT2 tyrosine phosphorylation. Addition of synovial fibroblasts to these transwell co-cultures reduced TNF-induced phosphorylation of STAT1 and STAT2, as well as total protein levels for STAT1 and STAT2 (Fig. 4A). These data indicate that synovial fibroblasts suppress induction of an IFN response in macrophages by inhibiting an IFN-β-mediated autocrine loop and downstream Jak-STAT signalling. This inhibition was biologically relevant, as synovial fibroblasts suppressed expression of several ISGs (Fig. 1) and strongly inhibited TNF-induced production of the potent pro-inflammatory chemokine CXCL10 when co-cultured with macrophages (Fig. 4B). Together these data suggest that fibroblast factors interfere with the potential of TNF to induce an effective IFN response program.
FIGURE 4.

Synovial fibroblast products suppress the production of type I IFN response protein effectors in the presence of TNF. (A) Western blot analysis of human macrophages stimulated by TNF for 16h in the presence of synovial fibroblasts suspended in porous transwell chambers. Mϕ (Fib), transwell cultures of macrophages with synovial fibroblasts, in which only the macrophage lysate was analyzed. Blots represent one of three experiments, with the average densitometry value and SE for phospho-STAT1 from all three experiments plotted to the right. (B) ELISA protein measurements for the CXCL10/IP10 chemokine found in supernatants from human macrophages and synovial fibroblasts cultured alone or in transwell cultures (as in part A) and treated with TNF at Day 0. Data points represent the average of 3 independent experiments, with error bars representing the standard error.
TNF precisely controls the macrophage IFN response by both inducing and restricting IFN responsiveness
The results in Figure 3B suggested that overnight exposure to exogenous TNF cooperates with soluble factors secreted by fibroblasts to suppress responses to exogenous IFN-β. We then tested whether TNF alone could partially induce hyporesponsiveness to type I IFNs in macrophages in the absence of fibroblast-derived factors. We performed a time course of TNF stimulation of macrophages in the absence of fibroblasts, followed by recombinant IFN-β treatment. As expected (37), after TNF exposure the expression of ISGs peaked between 6–12 hours depending on the donor and returned to baseline approximately 18 hours after TNF stimulation, consistent with a feedback mechanism that limits the IFN response (Fig. 5A and data not shown). To determine if TNF pre-treatment suppresses macrophage responsiveness to IFNs, we added exogenous IFN-β at this 18 hr time point. As would be expected in the absence of TNF pre-treatment, IFN-β strongly induced CXCL10 and IFIT1 expression in control cells (Fig. 5A, dotted lines). In contrast, the response to exogenous IFN-β was attenuated in TNF-pre-treated macrophages (Fig. 5A, solid lines). Furthermore, while exogenous IFN-β induced relatively high levels of STAT1 and STAT2 phosphorylation in control cells (Fig. 5B, lane 3), TNF pre-treatment for one day dampened this response (Fig. 5B, lane 4). This attenuation is consistent with negative feedback of IFNAR signalling, which is mediated by signalling inhibitors such as suppressor of cytokine signaling (SOCS) proteins or other mechanisms induced by inflammatory cytokines such as TNF or IL-1 (36, 43, 44). Thus in macrophages while TNF initially induces an autocrine IFN-β response it also overtime limits IFN signalling to induce a tempered response. Our co-culture studies demonstrate that synovial fibroblasts can inhibit the early and direct induction of IFNB1 expression by TNF and also enhance the late phase TNF-effect of dampening IFNAR signalling.
FIGURE 5.
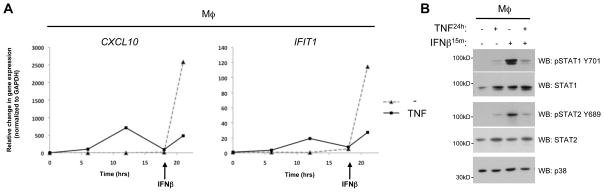
In macrophages, TNF induces an IFN signature but also limits IFN responsiveness. (A) qPCR mRNA analysis of macrophages cultured with and without TNF for 18h and subsequently stimulated with IFN-β for 3h. Graphs depict a representative experiment from 3 independent donors. Gene expression changes were normalized to GAPDH and calculated relative to uninduced. (B) Western blot analysis of macrophages treated with or without TNF for 24h followed by IFN-β for 15min. Blots represent one of three experiments.
Transcriptome-wide analysis of the synovial fibroblast-mediated modulation of the macrophage TNF-response
To address whether synovial fibroblasts selectively regulate macrophage IFN responses or whether they more broadly impact the TNF-induced macrophage phenotype, we performed genome-wide transcriptomic analysis using high throughput RNA sequencing (RNAseq). Human primary macrophages were stimulated with TNF and cultured either alone or in the presence of synovial fibroblasts. Transwell cultures were used to allow for isolation of pure macrophage populations (absence of significant fibroblast cell contamination was verified by monitoring expression of fibroblast-specific genes such as those that encode type I collagens). For macrophages cultured alone, the RNAseq analysis demonstrated that TNF induced or repressed expression of 1629 and 1628 genes by greater than 2-fold, respectively. The RNA-seq results for >220 genes regulated by TNF was confirmed by quantitative real time PCR and Nanostring nCounter mRNA expression analyses (data not shown). Importantly, amongst the 1629 TNF-inducible macrophage genes, co-culturing with fibroblasts attenuated expression for 366 of these genes by greater than 50% (Figure 6A, left panel). Fibroblast factors also upregulated (by greater than 50%) 557 of the 1628 genes repressed by TNF (Figure 6A, right panel). Thus, synovial fibroblasts opposingly regulated expression of approximately 28% of the genes that TNF controls in macrophages.
To gain insight into the pathways responsible for the gene expression changes, the Ingenuity Pathway Analysis (IPA) program was used to analyze the genes in TNF-induced macrophages that were differentially regulated by synovial fibroblasts more than 2-fold; the heat maps in Fig. 6B depict the differential pattern and levels of expression for genes repressed (left panel) or upregulated (right panel) by fibroblasts in TNF conditions. The IPA Upstream Regulator analytic was used to predict upstream molecules whose change in expression or function could be responsible for a subset of the observed gene expression changes (Fig. 6C). Reassuringly, two out of the three highly significant upstream regulators predicted to be inhibited by synovial fibroblasts under TNF-stimulated conditions were related to type I IFN activity (IFNA2; z-score=−4.7, p=4x10−17 and IFNβ; z-score=−2.4, p=3x10−12) (Fig. 6C, left table), thus confirming that fibroblasts broadly suppressed IFN responses in macrophages. Interestingly, the only other upstream regulator predicted to be significantly inhibited due to the presence of fibroblasts was MYC (z-score = −3.5, p = 2x10−17) (Fig. 6C, left table), a transcription factor that contributes to an alternatively activated macrophage phenotype (45). The network of genes known to be regulated by MYC and differentially expressed more than 4-fold in the presence of fibroblasts is shown in Fig. 6D (left panel). Conversely, upstream regulatory factors predicted to exhibit increased activity in TNF-induced macrophages due to the presence of fibroblasts included the growth factors EGF, hepatocyte growth factor (HGF), TGFβ, CSF1 (M-CSF), CSF2 (GM-CSF) and platelet-derived growth factor (PDGF); the small molecules cholesterol, prostaglandin E2, tretinoin (retinoic acid); and the cytokines IL-4 and IL-13 (Fig. 6C, right table). The network of genes known to be regulated by EGF and differentially expressed more than 4-fold in the presence of fibroblasts is shown in Fig. 6D (right panel). Cumulatively, these factors and downstream signalling pathways would contribute to increased macrophage survival and thereby potentially contribute to pathogenesis, with a subset (M-CSF, IL-4, IL-13) promoting features of alternative (M2) activation.
To assess whether the fibroblast-regulated pathways identified in the coculture experiments corresponded to pathways regulated in RA synovial macrophages, we compared our data to that from a parallel study in which we analyzed gene expression patterns specific to macrophages from the synovium of RA patients. Interestingly, among these RA macrophage-specific genes more than a quarter (27%) are also regulated by RA fibroblasts in TNF-induced macrophages. These data support the notion that in the RA synovium, synovial fibroblasts influence a significant portion of the cellular program of macrophages. Furthermore, through an Ingenuity Pathway Analysis of the RA macrophage data set, EGF (z-score=3.6, p=1x10−19) and CSF2 (z-score=2.7, p=2x10−29) are also predicted to be activated upstream regulators. Importantly, close to half (~40%) of the EGF and CSF2 target genes affected in RA macrophages were also affected by fibroblasts in our TNF-induced macrophages. Thus it will be interesting to determine whether the EGF- and CSF2-signatures in RA macrophages are derived from RA synovial fibroblast factors, as it is in macrophages from our co-culture experiments and ultimately whether these responses contribute to the pathologic state.
Discussion
In this study we explored how the two predominant synovial lining cell types, macrophages and synovial fibroblasts, co-ordinately respond to the pathogenic RA cytokine TNF. We demonstrate that synovial fibroblasts strongly suppress induction of an IFN response in macrophages by TNF, which recapitulates the suppressive activity of RA synovial fluids (31). Suppression of macrophage IFN responses was mediated by soluble fibroblast factors whose production was induced by TNF. These fibroblast-derived factors suppressed TNF-induced expression of ISGs in macrophages by two mechanisms: (1) suppression of TNF-induced production of IFN-β by macrophages, and (2) cooperation with TNF itself to limit macrophage responsiveness to IFNs by suppressing activation of the Jak-STAT signalling pathway. Thus, macrophages exposed to TNF in the presence of synovial fibroblasts are restricted both in their capacity to produce and respond to IFN. Transcriptomic analysis revealed that synovial fibroblasts regulated approximately 28% of the macrophage TNF response, including the IFN response and pathways downstream of important regulators of macrophage polarization and survival. Our findings identify a new function for synovial fibroblasts that may be relevant for inflammatory arthritis pathogenesis, namely the modulation of synovial macrophage phenotype.
Investigation of the pathogenic role of synovial fibroblasts in RA has focused on their ability to produce inflammatory cytokines, chemokines, and tissue destructive enzymes, their attachment to and invasion of synovial tissues, and their ability to present antigens and activate T cells and promote lymphocyte survival (10, 11, 46–48). Previous work on synovial fibroblast-macrophage interactions in the absence of inflammatory factors showed that these cells interact (“cocompact”) to form a lining-like structure that resembles that of the synovium, supporting the biological relevance of the co-culture approach (49, 50). Experiments using this system showed that synovial fibroblasts promote macrophage survival, and that stimulation with TNF results in lining hyperplasia and inflammatory mediator production (49); however, the effects of cell crosstalk in an inflammatory setting were not examined. Our findings extend this approach to show substantial crossregulation between these two cell types in the setting of inflammation as modelled by TNF stimulation. One outcome of this crossregulation is suppression of IFN responses in macrophages. Importantly, RA synovial fluids similarly suppress TNF-induced IFN responses in macrophages (31), suggesting that the inflammatory synovial environment, which often includes TNF, stimulates synovial fibroblasts to secrete factors that suppress IFN responses into synovial fluids, similar to the factors secreted into culture supernatants in our model. Regulation of IFN responses by synovial fibroblasts has a homeostatic component, as suppression of IFN-induced M1 chemokines such as CXCL10 would attenuate inflammation. However, type I IFNs also have anti-inflammatory functions, such as suppression of angiogenesis and proliferation (36). Thus, the overall functional consequences of fibroblast-mediated regulation of IFN responses would depend on the balance between the pathogenic and protective functions of type I IFNs in the context of RA. The role of type I IFNs in RA has not yet been clarified, but the importance of understanding IFN responses in RA is underscored by a recent study showing that 24% of SNPs associated with autoimmune diseases, including RA, fall within regulatory elements targeted by IFN-Jak-STAT signalling in immune cells (51).
One mechanism by which synovial fibroblasts suppressed macrophage IFN responses was to inhibit induction of autocrine IFN-β by TNF. Several signalling molecules have been previously shown to suppress IFNB expression, namely the glycogen synthase kinase 3 (GSK3) that is regulated by PI3K-Akt signalling, and the prostaglandin endoperoxide synthase 1 (PTGS1, also known as cyclooxygenase-1 or COX-1), possibly via production of nonprostaglandin lipid mediators and engagement of G protein-coupled receptors (52, 53). However, preliminary experiments using inhibitors of GSK3, G proteins, and COX proteins suggested that synovial fibroblast factors inhibit IFN-β production independently of these pathways (data not shown). Insight into which receptors and pathways are important will be provided by further biochemical characterization of fibroblast supernatants, particularly in identification of the > 50kD factor, likely a protein, that exhibited inhibitory activity, but which will be challenging given the complexity and limited quantities of culture supernatants. A second mechanism by which synovial fibroblasts suppressed macrophage IFN responses was to cooperate with the late-phase of the TNF response, which attenuates activation of Jak-STAT signalling. Overall, our findings suggest a tight functional coupling between synovial fibroblasts and macrophages in which the fibroblasts target different aspects of the TNF-induced autocrine IFN-β loop to effectively suppress ISG expression.
In addition to regulating IFN responses induced downstream of TNF, a pathway analysis of our transcriptomic data revealed that under TNF-stimulated conditions fibroblasts inhibit expression of genes downstream of MYC and augment expression of genes induced by multiple growth factors, including EGF, HGF, TGFβ, CSF1 (M-CSF), CSF2 (GM-CSF), PDGF, IL-4 and IL-13 in nonproliferating primary human macrophages. This suggests that under TNF-stimulated co-culture conditions synovial fibroblasts produce growth factors that regulate the macrophage phenotype; accordingly increased expression of vascular endothelial growth factor A (VEGFA), heparin-binding EGF-like growth factor (HBEGF) and insulin-like growth factor 1 (IGF1) was observed in the co-culture RNAseq data. Interestingly, MYC is not involved in proliferation or survival of human macrophages but instead has been implicated in the induction of a subset of genes in alternatively activated macrophages that are important for resolution of inflammation and tissue repair (45). Thus, suppression of MYC-dependent pathways and genes in synovial macrophages will alter and may partially compromise development of pro-resolution macrophage phenotype. Although primary human macrophages have minimal proliferative capacity, activation of growth factor pathways will promote cell survival, thereby promoting disease pathogenesis (54, 55). In addition, other macrophage pathways predicted be upregulated in the presence of fibroblasts, in particular IL-4 and IL-13 and to some extent CSF1 (M-CSF), promote a global alternative activation phenotype (also termed M2), which is associated with tissue remodelling and resolution of inflammation (56, 57). In many inflammatory settings M2 polarization would be considered beneficial as it can promote resolution of classical M1-mediated inflammation. However, RA pathogenesis is characterized by an expanding inflammatory mass of synovial fibroblasts and macrophages (termed pannus), and expression of subsets of M2 genes may actually contribute to pathogenesis by promoting growth and dysregulated tissue repair. As synovial fibroblasts simultaneously suppress expression of IFN-inducible genes such as CXCL9 and CXCL10 that are part of the classic activation (M1) phenotype, suppress MYC, but augment growth factor-mediated gene expression, the data argue that synovial fibroblasts partially shift the polarization pattern of macrophages towards an M2 phenotype, distinctively lacking the MYC-mediated component of M2 and the IFN-mediated component of M1 phenotypes. Mixed macrophage phenotypes simultaneously expressing subsets of M1 and M2 genes are common in complex inflammatory settings in vivo (18, 20, 58) and indeed we found a quarter of the genes uniquely regulated in macrophages from RA patients are also controlled by fibroblasts in TNF-induced macrophages, suggesting the presence of fibroblast-controlled M2 components in RA macrophages. Thus our results provide insights into mixed macrophage responses characteristic of RA and how such complex phenotypes can be generated.
In summary, we discovered a complex interplay between synovial fibroblasts and macrophages where fibroblasts modify subsets of macrophage responses to TNF, including suppression of ISG expression and modulation of select polarization features along the spectrum of classical inflammatory (M1) and alternative (M2) activation. Seminal previous work analyzing cytokine networks in co-cultures of cells including fibroblasts, macrophages and lymphocytes obtained from disaggregated RA synovial tissues led to the identification of TNF as an upstream regulator of cytokine production and therapeutic target in RA (22, 59). Similarly we propose that our work with a simplified synovial fibroblast-macrophage interaction system, which recapitulates the activity of RA synovial fluids, will yield not only mechanistic insights into regulation of TNF responses, but may identify new pathways and regulators that can be therapeutically targeted.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health, National Institutes of Arthritis and Musculoskeletal Disease (RO1 AR046713 (LBI) and K01 AR066063 (LTD)), the Arthritis National Research Foundation and the Sontag Foundation (GDK).
Our gratitude to Dr. Mark Figgie and his patients for providing the synovial tissues and Angela Lee and Christopher Sohn for generating the synovial fibroblast cell lines (Hospital for Special Surgery).
Abbreviations used in this article
- EGF
epidermal growth factor
- GSK3
glycogen synthase kinase 3
- HBEGF
heparin-binding EGF-like growth factor
- HGF
hepatocyte growth factor
- IFIT
Interferon-induced protein with tetratricopeptide repeats 1
- IFNAR
IFNα receptor
- IGF1
insulin-like growth factor 1
- ISG
Interferon-stimulated gene
- MX1
Interferon-induced GTP-binding protein Mx1
- NKG7
Natural Killer cell granule protein 7
- PDGF
platelet-derived growth factor
- PTGS1
prostaglandin endoperoxide synthase 1
- SLE
systemic lupus erythematosus
- SOCS
suppressor of cytokine signaling
- VEGFA
vascular endothelial growth factor A
References
- 1.Frenette PS, Pinho S, Lucas D, Scheiermann C. Mesenchymal Stem Cell: Keystone of the Hematopoietic Stem Cell Niche and a Stepping-Stone for Regenerative Medicine. Annu Rev Immunol. 2013;31:285–316. doi: 10.1146/annurev-immunol-032712-095919. [DOI] [PubMed] [Google Scholar]
- 2.Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol. 2012;12:383–396. doi: 10.1038/nri3209. [DOI] [PubMed] [Google Scholar]
- 3.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 4.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 5.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, Ribas A, Lo RS, Flaherty KT, Ogino S, Wargo JA, Golub TR. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2013;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones S, Horwood N, Cope A, Dazzi F. The antiproliferative effect of mesenchymal stem cells is a fundamental property shared by all stromal cells. J Immunol. 2007;179:2824–2831. doi: 10.4049/jimmunol.179.5.2824. [DOI] [PubMed] [Google Scholar]
- 8.Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell. 2012;10:709–716. doi: 10.1016/j.stem.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 9.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2012;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 10.Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. 2008;223:252–270. doi: 10.1111/j.1600-065X.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- 11.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naylor AJ, Filer A, Buckley CD. The role of stromal cells in the persistence of chronic inflammation. Clin Exp Immunol. 2012;171:30–35. doi: 10.1111/j.1365-2249.2012.04634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee A, Qiao Y, Grigoriev G, Chen J, Park-Min KH, Park SH, Ivashkiv LB, Kalliolias GD. Tumor Necrosis Factor αInduces Sustained Signaling and a Prolonged and Unremitting Inflammatory Response in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis & Rheumatism. 2013;65:928–938. doi: 10.1002/art.37853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lefevre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, Korb A, Schnaker EM, Tarner IH, Robbins PD, Evans CH, Sturz H, Steinmeyer J, Gay S, Scholmerich J, Pap T, Muller-Ladner U, Neumann E. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–1420. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, Gay S. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149:1607–1615. [PMC free article] [PubMed] [Google Scholar]
- 16.Filer A, Parsonage G, Smith E, Osborne C, Thomas AM, Curnow SJ, Rainger GE, Raza K, Nash GB, Lord J, Salmon M, Buckley CD. Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils. Arthritis Rheum. 2006;54:2096–2108. doi: 10.1002/art.21930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran CN, Thacker SG, Louie DM, Oliver J, White PT, Endres JL, Urquhart AG, Chung KC, Fox DA. Interactions of T cells with fibroblast-like synoviocytes: role of the B7 family costimulatory ligand B7-H3. J Immunol. 2008;180:2989–2998. doi: 10.4049/jimmunol.180.5.2989. [DOI] [PubMed] [Google Scholar]
- 18.Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2012;34:216–223. doi: 10.1016/j.it.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 20.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park SH, Park-Min KH, Chen J, Hu X, Ivashkiv LB. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat Immunol. 2011;12:607–615. doi: 10.1038/ni.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feldmann M. Translating molecular insights in autoimmunity into effective therapy. Annu Rev Immunol. 2009;27:1–27. doi: 10.1146/annurev-immunol-082708-100732. [DOI] [PubMed] [Google Scholar]
- 23.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Crow MK. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res Ther. 2010:S5. doi: 10.1186/ar2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. Journal of Experimental Medicine. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 27.Cantaert T, Baeten D, Tak PP, van Baarsen LG. Type I IFN and TNFalpha cross-regulation in immune-mediated inflammatory disease: basic concepts and clinical relevance. Arthritis Res Ther. 2010;12:219. doi: 10.1186/ar3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivashkiv LB. Type I interferon modulation of cellular responses to cytokines and infectious pathogens: potential role in SLE pathogenesis. Autoimmunity. 2003;36:473–479. doi: 10.1080/08916930310001605882. [DOI] [PubMed] [Google Scholar]
- 29.Williams EL, Gadola S, Edwards CJ. Anti-TNF-induced lupus. Rheumatology (Oxford) 2009;48:716–720. doi: 10.1093/rheumatology/kep080. [DOI] [PubMed] [Google Scholar]
- 30.Kalliolias GD, Kirou KA. Type I interferons as biomarkers in autoimmune diseases. Biomark Med. 2012;6:137–140. doi: 10.2217/bmm.12.8. [DOI] [PubMed] [Google Scholar]
- 31.Gordon RA, Grigoriev G, Lee A, Kalliolias GD, Ivashkiv LB. The interferon signature and STAT1 expression in rheumatoid arthritis synovial fluid macrophages are induced by tumor necrosis factor alpha and counter-regulated by the synovial fluid microenvironment. Arthritis Rheum. 2012;64:3119–3128. doi: 10.1002/art.34544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Pouw Kraan TC, van Gaalen FA, Kasperkovitz PV, Verbeet NL, Smeets TJ, Kraan MC, Fero M, Tak PP, Huizinga TW, Pieterman E, Breedveld FC, Alizadeh AA, Verweij CL. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum. 2003;48:2132–2145. doi: 10.1002/art.11096. [DOI] [PubMed] [Google Scholar]
- 33.Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis: pathogenic or protective? Arthritis Rheum. 2003;48:2092–2096. doi: 10.1002/art.11095. [DOI] [PubMed] [Google Scholar]
- 34.van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, Voskuyl AE, Rustenburg F, Baggen JM, Ibrahim SM, Fero M, Dijkmans BA, Tak PP, Verweij CL. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007;66:1008–1014. doi: 10.1136/ard.2006.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 36.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon–response genes. Nat Immunol. 2008;9:378–387. doi: 10.1038/ni1576. [DOI] [PubMed] [Google Scholar]
- 38.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 39.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosengren S, Corr M, Firestein GS, Boyle DL. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: autocrine role of type I interferon. Ann Rheum Dis. 2012;71:440–447. doi: 10.1136/ard.2011.150284. [DOI] [PubMed] [Google Scholar]
- 42.Hu X, Herrero C, Li WP, Antoniv TT, Falck-Pedersen E, Koch AE, Woods JM, Haines GK, Ivashkiv LB. Sensitization of IFN-gamma Jak-STAT signaling during macrophage activation. Nat Immunol. 2002;3:859–866. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- 43.Huynh L, Wang L, Shi C, Park-Min KH, Ivashkiv LB. ITAM-coupled receptors inhibit IFNAR signaling and alter macrophage responses to TLR4 and Listeria monocytogenes. J Immunol. 2012;188:3447–3457. doi: 10.4049/jimmunol.1102211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huangfu WC, Qian J, Liu C, Liu J, Lokshin AE, Baker DP, Rui H, Fuchs SY. Inflammatory signaling compromises cell responses to interferon alpha. Oncogene. 2011;31:161–172. doi: 10.1038/onc.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, Doni A, Nebuloni M, Swigart LB, Evan GI, Mantovani A, Locati M. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood. 2012;119:411–421. doi: 10.1182/blood-2011-02-339911. [DOI] [PubMed] [Google Scholar]
- 46.Buckley CD. Why does chronic inflammation persist: An unexpected role for fibroblasts. Immunol Lett. 2011;138:12–14. doi: 10.1016/j.imlet.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran CN, Davis MJ, Tesmer LA, Endres JL, Motyl CD, Smuda C, Somers EC, Chung KC, Urquhart AG, Lundy SK, Kovats S, Fox DA. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. 2007;56:1497–1506. doi: 10.1002/art.22573. [DOI] [PubMed] [Google Scholar]
- 48.Pilling D, Akbar AN, Girdlestone J, Orteu CH, Borthwick NJ, Amft N, Scheel-Toellner D, Buckley CD, Salmon M. Interferon-beta mediates stromal cell rescue of T cells from apoptosis. Eur J Immunol. 1999;29:1041–1050. doi: 10.1002/(SICI)1521-4141(199903)29:03<1041::AID-IMMU1041>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 49.Kiener HP, Watts GF, Cui Y, Wright J, Thornhill TS, Skold M, Behar SM, Niederreiter B, Lu J, Cernadas M, Coyle AJ, Sims GP, Smolen J, Warman ML, Brenner MB, Lee DM. Synovial fibroblasts self-direct multicellular lining architecture and synthetic function in three-dimensional organ culture. Arthritis Rheum. 2010;62:742–752. doi: 10.1002/art.27285. [DOI] [PubMed] [Google Scholar]
- 50.Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M, Brenner MB. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–1010. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- 51.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S, Natoli G. PNAS Plus: Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci USA. 2012:1–10. doi: 10.1073/pnas.1121131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H, Brown J, Garcia CA, Tang Y, Benakanakere MR, Greenway T, Alard P, Kinane DF, Martin M. The role of glycogen synthase kinase 3 in regulating IFN-beta-mediated IL-10 production. J Immunol. 2011;186:675–684. doi: 10.4049/jimmunol.1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34:81–89. doi: 10.1016/j.it.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 55.Hamilton JA, Tak PP. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum. 2009;60:1210–1221. doi: 10.1002/art.24505. [DOI] [PubMed] [Google Scholar]
- 56.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 58.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244–247. doi: 10.1016/s0140-6736(89)90430-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.