

Abstract
Abnormal autophagy has become a central thread linking neurodegenerative diseases, particularly of the motor neuron. One such disease is spinal muscular atrophy (SMA), a genetic neuromuscular disorder caused by mutations in the SMN1 gene resulting in low levels of Survival Motor Neuron (SMN) protein. Despite knowing the causal protein, the exact intracellular processes that are involved in the selective loss of motor neurons remains unclear. Autophagy induction can be helpful or harmful depending on the situation, and we sought to understand the state of the autophagic response in SMA. We show that cell culture and animal models demonstrate induction of autophagy accompanied by attenuated autophagic flux, resulting in the accumulation of autophagosomes and their associated cargo. Expression of the SMN-binding protein a-COP, a known modulator of autophagic flux, can ameliorate this autophagic traffic jam.
Keywords: Spinal Muscular Atrophy, autophagy, motor neuron, Survival Motor Neuron
Introduction
Spinal Muscular Atrophy (SMA) is a devastating inherited disorder caused by the loss of the SMN1 gene responsible for production of the Survival Motor Neuron (SMN) protein (1) (2). Low levels of this protein lead to dysfunction and degeneration of alpha motor neurons, muscle wasting and eventually death in the more severe forms. Understanding the processes by which these motor neurons fail and the timing of that failure increases our understanding of the disease state. Such information may educate therapeutic decisions. One cellular process, macroautophagy, has come to the forefront in motor neuron diseases as a common pathology present in degenerating motor neurons. Further study of the onset of autophagy and the dynamics of the autophagic pathway in SMA will allow for the potential therapeutic intervention, as pharmacological manipulation of this system is well understood.
Macroautophagy, hereafter referred to as autophagy, is responsible for the degradation of cytoplasmic proteins and organelles by incorporation into a double-membrane vesicle that is transported to the lysosome for degradation (3). This process appears to be dysregulated in a number of neurodegenerative conditions; typically becoming hyper-activated in an attempt to preserve cellular function or remove damaged cellular material such as at the axon terminal following injury. In other cases such as Alzheimer’s disease, Huntington’s disease or aging, the normal autophagic flux slows, resulting in a failure to clear the cell of obsolete proteins and organelles (4). Tissue-specific knockout experiments in mice have highlighted the importance of this pathway in neurons, demonstrating that loss of critical genes involved in the execution of the autophagic pathway in the central nervous system results in profound neurodegenerative disease (5, 6). Induction of autophagy has been reported in numerous models of neurodegenerative disease, and may be a component of the cellular response to depleted SMN levels (7). A recent report describes the accumulation of autophagosomes in motor neuron cultures following depletion of SMN and in motor neurons cultures from SMA mice (8). However, this study did not examine the dynamics of the autophagic pathway in SMA model cells. Previous work in cultured cells has shown that depletion of the SMN binding partner α-COP impaired autophagy resulting in an accumulation of autophagosomes that failed to complete fusion with the lysosome (9). Autophagosome accumulation can be a hallmark of induction of autophagy in response to general cellular stress but can also result from ineffective intracellular trafficking and slow delivery of autophagic vesicles to the lysosomal compartment. In fact, a recent publication demonstrated that SMN-depleted NSC-34 cells, a motor neuron-like cell line, showed impaired microtubule assembly and accumulation of mitochondria, both signs that normal intracellular trafficking is disturbed in the absence of SMN (10).
In this report, we demonstrate that autophagic vesicles are present both in an inducible NSC-34 cell culture model of SMA and in fibroblasts isolated from SMA patients. Using live-cell imaging, we find that cells display a reduced autophagic flux when challenged following SMN depletion. A nearly identical phenotype is seen in SH-SY5Y cells following knockdown of the SMN binding partner α-COP., and expression of α-COP reduced the number of autophagosomes in SMN-depleted NSC-34 cells. Finally, analysis of spinal cord lysates from a severe mouse model of SMA shows accumulation of LC3-II protein as well as an increase in p62 protein levels, indicating that dysregulated autophagy is a component of SMA disease pathology in these animals.
Materials and Methods
Cell Culture
NSC-34 clone 4#56 (dox-inducible SMN shRNA) was described previously (Custer et al, 2013). Cells were maintained in DMEM with 10% Tet-system FBS and Pen/Strep. SMN knockdown was induced with 2ug/ml doxycycline for 72hrs. Inducible alpha-COP knockdown SH-SY5Y cells were described previously (11) and were maintained with DMEM/F12 with 15% Tet-system FBS and Pen/Strep. Alpha-COP knockdown was induced with 2ug/ml doxycycline for 72 hrs. Tert-immortalized SMA patient fibroblasts (3814T and 3813T (13)) were maintained in SensiCell DMEM/F12 with 20% FBS supplemented with Penn/Strep and 0.1ug/ml puromycin.
Lentiviral expression of FLAG-hSMN
The FLAG-SMN lentiviral vector was a gift from Dr. Xue-Jun Li (12). Virus was produced in HEK-293TT cells co-transfected with Pax2 and MD2.G and supernatant was harvested at 48 and 72 hours. To concentrate the virus, supernatant was centrifuged at 20,000 rpm on a 20% sucrose cushion and resuspended in 1/10 the volume DMEM/10% FBS. Virus was immediately added to NSC-34 cell cultures and cells were lysed after 48 hours for Western blot analysis.
Immunofluorescence
eGFP-tagged rat LC3 was a gift from Dr T Yoshimori (13). The tandem-tagged LC3 flux reporter was a gift from Dr. Paul Taylor(14). All cells were transfected using Lipofectamine 2000 (Invitrogen) at a ratio of 1:3. For staining of endogenous LC3 and p62, cultures were grown on glass coverslips coated with either PDL (NSC-34 and SH-SY5Y) or collagen (SMA fibroblasts). Cultures were fixed in 4% paraformaldehyde for 10 minutes, then blocked/permeabilized in PBS with 5% normal goat serum and 1% TritonX-100 for 30 minutes. Rabbit polyclonal LC3 and p62 antibodies were purchases from Sigma and used at 1:300 and 1:1000 respectively at 4°C overnight. Anti-rabbit Alexa-488 was used at room temperature for 1 hour at 1:1000. Coverslips were sealed with VectaShield hard-set mounting media with DAPI to visualize nuclei. For LC3 puncta counts, images were captured at 100X, and the number of puncta per cell was determined in 50 cells per experimental condition from 3 biological replicates for a total n=150 per condition using the GFP-LC3 macro for ImageJ (created by Ruben Dagda). Puncta per cell were compared by two-tailed t-test. The experiment was repeated in triplicate with SMA patient fibroblasts (3813T) compared to cells from an unaffected carrier (3814T) For live-cell imaging, cells were plated onto glass-bottom 35 mm dishes. Following 48 hours of SMN knockdown, cells were transfected with the tandem-tagged LC3 using Lipofectamine 2000. 20 hours post-transfection, cultures were treated with rapamycin (0.5ug/ml) and switched to live-cell imaging medium (5X stock pH 7.4 750 mM NaCl, 100 mM HEPES, 5 mM CaCl2, 25 mM KCl, 5 mM Mg. On the day of use, this stock was diluted to 1X, supplemented with 95 mg sucrose and 95 mg of albumin and sterile filtered. This experiment was performed on three separate days with three different cultures, and a total of 45 controls and 52 SMN-depleted cells were imaged using an Olympus FV1000 MPE with a Spectra-Physics 2-photon MaiTai DeepSee laser, and the number of red and yellow puncta were counted. The percentage of yellow puncta in control versus SMN-depleted cells was compared by one-tailed t-test. For α-COP rescue experiments, NSC-34 cells were transfected with mCherry-tagged α-COP (a gift from Dr. Wilfried Rossoll) and LC3-GFP and the number of puncta per cell was quantified from three replicates.
Western Blotting
Cells and tissues were lysed in 8M urea lysis buffer (150 mM NaCl, 50 mM Tris, 1% NP40, 8M Urea supplemented with EDTA-free SigmaFast protease inhibitor cocktail). 20ug total protein per lane was run on 4–15% TGX gradient gels (Biorad) and transferred to PVDF membranes. Rabbit polyclonal p62, and LC3 antibodies (Sigma) were used at 1:3000 and 1:10,000 respectively. Rabbit polyclonal SMN antibody (SantaCruz) was used at 1:1000. Mouse monoclonal FLAG (M2) antibody and mouse monoclonal tubulin and actin antibodies (Sigma) were used at 1:10,000 or 1:5000.
SMA model mice
SMA model mice (Jax stock number 005058) were bred following the scheme outlined by the Didonato lab to produce litters that were 50% severe SMA and 50% phenotypically normal (15). SMA pups were euthanized at postnatal day 7 and the spinal cords were removed and rapidly homogenized in ice-cold 8M-urea lysis buffer. For western blot analysis of p62, SMN and LC3 in PND7 animals, spinal cords were removed from n=6 normal and n=5 SMA pups from two separate litters.
Results
SMN depleted NSC-34 cells accumulate autophagic puncta
We selected the motor neuron-like hybrid cell line NSC-34 to model cellular pathology in SMA. As we previously reported, we used a clonal line (NSC-34 4#56) expressing both the reverse Tet transactivator and a Tet-responsive construct containing shRNA specific to murine SMN (16). Following the addition of doxycycline, there is a robust knockdown of SMN to an average of 47.4 +/− 10.9% of normal within 48 hours (Figure 1c). The cell line quickly recovers normal levels of SMN protein following removal of the doxycycline. For comparison and to determine any off-target effects of doxycycline, we maintain the NSC-34 clone #4 which expresses only the rTta plasmid but not the shRNA. This cell line is hereafter referred to as #4. These tools allowed us to perform tightly controlled experiments to analyze the consequences of SMN depletion. When viewed with light microscopy, we observed that following SMN depletion, these cells became heavily vacuolated compared to control cultures (not shown), and we hypothesized that the cells were undergoing autophagy. To evaluate this, we transfected doxycycline-treated and control cultures with GFP-LC3. Prior to incorporation into autophagosomes, GFP-LC3 displays diffuse cytoplasmic staining, however after LC3I is lipidated to form LC3II and is incorporated in the autophagic vesicle membrane, it appears as discrete puncta. Following fixation and fluorescent microscopy, the number of puncta per cell was determined using the imageJ GFP-LC3 macro that was created and characterized for the quantitative assessment of autophagic puncta in fluorescent micrographs (17) (18). We could clearly visualize that SMN-depleted cultures had an increase in GFP-LC3-positive puncta compared to control cultures in which LC3 staining was mostly diffuse (Figure 1A, quantified in 1B). Puncta counts from 3 separate experiments showed a significant increase in the number of LC3 puncta per cell in SMN-depleted cultures by student’s t-test (54 +/− 14 puncta per cell compared to 9 +/− 6 in control cultures, p<0.01). These changes are specific to SMN knockdown as doxycycline-treated cultures from NSC-34 clone #4, which expresses the rTtA but not the SMN shRNA, show no significant increase in the number of GFP positive puncta per cell (Figure 1b). LC3 conversion can also be visualized by Western blot as LC3-II migrates faster than LC3I. Western blots of doxycycline-treated cultures show increased LC3-II compared to untreated controls. These levels return to normal upon removal of doxycycline and resumption of normal SMN protein production (Figure 1c). Quantification of western blots from 3 separate knockdowns followed by a 48 hour doxycycline washout shows that on average, p62 increased to 2.4 fold +/− 0.19 compared to controls (p<0.01 by student’s t-test) and LC3-II increased to 1.46 fold +/− 0.08 compared to controls (p<0.05) (Figure 1d).
Figure 1. SMN knockdown in NSC-34 cells induces autophagy.
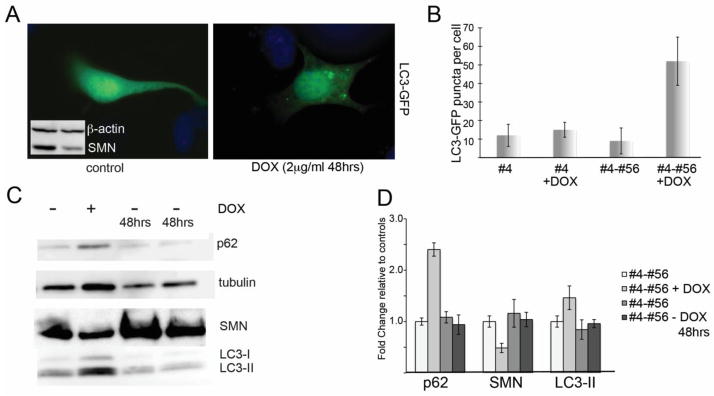
Clonal NSC-34 cells expressing a doxycycline-inducible shRNA against murine SMN (clone #4-56) were treated with doxycycline (2ug/ml) for 48 hours to deplete SMN levels to 47.4 +/− 10.9% relative to controls (inset in A shows Western blot of SMN levels following 48hr knockdown) and then transfected with GFP-LC3 to visualize autophagosomes. A) Control cultures show mostly diffuse LC3-GFP fluorescence with very few autophagic puncta per cell compared with Dox-treated cultures, which accumulate numerous GFP-positive autophagic puncta. B) Quantification of the number of LC3-GFP puncta per cell shows a that SMN-depleted cells have a statistically significant increase in puncta by Student’s t-test compared to control cultures (p<0.05). No increase in autophagic puncta was observed when cells from clone #4, which express only the rTta plasmid, were treated with doxycycline. C) Western blotting for LC3 shows that following SMN knockdown, the ration of LC3-II/LC3-I increases. Removal of doxycycline for 48 hrs allows SMN levels and LC3 ratios to return to normal. D) Quantitative analysis of repeated Western blots shows a significant increase in LC3-II protein following SMN depletion (p<0.05 by Student’s t-test).
Reduced autophagic flux in SMN-depleted NSC-34 cells
The accumulation of LC3 positive puncta and the increase in LC3II protein indicate induction of autophagy. However, we were interested in whether the reports of compromised microtubule dynamics following knockdown of SMN would permit proper progression through the autophagic pathway (10). LC3-II can also accumulate in cells where the progression through the autophagic pathway and eventual fusion with the lysosome is compromised. To distinguish between increase in autophagosome production and diminished autophagic flux, we infected the cultures with retrovirus expressing dual-tagged mRFP-LC3-GFP (19). This vector takes advantage of the fact that GFP fluorescence is quenched in the acidic lysosomal compartment to separate early autophagosomes, which have both red and green fluorescence and thus appear yellow, and autophagosome that have successfully fused with the lysosome and appear red. As basal autophagy in the control NSC-34 #4-#56 cultures is relatively low, all cultures were treated with the mTOR antagonist rapamycin to induce maximal levels of autophagosome production. Using live-cell confocal imaging, we observed that rapamycin-treated SMN knockdown cultures contained an increased percentage of yellow puncta (55 +/− 14% compared to 27 +/− 10% in control, p=0.027), indicating a failure to properly fuse with the lysosome (Figure 2A, quantified in 2B). These data support our conclusion that altered microtubule dynamics result in reduced autophagic flux in SMN-depleted NSC-34 cells.
Figure 2. SMN depletion reduces autophagic flux.

NSC-34 #4-#56 cells were treated with doxycycline for 48 hrs to induce SMN knockdown, and then transfected with mCherry-eGFP-LC3. Control and SMN-depleted cultures were treated with rapamycin to induce autophagy and live-cell confocal micrographs were captured to visualize total autophagosomes (yellow) compared to autophagosomes that had successfully fused with the lysosome (red). A) SMN knockdown cells display significantly increased number of yellow puncta (merged image) compared to controls (p=0.027 by Student’s t-test), indicating a reduced rate of autophagic flux (quantified in B). C) Western blot analysis demonstrates increased p62 and LC3-II, which is restored to normal by infection with lentivirus expressing FLAG-hSMN. D) Quantitative analysis of repeated Western blots shows a statistically significant increase in p62 and LC3-II protein levels by Student’s t-test following SMN knockdown. E) Staining for endogenous p62 in SMN-depleted cultures shows an increase in the number of cells with bright p62 puncta (quantified in F, p<0.05 by Student’s t-test).
Another method of examining autophagic flux is to assay for levels of the protein p62/SQSTM1, which is degraded exclusively by autophagy (20). Accumulation of p62 indicates reduced autophagic flux. Immunofluorescent labeling of NSC-34 #4-#56 cultures revealed a statistically significant increase in the number of cells with p62-positive puncta following SMN depletion (Figure 2E, quantified in 2F, p<0.05 by Student’s t-test). Western blot analysis shows an increase in p62 protein levels in doxycycline-treated NSC-45 #4-#56 cultures accompanied by an increase in LC3-II. This profile returned to baseline when cultures were infected with lentivirus expressing FLAG-tagged human SMN (21), which is immune to the murine-specific shRNA construct in these cells (Figure 2C). D) Quantification of Western blot analysis from repeated experiments shows that p62 increased by 1.38 fold +/− 0.13 in SMN-depleted cultures compared to controls and returned to normal (1.04 fold +/− 0.14) following the addition of FLAG-hSMN. LC3-II increased by 3.35 fold +/− 0.57 in SMN-depleted cultures and rescue with FLAG-hSMN significantly reduced it to 1.82 fold +/− 0.32 compared to control cells (p<0.01 compared to doxycycline-treated cells alone).
LC3-positive puncta accumulate in SMA patient fibroblasts
To determine whether our findings were a more general result of SMN depletion and not unique to our NSC-34 model, we transfected telomerase-immortalized SMA-derived human fibroblasts (line 3813T) (22) and with LC3-GFP and compared the number of GFP-positive puncta to cells from an unaffected heterozygous parent (line 3814T). As shown in Figure 3, we observe an increase in LC3-positive puncta compared to controls (Figure 3A). Quantification of the number of GFP-positive puncta per cell from three separate fibroblast cultures show that SMA cells have a statistically significant increase in the number of puncta per cell (p<0.01 by Student’s t-test). Western blot analysis from these cells shows an increase in p62 and LC3-II proteins, consistent with the poor flux seen in our NSC-34 cell model although these cells were not amenable to live-cell imaging with the flux reporter construct. Quantification of repeated Western blots shows that p62 levels increase to 1.5 fold +/− 0.13 compared to controls and LC3-II increased to 2.32 fold +/− 0.59 (p<0.05 for both by Student’s t-test).
Figure 3. Autophagy is disturbed in SMA patient fibroblasts.

SMA-derived fibroblasts (3813T) were compared to fibroblasts from an unaffected parent (3814T). A) Transfection with LC3-GFP shows increased GFP-positive autophagic puncta per cell in SMA cells compared to controls (quantified in B). C) Western blot analysis reveals a profile similar to SMN-depleted NSC-34 cells with accumulation of both p62 and LC3-II. D) Quantitative analysis of repeated Western blots shows the increases in p62 and LC3-II in SMA fibroblasts are statistically significant compared by Student’s t-test (p<0.05).
Markers of autophagy in a severe mouse model of SMA
We moved on from these cell culture models to examine the profile of autophagy markers in the so-called “Taiwanese” mouse model of SMA. These animals carry the ‘Hung’ targeted deletion of murine SMN as well as a human SMN2 transgene (23). In this model, animals that are SMN null and heterozygous for SMN2 die around 12 days with profound muscle wasting, weakness and loss of righting reflex. We chose to examine autophagy markers at postnatal day 7. At this age, the SMA pups are significantly smaller than their phenotypically normal siblings (2.5g +/− 0.7 compared to 3.8g +/− 0.5). The animals are still mobile but show a delayed righting reflex compared to their normal littermates (not shown). Their relative mobility insures that they are still gaining access to milk, preventing any potential autophagy induction due to poor nursing following the onset of major motor coordination losses. Similar to what we detect in the NSC-34 cells, spinal cord lysates from SMA pups show increases in p62 and in LC3-II relative to LC3-1 by Western blotting (Figure 5A). Quantification of Western analysis from multiple spinal cords isolated from two separate litters shows that in SMA mice p62 increase 2.09 fold +/− 0.41 compared to heterozygous littermates and LC3-II increased to 2.21 fold +/− 0.25 (Figure 5B, p<0.01 by Student’s t-test for both p62 and LC3-II). Interestingly, we found that this was restricted to the spinal cord. Muscle had a very modest increase in the ratio of LC3-II/I but no change in the p62 levels and brain showed no change in either marker (not shown). These results support our choice of the early time point, as we would expect starvation-induced autophagy to be present in all three tissues if it were a confounding factor at this age.
Figure 5. α-COP modulates autophagic flux.
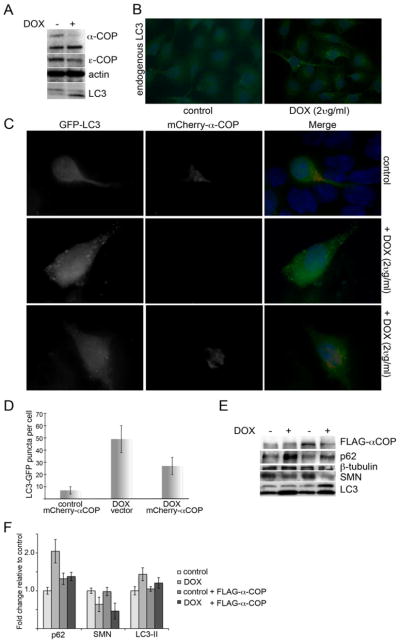
A) Western blotting after 48 hours of doxycycline treatment reveals robust reduction of α-COP and εCOP and an increase in LC3-II/I. B) Staining for endogenous LC3 in SH-SY5Y cells after doxycycline-induced α-COP knockdown shows an increase in the number of cells with LC3-positive puncta compared to untreated control cultures. C) Transfection of mCherry α-COP reduced the number of LC3-GFP puncta per cell in SMN knockdown cultures compared to SMN knockdown alone (quantified in D, p<0.05 by Student’s t-test). E) Western blot analysis of SMN-depleted NSC-34 cultures with and without FLAG-α-COP shows a reduction in p62 accumulation and LC3II/I ratios to normal levels. F) Quantitation of repeated Western blots shows that FLAG-α-COP restores p62 and LC3-ii protein levels to normal (1.38 fold +/− 0.11 for p62 relative to untreated controls and 1.21 +/− 0.14 for LC3-II).
The SMN binding partner α-COP regulates autophagy in SMN knockdown cells
We have shown that the COPI vesicle protein α-COP is an SMN binding protein and is able to stimulate neurite outgrowth in SMN-depleted NSC-34 cells (11, 16). Loss of COPI components (α, β or β′) in HeLa cells stably expressing the LC3-GFP autophagy reporter resulted in accumulation of GFP positive puncta and failure to properly fuse with the lysosome (9). We previously published an inducible α-COP knockdown line of SH-SY5Y cells (11). Following induction of α-COP knockdown with doxycycline (2ug/ml), we confirm a robust decrease in the levels of α-COP protein and a modest decrease in ε-COP, most likely due to destabilization of the α/β/ε subcomplex (Figure 5A). Immunofluorescent staining for endogenous LC3 in these cultures shows a significant increase in the number of cells with LC3 puncta (Figure 5B). Western blots reveal an increase in the accumulation of LC3-II compared to LC3-1 in knockdown cultures consistent with the immunofluorescent findings (Figure 5A). We previously reported that over-expression of α-COP rescues neurite outgrowth in SMN-depleted NSC-34 cells (16). To determine whether over-expression of α-COP would restore normal autophagic trafficking, we co-transfected mCherry-tagged α-COP with LC3-GFP into NSC-34 4#56 cultures with and without doxycycline. Immunofluorescence analysis shows that cells containing α-COP have significantly fewer GFP-positive puncta than SMN-depleted cells (28 +/− 9 puncta per cell compared to 49 +/− 11 with doxycycline alone, p=0.033 by Student’s t-test), but do not fully restore levels to normal (Figure 5C, D). Western blotting shows that expression of α-COP does not interfere with the SMN knockdown, but does shift the levels of p62 and the ratio of LC3-II/I towards normal although it does not completely rescue (Figure 5E). Quantitation of multiple Western blots showed that expression of FLAG-α-COP significantly reduces the increase of p62 from 2.04 fold +/− 0.31 in cells treated with doxycycline alone to 1.38 fold +/− 0.11 (p=0.026 by Student’s t-test).
Discussion
The autophagic pathway is an important part of neuronal homeostasis that provides cells with an opportunity to restore their cellular environment by removing aged protein and organelles. Under routine culture conditions, autophagy is constitutively active in neuronal cells to maintain a healthy micro-environment (24). Perturbations in the smooth progression through this pathway may accelerate accumulation of autophagosomes and their associated cargoes. The importance of proper autophagic clearance in motor neuron diseases has recently been highlighted by work in amyotrophic lateral sclerosis (ALS). Autophagosomes were detected in spinal cords of sporadic ALS patients (25) as well as in mouse models of hereditary ALS caused by mutations in superoxide dismutase 1 (SOD1) (26). In mice, these accumulations begin long before the onset of measurable symptoms (27). Evidence in NSC-34 cells indicated that SOD1 aggregates could be cleared by activation of autophagy, giving rise to the idea that the motor neurons could be rescued by forced clearance of SOD1 (28).
Whether autophagy is a viable therapeutic target in motor neuron disease remains unclear. The relatively well-tolerated drugs rapamycin and hydroxychloroquine have been shown to induce and inhibit autophagy respectively, but whether pharmacological manipulation of autophagy in SMA would be beneficial is unclear. In mouse models of ALS, genetic alterations that modulate autophagy lead to clearing of SOD1 aggregates and delay of symptom onset (29, 30). Similarly, expression of BCL-XL rescued motor neurons from SMA mice, possibly by interactions with Beclin-1 (31). Unfortunately, ALS mouse models treated with rapamycin actually progressed much more rapidly, most likely due to an intracellular traffic jam created by forced induction of autophagy in the face of reduced autophagic flux, which would account for the increased levels of p62 protein that were detected in these animals (32). In a very small pilot study, mice from the SMN delta7 model (33) were treated with rapamycin beginning at postnatal day 1. These animals died before vehicle treated siblings (C. Lorson and E. Osman, personal communication). Additionally, although it has been reported that SMN is normally cleared by the proteasome following ubiquitination (34), in our HEK293 cell based SMN-luciferase reporter assay we find a decrease in SMN2-derived full-length SMN protein in cells treated with rapamycin (not shown). Therefore, we may be seeking an elusive autophagic “sweet spot” that encourages proper cellular clearance without depleting the minimal amounts of SMN protein provided by the SMN2 gene.
Recent advances have developed a number of rapamycin analogues with improved bioavailability and solubility profiles (35, 36). These new compounds are able to specifically inhibit both mTORC1 and 2 and produce a more robust activation of the autophagic pathway. These newer compounds may be able to stimulate sufficient levels of autophagy to overcome the apparent deficits in flux and allow for motor neurons to clear accumulated and potentially toxic damaged organelles as well as use these recycled cellular components as an additional energy source in the face of SMA-induced stress. Another inducer of autophagy is sodium valproate, which acts on the autophagy pathway by reducing inositol and IP3 levels (37). Although the results in clinical trials have been inconclusive, valproic acid is known to increase survival in SMA mouse models (38).
The more we learn about the nature of the autophagic dysregulation in SMA, its onset and pathology, the more we can gauge whether the pharmacological tools we have available for modulating autophagy in the clinical setting may have beneficial effects.
Figure 4. Autophagy in a mouse model of SMA.
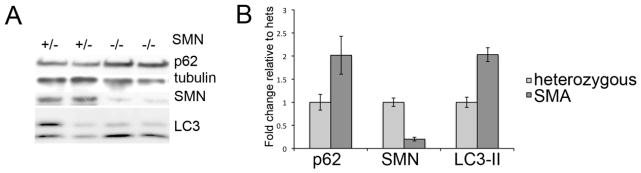
A) Western blot analysis of spinal cords from the severely affected Taiwanese SMA mouse model taken at post-natal day 7 shows accumulation of p62 and an increase in LC3-II/I ratios. B) Quantitative analysis of repeated Western blots from multiple SMA litters shows a statistically significant increase in both p62 and LC3-II protein levels in SMA pups compared to heterozygous littermates (p<0.01 by Student’s t-test).
Acknowledgments
Many thanks to Dr. Neil Cashman (Brain Research Center, University of British Columbia), Dr. T. Yoshimori (Research Institute for Microbial Disease, Osaka University), Dr. J Paul Taylor (St. Jude Children’s Research Hospital) Dr. Xue-Jun Li (University of Connecticut) and Wilfried Rossoll (Emory University) for providing reagents. Live-cell imaging was performed with assistance from the Indiana Center for Biological Microscopy. This work was supported by funding from Families with SMA (FSMA.org) (SKC), and the National Institutes of Health (NIH/NINDS R01NS082284) (EJA).
Abbreviations
- SMA
Spinal Muscular Atrophy
- SMN
Survival Motor Neuron
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- GFP
Green Fluorescent Protein
- RFP
Red Fluorescent Protein
- ALS
Amyotrophic Lateral Sclerosis
- α-COP
Coatomer Vesicle Protein alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clermont O, Burlet P, Lefebvre S, Burglen L, Munnich A, Melki J. SMN gene deletions in adult-onset spinal muscular atrophy. Lancet. 1995;346:1712–1713. doi: 10.1016/s0140-6736(95)92881-2. [DOI] [PubMed] [Google Scholar]
- 2.Rodrigues NR, Owen N, Talbot K, Ignatius J, Dubowitz V, Davies KE. Deletions in the survival motor neuron gene on 5q13 in autosomal recessive spinal muscular atrophy. Hum Mol Genet. 1995;4:631–634. doi: 10.1093/hmg/4.4.631. [DOI] [PubMed] [Google Scholar]
- 3.Moreau K, Luo S, Rubinsztein DC. Cytoprotective roles for autophagy. Curr Opin Cell Biol. 2010;22:206–211. doi: 10.1016/j.ceb.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menzies FM, Ravikumar B, Rubinsztein DC. Protective roles for induction of autophagy in multiple proteinopathies. Autophagy. 2006;2:224–225. doi: 10.4161/auto.2696. [DOI] [PubMed] [Google Scholar]
- 5.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 6.Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy. 2007;3:591–596. doi: 10.4161/auto.4964. [DOI] [PubMed] [Google Scholar]
- 7.Son JH, Shim JH, Kim KH, Ha JY, Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med. 2012;44:89–98. doi: 10.3858/emm.2012.44.2.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcera A, Bahi N, Periyakaruppiah A, Arumugam S, Soler RM. Survival motor neuron protein reduction deregulates autophagy in spinal cord motoneurons in vitro. Cell Death Dis. 2013;4:e686. doi: 10.1038/cddis.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol. 2009;185:305–321. doi: 10.1083/jcb.200810098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen HL, Lin YT, Ting CH, Lin-Chao S, Li H, Hsieh-Li HM. Stathmin, a microtubule-destabilizing protein, is dysregulated in spinal muscular atrophy. Hum Mol Genet. 2010;19:1766–1778. doi: 10.1093/hmg/ddq058. [DOI] [PubMed] [Google Scholar]
- 11.Peter CJ, Evans M, Thayanithy V, Taniguchi-Ishigaki N, Bach I, Kolpak A, Bassell GJ, Rossoll W, Lorson CL, Bao ZZ, et al. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum Mol Genet. 2011;20:1701–1711. doi: 10.1093/hmg/ddr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang ZB, Zhang X, Li XJ. Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res. 2013;23:378–393. doi: 10.1038/cr.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6:217–227. doi: 10.4161/auto.6.2.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gogliotti RG, Hammond SM, Lutz C, Didonato CJ. Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 2010;391:517–522. doi: 10.1016/j.bbrc.2009.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Custer SK, Todd AG, Singh NN, Androphy EJ. Dilysine motifs in exon 2b of SMN protein mediate binding to the COPI vesicle protein alpha-COP and neurite outgrowth in a cell culture model of spinal muscular atrophy. Hum Mol Genet. 2013;22:4043–4052. doi: 10.1093/hmg/ddt254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy. 2008;4:770–782. doi: 10.4161/auto.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra SJ, 3rd, Clark RS. Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Methods Enzymol. 2009;453:217–249. doi: 10.1016/S0076-6879(08)04011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tresse E, Giusti C, Kosta A, Luciani MF, Golstein P. Autophagy and autophagic cell death in Dictyostelium. Methods Enzymol. 2008;451:343–358. doi: 10.1016/S0076-6879(08)03223-0. [DOI] [PubMed] [Google Scholar]
- 20.Larsen KB, Lamark T, Overvatn A, Harneshaug I, Johansen T, Bjorkoy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy. 2010;6:784–793. doi: 10.4161/auto.6.6.12510. [DOI] [PubMed] [Google Scholar]
- 21.Wang ZB, Zhang X, Li XJ. Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res. 2012 doi: 10.1038/cr.2012.166. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 24.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto N, Nagai M, Ohta Y, Miyazaki K, Kurata T, Morimoto M, Murakami T, Takehisa Y, Ikeda Y, Kamiya T, et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007;1167:112–117. doi: 10.1016/j.brainres.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Zhang X, Le W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy. 2008;4:290–293. doi: 10.4161/auto.5524. [DOI] [PubMed] [Google Scholar]
- 27.Tian F, Morimoto N, Liu W, Ohta Y, Deguchi K, Miyazaki K, Abe K. In vivo optical imaging of motor neuron autophagy in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7:985–992. doi: 10.4161/auto.7.9.16012. [DOI] [PubMed] [Google Scholar]
- 28.Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, Galbiati M, Fontana E, Marino M, Carra S, et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS) Hum Mol Genet. 2010;19:3440–3456. doi: 10.1093/hmg/ddq257. [DOI] [PubMed] [Google Scholar]
- 29.Lee JK, Shin JH, Hwang SG, Gwag BJ, McKee AC, Lee J, Kowall NW, Ryu H, Lim DS, Choi EJ. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc Natl Acad Sci U S A. 2013;110:12066–12071. doi: 10.1073/pnas.1300894110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcera A, Mincheva S, Gou-Fabregas M, Caraballo-Miralles V, Llado J, Comella JX, Soler RM. A new model to study spinal muscular atrophy: neurite degeneration and cell death is counteracted by BCL-X(L) Overexpression in motoneurons. Neurobiol Dis. 2011;42:415–426. doi: 10.1016/j.nbd.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, Wang Z, Le W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7:412–425. doi: 10.4161/auto.7.4.14541. [DOI] [PubMed] [Google Scholar]
- 33.Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 34.Chang HC, Hung WC, Chuang YJ, Jong YJ. Degradation of survival motor neuron (SMN) protein is mediated via the ubiquitin/proteasome pathway. Neurochem Int. 2004;45:1107–1112. doi: 10.1016/j.neuint.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009;421:29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feldman ME, Shokat KM. New inhibitors of the PI3K-Akt-mTOR pathway: insights into mTOR signaling from a new generation of Tor Kinase Domain Inhibitors (TORKinibs) Curr Top Microbiol Immunol. 2010;347:241–262. doi: 10.1007/82_2010_64. [DOI] [PubMed] [Google Scholar]
- 37.Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai LK, Tsai MS, Ting CH, Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med (Berl) 2008;86:1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]