

Abstract
The pathophysiology of degenerative, infectious, inflammatory and traumatic diseases of the central nervous system includes a significant immune component. As to the latter, damage to the cerebral vasculature and neural cell bodies, caused by traumatic brain injury (TBI) activates innate immunity with concomitant infiltration of immunocytes into the damaged nervous system. This leads to pro-inflammatory cytokine and prostaglandin production and lost synaptic integrity and more generalized neurotoxicity. Engagement of adaptive immune responses follows including the production of antibodies and lymphocyte proliferation. These affect the tempo of disease along with tissue repair and as such provide a number of potential targets for pharmacological treatments for TBI. However, despite a large body of research, no such treatment intervention is currently available. In this review we will discuss the immune response initiated following brain injuries, drawing on knowledge gained from a broad array of experimental and clinical studies. Our discussion seeks to address potential therapeutic targets and propose ways in which the immune system can be controlled to promote neuroprotection.
Keywords: Traumatic brain injury, neuroimmunity, mononuclear phagocytes, astrocytes, neurodegeneration, inflammation
Introduction
Bridges between immunity and neurodegeneration have been widely studied in brain diseases that have infectious, degenerative, immune and traumatic origins [1–3]. Indeed, neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases (AD and PD), amyotrophic lateral sclerosis (ALS), stroke and Huntington’s disease (HD) show a significant inflammatory component that influences neuronal injuries [1]. Such linkages also prove important to the pathophysiology of acute neurological injury that occurs from traumatic brain injury (TBI) [2].
TBI has recently entered the public consciousness as a significant brain disorder. The return of wounded service members following United States military operations in Iraq and Afghanistan is now considered a major issue for military medicine [3]. Likewise, injuries leading to neurological impairments have led to long-term impairments and subsequent high-profile lawsuits against the National Football League. These were initiated by former players as a result of head injuries sustained during football games and allegedly resulted in delayed onset cognitive and motor dysfunctions [4]. Notwithstanding such recent emerging events, TBI has remained a significant and long-standing public health concern as it annually affects up to 1.7 million Americans [5].
The pathobiology of TBI is comprised of two parts. The first is the mechanical impact (primary injury) that initiates a cascade of biochemical reactions. This latter secondary injury produces a robust immune response [6] and neurodegeneration with secondary repair [7–9]. To reflect both injury types preclinical models were developed that reflect the pathophysiology of disease (Figure 1). These include focal injury (controlled cortical impact, weight-drop and others) and diffuse injury (lateral fluid percussion, Marmarou, for example) [10]. Notably, while immune based inflammatory responses were observed, no immune modulatory therapies have yielded a single therapeutic agent approved by the Food and Drug Administration [11, 12]. A review of immune system components implicated in TBI pathology that include both damage and repair are included herein. Moreover, we discuss the means available that could ameliorate neurotoxic inflammation and as such potentially improve neuronal function as well as the limitations seen in moving these from experimental model systems to humans. The discussions draw on work previously conducted with TBI models. Additionally, the designs of pre-clinical works are discussed as they can influence human translational studies [13]. To this end we seek to present a balanced vision of the role of neuroimmune pharmacology in TBI. This included noted obstacles that lay in the path of translated immune modulation to affecting positive disease outcomes.
Figure 1.
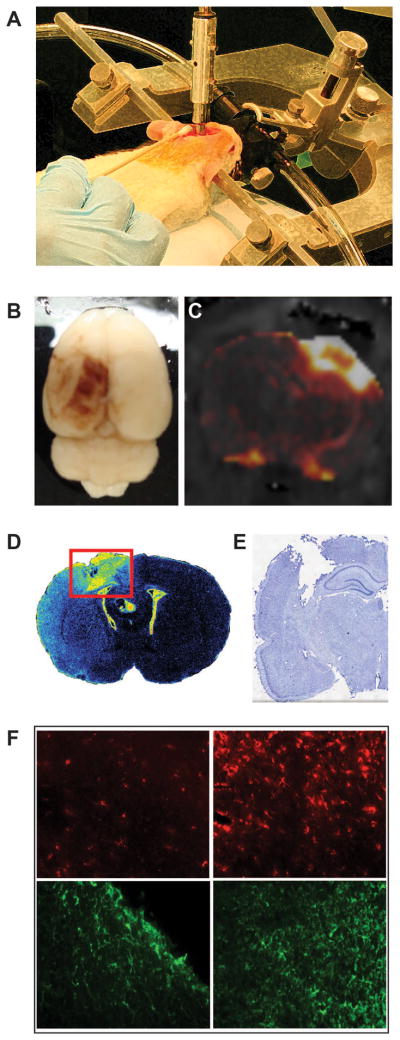
Acute and chronic pathology resulting from experimental traumatic brain injury. A. Representation of the controlled cortical impact (CCI), one of the most widely used models of experimental TBI. The injury is administered directly to the brain of an anesthetized animal with a computer-controlled, air-driven piston. This injury results in breakdown of the BBB and the extravasation of blood components into the brain parenchyma that can be observed through gross evaluation (B) and gadolinium-enhanced magnetic resonance imaging (C). A robust immune response results over a period of weeks (D) observed through binding of [3H]-PK11195 to the translocator protein (18 kDa) expressed on the outer mitochondrial membrane of activated microglia) that ultimately leads to tissue loss and cavity formation (E). (F) Further evidence of this response can be observed through immunohistochemistry. The uninjured hemisphere (left column) has relatively low expression of microglia/macrophage marker ED1 (top row) or astrocyte marker GFAP (bottom row) compared to the lesion site (right column).
Innate immunity
Neutrophils
The innate immune system is a complex network of cells and signaling mediators that serves as the first-line of defense against invading pathogens and injuries. Indeed, in the context of TBI where the initial mechanical trauma causes direct damage, it is the endogenous glia (microglia and astrocytes) that begins a cascade of immune events heralded by the release of damage-associated molecules [14, 15]. The stimulation of nonspecific immunes ensues that involves inflammation produced from brain attracted mononuclear phagocytes (MP; blood borne monocyte-derived macrophages and neutrophils) from blood while also activating resident glia. These cells produce an array of pro-inflammatory mediators, reactive oxygen species, and pro-apoptotic proteins that perpetuate neural injury. The complexity of the innate immune response following TBI was recently demonstrated by the demonstration that > 100 genes related to inflammation were significantly upregulated following controlled cortical impact (CCI) [16].
Neutrophils are the most prevalent leukocytes in blood, generated at a rate of up to 2 × 1011 cells/day [17] by the bone marrow. Upon the initiation of inflammation, they are attracted to the endothelium, activated and express integrins. In response to proinflammatory cytokines, integrins bind adhesion molecules on the surface of the neutrophil to form contact. Following this contact β2 integrins expressed on neutrophils, including Mac-1 and LAF-1, bind to intracellular adhesion molecule (ICAM)-1 and −2 to form a firm adhesion [18, 19]. Once firm adhesion is established, transendothelial migration occurs which allows entry of the neutrophils into the damaged tissue where these cells perform phagocytic and clearance functions [20].
Using intravital microscopy, leukocytes were observed to “roll” in the venular endothelium near the injury site immediately following TBI [21]. The earliest cells that enter the site of neural injury are neutrophils that appear within minutes and peak two hours later in the subarachnoid and subdural spaces as shown by immunohistochemistry [18, 22]. Infiltration into the brain parenchyma peaks 24 to 48 hours in the course of the inflammatory cascade after cortical injury [18, 23] then decreases substantially over an ensuing 7 days [24–26]. Several groups identified neutrophil infiltration in subcortical structures such as the hippocampus [22, 23], whereas others do not report infiltration anywhere but the damaged cortex [20]. However, several variables including trauma model, animal species, and injury intensity could account for such discrepancies. The recruitment of neutrophils to the area of damage is dependent on ICAM-1 [18] and the cell surface integrin Mac-1 [19]. Once in the brain tissue, neutrophils may perpetuate infiltration of other cells by secreting vascular endothelial growth factor (VEGF) [27], a mediator of blood-brain barrier (BBB) disruption and worsening edema as well as chemokines. Some evidence exists suggesting that early BBB disruption occur independent of neutrophil infiltration [28]. However, this may be a function of the marker size. A recent study using both the wet-dry method of determining brain water content and Evans blue extravasation found that induced neutropenia reduced the amount of water in the injured hemisphere while there was no significant difference in dye content [20]. Also of significance in this study, neutropenia reduced lesion size, reduced caspase-3 staining, and microglia and blood borne macrophage staining.
Few studies were conducted investigating neutrophil dynamics in clinical TBI as the cells are transient. Histopathological analyses have shown they are also few in number and present only at the earliest stages of brain injuries. Notably, in brain tissue collected from postmortem TBI, neutrophils can be regularly observed in the first hours following injury, though there is evidence that the cells can enter the brain in minutes [29]. A comprehensive study including brains from 305 post-traumatic autopsy cases found evidence of neutrophil infiltration in approximately 43% of samples from five minutes after injury [30]. These data suggest that neutrophil recruitment following injury is a significant early event in TBI pathogenesis.
Blood borne macrophages and microglia
Blood borne macrophages and microglia are a first-line innate immune cell of the brain. These cells are derived from embryonic yolk-sac macrophages that migrate to the brain upon development of the cerebral vasculature [31] and make up 10% of total brain cells [32]. Under resting conditions, these cells have elongated, ramified processes protruding from a small cell body. While largely considered to be “resting” under physiological conditions, this is not an accurate representation of their complex role in the brain. The normal function of these cells is to promote homeostasis of the brain by rapidly extending high-order processes off of the main branches to engulf metabolic by-products and cellular debris [14, 15]. These cells can be identified by immunostaining surface markers including receptors for chemokines, complement, cytokines, antibodies, pattern recognition, advanced glycation end products, lectins and adhesion molecules [33].
However, following central nervous system (CNS) damage, these cells become quickly activated by purinergic signaling through the P2Y family of receptors. This leads to cell migration to the area of injury (< 75 μm) [15]. Leukocytes migrate towards the damage while adjacent cells move further away and extend their processes. In addition to the release of ATP from damaged cells, other damage-associated molecular patterns (DAMPs) and activation pathways influence immune activation after trauma. High mobility group box-1 (HMGB1) protein was found to translocate from the neuronal nucleus to the cytoplasm in the early hours following TBI and localized to microglia at a later stage [34]. Antibodies against HMGB1, which can interfere with this translocation, have recently been tested as a therapeutic agent in preclinical models with promising results [35]. Heat shock proteins such as Hsp70 are also highly expressed in the lesion area within days following injury [36]. Likewise, S100B is known to be released at high levels following injury and has been extensively studied for use as a biomarker to predict the clinical course of TBI victims [37]. However, evidence supporting its utility as a biomarker is variable [38, 39]. These and other DAMPs activate toll-like receptors (TLR) and intracellular signaling pathways that promote an immune response [40] and have been observed in the injured cortex and subcortical structures by one day post-injury [36].
Additionally, the release of excitatory neurotransmitters, a significant pathological mechanism of TBI [41, 42] also affects cellular activation. The α7 nicotinic acetylcholine receptor (nAChR) [43, 44] and the n-methyl-D-aspartate receptor (NMDAR) [45], have similar biophysical properties and are present on microglia, but have divergent signaling pathway that affect cellular outcomes. Both receptors are ligand-gated ion channels with high calcium permeability and affecting large numbers of neuronal populations by the TBI [46–48]. Activation of the α7 nAChR on microglia induces a neuroprotective phenotype [43, 44] whereas NMDAR activation on microglia leads to an opposite, neurotoxic phenotype [45]. In fact, others have hypothesized that increased activity from the vagus nerve following TBI acts on the α7 nAChR on peripheral blood borne macrophages [49], promoting an anti-inflammatory state. This commonly leads to an increased risk of infection and poor clinical outcomes [50].
Due to parallel lineages, it is difficult to discern the presence and functions of activated microglia versus peripheral blood borne macrophages that infiltrate the brain following trauma. Jin et al. [26] identified two subsets of cells based on the degree of binding to CD45; CD45High/CD11b+ “macrophages” and CD45Low/CD11b+ “microglia.” Based on these criteria, CD45High/CD11b+ cells peaked within one day following controlled cortical impact (CCI) and remained elevated in numbers at three days before reaching control levels. Microglia, on the other hand, increase in numbers to a peak at day 7 then decrease over the next week before increasing again through 28 days [26]. This profile is similar to what investigators observed through immunohistochemical analyses of ionized calcium-binding adapter molecule 1 (Iba1). Yet additional groups of investigators have also observed this profile of prolonged Iba1+ cells to 28 days along with increased binding of [3H]-PK11195, a marker for the 18 kDa translocator protein (TPSO) present on the activated glial cell outer mitochondrial membrane [51]. Staining is especially evident in the thalamic regions at later time points [52, 53] reflective of progressive neurodegeneration. These dynamics for microglia activation are similar to those proposed by Loane and Byrnes [54], who also cite evidence that a secondary peak occurs between 30 – 60 days.
Using the TPSO ligand [11C]PK11195 for microglia positron emission tomography (PET) was used to determine long-term inflammatory responses following TBI [55]. This study enrolled 10 patients, all of whom were ≥ 11 months post-injury. The investigators reported an increase of [11C]PK11195 binding in regions away from the point of focal injury including the thalami, putamen, occipital lobes, and posterior limb of the internal capsules. Interestingly, the authors found no correlation between [11C]PK11195 binding and length of time after trauma. In fact, the subject with the highest binding values was also the subject with the longest time since the injury. However, it was shown that several aspects of the report will require substantiation before this report can be accepted as definitive proof of long-term microglia activation [55].
Nonetheless and assuming microglia are activated for years following traumatic injury, what role these cells may have in a chronic state is far from certain. It has been hypothesized that microglia and its associated neuroinflammation may play a deleterious or protective role following insult. These divergent roles are due to subsets of activated microglia with seemingly opposite functions. Microglia and infiltrating macrophages can assume neurotoxic (M1) or neuroprotective and restorative phenotypes (M2) following traumatic injury [56]. Recently, it was suggested that the brain experiences a bimodal increase in the number of microglia present after TBI [26]. This study further went on to classify the microglia phenotype out to 4 weeks post-injury. Using flow cytometry to determine cells co-expressing CD86/CD11b (M1) or CD206/CD11b (M2), Jin et al. [26] reported that the anti-inflammatory M2-like microglia were increased at seven days after injury and decreased thereafter. The proinflammatory M1 phenotype however was not increased until 28 days. This suggests a chronic neurotoxic inflammation, though the study did not determine any time points beyond 28 days. Additional investigations are required to determine if this pattern continues or if M2-like microglia resurge during later time periods.
Improvement in behavioral and histological outcomes through inhibition of microglia activation with the use of minocycline, a semisynthetic antibiotic in the tetracycline family, has been controversial. Initial work with minocycline showed promising effects in experimental stroke models [57, 58], leading to clinical trials for stroke therapy [59, 60]. Minocycline has since been studied for other neurodegenerative disease models including PD [61], AD [62, 63], HD [64] and spinal cord injury [65]. However, conflicting results on the effectiveness of minocycline as a neurotherapeutic agent were reported [66]. Likewise, preclinical TBI studies have been inconsistent between laboratories [Table 1]. Clinical trials using minocycline have also shown mixed treatment outcomes. While demonstrating safety and efficacy when used as an acute neuroprotective agent in investigations of ischemic stroke [59, 60, 67]; HD [68], PD and ALS [69] longer term clinical trials have failed to show positive therapeutic outcomes. Nonetheless, the well-known safety profile of minocycline supports its further study for TBI. To this end, a single trial to evaluate minocycline for the treatment of TBI is listed as “recruiting” at www.clinicaltrials.gov [70].
Table 1.
Summary of studies using minocycline as a treatment for TBI.
| TBI Model | Minocycline Tx | Outcome | Ref |
|---|---|---|---|
| CCI - Rat | 40 mg/kg ip daily × 5 doses, beginning post- op day 1 | ↔ Morris water maze ↔ lesion size ↔ [3H]-PK11195 autoradiography |
[171] |
| Blast - Rat | 50 mg/kg ip × 4 doses, beginning 4 h post- injury | ↓ 9 different serum markers ↓ 9 different brain markers in various regions |
[172] |
| Shohami Weight Drop - Mouse | 90 mg/kg @ 5min post- injury 45 mg/kg @ 3 h and 9 h post injury |
↑ recognition index in the Novel Object Recognition Test at 4 and 13 weeks post-TBI | [173] |
| Shohami’s Weight drop - Mouse | 45 mg/kg every 12 h | ↓ NSS score Days 3–7, ↔ thereafter to 6 weeks ↓ activated microglia/macrophages (F4/80+ cells) ↔ BrdU+ cells in SGZ or GCL ↔/↓ DCX+ cells in SGZ or GCL ↔ NeuN or GFAP labeling |
[174] |
| CCI – AQP4- wildtype and null mice | 45 mg/kg ip 12 h before TBI; 90 mg/kg ip 30 min after TBI every 12 h for 2 doses; 45 mg/kg ip daily thereafter | ↓ post-traumatic seizure phenotype ↔ Lesion size ↔ AQP4 immunoreactivity ↔ GFAP immunoreactivity ↓ CD11b+ immunoreactivity |
[175] |
| Shohami’s Weight drop - Mouse | 90 mg/kg @ 5min post- injury 45 mg/kg @ 3 h and 9 h post injury |
↑ aversion to acetic acid olfaction ↓ lesion size in olfactory bulb |
[176] |
| Shohami’s Weight drop - Mouse | 90 mg/kg @ 5min post- injury 45 mg/kg @ 3 h and 9 h post injury |
↑ sAPPα levels ↑ corpus callosum and striatum volume ratio ↓ GFAP immunoreactivity ↓ CD-11b immunoreactivity |
[177] |
| CCI - Rats | 45 mg/kg ip daily beginning 1 h post- injury | ↑ Behavior ↓ myelin loss ↔ Lesion size ↔ IL-1β production | [178] |
| Shohami’s Weight drop - Mouse | 90 mg/kg @ 5min post- injury 45 mg/kg @ 3 h and 9 h post injury |
↓ TBI-induced locomotor hyperactivity (2 doses) ↓ CD11b immunoreactivity ↓ Lesion size |
[179] |
| Shohami’s Weight drop - Mouse | Protocol 1: 45 or 90 mg/kg ip @ 5 min post- injury OR Protocol 2: Protocol 1 + 45 mg ip @ 3 h post- injury OR Protocol 3: Protocol 2 + 45 mg/kg ip @ 9 h post-injury |
Protocol 1: ↓ IL-1β expression (90 mg/kg) ↓ proMMP-9 ↔ edema Protocol 2: ↓ IL-1β expression (45 and 90 mg/kg) ↓ edema (90 mg/kg) ↔ GSSG/Gsx or GSH/Gsx Protocol 3: ↓ edema ↑ String test performance |
[180] |
| Marmarou Weight drop - Rat | 1 mg/kg IV 30 min after injury | ↑ synaptophysin expression (mRNA and protein) ↓ MMP-2 expression (mRNA and protein) and function ↓ MMP-9 expression (mRNA and protein) and function |
[181] |
| Shohami’s Weight drop - Mouse | 45 mg/kg every 12 h | DNA microarray experiment found 235 genes affected involved in cellular signaling and transduction, metabolism, chemotaxis, immune response, and growth factors. | [182] |
| Shohami’s Weight drop - Mouse | 45 mg/kg every 12 h | ↔ Neurological Severity Score ↓ errors Day 1 of Ledged beam test, ↔ Day 2–4. ↓ Lesion size Day 1, ↔ Day 4 ↔ neutrophil infiltration ↓ activated microglia/macrophages (F4/80+ cells) ↔ TUNEL+ cells – cortex |
[183] |
| Feeney’s Weight drop - Mouse | Pre: 45 mg/kg ip 12 h before TBI; 90 mg/kg ip 30 min after TBI every 12 h for 2 doses; 45 mg/kg ip daily thereafter OR Post: same paradigm without pre injection |
↑ Rotarod Performance ↓ Lesion size ↓ IL-1β |
[184] |
| Marmarou Weight drop - Rat | 1 mg/kg IV 30 min after injury | ↓ brain edema ↓ FITC-dextran extravasation ↓ AQP-4, MMP-9 ↔ HIF-1α, ZO-1 |
[185] |
Astrocytes
Originally described as “brain glue”, the astrocyte was long thought to have merely a supportive role for brain homeostasis. Now it is understood that astrocytes have broader functional role, being responsible for maintaining ionic balance, water balance, BBB integrity, and maintenance of neurotransmitter levels. A defining characteristic of these cells is the expression of glial fibrillary acidic protein (GFAP), though recent studies demonstrated that approximately 15% of the cell volume actually stains positive for GFAP [71]. The human cortex contains four morphologic subsets of GFAP cells; interlaminar astrocytes in layer 1, protoplasmic astrocytes in layers 2–6, varicose projection astrocytes in layers 5–6, and fibrous astrocytes in the white matter [72]. Rodents, on the other hand, possess only protoplasmic and fibrous astrocytes. Differences between the species also exist in the morphology of protoplasmic astrocytes with human cells being 2.55-fold larger with 10-fold more GFAP processes extending from the cell body [72]. Oberheim et al. [72] estimated that this enlarged volume allows a single astrocytic domain to cover approximately 270,000 to 2 million synapses, considering there are approximately 1094 million synapses/mm3 in the human cortex [73].
Following TBI, astrocytes become reactive which entails a phenotypic change including increased expression of GFAP, cellular enlargement and extension of its processes. While astrocytes perform critical functions in the uninjured brain, it is unknown if this reactive astrocytosis, is beneficial or detrimental to TBI pathology [74]. Some of the strongest evidence supporting a neuroprotective role for reactive astrocytes following TBI includes decreased neuronal staining with NeuN or cresyl violet that corresponded to areas lacking GFAP+ cells when reactive astrocytes were eliminated using gancyclovir administration to a transgenic mouse model that targeted the herpes simplex virus thymidine kinase gene to astrocytes using the GFAP promoter [75]. This study also found in regions lacking reactive astrocytes there was a significant increase in CD45+ cells, suggesting a significant infiltration of inflammatory cells that had a round, phagocytic morphology.
One of the primary functions of astrocytes is the uptake and control of glutamate. Regulation of extracellular glutamate is performed primarily by uptake through astrocytic EAATs GLT-1 (rat homolog of human EAAT2) and GLAST (rat homolog of human EAAT1) [76]. These two transporter subtypes are distributed widely throughout the brain [77, 78], including the hippocampus where they account for 0.32% and 1.3% (for GLAST and GLT1, respectively) of the total protein in the CA1 region [79]. Additionally, three other subtypes of glutamate transporter exist; EAAC (rat homolog of EAAT3) and EAAT4-5 complete the family of high-affinity glutamate transporters. Of these, EAAC was the only other subtype that has shown to be localized in the hippocampus, though its expression is much lower than that of GLT-1 or GLAST [80] and is thought to have minimal contribution to glutamate uptake [81]. However, evidence has been reported suggesting the presence of EAAT4 in astrocytes of the hippocampus [82, 83]. As reviewed by Floyd and Lyeth [84], the reduced ability of astrocytes to recycle glutamate through these transporters is dependent on several mechanisms that are altered during the course of TBI pathophysiology: increased energy demands from overstimulation of Na+-K+ATPase and excess glutamate uptake; astrocytic swelling due to acidosis and increased intracellular [Na+]; and astrocytic loss from apoptosis. Additionally, the expression of EAATs also undergoes significant TBI-induced changes. Decreases in expression of both GLT-1 and GLAST have been observed as early as 15 minutes in the hippocampus following CCI [85] and remained decreased through 72 hours [86]. The reduction in EAAT expression was found to coincide with a decrease in d-[3H]aspartate binding [86] suggesting an inhibited functional capacity of this transporter to remove glutamate from the synapse and therefore sustaining excitotoxicity. Evidence of this effect was seen [87] after increased neuronal damage following TBI was induced in rats who were administered antisense oligonucleotides used to lower GLT-1 expression.
Pharmacological modulation of GLT-1 has shown neuroprotective potential in several models of neurodegeneration. Rothstein et al. [88] was the first to identify GLT-1 upregulation following ceftriaxone exposure and found significant neuroprotection when used in models of ALS and ischemia. Likewise, other labs have found similar results for ischemia [89–91]. Here, ceftriaxone upregulates GLT-1 expression, presumably increasing the functional capacity of the protein [88]. This work has now been expanded to models of HIV dementia [92] and a clinical study for ALS [93]. However, Lipski et al. [89] caution that the neuroprotective effects of ceftriaxone are not broadly applicable.
Further evidence of the neuroprotective effects of the glutamate transporters were made through pharmacological studies conducted using inhibitors of glutamate transport. DL-threo-β-Benzyloxyaspartate (DL-TBOA), a non-selective, non-transportable inhibitor of GLT-1, GLAST, and EAAC1 are neurotoxic under normal conditions used during in vivo tests [94]. Recently, a report showing a neuroprotective effect of ceftriaxone in a preclinical TBI model was seen [95]. This study found that a single intravenous dose of ceftriaxone not only improved cognitive functioning but decreased edema and proinflammatory cytokine production out to at least 3 days post-injury [95]. Surprisingly, the authors found that ceftriaxone only increased GLT-1 expression at 48 hours post-injury, which is apparently at odds with other neurodegeneration models and did not correspond to the anti-edema and anti-inflammatory effects that were reported [89–91]. However, another group recently found that ceftriaxone improved GLT-1 expression at seven days following injury which corresponded to a decrease in GFAP expression and a decrease in post-traumatic seizure activity [96]. Ceftriaxone has several benefits including a well-defined safety profile obtained from its wide clinical use and low cost. Future studies should be conducted to determine its ultimate efficacy.
Immune secretory products
Cytokines are inflammatory regulators that are produced by blood-borne leukocytes, glial cells, and possibly neurons [97]. These proteins can be proinflammatory or anti-inflammatory in action and are generally classified into several families based on their receptor interactions [98; Table 2]. Many of these cytokines have been shown to have a role in TBI and have been recently reviewed [99]. We highlight a few below that are important for understanding work aimed to control immunity to mitigate neurodegenerative disease.
Table 2.
Cytokine family classification.
| Cytokine Family | Cytokines |
|---|---|
| Type I | Interleukin (IL)-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-9, IL-12, granulocyte-colony stimulating factor(G-CSF), granulocyte macrophage-colony stimulating factor (GM-CSF) |
| Type II | IL-10, IL-19, IL-20, IL-22, interferon (IFN)-α, IFN-β, IFN-ε, IFN-κ, IFN-ω, IFN-δ, IFN-τ, IFN-γ |
| Tumor necrosis factor (TNF) | TNF-α, TNF-β, Fas ligand, CD40 ligand, TNF-related apoptosis- inducing ligand |
| IL-1 | IL-1α, IL-1β, IL-18, IL-1Ra |
| Transforming growth factor (TGF)-β | Activins, inhibins, bone morphogenetic proteins, glial cell line- derived neurotrophic factor (GDNF) |
Interleukin-10 (IL-10)
IL-10 is an established anti-inflammatory cytokine with potent immunomodulatory effects. Knoblach and Faden [100] tested the effect of IL-10 using differing administration paradigms. When administered to rats as an IV injection at 30 minutes before and 1 hour after a lateral fluid percussion (LFP) model, IL-10 improved neurological recovery out to 14 days after injury. This administration strategy also decreased the amount of interleukin-1beta (IL-1β) and tumor necrosis factor alpha (TNFα) in the brain. When administered as a subcutaneous injection at 10 minutes, 1, 3, 6, 9, and 12 hours after trauma, neurological recovery was improved at 7 days, but this effect was gone at 14 days. When IL-10 was administered by intracerebroventricular injection, there was no effect. Interestingly, Kline et al. [101] found that while systemic administration of IL-10 following CCI decreased the number of neutrophils accumulating in the parenchyma, it did not improve behavioral outcomes and decreased the neuroprotective effect of hypothermia.
Granulocyte macrophage colony stimulating factor (GM-CSF)
GM-CSF is a well-established hematopoietic cell growth and differentiation factor released from macrophages, T cells, mast cells, endothelial cells, and fibroblasts, which stimulates the mobilization of hematopoietic progenitor cells. Receptors for GM-CSF have been found in both the central and peripheral immune systems [102] and neurons [103]. Both in vitro and in vivo evidence shows that GM-CSF can induce an expansion of the regulatory T cell (Treg) population that can attenuate immune activation leading to neuroprotection [104–106].
Recently, it was demonstrated that delayed administration of GM-CSF could have long-lasting neuroprotective effects. Subcutaneous injections of GM-CSF (10 μg/kg) when combined with IL-3 and administered beginning two days following a cortical stab injury and continued daily for seven days [107]. Although GM-CSF administration alone did not produce effects different from vehicle, the two cytokines together did attenuate tissue loss and improved motor function when assessed two months after injury.
Arachidonic acid metabolites
The cyclooxygenase (COX) family of enzymes, which is primarily comprised of two isoforms (COX-1 and COX-2), are responsible for the oxidation of arachidonic acid to prostaglandin (PG) G2 and the reduction of PGG2 to PGH2. PGH2 is converted through enzymatic and non-enzymatic pathways to PGD2, PGE2, PGF2α, PGI2, or thromboxane (Tx) A2. COX-1 is constitutively expressed in most tissues whereas COX-2 is inducible in response to cellular stress [108]. However, both enzymes are normally expressed in the mammalian brain [108–110]. Found mostly in glutamate neurons, COX-2-derived PGE2 has been shown to be a crucial signaling molecule for synaptic plasticity [111, 112]. Following brain injury, COX-2 expression is further induced over basal levels within three hours after injury [113–116] and COX-1+ cells accumulate around the lesion site and injured vasculature [117, 118]. Infiltration of neutrophils has been shown to be dependent, at least in part on the upregulation of these enzymes with some evidence suggesting an inhibitory role for COX-1 and a permissive role for COX-2 [119].
However, COX inhibition for the treatment of TBI has shown limited success. Multiple preclinical studies have not demonstrated an effect of COX-2 selective inhibition on behavioral and/or neurological tasks such as the Morris water maze, the Barnes maze, or neurological severity scoring [113, 114, 120] though other groups have demonstrated and improvement in outcomes [116, 121]. Biochemical and histological endpoints have similarly been variable. While decreased PGE 2 production is a consistent finding following COX-2 inhibition, some studies have found it associated with a decrease in lesion size [120] while others found no such association [114]. Other groups have found no evidence of COX-2 mediated neuroprotection as evidenced by fluoro-jade B labeling and TUNEL staining that was not significantly difference compared to control [122]. Likewise, there were no significant differences in behavioral or histological outcomes between COX-1 null or COX-2 null animals following TBI compared with their respective wildtype animals. [123, 124].
The use of NSAIDs and COX-2 inhibitors largely fell out of favor around 2004 when the COX-2 inhibitor Vioxx ® (rofecoxib) was withdrawn from the market following randomized clinical trials showing an increased relative risk of cardiovascular events following chronic daily use [125]. Other COX-2 inhibitors showed a similar increase in cardiovascular events [126]. However, studies continue showing the neuroprotective effects of nonselective COX inhibition [127] and selective COX-2 inhibition [128].
An emerging strategy is to modulate the pathways downstream of COX activity. The balance between thromboxane A2 (TXA2) and prostacyclin (PGI2) is a well-known regulatory mechanism responsible for cardiovascular health with PGI2 acting as a vasodilator as well as an inhibitor of leukocyte activation and adhesion. Following TBI, this balance is shifted towards TXA2 as evidenced by the increased production of its metabolite TXB2 [129, 130]. However, clinical studies using PGI2 administration for the treatment of severe TBI have not improved outcomes compared to placebo [131, 132]. Another strategy is directly targeting PGE2 receptors in the brain. Mammalian cells throughout the CNS constitutively express the G-protein coupled PGE2 receptor EP1. Antagonism of this receptor with the compound SC51089 was shown to be neuroprotective following middle cerebral artery occlusion [133] and in a surgical excision model of brain injury [134].
Adaptive Immunity
The adaptive immune response consists of two parts; humoral immunity and cell-mediated immunity. Humoral immunity is mediated by B-lymphocytes that are responsible for the production of antibodies following activation by an antigen. Cell-mediated immunity is primarily directed by activated memory effector T-lymphocytes or regulatory cells and can have destructive or homeostatic effects. These responses can be activated as a result of protein modifications that result from a TBI [135, 136] and lead to chronic neurodegeneration.
B cells
B-lymphocytes are produced from progenitors in the bone marrow, which enter the circulation and ultimately reside in the spleen and lymph nodes. Upon differentiation to a plasma cell following activation by a pathogen, the primary function of these cells is the production of antibodies. Evidence of B cell involvement following TBI has been little studied relative to other aspects of immunity. Cells expressing the B cell marker OX33 have only been reported 4 – 6 days following experimental injury [137]. CCL20, a chemokine that has been shown to have a role in B cell chemotaxis [138] is elevated within four hours after injury and persists out to three days following CCI [139]. However, mice lacking the Rag1 gene, leading to a deficiency of mature B and T cells, were not found to be significantly different from wild type controls in neurological functioning or evidence of histological damage as evidenced by lesion size, TUNEL staining, and staining for the neuronal marker NeuN, following a weight drop injury [140]. Rag1−/− mice were protected against aseptic cerebral injury produced by the application of a liquid nitrogen-chilled rod the intact skull for six seconds. When compared to C57BL/6 mice, Rag1−/− mice had a 50% reduction in lesion volume and a reduction in the number of apoptotic cells [141], though it is not known if this result is due to a lack of T cells, B cells, or both. Brain-reactive antibodies have been found in the serum out to one month following weight-drop injury to rats, but largely disappear by three months [142]. These antibodies primarily showed a similar pattern of labeling to anti-NeuN with weak co-localization to anti-GFAP (astrocytes).
Few clinical studies have been conducted to determine the effect of TBI on B cell populations. One such study investigated B cell (CD5+/CD19+) populations in the peripheral blood of 20 patients admitted with a severe TBI and found no significant difference when compared to healthy controls [143].
T cells
T cells are lymphocytes that have origins in the thymus and are the primary mediators of the adaptive immunity, acting in a cytotoxic, helper, or regulatory manner. Following a CCI to C57BL/6 mice Clausen et al. [24] found that a significant number of T-cells infiltrated the pericontusional cortex, but not the hippocampus or thalamus, and continued to increase in number until at least 7 days. T cells were identified by immunohistochemical staining with an anti-T-lymphocyte receptor (TcR) antibody so the phenotype of these cells is unknown. Interestingly, this same group reported a different expression profile in rats [144] which the authors suggest is an interspecies variation. In that study, TcR+ cells peaked at 24 hours and decreased thereafter. A continued presence was observed in the white matter at 72 hours and 7 days post-injury, but this was not observed in all animals.
Jin et al. [26] performed an extensive characterization of inflammatory cell dynamics following CCI, including not only the brain but the cervical lymph nodes and spleen. CD45+/CD3+ T cells were reduced in the cervical lymph nodes as early as one day following injury followed by a return to baseline until 28 days when cell numbers increased. In the spleen, T cell populations also decreased at one day following injury but remained decreased until day 21. In the brain, T cells increased to a peak at three days, but then decreased to baseline levels by day 14 before rising again out to 28 days. Although these cells were significantly increased in the brain after trauma, their numbers were fewer than neutrophils, macrophages, and microglia. Further research characterizing specific T cell subsets after injury would be of great interest.
However, investigations into the infiltration of T cells into the brain after injury are complicated with potential opposing effects. Cytotoxic T cells are historically identified by the expression of CD8, a transmembrane glycoprotein expressed on the T cell surface and acts as a co-receptor for the TCR. However, Zhang et al. [145] suggested that the majority of CD8+ cells in the rat brain parenchyma following injury are of the mononuclear phagocyte lineage rather than T cells. The authors performed double-labeling immunohistochemical studies of rat brains at various time points following trauma. While CD8+ cells were increased early following injury with a progressive increase out to 96 hours, the majority of these cells also expressed ED-1 and relatively few expressed the T cell marker W3/13.
As further evidence of the role of T cells, 1×106 CD4+ T cells were adoptively transferred into Rag1−/− mice, 24 hours before aseptic cerebral injury exacerbated the lesion size and number of apoptotic cells [141]. When T cells were further sorted into naïve or effector groups and adoptively transferred to Rag1−/− mice, the animals receiving the effector cells had a more extensive injury than did the naïve animals.
The role of attenuating immunity thereby preventing “autoimmune-like” responses is primarily performed by regulatory T cells (Treg). These cells are characteristic in their expression of the transcription factor Foxp3 [146]. Using the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication model of Parkinson’s disease, our group has previously shown that Treg can attenuate the proinflammatory effects of Th17 cells [147]. Th17 cells are a subset of helper T cells that produce IL-17 and are known for promoting cellular migration across the BBB and having a role in autoimmune diseases of the CNS such as multiple sclerosis [148]. Recent work has suggested that Treg amplify a negative feedback loop present in Th17 cells through an IL-10 dependent mechanism [149, 150]. It is possible that neurons directly influence the production of Treg through signaling of cytotoxic T lymphocyte antigen 4 and transforming growth factor (TGF)-β [97], the latter having been shown to increase following experimental [151] and clinical TBI [152]. Likewise, it has been suggested that astrocyte interactions with T cells can induce a regulatory phenotype [153].
Human studies have generally supported preclinical evidence of T cell recruitment following injury. Holmin et al. [154] performed an analysis of brain biopsy tissue collected from TBI victims at different time points post-injury. This study evaluated the percentage of patients who had immunohistochemical evidence of inflammatory cells present in the tissue sample. The authors report no evidence of CD3+, CD4+, or CD8+ T cells in brain biopsies collected within 24 hours following trauma. Comparatively, all brain samples collected 3 – 5 days following trauma had some expression of CD3+ and CD4+ cells, and 75% of the samples had CD8+ cells. While this pattern supports preclinical evidence of delayed involvement, it should be noted that the injuries did not occur in the same region for all patients and that this study included only 12 patients, four of which were in the 3 – 5 day group. A more recent study investigating 56 postmortem TBI cases found CD3+ T cells in the lesion area after 4 days post-injury and persisted for at least 26 days [29], but did not assess T cell subsets by identifying any other markers.
Harnessing the natural ability of Treg function to limit autoimmune responses may be a highly efficacious method of producing a neuroprotective response. Increasing the population and/or function of Tregs for the treatment of neurodegenerative disease is a highly promising strategy. Our group has previously demonstrated a neuroprotective role of Tregs in protection from MPTP intoxication. Adoptive transfer of Treg at 12 hours following the last MPTP injection provided over 90% protection of tyrosine hydroxylase-immunoreactive neurons of the substantia nigra [155]. Using this strategy, our own group also found that Treg responses provided neuroprotection in a model of HIV-1 encephalitis [156].
In addition to adoptive transfer strategies, our group and others are investigating the potential of GM-CSF administration, which can elicit an increased Treg response. Increased splenic Treg frequencies from mice treated with 50 μg/kg GM-CSF are neuroprotective in models of both myasthenia gravis [157] and PD [158]. Furthermore, in an acute experimental stroke model, GM-CSF administered immediately following middle cerebral artery occlusion followed by daily injections for five days decreased infarct volume, improved neurological and motor outcomes, and decreased the presence of proapoptotic mediators [159]. While this research group found a proliferative effect of GM-CSF treatment on blood mononuclear cells, others have identified Treg as the mediator of neuroprotection, exerting an effect via suppression of Iba-1+ microglia and proinflammatory cytokines by the secretion of IL-10 following middle cerebral artery occlusion [160].
Harnessing Immune Response for Neural Repair
At the interface of neuroscience, immunology, and pharmacology is the emerging field of neuroimmune pharmacology. Researchers whose work encompasses these disciplines have come to appreciate the complexity of the interaction between the three, specifically how the immune system interacts and affects the brain during both normal and pathological states. Pharmacological treatment of the diseased brain can influence how the immune system affects the brain or vice versa, sometimes leading to less than optimal outcomes. Exciting new research is constantly challenging our paradigms and hypotheses as to how these pieces fit together and what is the appropriate strategy for mitigating the devastating disease states of the nervous system.
The field has grown exponentially in recent years. Its origins are from the early 1980’s when exciting research emerged on the effect of drugs of abuse on immunological function. The field quickly gained traction with the discovery of HIV. The discipline now has an active society with a dedicated membership, a peer-reviewed journal that reports exciting new research, a textbook that defines the important concepts that serve as the basis of the discipline, and numerous academic departments and centers contain leaders who carry on important work [161]. As the field develops, more diseases of the brain have been found to have a robust immunological component, ensuring that expansion will continue.
One of the principles that governs neuroimmune pharmacology is that the brain is an immune privileged site, though this concept has evolved significantly since the first observations were reported in 1921 when it was shown that rat sarcoma did not proliferate when implanted into the mouse periphery, but did grow when implanted into the mouse brain parenchyma (reviewed in [162]). This finding has since been expanded to include viruses and bacterial lysates. It is now understood that the concept of immune privilege is relative. The brain parenchyma lacks dendritic cells (DC) which are responsible for introducing antigen to T cells in the lymph organs, though other structures in the brain such as the meninges and choroid plexus have populations of cells that express DC markers [163]. This regulation was once thought to be attributed wholly to an intact blood-brain barrier [164] but it is now understood that regulation of the immune privileged state is much more complicated. The brain hosts a robust innate immune system, consisting primarily of microglia, which is the first line of defense against infection or insult. As these cells become activated in response to the trauma, the adaptive immune response is signaled, producing autoantibodies to CNS proteins and activating T cell subsets and recruiting migration to the area of injury where they may have neurotoxic or neuroprotective actions (Figure 2).
Figure 2.

The stages of the inflammatory response following TBI. A. The person sustains a traumatic brain injury. B. Blood components such as neutrophils and monocytes pass through the damaged blood brain barrier and enter the brain parenchyma. C. The reactive phase is activated as macrophages begin to phagocytize cellular debris. Effector T cells are increased while neuroprotective regulatory T cell populations are low. Proliferation of astrocytes and microglia is present. Microglia undergo a change from a ramified resting state to an activated state where they produce inflammatory mediators such as cytokines and prostaglandins. These mediators are toxic to neurons that have already been subjected to toxic concentrations of excitatory neurotransmitters. D. The resolution phase is characterized by an increase in the production of anti-inflammatory mediators such as TGF-β, IL-10, and IL-4. An expansion of the regulatory T cell population corresponds with a change in microglia phenotype to a neuroprotective (M2) state.
Manipulation of the immune system holds enormous promise as a therapeutic strategy (Figure 3). An exciting new avenue of research has emerged in which the immune system is used to deliver neuroprotective agents to the injured brain. This possibility has been made possible through advances in polymer chemistry and nanomedicine, allowing the drug to escape normal routes of degradation and cross the BBB. Our group has previously demonstrated the utility of formulation and stabilization of protein therapeutics in polymer complexes [165–167] and have shown the utility of cell-based drug delivery for the treatment of neurological disorders. The ex vivo loading of the complex (termed nanozyme) formed with the antioxidant enzyme catalase and a cationic block copolymer into bone marrow-derived macrophages (BMM) decreased microglia activation [168]. Likewise, this nanozyme increased brain penetration over free nanozyme following MPTP or 6-hydroxydopamine intoxication [167].
Figure 3.

Immune system strategies for treatment of TBI. The immune response following a TBI is multifaceted and involves the innate and adaptive immune system. Anti-inflammatory therapies including NSAIDs, COX-2 inhibitors, and minocycline have been tried with varying degrees of success. Current strategies are focused on promoting and anti-inflammatory reaction through the use of immunomodulators, such as GM-CSF, which have shown promise in treating other neurodegenerative conditions. Figure modified from Stone DK, Reynolds AD, Mosley RL, and Gendelman HE. (2009) Innate and adaptive immunity for the pathobiology of Parkinson’s disease. Antioxidants & Redox Signaling. 11(9): 2151–2166.
However, this strategy is not without potential problems. Macrophages can become activated upon entry to the injured brain and can take on a cytotoxic phenotype. Indeed, when nanozyme-loaded BMM, free nanozyme, or BMM only were administered to MPTP-intoxicated mice, only nanozyme-loaded BMM or free nanozyme decreased microglia activation, whereas BMM-only was comparable to intoxicated animals injected with saline [168]. In light of recent evidence from us and others that Treg can infiltrate damaged areas of the brain and produce a neuroprotective response in models of stroke [160], PD [155], HIV-associated neurocognitive disorders [169], and ALS [170], Treg would make a logical choice to provide a synergistic effect if used as a drug carrier.
Conclusions
In contrast to other diseases, TBI is not acquired or slowly developed over a period of years. There are no warning signs for a victim to take notice of, and while lifestyle does have some role, it mostly occurs at random. These devastating injuries occur within milliseconds and can severely affect the survivor for the rest of his/her life. Although research has been progressing for over the past 25 years, there is still no therapy available that can offer a proven therapeutic benefit or help the victim regain lost functional ability or cognition.
The development of new pharmacological agents holds great promise for ameliorating some of the most devastating neurologic and neurodegenerative diseases, including TBI. The robust immune response that is initiated following injury provides a host of potential targets that may dampen progressive neurodegeneration. The challenge lies in selecting the potential target. Many strategies have shown great promise in the laboratory setting, only to collapse when moved to a clinical study. Many times, inhibiting one pathway only leads to more problems. We suggest another strategy: influence the body to produce an anti-inflammatory response. We hypothesize that this strategy will dampen the pro-inflammatory and neurotoxic effects of an activated immune system without artificially suppressing pathways that may be necessary for repair and/or regeneration. Another benefit of this strategy is that it may facilitate the delivery of therapeutic agents to areas of damage. By packaging therapeutic molecules or proteins in carriers and targeting normally expressed proteins on these inflammatory cells, drug can be taken up and delivered to the site where it is needed, possibly eliminating many side effects and/or dose-limiting toxicities. In closing, bridges between immunity and traumatic injury of the nervous system are constantly evolving to reveal new pathways and targets that may facilitate improvements in clinical outcomes for TBI.
Acknowledgments
The authors would like to acknowledge Mrs. Robin Taylor for her assistance with the figures and Dr. JoEllyn McMillan for her editorial comments. This work was supported by the Carol Swarts Neuroscience Research Laboratory, the Frances and Louie Blumkin Foundation, Department of Defense Grant W81XWH11-1-0700, and National Institutes of Health grants P20 GM103480, P01 DA028555, R01 NS36126, P01 NS31492, 2R01 NS034239, P01 MH64570 and P01 NS43985.
References
- 1.Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Molecular neurodegeneration. 2009;4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hellewell SC, Morganti-Kossmann MC. Guilty molecules, guilty minds? The conflicting roles of the innate immune response to traumatic brain injury. Mediators of inflammation. 2012;2012:356494. doi: 10.1155/2012/356494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoge CW, McGurk D, Thomas JL, et al. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. The New England journal of medicine. 2008 Jan 31;358(5):453–63. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- 4.NFL Concussion Lawsuits. [Internet] The Washington Times; [updated 3/20/2013; cited 2013 3/21/2013]; Available from: http://www.washingtontimes.com/footballinjuries/ [Google Scholar]
- 5.Faul M, Xu L, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- 6.Das M, Mohapatra S, Mohapatra SS. New perspectives on central and peripheral immune responses to acute traumatic brain injury. Journal of neuroinflammation. 2012;9:236. doi: 10.1186/1742-2094-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson KJ, Miller KM, Fugaccia I, Scheff SW. Regional distribution of fluoro-jade B staining in the hippocampus following traumatic brain injury. Exp Neurol. 2005 May;193(1):125–30. doi: 10.1016/j.expneurol.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 8.Hall ED, Sullivan PG, Gibson TR, et al. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma. 2005 Feb;22(2):252–65. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- 9.Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de olmos silver and fluorojade staining methods. J Neurotrauma. 2008 Mar;25(3):235–47. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- 10.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nature reviews. 2013 Feb;14(2):128–42. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narayan RK, Michel ME, Ansell B, et al. Clinical trials in head injury. J Neurotrauma. 2002 May;19(5):503–57. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marklund N, Bakshi A, Castelbuono DJ, Conte V, McIntosh TK. Evaluation of pharmacological treatment strategies in traumatic brain injury. Current pharmaceutical design. 2006;12(13):1645–80. doi: 10.2174/138161206776843340. [DOI] [PubMed] [Google Scholar]
- 13.Landis SC, Amara SG, Asadullah K, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012 Oct 11;490(7419):187–91. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nimmerjahn A, Kirchhoff F, Helmchen F. Restin microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005 May 27;308(5726):1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 15.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005 Jun;8(6):752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 16.Lagraoui M, Latoche JR, Cartwright NG, et al. Controlled cortical impact and craniotomy induce strikingly similar profiles of inflammatory gene expression, but with distinct kinetics. Frontiers in neurology. 2012;3:155. doi: 10.3389/fneur.2012.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010 Nov 24;33(5):657–70. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. Journal of leukocyte biology. 1997 Mar;61(3):279–85. doi: 10.1002/jlb.61.3.279. [DOI] [PubMed] [Google Scholar]
- 19.Clark RS, Carlos TM, Schiding JK, et al. Antibodies against Mac-1 attenuate neutrophil accumulation after traumatic brain injury in rats. J Neurotrauma. 1996 Jun;13(6):333–41. doi: 10.1089/neu.1996.13.333. [DOI] [PubMed] [Google Scholar]
- 20.Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. Journal of neuroinflammation. 2012;9:17. doi: 10.1186/1742-2094-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarzmaier SM, Kim SW, Trabold R, Plesnila N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J Neurotrauma. 2010 Jan;27(1):121–30. doi: 10.1089/neu.2009.1114. [DOI] [PubMed] [Google Scholar]
- 22.Soares HD, Hicks RR, Smith D, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci. 1995 Dec;15(12):8223–33. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Royo NC, Wahl F, Stutzmann JM. Kinetics of polymorphonuclear neutrophil infiltration after a traumatic brain injury in rat. Neuroreport. 1999 Apr 26;10(6):1363–7. doi: 10.1097/00001756-199904260-00038. [DOI] [PubMed] [Google Scholar]
- 24.Clausen F, Hanell A, Bjork M, et al. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009 Aug;30(3):385–96. doi: 10.1111/j.1460-9568.2009.06820.x. [DOI] [PubMed] [Google Scholar]
- 25.Clark RS, Schiding JK, Kaczorowski SL, Marion DW, Kochanek PM. Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J Neurotrauma. 1994 Oct;11(5):499–506. doi: 10.1089/neu.1994.11.499. [DOI] [PubMed] [Google Scholar]
- 26.Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One. 2012;7(7):e41892. doi: 10.1371/journal.pone.0041892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chodobski A, Chung I, Kozniewska E, et al. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience. 2003;122(4):853–67. doi: 10.1016/j.neuroscience.2003.08.055. [DOI] [PubMed] [Google Scholar]
- 28.Whalen MJ, Carlos TM, Kochanek PM, et al. Neutrophils do not mediate blood-brain barrier permeability early after controlled cortical impact in rats. J Neurotrauma. 1999 Jul;16(7):583–94. doi: 10.1089/neu.1999.16.583. [DOI] [PubMed] [Google Scholar]
- 29.Dressler J, Hanisch U, Kuhlisch E, Geiger KD. Neuronal and glial apoptosis in human traumatic brain injury. International journal of legal medicine. 2007 Sep;121(5):365–75. doi: 10.1007/s00414-006-0126-6. [DOI] [PubMed] [Google Scholar]
- 30.Oehmichen M, Walter T, Meissner C, Friedrich HJ. Time course of cortical hemorrhages after closed traumatic brain injury: statistical analysis of posttraumatic histomorphological alterations. J Neurotrauma. 2003 Jan;20(1):87–103. doi: 10.1089/08977150360517218. [DOI] [PubMed] [Google Scholar]
- 31.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010 Nov 5;330(6005):841–5. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013 Jan 11;339(6116):156–61. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghorpade A, Gendelman HE, Kipnis J. Macrophages, Microglia, and Dendritic Cells. In: Ikezu T, Gendelman HE, editors. Neuroimmune Pharmacology. New York, NY: Springer; 2008. pp. 89–104. [Google Scholar]
- 34.Gao TL, Yuan XT, Yang D, et al. Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. The journal of trauma and acute care surgery. 2012 Mar;72(3):643–9. doi: 10.1097/TA.0b013e31823c54a6. [DOI] [PubMed] [Google Scholar]
- 35.Okuma Y, Liu K, Wake H, et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann Neurol. 2012 Sep;72(3):373–84. doi: 10.1002/ana.23602. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Zhang ZY, Wu Y, Schluesener HJ. Immunolocalization of Toll-like receptors 2 and 4 as well as their endogenous ligand, heat shock protein 70, in rat traumatic brain injury. Neuroimmunomodulation. 2012;19(1):10–9. doi: 10.1159/000326771. [DOI] [PubMed] [Google Scholar]
- 37.Kleindienst A, Hesse F, Bullock MR, Buchfelder M. The neurotrophic protein S100B: value as a marker of brain damage and possible therapeutic implications. Progress in brain research. 2007;161:317–25. doi: 10.1016/S0079-6123(06)61022-4. [DOI] [PubMed] [Google Scholar]
- 38.Honda M, Tsuruta R, Kaneko T, et al. Serum glial fibrillary acidic protein is a highly specific biomarker for traumatic brain injury in humans compared with S-100B and neuron-specific enolase. J Trauma. 2010 Jul;69(1):104–9. doi: 10.1097/TA.0b013e3181bbd485. [DOI] [PubMed] [Google Scholar]
- 39.Metting Z, Wilczak N, Rodiger LA, Schaaf JM, van der Naalt J. GFAP and S100B in the acute phase of mild traumatic brain injury. Neurology. 2012 May 1;78(18):1428–33. doi: 10.1212/WNL.0b013e318253d5c7. [DOI] [PubMed] [Google Scholar]
- 40.Kong Y, Le Y. Toll-like receptors in inflammation of the central nervous system. International immunopharmacology. 2011 Oct;11(10):1407–14. doi: 10.1016/j.intimp.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 41.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989 May 19;244(4906):798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 42.Palmer AM, Marion DW, Botscheller ML, et al. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. Journal of neurochemistry. 1993 Dec;61(6):2015–24. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- 43.Shytle RD, Mori T, Townsend K, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. Journal of neurochemistry. 2004 Apr;89(2):337–43. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- 44.De Simone R, Ajmone-Cat M, Carnevale D, Minghetti L. Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. Journal of neuroinflammation. 2005;2(1):4. doi: 10.1186/1742-2094-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaindl AM, Degos V, Peineau S, et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol. 2012 Oct;72(4):536–49. doi: 10.1002/ana.23626. [DOI] [PubMed] [Google Scholar]
- 46.Verbois SL, Scheff SW, Pauly JR. Time-dependent changes in rat brain cholinergic receptor expression after experimental brain injury. J Neurotrauma. 2002 Dec;19(12):1569–85. doi: 10.1089/089771502762300238. [DOI] [PubMed] [Google Scholar]
- 47.Verbois SL, Sullivan PG, Scheff SW, Pauly JR. Traumatic brain injury reduces hippocampal alpha7 nicotinic cholinergic receptor binding. J Neurotrauma. 2000 Nov;17(11):1001–11. doi: 10.1089/neu.2000.17.1001. [DOI] [PubMed] [Google Scholar]
- 48.Biegon A, Fry PA, Paden CM, et al. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc Natl Acad Sci U S A. 2004 Apr 6;101(14):5117–22. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003 Jan 23;421(6921):384–8. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 50.Kox M, Pompe JC, Pickkers P, et al. Increased vagal tone accounts for the observed immune paralysis in patients with traumatic brain injury. Neurology. 2008 Feb 5;70(6):480–5. doi: 10.1212/01.wnl.0000279479.69502.3e. [DOI] [PubMed] [Google Scholar]
- 51.Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. 2012 Dec 6;225:65–75. doi: 10.1016/j.neuroscience.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Exp Neurol. 2003 Jul;182(1):87–102. doi: 10.1016/s0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- 53.Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Exp Neurol. 2000 Jan;161(1):102–14. doi: 10.1006/exnr.1999.7269. [DOI] [PubMed] [Google Scholar]
- 54.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010 Oct;7(4):366–77. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol. 2011 Sep;70(3):374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 56.Kigerl KA, Gensel JC, Ankeny DP, et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009 Oct 28;29(43):13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15769–74. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yrjanheikki J, Tikka T, Keinanen R, et al. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999 Nov 9;96(23):13496–500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lampl Y, Boaz M, Gilad R, et al. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007 Oct 2;69(14):1404–10. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- 60.Padma Srivastava MV, Bhasin A, Bhatia R, et al. Efficacy of minocycline in acute ischemic stroke: a single-blinded, placebo-controlled trial. Neurology India. 2012 Jan-Feb;60(1):23–8. doi: 10.4103/0028-3886.93584. [DOI] [PubMed] [Google Scholar]
- 61.Wu DC, Jackson-Lewis V, Vila M, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002 Mar 1;22(5):1763–71. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi Y, Kim HS, Shin KY, et al. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology. 2007 Nov;32(11):2393–404. doi: 10.1038/sj.npp.1301377. [DOI] [PubMed] [Google Scholar]
- 63.Ferretti MT, Allard S, Partridge V, Ducatenzeiler A, Cuello AC. Minocycline corrects early, pre-plaque neuroinflammation and inhibits BACE-1 in a transgenic model of Alzheimer’s disease-like amyloid pathology. Journal of neuroinflammation. 2012;9:62. doi: 10.1186/1742-2094-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Menalled LB, Patry M, Ragland N, et al. Comprehensive behavioral testing in the R6/2 mouse model of Huntington’s disease shows no benefit from CoQ10 or minocycline. PLoS One. 2010;5(3):e9793. doi: 10.1371/journal.pone.0009793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Festoff BW, Ameenuddin S, Arnold PM, et al. Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. Journal of neurochemistry. 2006 Jun;97(5):1314–26. doi: 10.1111/j.1471-4159.2006.03799.x. [DOI] [PubMed] [Google Scholar]
- 66.Diguet E, Gross CE, Tison F, Bezard E. Rise and fall of minocycline in neuroprotection: need to promote publication of negative results. Exp Neurol. 2004 Sep;189(1):1–4. doi: 10.1016/j.expneurol.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 67.Fagan SC, Waller JL, Nichols FT, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. 2010 Oct;41(10):2283–7. doi: 10.1161/STROKEAHA.110.582601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huntington Study Group DI. A futility study of minocycline in Huntington’s disease. Mov Disord. 2010 Oct 15;25(13):2219–24. doi: 10.1002/mds.23236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gordon PH, Moore DH, Miller RG, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet neurology. 2007 Dec;6(12):1045–53. doi: 10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- 70. NCT01058395. [accessed 23 March 2013];Safety and Feasibility of Minocycline in the Treatment of Traumatic Brain Injury. Available from: http://clinicaltrials.gov/ct2/show/NCT01058395.
- 71.Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002 Jan 1;22(1):183–92. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oberheim NA, Takano T, Han X, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009 Mar 11;29(10):3276–87. doi: 10.1523/JNEUROSCI.4707-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DeFelipe J, Alonso-Nanclares L, Arellano JI. Microstructure of the neocortex: comparative aspects. Journal of neurocytology. 2002 Mar-Jun;31(3–5):299–316. doi: 10.1023/a:1024130211265. [DOI] [PubMed] [Google Scholar]
- 74.Laird MD, Vender JR, Dhandapani KM. Opposing roles for reactive astrocytes following traumatic brain injury. Neuro-Signals. 2008;16(2–3):154–64. doi: 10.1159/000111560. [DOI] [PubMed] [Google Scholar]
- 75.Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006 Oct;129(Pt 10):2761–72. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- 76.Danbolt NC. Glutamate uptake. Progress in neurobiology. 2001 Sep;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 77.Rothstein JD, Martin L, Levey AI, et al. Localization of neuronal and glial glutamate transporters. Neuron. 1994 Sep;13(3):713–25. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 78.Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995 Mar;15(3 Pt 1):1835–53. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998 Nov 1;18(21):8751–7. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haugeto O, Ullensvang K, Levy LM, et al. Brain glutamate transporter proteins form homomultimers. The Journal of biological chemistry. 1996 Nov 1;271(44):27715–22. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- 81.Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996 Mar;16(3):675–86. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 82.Hu WH, Walters WM, Xia XM, Karmally SA, Bethea JR. Neuronal glutamate transporter EAAT4 is expressed in astrocytes. Glia. 2003 Oct;44(1):13–25. doi: 10.1002/glia.10268. [DOI] [PubMed] [Google Scholar]
- 83.Yi JH, Herrero R, Chen G, Hazell AS. Glutamate transporter EAAT4 is increased in hippocampal astrocytes following lateral fluid-percussion injury in the rat. Brain Res. 2007 Jun 18;1154:200–5. doi: 10.1016/j.brainres.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 84.Floyd CL, Lyeth BG. Astroglia: important mediators of traumatic brain injury. Prog Brain Res. 2007;161:61–79. doi: 10.1016/S0079-6123(06)61005-4. [DOI] [PubMed] [Google Scholar]
- 85.van Landeghem FK, Stover JF, Bechmann I, et al. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia. 2001 Sep;35(3):167–79. doi: 10.1002/glia.1082. [DOI] [PubMed] [Google Scholar]
- 86.Rao VL, Baskaya MK, Dogan A, Rothstein JD, Dempsey RJ. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. Journal of neurochemistry. 1998 May;70(5):2020–7. doi: 10.1046/j.1471-4159.1998.70052020.x. [DOI] [PubMed] [Google Scholar]
- 87.Rao VL, Dogan A, Bowen KK, Todd KG, Dempsey RJ. Antisense knockdown of the glial glutamate transporter GLT-1 exacerbates hippocampal neuronal damage following traumatic injury to rat brain. Eur J Neurosci. 2001 Jan;13(1):119–28. [PubMed] [Google Scholar]
- 88.Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005 Jan 6;433(7021):73–7. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 89.Lipski J, Wan CK, Bai JZ, et al. Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience. 2007 May 11;146(2):617–29. doi: 10.1016/j.neuroscience.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 90.Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007 Apr 18;27(16):4253–60. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chu K, Lee ST, Sinn DI, et al. Pharmacological Induction of Ischemic Tolerance by Glutamate Transporter-1 (EAAT2) Upregulation. Stroke. 2007 Jan;38(1):177–82. doi: 10.1161/01.STR.0000252091.36912.65. [DOI] [PubMed] [Google Scholar]
- 92.Rumbaugh JA, Li G, Rothstein J, Nath A. Ceftriaxone protects against the neurotoxicity of human immunodeficiency virus proteins. Journal of neurovirology. 2007 Apr;13(2):168–72. doi: 10.1080/13550280601178218. [DOI] [PubMed] [Google Scholar]
- 93.Berry JD, Shefner JM, Conwit R, et al. Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS One. 2013;8(4):e61177. doi: 10.1371/journal.pone.0061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Montiel T, Camacho A, Estrada-Sanchez AM, Massieu L. Differential effects of the substrate inhibitor l-trans-pyrrolidine-2,4-dicarboxylate (PDC) and the non-substrate inhibitor DL-threo-beta-benzyloxyaspartate (DL-TBOA) of glutamate transporters on neuronal damage and extracellular amino acid levels in rat brain in vivo. Neuroscience. 2005;133(3):667–78. doi: 10.1016/j.neuroscience.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 95.Wei J, Pan X, Pei Z, et al. The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. The journal of trauma and acute care surgery. 2012 Sep;73(3):654–60. doi: 10.1097/TA.0b013e31825133c0. [DOI] [PubMed] [Google Scholar]
- 96.Goodrich GS, Kabakov AY, Hameed MQ, et al. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter GLT-1, reduces regional gliosis, and reduces posttraumatic seizures in the rat. J Neurotrauma. 2013;30(16):1434–41. doi: 10.1089/neu.2012.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006 May;12(5):518–25. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 98.Huang Y, Erdmann N, Hexum TD, Zheng J. Cytokines and Chemokines. In: Ikezu T, Gendelman HE, editors. Neuroimmune Pharmacology. New York, NY: Springer; 2008. pp. 183–206. [Google Scholar]
- 99.Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Frontiers in neurology. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knoblach SM, Faden AI. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Exp Neurol. 1998 Sep;153(1):143–51. doi: 10.1006/exnr.1998.6877. [DOI] [PubMed] [Google Scholar]
- 101.Kline AE, Bolinger BD, Kochanek PM, et al. Acute systemic administration of interleukin-10 suppresses the beneficial effects of moderate hypothermia following traumatic brain injury in rats. Brain Res. 2002 May 24;937(1–2):22–31. doi: 10.1016/s0006-8993(02)02458-7. [DOI] [PubMed] [Google Scholar]
- 102.Hercus TR, Broughton SE, Ekert PG, et al. The GM-CSF receptor family: mechanism of activation and implications for disease. Growth Factors. 2012 Apr;30(2):63–75. doi: 10.3109/08977194.2011.649919. [DOI] [PubMed] [Google Scholar]
- 103.Ridwan S, Bauer H, Frauenknecht K, von Pein H, Sommer CJ. Distribution of granulocyte-monocyte colony-stimulating factor and its receptor alpha-subunit in the adult human brain with specific reference to Alzheimer’s disease. Journal of neural transmission. 2012 Nov;119(11):1389–406. doi: 10.1007/s00702-012-0794-y. [DOI] [PubMed] [Google Scholar]
- 104.Zou T, Satake A, Ojha P, Kambayashi T. Cellular therapies supplement: the role of granulocyte macrophage colony-stimulating factor and dendritic cells in regulatory T-cell homeostasis and expansion. Transfusion. 2011 Nov;51( Suppl 4):160S–8S. doi: 10.1111/j.1537-2995.2011.03379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sheng JR, Muthusamy T, Prabhakar BS, Meriggioli MN. GM-CSF-induced regulatory T cells selectively inhibit anti-acetylcholine receptor-specific immune responses in experimental myasthenia gravis. J Neuroimmunol. 2011 Dec 15;240–241:65–73. doi: 10.1016/j.jneuroim.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schabitz WR, Kruger C, Pitzer C, et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF) J Cereb Blood Flow Metab. 2008 Jan;28(1):29–43. doi: 10.1038/sj.jcbfm.9600496. [DOI] [PubMed] [Google Scholar]
- 107.Nishihara T, Ochi M, Sugimoto K, et al. Subcutaneous injection containing IL-3 and GM-CSF ameliorates stab wound-induced brain injury in rats. Exp Neurol. 2011 Jun;229(2):507–16. doi: 10.1016/j.expneurol.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 108.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993 Aug;11(2):371–86. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 109.Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A. 1996 Mar 19;93(6):2317–21. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Breder CD, Dewitt D, Kraig RP. Characterization of inducible cyclooxygenase in rat brain. J Comp Neurol. 1995 May 1;355(2):296–315. doi: 10.1002/cne.903550208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen C, Bazan NG. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005 Feb;93(2):929–41. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- 112.Chen C, Magee JC, Bazan NG. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 2002 Jun;87(6):2851–7. doi: 10.1152/jn.2002.87.6.2851. [DOI] [PubMed] [Google Scholar]
- 113.Dash PK, Mach SA, Moore AN. Regional expression and role of cyclooxygenase-2 following experimental traumatic brain injury. J Neurotrauma. 2000 Jan;17(1):69–81. doi: 10.1089/neu.2000.17.69. [DOI] [PubMed] [Google Scholar]
- 114.Hickey RW, Adelson PD, Johnnides MJ, et al. Cyclooxygenase-2 Activity Following Traumatic Brain Injury in the Developing Rat. Pediatr Res. 2007 Jul 6; doi: 10.1203/PDR.0b013e3180db2902. [DOI] [PubMed] [Google Scholar]
- 115.Strauss KI, Barbe MF, Marshall RM, et al. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000 Aug;17(8):695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cernak I, O’Connor C, Vink R. Inhibition of cyclooxygenase 2 by nimesulide improves cognitive outcome more than motor outcome following diffuse traumatic brain injury in rats. Exp Brain Res. 2002 Nov;147(2):193–9. doi: 10.1007/s00221-002-1245-z. [DOI] [PubMed] [Google Scholar]
- 117.Schwab JM, Beschorner R, Meyermann R, Gozalan F, Schluesener HJ. Persistent accumulation of cyclooxygenase-1-expressing microglial cells and macrophages and transient upregulation by endothelium in human brain injury. Journal of neurosurgery. 2002 May;96(5):892–9. doi: 10.3171/jns.2002.96.5.0892. [DOI] [PubMed] [Google Scholar]
- 118.Schwab JM, Seid K, Schluesener HJ. Traumatic brain injury induces prolonged accumulation of cyclooxygenase-1 expressing microglia/brain macrophages in rats. J Neurotrauma. 2001 Sep;18(9):881–90. doi: 10.1089/089771501750451802. [DOI] [PubMed] [Google Scholar]
- 119.Choi SH, Aid S, Choi U, Bosetti F. Cyclooxygenases-1 and -2 differentially modulate leukocyte recruitment into the inflamed brain. Pharmacogenomics J. 2010 Oct;10(5):448–57. doi: 10.1038/tpj.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Koyfman L, Kaplanski J, Artru AA, et al. Inhibition of cyclooxygenase 2 by nimesulide decreases prostaglandin E2 formation but does not alter brain edema or clinical recovery after closed head injury in rats. Journal of neurosurgical anesthesiology. 2000 Jan;12(1):44–50. doi: 10.1097/00008506-200001000-00009. [DOI] [PubMed] [Google Scholar]
- 121.Gopez JJ, Yue H, Vasudevan R, et al. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005 Mar;56(3):590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kunz T, Marklund N, Hillered L, Oliw EH. Effects of the selective cyclooxygenase-2 inhibitor rofecoxib on cell death following traumatic brain injury in the rat. Restorative neurology and neuroscience. 2006;24(1):55–63. [PubMed] [Google Scholar]
- 123.Kelso ML, Scheff SW, Pauly JR, Loftin CD. Effects of genetic deficiency of cyclooxygenase-1 or cyclooxygenase-2 on functional and histological outcomes following traumatic brain injury in mice. BMC Neurosci. 2009 Aug 31;10(1):108. doi: 10.1186/1471-2202-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ahmad M, Rose ME, Vagni V, et al. Genetic disruption of cyclooxygenase-2 does not improve histological or behavioral outcome after traumatic brain injury in mice. Journal of neuroscience research. 2008 Dec;86(16):3605–12. doi: 10.1002/jnr.21809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. The New England journal of medicine. 2005 Mar 17;352(11):1092–102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 126.Nussmeier NA, Whelton AA, Brown MT, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. The New England journal of medicine. 2005 Mar 17;352(11):1081–91. doi: 10.1056/NEJMoa050330. [DOI] [PubMed] [Google Scholar]
- 127.Girgis H, Palmier B, Croci N, et al. Effects of selective and non-selective cyclooxygenase inhibition against neurological deficit and brain oedema following closed head injury in mice. Brain Res. 2013 Jan 23;1491:78–87. doi: 10.1016/j.brainres.2012.10.049. [DOI] [PubMed] [Google Scholar]
- 128.Thau-Zuchman O, Shohami E, Alexandrovich AG, Trembovler V, Leker RR. The anti-inflammatory drug carprofen improves long-term outcome and induces gliogenesis after traumatic brain injury. J Neurotrauma. 2012 Jan 20;29(2):375–84. doi: 10.1089/neu.2010.1673. [DOI] [PubMed] [Google Scholar]
- 129.Dewitt DS, Kong DL, Lyeth BG, et al. Experimental traumatic brain injury elevates brain prostaglandin E2 and thromboxane B2 levels in rats. J Neurotrauma. 1988;5(4):303–13. doi: 10.1089/neu.1988.5.303. [DOI] [PubMed] [Google Scholar]
- 130.Shohami E, Shapira Y, Cotev S. Experimental closed head injury in rats: prostaglandin production in a noninjured zone. Neurosurgery. 1988 May;22(5):859–63. [PubMed] [Google Scholar]
- 131.Olivecrona M, Rodling-Wahlstrom M, Naredi S, Koskinen LO. Prostacyclin treatment and clinical outcome in severe traumatic brain injury patients managed with an ICP-targeted therapy: a prospective study. Brain Inj. 2012;26(1):67–75. doi: 10.3109/02699052.2011.635351. [DOI] [PubMed] [Google Scholar]
- 132.Olivecrona M, Rodling-Wahlstrom M, Naredi S, Koskinen LO. Prostacyclin treatment in severe traumatic brain injury: a microdialysis and outcome study. J Neurotrauma. 2009 Aug;26(8):1251–62. doi: 10.1089/neu.2008.0605. [DOI] [PubMed] [Google Scholar]
- 133.Kawano T, Anrather J, Zhou P, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006 Feb;12(2):225–9. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- 134.Khatibi NH, Jadhav V, Matus B, et al. Prostaglandin E2 EP1 receptor inhibition fails to provide neuroprotection in surgically induced brain-injured mice. Acta Neurochir Suppl. 2011;111:277–81. doi: 10.1007/978-3-7091-0693-8_46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free radical biology & medicine. 2008 Aug 15;45(4):443–52. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma. 2008 May;25(5):513–26. doi: 10.1089/neu.2007.0451. [DOI] [PubMed] [Google Scholar]
- 137.Holmin S, Mathiesen T, Shetye J, Biberfeld P. Intracerebral inflammatory response to experimental brain contusion. Acta neurochirurgica. 1995;132(1–3):110–9. doi: 10.1007/BF01404857. [DOI] [PubMed] [Google Scholar]
- 138.Meissner A, Zilles O, Varona R, et al. CC chemokine ligand 20 partially controls adhesion of naive B cells to activated endothelial cells under shear stress. Blood. 2003 Oct 15;102(8):2724–7. doi: 10.1182/blood-2003-01-0007. [DOI] [PubMed] [Google Scholar]
- 139.Dalgard CL, Cole JT, Kean WS, et al. The cytokine temporal profile in rat cortex after controlled cortical impact. Frontiers in molecular neuroscience. 2012;5:6. doi: 10.3389/fnmol.2012.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Weckbach S, Neher M, Losacco JT, et al. Challenging the role of adaptive immunity in neurotrauma: Rag1(−/−) mice lacking mature B and T cells do not show neuroprotection after closed head injury. J Neurotrauma. 2012 Apr 10;29(6):1233–42. doi: 10.1089/neu.2011.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fee D, Crumbaugh A, Jacques T, et al. Activated/effector CD4+ T cells exacerbate acute damage in the central nervous system following traumatic injury. J Neuroimmunol. 2003 Mar;136(1–2):54–66. doi: 10.1016/s0165-5728(03)00008-0. [DOI] [PubMed] [Google Scholar]
- 142.Rudehill S, Muhallab S, Wennersten A, et al. Autoreactive antibodies against neurons and basal lamina found in serum following experimental brain contusion in rats. Acta neurochirurgica. 2006 Feb;148(2):199–205. doi: 10.1007/s00701-005-0673-5. discussion. [DOI] [PubMed] [Google Scholar]
- 143.Mrakovcic-Sutic I, Tokmadzic VS, Laskarin G, et al. Early changes in frequency of peripheral blood lymphocyte subpopulations in severe traumatic brain-injured patients. Scandinavian journal of immunology. 2010 Jul;72(1):57–65. doi: 10.1111/j.1365-3083.2010.02407.x. [DOI] [PubMed] [Google Scholar]
- 144.Clausen F, Lorant T, Lewen A, Hillered L. T lymphocyte trafficking: a novel target for neuroprotection in traumatic brain injury. J Neurotrauma. 2007 Aug;24(8):1295–307. doi: 10.1089/neu.2006.0258. [DOI] [PubMed] [Google Scholar]
- 145.Zhang Z, Artelt M, Burnet M, Trautmann K, Schluesener HJ. Early infiltration of CD8+ macrophages/microglia to lesions of rat traumatic brain injury. Neuroscience. 2006 Aug 25;141(2):637–44. doi: 10.1016/j.neuroscience.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 146.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003 Feb 14;299(5609):1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 147.Reynolds AD, Stone DK, Hutter JA, et al. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol. 2010 Mar 1;184(5):2261–71. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007 Oct;13(10):1173–5. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chaudhry A, Samstein RM, Treuting P, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011 Apr 22;34(4):566–78. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huber S, Gagliani N, Esplugues E, et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(−) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011 Apr 22;34(4):554–65. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Logan TT, Villapol S, Symes AJ. TGF-beta superfamily gene expression and induction of the Runx1 transcription factor in adult neurogenic regions after brain injury. PLoS One. 2013;8(3):e59250. doi: 10.1371/journal.pone.0059250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Morganti-Kossmann MC, Hans VH, Lenzlinger PM, et al. TGF-beta is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. J Neurotrauma. 1999 Jul;16(7):617–28. doi: 10.1089/neu.1999.16.617. [DOI] [PubMed] [Google Scholar]
- 153.Trajkovic V, Vuckovic O, Stosic-Grujicic S, et al. Astrocyte-induced regulatory T cells mitigate CNS autoimmunity. Glia. 2004 Aug 1;47(2):168–79. doi: 10.1002/glia.20046. [DOI] [PubMed] [Google Scholar]
- 154.Holmin S, Soderlund J, Biberfeld P, Mathiesen T. Intracerebral inflammation after human brain contusion. Neurosurgery. 1998 Feb;42(2):291–8. doi: 10.1097/00006123-199802000-00047. discussion 8–9. [DOI] [PubMed] [Google Scholar]
- 155.Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. Journal of leukocyte biology. 2007 Nov;82(5):1083–94. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- 156.Liu J, Gong N, Huang X, et al. Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. J Immunol. 2009 Mar 15;182(6):3855–65. doi: 10.4049/jimmunol.0803330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sheng JR, Li LC, Ganesh BB, Prabhakar BS, Meriggioli MN. Regulatory T cells induced by GM-CSF suppress ongoing experimental myasthenia gravis. Clin Immunol. 2008 Aug;128(2):172–80. doi: 10.1016/j.clim.2008.03.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mosley RL, Benner EJ, Kadiu I, et al. Neuroinflammation, Oxidative Stress and the Pathogenesis of Parkinson’s Disease. Clin Neurosci Res. 2006 Dec 6;6(5):261–81. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kong T, Choi JK, Park H, et al. Reduction in programmed cell death and improvement in functional outcome of transient focal cerebral ischemia after administration of granulocyte-macrophage colony-stimulating factor in rats. Laboratory investigation. J Neurosurg. 2009 Jul;111(1):155–63. doi: 10.3171/2008.12.JNS08172. [DOI] [PubMed] [Google Scholar]
- 160.Liesz A, Suri-Payer E, Veltkamp C, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009 Feb;15(2):192–9. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 161.Cosentino M, Gendelman HE. The first Insubria autumn school on neuroimmunopharmacology: challenging paradigms beyond boundaries. J Neuroimmune Pharmacol. 2013 Mar;8(1):1–3. doi: 10.1007/s11481-013-9440-1. [DOI] [PubMed] [Google Scholar]
- 162.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends in immunology. 2007 Jan;28(1):12–8. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 163.Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996 Sep;74(2):599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- 164.Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends in immunology. 2007 Jan;28(1):5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 165.Klyachko NL, Manickam DS, Brynskikh AM, et al. Cross-linked antioxidant nanozymes for improved delivery to CNS. Nanomedicine. 2012 Jan;8(1):119–29. doi: 10.1016/j.nano.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rosenbaugh EG, Roat JW, Gao L, et al. The attenuation of central angiotensin II-dependent pressor response and intra-neuronal signaling by intracarotid injection of nanoformulated copper/zinc superoxide dismutase. Biomaterials. 2010 Jul;31(19):5218–26. doi: 10.1016/j.biomaterials.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhao Y, Haney MJ, Mahajan V, et al. Active Targeted Macrophage-mediated Delivery of Catalase to Affected Brain Regions in Models of Parkinson’s Disease. J Nanomed Nanotechnol. Sep 10;:S4. doi: 10.4172/2157-7439.S4-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brynskikh AM, Zhao Y, Mosley RL, et al. Macrophage delivery of therapeutic nanozymes in a murine model of Parkinson’s disease. Nanomedicine (Lond) Apr;5(3):379–96. doi: 10.2217/nnm.10.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gong N, Liu J, Reynolds AD, et al. Brain ingress of regulatory T cells in a murine model of HIV-1 encephalitis. J Neuroimmunol. Jan;230(1–2):33–41. doi: 10.1016/j.jneuroim.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Banerjee R, Mosley RL, Reynolds AD, et al. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS One. 2008;3(7):e2740. doi: 10.1371/journal.pone.0002740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kelso ML, Scheff NN, Scheff SW, Pauly JR. Melatonin and minocycline for combinatorial therapy to improve functional and histopathological deficits following traumatic brain injury. Neurosci Lett. 2011 Jan 13;488(1):60–4. doi: 10.1016/j.neulet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kovesdi E, Kamnaksh A, Wingo D, et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Frontiers in neurology. 2012;3:111. doi: 10.3389/fneur.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Siopi E, Llufriu-Daben G, Fanucchi F, et al. Evaluation of late cognitive impairment and anxiety states following traumatic brain injury in mice: the effect of minocycline. Neurosci Lett. 2012 Mar 9;511(2):110–5. doi: 10.1016/j.neulet.2012.01.051. [DOI] [PubMed] [Google Scholar]
- 174.Ng SY, Semple BD, Morganti-Kossmann MC, Bye N. Attenuation of microglial activation with minocycline is not associated with changes in neurogenesis after focal traumatic brain injury in adult mice. J Neurotrauma. 2012 May 1;29(7):1410–25. doi: 10.1089/neu.2011.2188. [DOI] [PubMed] [Google Scholar]
- 175.Lu DC, Zador Z, Yao J, Fazlollahi F, Manley GT. Aquaporin-4 Reduces Post-Traumatic Seizure Susceptibility by Promoting Astrocytic Glial Scar Formation in Mice. J Neurotrauma. 2011 Sep 22; doi: 10.1089/neu.2011.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Siopi E, Calabria S, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M. Minocycline restores olfactory bulb volume and olfactory behavior after traumatic brain injury in mice. J Neurotrauma. 2012 Jan 20;29(2):354–61. doi: 10.1089/neu.2011.2055. [DOI] [PubMed] [Google Scholar]
- 177.Siopi E, Cho AH, Homsi S, et al. Minocycline restores sAPPalpha levels and reduces the late histopathological consequences of traumatic brain injury in mice. J Neurotrauma. 2011 Oct;28(10):2135–43. doi: 10.1089/neu.2010.1738. [DOI] [PubMed] [Google Scholar]
- 178.Abdel Baki SG, Schwab B, Haber M, Fenton AA, Bergold PJ. Minocycline synergizes with N-acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PLoS One. 2010;5(8):e12490. doi: 10.1371/journal.pone.0012490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Homsi S, Piaggio T, Croci N, et al. Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J Neurotrauma. 2010 May;27(5):911–21. doi: 10.1089/neu.2009.1223. [DOI] [PubMed] [Google Scholar]
- 180.Homsi S, Federico F, Croci N, et al. Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res. 2009 Sep 29;1291:122–32. doi: 10.1016/j.brainres.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 181.Ding JY, Kreipke CW, Speirs SL, et al. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett. 2009 Mar 27;453(1):68–72. doi: 10.1016/j.neulet.2009.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Crack PJ, Gould J, Bye N, et al. The genomic profile of the cerebral cortex after closed head injury in mice: effects of minocycline. Journal of neural transmission. 2009 Jan;116(1):1–12. doi: 10.1007/s00702-008-0145-1. [DOI] [PubMed] [Google Scholar]
- 183.Bye N, Habgood MD, Callaway JK, et al. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol. 2007 Mar;204(1):220–33. doi: 10.1016/j.expneurol.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 184.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001 Jun;48(6):1393–9. doi: 10.1097/00006123-200106000-00051. discussion 9–401. [DOI] [PubMed] [Google Scholar]
- 185.Higashida T, Kreipke CW, Rafols JA, et al. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. Journal of neurosurgery. 2011;114(1):92–101. doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]