

Abstract
Despite scores of investigations, the actual impact of resveratrol (3,5,4′-trihydroxy-trans-stilbene) on human health, as a dietary component or supplement, remains moot. This is due to many factors, such as relatively low potency, pleiotropic mechanisms, and rapid metabolism. Nonetheless, as a promiscuous molecule that interacts with numerous targets, resveratrol can be viewed as a scaffold for designing structural relatives potentially capable of mediating more intense responses with greater mechanistic stringency. We currently report the synthesis and biological evaluation of 92 stilbene analogs. The compounds were tested with in vitro assays for activation of quinone reductase 1, inhibition of QR2, nitric oxide production, aromatase, NFκB, TPA-induced ornithine decarboxylase, or cyclooxygenase-1 and -2, quenching of 2,2-diphenyl-1-picrylhydrazyl free radical, interaction with estrogen receptors, and as antiproliferative agents. Several compounds were found to mediate responses with much greater potency than resveratrol; some mediated pleiotropic responses, as is the case with the parent molecule, but others were highly specific or totally inactive. When administered to rats, higher serum concentrations and greater stability was demonstrated with prototype lead molecules. Due to structural simplicity, facile syntheses are available for large-scale production. These data support the promise of more advanced development of novel resveratrol derivatives as drug entities.
Keywords: resveratrol, chemoprevention, pleiotropic effect, Caco-2 cells, absorption, mass spectrometry
Introduction
As a natural product, resveratrol (3,5,4′-trihydroxy-trans-stilbene; compound 1) is a phytoalexin produced by plant species in response to environmental stress or pathogenic attack. This compound was first reported by our group as a candidate for cancer chemoprevention [1] and extensive studies have followed [2, 3]. Resveratrol demonstrates a broad spectrum of biological activities potentially capable of inhibiting carcinogenesis at the stages of initiation, promotion and progression. Examples include down-regulation of the synthesis and release of proinflammatory mediators, inhibition of activated immune cells, aromatase, iNOS, COX-1 and COX-2, and effects on NFκB [3]. Activation of NAD(P)H:quinone reductase (QR1), a well-established mechanism for chemoprevention that correlates with the induction of other phase II detoxifying enzymes, is also facilitated by resveratrol [4–6]. The molecule is unusually promiscuous [7]. Although some high-affinity targets exist, such as quinone reductase 2 [8], it does not appear likely that resveratrol functions through one specific mechanism. Considering the structural simplicity of this stilbene, the intensity of research publications is phenomenal, indicating the molecule holds great potential [9–13].
In vivo studies suggest resveratrol is relatively safe and can be used for the prevention and therapy of cancer [13]. Studies with human beings suggest a daily dosage of 1–5 g may be acceptable [14]. Obviously, dosage levels in this range are not available through normal dietary consumption, so resveratrol is evolving more as a natural product drug than a natural dietary component that may affect human health. As such, it seems logical to investigate structural derivatives that may show greater efficacy and improved biophysical characteristics.
We currently report the synthesis and biological evaluation of a library of resveratrol derivatives. The derivatives include compounds differing in the number, position, and type of substituents, and the presence or absence of stilbenic double bond. Sulfate derivatives were tested as well, since serum concentrations of these metabolites are higher than those of resveratrol following treatment with the parent compound. A battery of in vitro assays was used to monitor the activity of all designed derivatives, and some preliminary absorption and metabolism studies have been performed with promising leads. Structure-activity relationships are beginning to emerge. In sum, based on information that has been gleaned from the extensive work that has been performed with resveratrol, it is relatively clear that derivatives with greater potency and specificity can be developed.
Background on resveratrol
Resveratrol, a naturally occurring polyphenol, is a phytoalexin found in grapes, mulberries, cranberries, blueberries, peanuts, and chocolate [1, 15, 16]. It is produced by these plants as a response to stress, such as a bacterial or fungal infection [17]. Resveratrol is produced from two precursor molecules, malonyl-coenzyme A and p-coumaroyl-coenzyme A, by the enzyme stilbene synthase [18]. The compound was first purified from white hellebore in 1939 by M. Takaoka [19]. In 1963, it was found to be a component of Japanese ironwood, which is currently used as the source of many resveratrol supplements [20]. The consumption of wine entered the mainstream media in 1992, when it was touted as the explanation for the “French paradox” because of biological activities which improved cardiovascular health [21]. The lower incidence of cardiovascular diseases among the French people, in spite of their diet high in saturated fats had been reported [22, 23], and epidemiological studies linked moderate consumption of red wine with a lower incidence of heart disease. A link to resveratrol, in particular, was established later, when the cancer chemopreventive potential was described [1]. Since grape skins are included in the fermentation of red wine, it typically contains a significantly higher amount of resveratrol (2–40 μM) [24] than white wine. Nonetheless, resveratrol concentration varies with the type of red wine, with Pinot noir sometimes containing the highest level of resveratrol [25]. The inverse correlation with heart disease was further strengthened when resveratrol was shown to decrease serum cholesterol [26], promote vasorelaxation [27,28], suppress atherosclerosis [29], and inhibite platelet aggregation [30].
In the area of chemoprevention, following our observation of the potential of resveratrol to block 7,12-dimethylbenz(a)anthracene (DMBA)-induced tumorigenesis and various stages of carcinogenesis [1], a great number of studies have been performed. It has been shown to act through varied mechanisms, including the regulation of cell cycle progression [31], apoptosis [32, 33], inhibition of tumor invasion and angiogenesis [34], prevention of inflammation [35], activation of adenosine monophosphate (AMP)-activated protein kinase (AMPK) [36], scavenging of reactive oxygen species (ROS), [37], and modulation of NFκB [38, 39]. This activation of multiple anti-cancer pathways (pleiotropism) can be viewed as an attractive feature of resveratrol, since it may help to overcome drug resistance [40, 41]. It has been suggested that resveratrol is capable of mediating a great number of other biological responses relevant to human health [9], such as protection against viral infections [42], caloric restriction mimicry [43], as well as effects on longevity [44].
Resveratrol is readily absorbed in humans and rapidly metabolized into its sulfate and glucuronide conjugates [45]. In addition, it is well tolerated and does not appear to be toxic in humans [14, 45]. However, pharmacokinetic studies have shown that the concentrations necessary to mediate activity observed using in vitro experiments cannot be achieved in animals [46], so identification of derivatives with greater bioavailability is desirable. Isomerization of the double bond that links the two phenolic rings of resveratrol creates two possible geometrical isomers. The trans form is more common in nature due to thermodynamic factors [47]. Much less is known about the pharmacological effects of cis-resveratrol [48].
Pleiotropic effect of resveratrol
Carcinogenesis occurs over a long period of time through consecutive stages [49] with alterations of phenotypic, cellular, genotypic, molecular, and drug-related biomarkers [50]. Resveratrol has been reported to prevent or inhibit carcinogenesis by acting on multiple targets at all stages.
Antioxidant activity of resveratrol
Oxidative damage in cells has been implicated as a causative factor in many diseases including cancer and Alzheimer’s, and has an impact on the aging process [51]. Resveratrol has demonstrated antioxidant potential by promoting various enzymes. We have shown that resveratrol can inhibit TPA–induced free radical formation with cultured HL-60 cells. Also, resveratrol exhibited 2,2-diphenyl-1-picrylhydrazyl (DPPH) free-radical scavenging activity (IC50 = 94.6 μg/mL) and hypoxanthine/xanthine oxidase enzyme inhibitory activity (IC50 = 59.1 μg/mL) [52, 53]. Resveratrol suppresses the formation of super-oxide (O2−) and H2O2 produced by lipopolysaccharide (LPS) or TPA-stimulated macrophages [54]. The compound inhibits H2O2-induced DNA damage in human lymphocytes by increasing glutathione levels, and induction of glutathione peroxidase, glutathione reductase, and glutathione S-transferase [55].
Inhibition of inflammatory enzymes
It has been reported that chronic inflammation can lead to various cancers [56]. Therefore, inhibition of inflammation is a promising approach for cancer chemoprevention. Resveratrol inhibits the activity of COX-1 [1] and COX-2 as reflected in decreased levels of prostaglandin E2 (IC50 = 32.2 μM) [57], suppresses inducible nitric oxide synthase (iNOS) in activated macrophages [58], and TANK-binding kinase 1 in TRIF signaling in the TLR3 and TLR4 inflammatory pathway [59]. In TPA-induced mouse skin, resveratrol significantly inhibited COX-2 expression and NFκB activation by inhibiting the activity of IκB kinase (IKK) [60].
Inhibition of cytochrome P450
Cytochrome (CYP) P450 isozymes are a large family of enzymes involved in the metabolism of xenobiotics [53]. Transcription of CYP1A1 is induced by the aryl hydrocarbon receptor (AhR). The aberrant expression of AhR has been reported in breast and prostate cancers [61]. Resveratrol inhibited TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin)-induced CYP1A1 expression by decreasing binding of AhR to the promoter region of CYP1A1 [62]. CYP1B1 was reported to function as an estrogen 4-hydroxylase [63] and activate many carcinogens including estrogens. Resveratrol exhibited competitive inhibition of human CYP1A1 as well as non-competitive inhibition of human CYP1B1 [64], and inhibited aromatase enzyme (CYP19) activity (IC50 12.8 μM) [65].
Protein kinase C (PKC)
Many studies have been performed to understand the function of PKC in carcinogenesis and to find inhibitors for cancer treatment or prevention [66]. Resveratrol has been reported to inhibit TPA-mediated induction of protein kinase C [67].
Ornithine decarboxylase (ODC)
Ornithine decarboxylase (ODC) is up-regulated in various tumor cells. Resveratrol significantly decreased the ODC activity in Caco-2 cells [68], and blocked N-nitrosodiethylamine (NDEA)-induced cytosolic ODC levels in the mice liver and lungs [69].
Quinone reductases 1 and 2
Phase II enzymes are known to detoxify carcinogens, including aldehyde reductase, glutathione reductase, epoxide hydrolase, glutathione S-transferase (GST), quinone reductase 1 (QR1), and UDP-glucuronosyltransferases (UGT) [70]. Resveratrol induces QR1 activity with a concentration required to double activity (CD) of 21 μM in cultured mouse hepatoma (Hepa 1c1c7) cells [71]. Resveratrol is also a potent inhibitor of quinone reductase 2, a cytosolic enzyme that catalyzes the reduction of quinones. It is hypothesized that inhibition of QR2 may lead to protection of cells against reactive species that are causing cellular damage [8].
Modulation of cancer cell cycle and apoptosis
Cell cycle mediators such as cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitors are involved in the deregulated cell cycle progression of cancer cells [72]. Resveratrol arrests various phases of the cell cycle, depending on cell types [73], and can induce apoptosis via enhancement of death receptor signaling [32, 74].
Absorption and metabolism
A notable characteristic of resveratrol is its relative lack of potency. Many potential health benefits have been suggested, but considerable controversy exists as to whether it is an active molecule in vivo [75]. Resveratrol is efficiently absorbed on oral administration and rapidly metabolized to its 3- and 4′-O-sulfate, and 3-O-glucuronide conjugates [76–78]. After a dietarily relevant 25 mg oral dose of resveratrol, the plasma concentration of resveratrol is only in the nanomolar range compared with the micromolar range of its metabolites [75, 77, 79]. Based on resveratrol metabolism studies, it is reasonable to suggest the in vitro data obtained using high concentrations of resveratrol lack direct in vivo relevance. Although large amounts of resveratrol can be administered, there is merit in searching for analogues with significantly greater potency, thereby reducing the need for large dosages and allowing comparisons with known modulators [80].
Methods
Preparation of compounds
Syntheses of the resveratrol derivatives 2–93 have been reported previously [8].
Aromatase assay
Aromatase activity was assayed as previously reported [81]. Briefly, test compounds at final concentration of 50 μM were preincubated with NADPH-regenerating system before the enzyme and substrate mixture were added, and the plate was incubated at 37°C before quenching with NaOH. Fluorescence was measured at 485 nm (excitation) and 530 nm (emission). IC50 values and dose-response curves were based on two independent experiments performed in duplicate using five concentrations of test substance. Naringenin (IC50 = 0.23 μM) was used as a positive control.
NFκB luciferase assay
Stably transfected human embryonic kidney cells 293 were used for monitoring any changes occurring along the NFκB pathway [82] with TNF-α (1 nM) as activator. After incubation with tested compounds, cells were lysed in reporter lysis buffer, and a luciferase assay was performed using the Luc assay system from Promega (Madison, WI). Luminescence was detected in a LUMIstar Galaxy BMG luminometer. Data for NFκB activity are expressed as IC50 values. As a positive control, two NFκB inhibitors were used: TPCK, IC50 = 3.8 μM and BAY-11, IC50 = 2.0 μM.
Quinone reductase 1 (QR1) assay
QR1 activity was assessed using Hepa 1c1c7 murine hepatoma cells as previously described [83]. Test compounds were added to a final concentration of 50 μM, and QR activity was measured as a function of the NADPH-dependent menadiol-mediated reduction of 3-(4,5-dimetylthiazo-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to a blue formazan. The induction ratio (IR) of QR activity represents the specific enzyme activity of agent-treated cells compared with a DMSO-treated control. The concentration to double activity (CD) was determined through a dose-response assay for active substances (IR >2). The chemopreventive index (CI) is calculated as IC50/CD. 4′-Bromoflavone (CD = 0.01 μM) was used as a positive control.
Quinone reductase 2 (QR2) assay
The QR2 assay was performed according the procedure described by Sun et al. [8]. Briefly, the assay mixture contained 12 nM QR2, 17.5 μM NMeH and 200 μM MTT in a reaction buffer containing 100 mM NaCl, 50 mM Tris and 0.1% Triton-X100 (Fisher Biotech 93004). All reactions were initiated by the addition of QR2. Activity of QR2 was determined using a SpectraMax Plus 384 UV-vis microplate reader by monitoring the increase in absorbance at 612 nm. IC50 values were determined using the same assay conditions except that inhibitor concentrations were varied from 0.2 to 100 μM. The percent inhibition data were then plotted as a function of inhibitor concentration, [I], and the data were then fit to the following equation: %I = %Imax/[(1 + [I]/IC50)] using nonlinear regression describing a simple binding isotherm. All data were globally fit to the equation using the Enzyme Kinetics Module of the program SIGMAPLOT from SPSS Scientific (Chicago, IL).
Measurement of nitric oxide (NO) production in LPS-stimulated macrophages
RAW 264.7 cells were treated with various concentrations of compounds followed by 1 μg/ml of LPS. Nitrite in cultured media, a stable end product of NO, was estimated by the addition of Griess reagent and the absorbance was measured at 540 nm. A standard curve was created by using known concentrations of sodium nitrite to convert the absorbance to the concentration of nitrite in cultured media [84].
Determination of TPA-induced ODC activity
Human epithelial urinary bladder cells T-24 were incubated in the absence of serum; resveratrol derivatives were added in duplicate before the induction of ODC activity with TPA. ODC activity was assayed by measuring the release of [14C]CO2 from L-[1-14C]-ornithine HCl using a Beckman Coulter LS 6500 counter. IC50 values were generated from the results of five serial dilutions of tested derivatives in duplicate. Standard inhibitors of ODC activity include apigenin, IC50 = 6.0 μM, menadione, IC50 = 8.3 μM, and deguelin, IC50 = 0.1 μM [85].
DPPH radical scavenging assay
To evaluate antioxidant capacity, 2,2-diphenyl-1-picrylhydrazyl (DPPH) free-radical scavenging was performed according to the method described by Lee et al. [52]. The absorbance was measured at 515 nm. The DPPH radical scavenging activity of each sample was evaluated by calculating % of inhibition as follows: % inhibition = (1 - Abssample/Abscontrol) × 100.
COX-1 and -2 assays
Assays were conducted in a 96-well format [86] with analyses performed as follows. Negative ion electrospray tandem mass spectrometric measurement of PGE2 was carried out using a Shimadzu (Columbia, MD) LCMS-8030 triple quadrupole mass spectrometer equipped with a Shimadzu (Columbia, MD) Nexera UHPLC system. A Waters (Milford, MA) Acquity UPLC® BEH C18 column (2.1 mm × 50 mm, 1.7 μm) was used for LC separation with a 0.5 min isocratic mobile phase consisting of acetonitrile/aqueous 0.1% formic acid (40:60; v/v) at a flow rate of 1.2 mL/min. The deprotonated molecules of m/z 351 and m/z 355 corresponding to PGE2 and the surrogate standard d4-PGE2, respectively, and the abundant product ions of m/z 271 and m/z 275, were measured using selected reaction monitoring.
Binding to human estrogen receptors using ultrafiltration LC-MS
An equimolar mixture of resveratrol analogues (0.2 μM each) was incubated for 2 h at room temperature with 100 pmol ERα or ERβ (human recombinant ER-α and ER-β produced using recombinant baculovirus-infected insect cells; Invitrogen, Carlsbad, CA) in binding buffer consisting of 50 mM Tris-HCl (pH 7.4), 10% glycerol, 50 mM KCl and 1 mM EDTA in a total volume of 150 μl. Identical negative control incubations in which ER was omitted were used to correct for nonspecific binding of the compounds to the ultrafiltration membrane. 8-Prenylnaringenin, a potent phytoestrogen, was used as a positive control. After incubation, each mixture was filtered through a Microcon YM-30 centrifugal filter by centrifugation at 10,000 g for 10 min at 4°C. The filter was washed three times by centrifugation with 150 μl aliquots of ammonium acetate buffer (pH 7.4) to remove the unbound compounds. The bound ligands were released by adding 400 μl of methanol/water (90:10; v/v) followed by centrifugation at 10,000 g for 10 min. The ultrafiltrate was evaporated to dryness under a stream of dry nitrogen and then reconstituted in 60 μl of methanol/water (50:50; v/v). Naringenin was added as an internal standard to normalize the ionization efficiency between experiments and controls. Aliquots (10 μl) were analyzed using LC-MS which consisted of a Shimadzu LC-20AD HPLC coupled to a Shimadzu IT-TOF mass spectrometer. HPLC separation were carried out using a Waters Xterra MS C18 (3.5μm, 2.1 X 100 mm) HPLC column.
Caco-2 cell monolayer permeability and plasma protein binding
Materials
The Caco-2 cell line, Eagle’s minimum essential medium (EMEM), and fetal bovine serum were purchased from the American Type Culture Collection (Rockville, MD). Trypsin-EDTA (0.25%), Hank’s balanced salt solution (HBSS), HEPES buffer solution, PBS, penicillin, and streptomycin were purchased from Life Technology (Grand Island, NY). Transwell polycarbonate inserts (24 mm diameter, 4.71 cm2 surface, 0.4 μm pore size) were purchased from Corning Costar (Cambridge, MA). Transepithelial electrical resistance (TEER) was measured using a Millicell-ERS instrument from Millipore (Bedford, MA). Pooled human plasma was purchased from Innovative Research (Novi, MI), and rapid equilibrium dialysis devices were purchased from Pierce Biotechnology (Rockford, IL).
Caco-2 cell monolayer model assay
Caco-2 cells were cultured in Eagle’s minimum essential medium with 10% fetal bovine serum, 100 unit/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere of 5% CO2 and 95% air at 37°C. When the cells (passage numbers 10–12) had reached approximately 70–80% confluence, they were removed using 0.25% trypsin/EDTA, seeded onto polycarbonate membranes fitted into Transwell 6-well plates (24 mm diameter, 4.71 cm2 surface area, 0.4 μm pore size) at a density of 2 x 105 cells/insert and cultured until late confluence. The cell culture medium was changed every other day after seeding. The integrity of each monolayer of differentiated cells was monitored by measuring the transepithelial electrical resistance (TEER). Only, monolayers with TEER values >300 Ω/cm2 were utilized.
In preparation for the Caco-2 cell monolayer assay, the cell culture medium was removed from both the apical (AP) and basolateral (BL) chambers. The cells were washed three times with PBS (pH 7.4) and pre-incubated with HBSS containing 20 mM HEPES, pH 7.4 for 30 min at 37°C on a shaker bath at 50 rpm. A 20 mM stock solution of resveratrol derivatives in DMSO was diluted to different concentrations in HBSS/HEPES buffer. The test solutions were added either to the apical (AP) side (for AP→BL measurement) or to the basolateral (BL) side (BL→AP measurement) of the monolayers, and blank HBSS/HEPES buffer was added to the other side. As control experiments, the low permeability standard sucrose (50 μM) and the high permeability standard propranolol (10 μM) were added to the AP chambers of parallel incubations. Aliquots from the recipient compartments were collected at 10, 20, 30, 40, and 60 min and then analyzed using LC-MS-MS. In some experiments, the effects of the p-glycoprotein inhibitor verapamil (50 μM) were investigated. All experiments were carried out in triplicate.
Binding to human plasma proteins
A rapid equilibrium device from Pierce Biotechnology (Rockford, IL) was used as directed by the manufacturer for the determination of binding of tested derivatives to human plasma proteins. Briefly, 200 μL of 10 μM compound in pooled human plasma was pipetted into the rapid equilibrium dialysis device sample chamber. The buffer chamber was then filled with 350 μL PBS consisting of 100 mM sodium phosphate and 150 mM sodium chloride. The device was sealed and placed in an orbital shaker at 37 °C set at 200 rpm for 5 h. After dialysis, 100 μL aliquots were removed from the buffer and plasma chambers and mixed with 100 μL of plasma or PBS, respectively. Proteins were precipitated by addition of 600 μL of ice-cold 90:10 (v/v) acetonitrile/water containing 0.1% formic acid followed by incubation on ice for 30 min. After centrifugation for 15 min at 14,000 g, the supernatants were removed, evaporated to dryness and then reconstituted in 100 μL of HPLC mobile phase containing ketoconazole as an internal standard at a final concentration of 0.5 μM. The concentration of compound in each sample was determined using LC-MS-MS.
Mass spectrometry
LC-MS-MS analyses was carried out using an Agilent (Santa Clara, CA) 6410 triple quadruple mass spectrometer equipped with positive ion electrospray and a Model 1200 HPLC. The solvent system consisted of a linear 3-min gradient from 30% to 100% acetonitrile in 0.1% aqueous formic acid and an Agilent XTerra C18 HPLC column (2.1 mm x 100 mm; 3.5 μm) at 28 °C. The flow rate was 200 μL/min. After collision-induced dissociation of the protonated molecule of compounds at m/z 256, selected reaction monitoring (SRM) was used to record the transition from m/z 256 to the abundant product ion of m/z 118 with a dwell time of 250 ms/ion. The SRM transition of m/z 531 to m/z 489 was monitored for the internal standard ketoconazole. Propranolol and sucrose were measured using LC-MS as described by Li et al. [87].
Animal studies
Animal studies were performed to investigate oral absorption and dose-tolerance of compound 5 (protocol 101008929). Following oral administration (40 mg/kg body weight) for three days, tissues and serum were examined for parent compound and metabolites. Animals were sacrificed 24 h after the final dose. Liver, mammary, gland, perirenal fat, and serum were collected and stored at −80°C until analyzed. Serum and tissue homogenates were analyzed via liquid chromatography-tandem mass spectrometry (LC-MS-MS). Analyses were carried out using a Shimadzu LC-20AD Prominence HPLC system interfaced with an Applied Biosystems API 4000 triple quadrapole mass spectrometer. A Waters XTerra MS C18 column (2.1 mm x 100 mm, 3.5 μm) was used for HPLC separation. Positive ion electrospray was used for ionization, and product ion mass spectra were recorded using collision-induced dissociation and selected reaction monitoring (SRM) of the transition of m/z 333 to 298, m/z 353 to 166, and m/z 483 to 130 (300 ms per transition) for measurement of compounds and internal standard [88,89].
Results
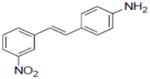
A total of 92 resveratrol derivatives were synthesized and tested. Biological responses and corresponding structure-function relationships of resveratrol and derivatives that functioned in a pleiotropic manner are summarized in Table 1. In a nitrite assay, the fundamental trans-stilbene structure (compound 75) showed 28.2% inhibition of nitric oxide (NO) production at a test concentration of 34 μM. Resveratrol (1) mediated moderate activity with an IC50 of 28.9 μM. Incorporation of a carboxyl group, nitro group, or carbonyl at 4′-position in ring B did not improve inhibitory activity. However, addition of an amino group in the para-position (2) showed the most potent activity with an IC50 of 3.7 μM. Changing the location of the amino group to the ortho- or meta- position yielded IC50 values of 7.2 and 10.2 μM, respectively (17 and 3). None of the derivatives tested showed selective inhibition of NO production.
Table 1.
Resveratrol derivatives with pleiotropic mechanisms of action.a
| Compounds tested | NOb IC50 | Arom IC50 | NFκB IC50 | QR1c IR | QR1d CD | QR2 IC50 | ODC IC50 | DPPHe IC50 | ERα | ERβ | COX-1f IC50 | COX-2f IC50 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
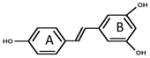
|
28.9±1.0 | 25±2.6 | 2.1±0.15 | 2.4±0.29 | 21±0.46 | 6.9±0.4 | 8.8±1.18 | 78.0±1.1 | 5.32 | 1.23 | 0.35±0.1 | 2.3±0.28 |
|
| |||||||||||||
| 2 |
|
3.7±0.9 | 22±0.54 | 0.9±0.23 | 2.6±0.17 | 6.5±0.24 | NTg | 18.1±2.1 | 147.9±0.3 | NAh | NA | 2.2±0.34 | 2.5±0.6 |
| 3 |
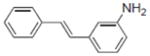
|
10.2±1.4 | 4.22±0.37 | 0.3±0.1 | NA | NA | NT | 6.4±0.8 | NA | NA | NA | 2.3±0.22 | NA |
| 4 |
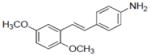
|
NA | 0.31±0.09 | 0.3±0.15 | NA | NA | 9.0±0.5 | 5.2±1.4 | 258.0±1.0 | 2.82 | 2.06 | 0.61±.22 | 2.3±0.32 |
| 5 |
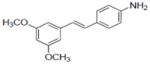
|
33.9±3.1 | 0.31±0.12 | 5.8±1.6 | 1.9±0.15 | NA | 1.7±0.06 | 1.3±0.4 | 227.7±0.5 | 1.65 | 1.33 | 0.76±0.14 | 12.4±5.1 |
| 6 |
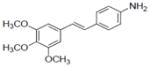
|
4.6±0.5 | 0.9±0.17 | 0.9±0.3 | NA | NA | 0.27±0.01 | 13.1±2.6 | 101.3+0.9 | NA | NA | 1.4±0.11 | NA |
| 7 |
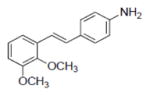
|
8.6±1.2 | 0.98±0.24 | 4.7±1.1 | NA | NA | NA | NA | 171.0±0.4 | 3.65 | 1.5 | 7.1±2.3 | NA |
| 8 |
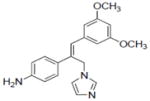
|
8.3±1.3 | 0.04±0.01 | 15.9±3.1 | NA | NA | 18.3±1.6 | NA | NA | NA | NA | 1.5±0.28 | NA |
| 9 |
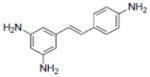
|
NA | 8.53±0.62 | 6.5±0.9 | NA | NA | 34 | 1.3±0.16 | 42.2±0.3 | 1.23 | 3.21 | 4.1±2.1 | NA |
| 10 |
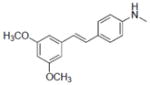
|
NA | NA | 11.6±1.9 | NA | NA | NT | NA | 135.8±0.6 | NA | NA | 3.8±1.5 | NA |
| 11 |
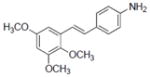
|
14.7±2.7 | 2.42±0.37 | 17.3±3.1 | NA | NA | 1.5±0.07 | NA | 50.5±0.2 | 3.5 | 5.25 | 2.3±0.28 | NA |
| 12 |
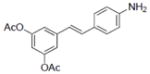
|
31.2±2.0 | 2.83±0.25 | 0.5±0.2 | 2.8±0.16 | 5.6±0.26 | 14.9 | 12.4±1.9 | NA | NA | NA | 6.9±3.4 | NA |
| 13 |
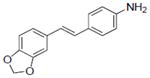
|
16.6±3.8 | 8.49±0.48 | 0.3±0.15 | NA | NA | >50 | NA | 87.9±0.4 | NA | NA | 0.97±0.21 | NA |
| 14 |
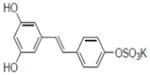
|
NA | NA | 18.2±3.0 | NA | NA | NA | 0.7±0.12 | NA | NA | NA | 0.46±0.11 | NA |
| 15 |
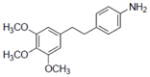
|
32.2±0.4 | 2.80±0.19 | 4.7±1.1 | NA | NA | 33.8 | 4.2±0.9 | NA | NA | NA | 5.0±1.2 | NA |
| 16 |
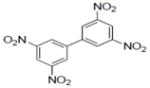
|
11.7±1.9 | 4.30±0.47 | 0.2±0.11 | 2.3±0.12 | 45.8±0.87 | NT | 2.1±1.3 | NA | NA | NA | NA | NA |
| 17 |
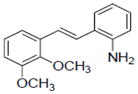
|
7.2±0.6 | NA | 6.1±1.9 | 3.7±0.16 | 2.1±0.3 | NT | 12.3±2.4 | 80.7±0.3 | NA | NA | NA | NA |
| 18 |
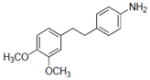
|
NA | 1.8±0.22 | 4.9±0.72 | NA | NA | 2.5±0.2 | 0.9±0.3 | 88.6±12 | 2.7 | 1.98 | 5.3±1.4 | NA |
| 19 |
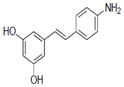
|
NA | 4.41±1.23 | 0.7±0.32 | NA | NA | 1.4±0.08 | NA | 54.4±8.2 | 2.17 | 2.91 | 1.1±0.05 | NA |
Compounds were classified as pleiotropic if activity was demonstrated in three or more of the assays; all IC50 values are in μM.
100% is with LPS as activator.
Induction ratio for treated vs. control cells.
Concentration to double the expression of QR1.
Sample concentration which caused 50% inhibition of DPPH, 100% activity is with scavenging DPPH free radical.
Concentration to inhibit COX-1 or -2 activity by 50%.
NT, not tested;
NA, not active, see Figure 1 for activity criteria.
Resveratrol has relatively weak activity as an inhibitor of aromatase (IC50 = 25 μM), but a number of resveratrol analogs displayed much greater inhibition (Table 1). Of the 92 compounds tested, 32 were at least moderately active against aromatase. Of these, eight had IC50 values less than 1 μM, and two imidazole analogs inhibited aromatase activity by 50% at concentrations less than 100 nM (8, 20). These analogs are obviously superior inhibitors relative to the parent molecule of resveratrol. In addition, they are more potent than the commercially available aromatase inhibitor, aminoglutethimide (IC50 = 0.2 μM), although less active than some other inhibitors such as anastrozole and letrozole. Additional inhibitors are shown in Table 2.
Table 2.
Selective inhibition of aromatase mediated by resveratrol derivatives.
| Compounds tested | IC50 (μM)a | |
|---|---|---|
| 20 |
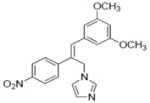
|
0.06±0.01 |
| 21 |

|
0.68 ± 0.07 |
| 22 |
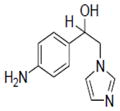
|
5.32±1.12 |
| 23 |
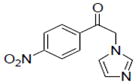
|
0.2±0.06 |
| 24 |

|
3.91±0.22 |
| 25 |
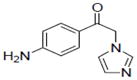
|
1.84±0.3 |
| 26 |
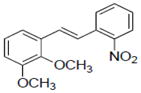
|
3.2±0.29 |
| 27c |
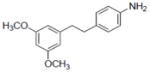
|
2.8±0.14 |
Sample concentration which caused 50% inhibition of aromatase; Naringenin (IC50 = 0.23 μM) was used as a positive control.
Compound 20 inhibited NFκB with and IC50 of 13.1±1.4 μM.
Compound 27 also inhibited COX-1 and -2 with IC50 values of 5.3±0.95 and 25.4±6.6 μM, respectively.
Based on a luciferase expression assay, resveratrol (1) is a moderate inhibitor of NFκB activity (IC50 = 2.1 μM). This result encouraged further exploration of different derivatives as possible inhibitors of NFκB. Among the 92 derivatives tested, 74 demonstrated weak to good inhibitory activity against NFκB with IC50 values in the 0.2–20 μM range. Within this group, eleven compounds were more potent than resveratrol, with IC50 values less than 2 μM. Compound 2, with a para-amino group in the trans-stilbene backbone structure, showed greater inhibition of NFκB than resveratrol. Some improvement was observed when an amino group in ring B was moved from the para- to the meta- position (3). Nevertheless, the majority of derivatives, with IC50 values in the low micromolar range, have an amino group in the para-position (4, 6, 7, 12, 13). Addition of hydroxyl, methoxy, acetoxy, or methylenedioxy groups did not significantly change potency against NFκB. The most active NFκB inhibitor in the series was compound 16, which contains four nitro groups. The compounds presented in Table 3 demonstrated stronger inhibitory activity than the positive controls: N-tosyl-L-phenylalaninyl-chlormethylketone (TPCK) (IC50 = 5 μM) and BAY-11-7082 (IC50 = 2 μM). Notably, these compounds mediated the inhibitory response with specificity.
Table 3.
Selective inhibition of NFκB mediated by resveratrol derivatives.
| Compounds tested | IC50 (μM)a | |
|---|---|---|
| 28b |
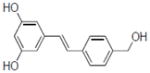
|
0.2±0.09 |
| 29 |
|
1.1±0.23 |
| 30b |
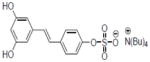
|
2.5±0.16 |
| 31b |
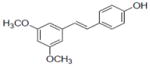
|
1.0±0.19 |
| 32 |
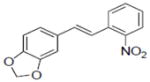
|
0.7±0.11 |
| 33b |
|
0.5±15 |
Sample concentration which caused 50% inhibition of TNF-induced NFκB activity treated with solvent (DMSO) only. N-Tosyl-L- phenylalanine-chloromethyl ketone (TPCK) and BAY-11 were used as a positive controls (IC50 = 3.8 and 2.0 μM, respectively).
Compounds 28, 30, 31, and 33 inhibited COX-1 with IC50 values of 0.87±0.04, 0.68±0.2, 2.8±0.67, and 2.0±0.57 μM, respectively. Compound 31 inhibited COX-2 with and IC50 value of 7.5±1.6 μM.
Resveratrol induced QR1 activity with an induction ratio (IR) of 2.4 and a CD of 21 μM (Table 1). It appears that substitution of larger groups (acetyl, methoxy) in place of the hydroxy substituents results in increased induction. This can be seen in 12 (IR 2.8). It is also apparent that a 5,6-methoxy substitution on ring A increases induction, e.g., 17, (IR 3.7, CD 2.1). An amino group on ring B yields a more active compound than a nitro group, and the 2-amino group is optimal. Of the derivatives tested, specific induction of QR1 without affecting other targets was not observed.
Induction of QR1 is considered a biomarker for cancer prevention with a main function to inactivate harmful xenobiotic compounds [90]. On the other hand, with a distinctly different function, the enzyme QR2 is one of the highest affinity targets of resveratrol that has been identified to date [91]. To expedite the search for new inhibitors of QR-2, we developed two different high-throughput screening assays, one based on enzymatic activity and the other based on ultrafiltration liquid chromatography-mass spectrometry [92]. Compounds 5, 6, 11, 18 and 19 demonstrated dose-dependent inhibition of QR2 (Table 1) as well as other activities. As shown in Table 4, three compounds were identified with high QR2 inhibitory specificity.
Table 4.
Selective inhibition of QR2 mediated by resveratrol derivatives.a
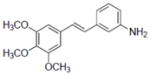
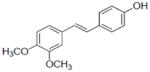
| Compounds tested | IC50 (μM) | |
|---|---|---|
| 34 |
|
4.1±0.6 |
| 35b |
|
2.1±0.2 |
| 36 |
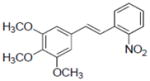
|
3.3±0.5 |
Human QR2 was purified from Escherichia coli BL21. Frozen QR2, previously stored at −80°C, was thawed on ice and then adjusted to the 4 mg/ml concentration for IC50 determination with different compounds.
Compound 35 inhibited COX-1 with and IC50 value of 2.0±1.4 μM.
Ornithine decarboxylase (ODC) is a key enzyme in synthesis of polyamines. It is activated in different types of tumors and discovery of highly specific inhibitors of ODC is a promising approach in the field of cancer chemoprevention. Of the 92 derivatives tested, 36 compounds were found to actively inhibit the induction of ODC. They can be divided into two groups: those with IC50 values in the range 10–20 μM (eight compounds) and those in the range of 0.7–10 μM (15 compounds). The most active derivative was resveratrol metabolite resveratrol 4′-sulfate (IC50 = 0.7 μM, 14); another six inhibitors with high potency and specificity toward ODC are presented in Table 5.
Table 5.
Selective inhibition of TPA-induced ODC activity by resveratrol derivatives.
| Compounds tested | IC50 (μM)* | |
|---|---|---|
| 37b |
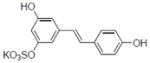
|
1.2±0.22 |
| 38 |
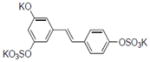
|
1.0±0.16 |
| 39 |
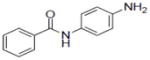
|
0.9±0.24 |
| 40b |
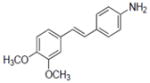
|
0.9±0.15 |
| 41 |
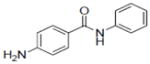
|
0.8±0.14 |
| 42b |
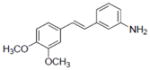
|
2.1±0.38 |
Concentration which caused 50% TPA-induced ODC activity. Apigenin (IC50 = 6.0 μM), menadione (IC50 = 8.3 μM), and deguelin (IC50 = 0.1 μM) were used as positive controls.
Compounds 37, 40, and 42 inhibited COX-1 with IC50 values of 1.19±0.18, 1.92±1.3, and 1.54±0.64 μM, respectively. Compound 37 inhibited COX-2 with and IC50 value of 1.41±0.51 μM.
As shown in Table 1, resveratrol selectively interacted with ERα, as did compound 7. Other derivatives mediating pleiotropic activities did not generally show selectivity for estrogen type. However, some of the derivatives devoid of other activities did interact with estrogen receptors (Table 6). Among these, compounds 46 and 47 were ERβ selective, whereas other compounds bound to both ERα and ERβ. The potent botanical estrogen 8-prenylnaringenin was included as a positive control, and it bound tightly to both ERα and ERβ.
Table 6.
Selective binding of resveratrol derivatives to human estrogen receptors.a
| Compounds tested | ERα Signal enrichment | ERβ Signal enrichment | |
|---|---|---|---|
| 43b |
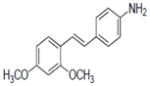
|
3.99 | 4.87 |
| 44 |
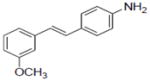
|
2.57 | NDc |
| 45b |
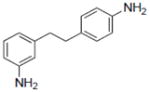
|
2.17 | 2.91 |
| 46b |
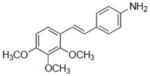
|
1.87 | 3.53 |
| 47b |
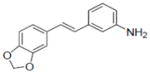
|
1.76 | 4.62 |
| 48b |
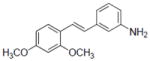
|
1.67 | 2.27 |
8-Prenylnaringenin yielded values of 21.07 and 15.54 for ERα and ERβ, respectively.
Compounds 43, 45, 46, 47 and 48 inhibited COX-1 with IC50 values of 0.8±0.01, 2.8±0.53, 0.9±0.42, 1.2±0.37 and 3.2±0.75 μM, respectively. Compounds 43, 46 and 48 inhibited COX-2 with and IC50 values of 1.7±0.29, 6.4±1.6 and 2.7±0.73 μM, respectively.
In the DPPH assay, compound 9, which has three –NH2 groups, showed the most active scavenging activity with an IC50 of 42.2 μM. Substituting an amino group with a methoxy group (10, 11) produced good scavenging activity, although the resulting compounds are less potent than 9. Resveratrol had an IC50 value of 78 μM in the DPPH assay.
As demonstrated previously, resveratrol inhibits COX-1 and -2, and this is considered a target of physiological relevance. Of the derivatives tested, several were found to inhibit these enzymes with potency roughly comparable to the parent compound. None of the derivatives was found to selectively inhibit COX-2. However, as summarized in Table 7, five derivatives were found to inhibit COX-1 without affecting the other targets described herein.
Table 7.
Selective inhibition of COX-1 mediated by resveratrol derivatives.
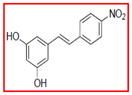
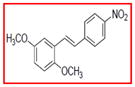
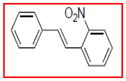
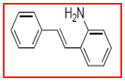
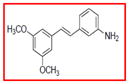
Sample concentration which caused 50% inhibition of COX-1. Resveratrol was used as a positive control (Table 1)
COX-2 IC50 = 19.5±1.4 μM.
All compounds were tested for anti-proliferation effects using three different cancer cell lines: MCF7, PC-3, and HL-60 (data not shown). Compound 10 showed the most potent activity in all three cancer cell lines: in MCF7 cells, the IC50 was 0.02 μM, and with PC- 3 and HL- 60 cells IC50 values were 2 and 0.5 μM, respectively. The second most active compound was 6, which has methoxy groups at the 3, 4, and 5 positions, and an amino group at the 4′ position. However, the cytotoxic effect is decreased when one –OCH3 is removed (5), the double bond between the two rings in the stilbene backbone structure is reduced (15), or the –OCH3 position is changed (11).
Preliminary animal studies
Substituted stilbene 5 was found to possess characteristics attributable to a chemopreventive agent via different pathways (Table 1). It inhibits ornithine decarboxylase induction (IC50 of 1.3 μM), modestly inhibits NFκB (IC50 of 5.8 μM), induces QR1 by 1.9-fold, and inhibits aromatase with IC50 value of 0.31 μM. Compound 5 has been synthesized on a multi-gram scale [8]. To determine the feasibility of chemopreventive studies with animals, large-scale synthesis has been completed [8], and preliminary absorption and metabolism studies were performed. As shown in Table 7, the apparent permeability coefficients (Papp) of sucrose across the Caco-2 cell monolayers was ~0.1 x 10−6 cm/sec, and the Papp of propranol was >30 x 10−6 cm/sec, indicating that the Caco-2 cell monolayers utilized in this study possessed suitable monolayer integrity and function. The Papp values of compound 5 in the apical to basolateral direction ranged from 8.6 to 12.6 x 10−6 cm/sec depending on the initial concentration. Since the permeability of 5 decreased as its initial concentration increased, participation of a saturable transporter was indicated. In the basolateral direction, the Papp values of 5 were 25 to 42% higher, which indicated that 5 is the substrate for an efflux protein. Treatment of the Caco-2 cells with the p-glycoprotein inhibitor verapamil enhanced the rate at which 5 crossed the Caco-2 monolayer in the apical to basolateral direction (Table 8). Therefore, 5 appears to be a substrate for p-glycoprotein.
Table 8.
Apparent permeability coefficients of compound 5 across Caco-2 cell monolayers.
| Test compound (initial conc., μM) | Apparent permeability coefficients (Papp)a Papp x 10−6 cm/sec |
||
|---|---|---|---|
| Papp(AP → BL) | Papp(BL → AP) | Ratio | |
| 5 (10) | 12.6 ± 0.9a | 15.8 ± 1.4 | 0.79 |
| 5 (25) | 10.5 ± 2.4 | 14.1 ± 0.2 | 0.74 |
| 5 (50) | 8.6 ± 1.0 | 12.2 ± 0.5 | 0.70 |
|
| |||
| 5 (10) + Verapamil (50) | 24.9 ± 0.3 | 14.1 ± 0.2 | 1.8 |
| 5 (25) + Verapamil (50) | 13.8 ± 0.2 | 15.1 ± 2.9 | 0.91 |
| 5 (50) + Verapamil (50) | 8.6 ± 0.8 | b | |
|
| |||
| Sucrose (50) | 0.14 ± 0.04 | ||
| Propranolol (10) | 32.1 ± 1.2 | ||
Mean ± S.D. (N = 3)
Since the rate at which 50 μM compound 2 crossed the Caco-2 cell monolayer in the basolateral to apical direction was non-linear in the presence of verapamil, the Papp value could not be calculated.
Following oral administration to rats, the concentrations of 5 in plasma and tissue samples were determined using LC-MS-MS (Table 9). The level of 5 in serum was high (3.21 ± 0.12 μg/mL) during the first day. After 3 days, this was reduced to 0.64 ± 0.034 μg/mL. Comparable concentrations were observed in mammary tissue and perirenal fat; the highest levels were in liver. These data indicate high serum and tissue levels can be achieved by oral administration, but dosing regimens should be further evaluated. In vitro studies with plasma proteins indicated 99.4 ± 0.11% (mean ± S.D; n = 3) of compound 5 was bound.
Table 9.
In vivo disposition of compound 5 in the rat.
| Compound 5 40 mg/kg | Serum 1-day (μg/mL) N=9 |
Serum 3-day (μg/mL) N=9 |
Liver (μg/g) N=9 |
Mammary gland (μg/g) Pooled, N=3 |
Fat (μg/g) Pooled, N=3 |
|---|---|---|---|---|---|
| Rat 1 | 2.99 ± 0.12 | 1.04 ± 0.005 | 4.68 ± 0.57 | -- | -- |
| Rat 2 | 2.90 ± 0.09 | 0.67 ± 0.005 | 9.88 ± 0.057 | -- | -- |
| Rat 3 | 4.00 ± 0.06 | 0.21 ± 0.001 | 5.61 ± 0.91 | -- | -- |
| Total | 3.21 ± 0.12 | 0.64 ± 0.034 | 6.28 ± 2.36 | 0.51 ± 0.01 | 0.69 ± 0.04 |
Discussion
As described herein, 92 resveratrol derivatives were synthesized and biological activity was assessed with various chemoprevention targets. As with the parent molecule, resveratrol, several of the derivatives mediated pleiotropic effects (Table 1). For example, compound 2, with a para-amino group in trans-stilbene backbone structure, affected several targets, but inhibition of NO production was particularly notable. On the other hand, when methoxylated to yield compound 5, pleiotropic responses were still observed, but inhibition of aromatase and QR2 were especially enhanced. It is notable that resveratrol interacts with QR2 with a Kd value of 40 nM [91] and mediates inhibitory activity with an IC50 value of 6.9 μM (Table 1). Compound 5 demonstrates greater inhibitory potential, and this is further enhanced by placement of an additional methoxy-group, as illustrated by compound 6. These compounds are calculated by molecular modeling to bind to the enzyme in a similar orientation to that of resveratrol, but interaction occurs with the side chain of Thr71, whereas resveratrol binds to this residue indirectly through a water molecule [8]. It may be suggested that these new analogs bind to QR2 with a Kd value in the 1 to 10 nM range.
Compound 5 was subjected to preliminary absorption and metabolism studies. High Caco-2 cell permeability suggests that intestinal absorption would be rapid and complete following oral administration. The in vivo data obtained using oral administration of 5 to rats are consistent with these in vitro Caco-2 cell monolayer permeability studies. Good absorption and stability were observed, unlike the parent molecule, which is known to be rapidly and extensively metabolized. In addition, a high level of binding with human plasma proteins was noted. Since only free drug would be available for metabolism or excretion, binding of this type might enhance the in vivo half-life which would be beneficial for chemoprevention agents. However, strong binding to plasma proteins might also increase the probability of drug-drug interactions due to competition between drugs for plasma protein binding sites.
In addition to compounds 5 and 6, several derivatives were found to inhibit aromatase activity in the sub-micromolar range, e.g., 4, 8, and 14. The most promising inhibitors of aromatase, however, are shown in Table 2, with 21 and 23 mediating a selective response in the nM concentration range. Analysis of structure-activity relationships revealed that the type and position of groups on ring B were more important for aromatase inhibitory activity than those on ring A. An amino group on ring B yielded more potent inhibitors than a nitro, halogen, hydroxyl, nitrile, aceto, or aminomethyl group. An amino group in the para-position resulted in greater inhibitory potency than the same group in an ortho- or meta-position. Although changing groups on ring A had less impact on enzyme activity, some conclusions could be drawn from the series regarding these groups. Methoxy groups on ring A yielded more inhibitory activity than hydroxyl or acetyl groups. A 3,5-arrangement of methoxy groups was found to be optimal. Thus, compound 5 [(E)-4-(3,5-dimethoxystyryl)aniline] was one of the most active compounds of the series, with an IC50 of 0.31 μM. Addition of an imidazole group increased inhibitory potency against aromatase significantly, and the most active compound in the series demonstrated an IC50 value of 36 nM (8). Molecular modeling indicated that one of the imidazole nitrogens coordinates with the iron atom of the aromatase heme, which plays a critical role in catalysis [8].
As demonstrated previously, NFκB inhibition requires the planar conformation of resveratrol, preferring large, hydrophobic rings and a catechol at the R3 position [80]. Table 1 illustrates active inhibitors of NFκB with pleiotropic effects. Interestingly, bond reduction between rings negatively impacted sensitivity towards iNOS, aromatase, NFκB, and growth inhibition with MCF-7 and HL-60 cells (compare 6 and 15). In general, chemical modification does not greatly enhance NFκB inhibitory potential (Table 1, refs. 80, 82). However, as summarized in Table 3, six derivatives were identified that selectively inhibited NFκB with IC50 values lower than that observed with resveratrol.
Resveratrol is a modest inducer of QR1. Of the derivatives tested, some were found to be approximately equipotent, but no selective inducers were found. Overall, compound 17 demonstrated the greatest promise. In contrast to the NADP-dependent QR1, which is upregulated by cancer chemopreventive agents via the ARE (antioxidant-responsive element), the inhibition of QR2 by resveratrol or derivatives might protect cells from harmful metabolically activated compounds that can promote DNA damage and therefore cancer [12]. Three selective inhibitors were identified (Table 4) that should be of interest in future work.
The strongest inhibitor of ODC induction was the natural metabolite resveratrol-4′-sulfate (15). We showed earlier that this metabolite inhibited NFκB induction, as well as COX-1 and COX-2 activities [82]. Rapid metabolism of resveratrol during oral administration leads to production sulfates and glucuronides [93]. The ability of sulfate metabolite to inhibit ODC further supports the potential importance of resveratrol metabolites, although other derivatives are selectively active in this process (Table 5).
Estrogen regulates growth, differentiation, and functioning of many target tissues. Resveratrol, being structurally similar to the synthetic estrogenic compound diethylstilbestrol, has been demonstrated to be a competitive inhibitor of binding to the estrogen receptor in human breast cancer cells MCF-7 [94]. The estrogenic activity of resveratrol as an agonist of estrogen receptors has been documented [95], although the relevance of such an effect is moot [7]. Estrogen receptors α and β (ER1 and ER2) are members of the nuclear receptor family, which controls many processes in cells, including cancer development. Binding of agonistic or antagonistic molecules to these receptors is a well-established approach in the search for anticancer agents. As shown in Table 1, resveratrol selectively interacts with ERα, as did compound 7. Other derivatives such as 46 and 47 were ERβ selective. It would be of value to study these types of interactions in greater detail by means of X-ray crystallographic analysis. These compounds should be of value in performing such studies.
With our experimental conditions, resveratrol inhibits COX-1 and COX-2, but greater activity was observed with COX-1 (Table 1). Similar to resveratrol, several derivatives active as COX inhibitors blocked both forms of the enzyme (2, 4, 5, 27, 31, 27, 43, 46, 48), but no COX-2 specific inhibitors were found. On the other hand, five compounds were identified as selective inhibitors of COX-1. The significance of this response remains to be explored. It may be noted, however, resveratrol mediates a significant anti-inflammatory response in a rat model [1], and, as noted above, greater activity is observed with COX-1.
The SARs that can be derived from the data set described herein are extensive. Some highlights are given in the text but certainly additional concepts can be gleaned from these results. Relative to resveratrol, it is clear that improvements in activity can be achieved. In some cases, as with resveratrol, pleiotropic activities can be observed with derivatives, albeit with greater potency. On the other hand, a total lack of activity can be generated, as shown in Figure 1, which can teach us something about SARs. Perhaps of greatest interest are chemical derivatives that demonstrate much greater specificity with much greater potency. In many cases, these novel compounds demonstrate activity even greater than the positive control compounds used for the respective assays, suggesting further exploration as therapeutic or preventative agents is reasonable.
Figure 1.

Compounds devoid of significant biological activity in the assays tested.a
aEach of the compounds shown in the figure were tested and found to be without significant activity by the following criteria (IC50 >n μM): NO (35); Aromatase (25); NFκB (20); QR1 (CD>50); QR2 (50);ODC (20); DPPH (340); COX-1 and -2 (68).
Acknowledgments
The authors are grateful to Ke Huang and Lian Chen for assistance with plasma protein binding and metabolic stability studies, respectively. This work was supported by program project grant P01 CA48112 awarded by the National Cancer Institute.
Abbreviations
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- DPPH
2,2-diphenyl-1-picrylhydrazyl
- ER
estrogen reseptor
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MTT
3-(4,5-dimetylthiazo-2-yl)-2,5-diphenyltetrazolium bromide
- ODC
ornithine decarboxylase
- QR1
quinone reductase-1
- QR2
quinone reductase-2
- ROS
reactive oxygen species
- TPCK
N-tosyl-L-phenylalaninyl-chlormethylketone
- TPA
12-O-tetradecanoylphorbol 13-acetate
Footnotes
Conflict of interest statement
The authors have declared no conflict of interest.
References
- 1.Jang M, Cai L, Udeani GO, Slowing KV, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 2.Francy-Guilford J, Pezzuto J. Mechanisms of cancer chemopreventive agents: a perspective. Planta Medica. 2008;74:1644–1650. doi: 10.1055/s-2008-1074553. [DOI] [PubMed] [Google Scholar]
- 3.Pezzuto JM. Resveratrol as an inhibitor of carcinogenesis. Pharm Biol. 2008;46:443–573. [Google Scholar]
- 4.Tong KI, Katoh Y, Kusunoki H, Itoh K, et al. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26:2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nho C, Jeffery E. The synergistic upregulation of phase II detoxification enzymes by glucosinolate breakdown products in cruciferous vegetables. Toxicol App Pharmacol. 2001;174:146–152. doi: 10.1006/taap.2001.9207. [DOI] [PubMed] [Google Scholar]
- 6.Kennelly E, Gerhäuser C, Song L, Graham J, et al. Induction of quinone reductase by withanolides isolated from Physalis philidelphica (tomatillos) J Agri Food Chem. 1997;45:3771–3777. [Google Scholar]
- 7.Pezzuto JM. The phenomenon of resveratrol: Redefining the virtues of promiscuity. Ann NY Acad Sci. 2011;1215:123–130. doi: 10.1111/j.1749-6632.2010.05849.x. [DOI] [PubMed] [Google Scholar]
- 8.Sun B, Hoshino J, Jermihov K, Marler L, et al. Design, synthesis, and biological evaluation of resveratrol analogues as aromatase and quinone reductase 2 inhibitors for chemoprevention of cancer. Bioorg Med Chem. 2010;18:5352–5366. doi: 10.1016/j.bmc.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pezzuto JM. In: Resveratrol in Health and Disease. Aggarwal BB, Shishodia S, editors. Marcel Dekker, Inc; New York, New York: 2006. pp. 233–383. [Google Scholar]
- 10.Pezzuto JM, Kondratyuk TP, Shalaev E. In: Carcinogenic and Anticarcinogenic Food Components. Baer-Dubowska W, Bartoszek A, Malejka-Giganti D, editors. CRC Press; Boca Raton, Florida: 2006. pp. 239–282. [Google Scholar]
- 11.Harikumar KB, Aggarval BB. Resveratrol: Amultitargeted agent for age-associated chronic diseases. Pleiotropic mechanisms facilitated by resveratrol and its metabolites. Cell Cycle. 2008;7:1020–1035. doi: 10.4161/cc.7.8.5740. [DOI] [PubMed] [Google Scholar]
- 12.Calamini B, Ratia K, Malkowski M, Cuendet M, et al. Pleiotropic mechanisms facilitated by resveratrol and its metabolites. Biochem J. 2010;429:273–282. doi: 10.1042/BJ20091857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aggarwal BA, Bhardwaj A, Aggarwal RS, Seeram NP, et al. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24:3–60. [PubMed] [Google Scholar]
- 14.Brown VA, Patel KR, Viskaduraki M, Crowell JA, Perloff M, et al. Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res. 2010;70:9003–9011. doi: 10.1158/0008-5472.CAN-10-2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burns J, Yokota T, Ashihara H, Lean M, Crozier A. Plant foods and herbal sources of resveratrol. J Agric Food Chem. 2002;50:3337–3340. doi: 10.1021/jf0112973. [DOI] [PubMed] [Google Scholar]
- 16.Rimando A, Kalt W, Magee J, Dewey J, Ballington J. Resveratrol, pterostilbene, and piceatannol in vaccinium berries. J Agri Food Chem. 2004;52:4713–4719. doi: 10.1021/jf040095e. [DOI] [PubMed] [Google Scholar]
- 17.Bhattacharya A, Sood P, Citovsky V. The roles of plant phenolics in defence and communication during Agrobacterium and Rhizobium infection. Mol Plant Pathol. 2010;11:705–719. doi: 10.1111/j.1364-3703.2010.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hain R, Bieseler B, Kindl H, Schroder G, Stocker R. Expression of a stilbene synthase gene inNicotiana tabacum results in synthesis of the phytoalexin resveratrol. Plant Mol Biol. 1990;15:325–335. doi: 10.1007/BF00036918. [DOI] [PubMed] [Google Scholar]
- 19.Takaoka M. Resveratrol, a new phenolic compound, from Veratrum grandiflorum. J Chem Soc Japan. 1939;60:1090–1100. [Google Scholar]
- 20.Nonomura S, Kanagawa H, Makimoto A. Chemical constituents of polygonaceous plants. I. Studies on the components of Ko-Jo-Kon. (Polygonum cuspidatum Sieb. et Zucc.) J Pharm Soc Japan. 1963;83:988–990. [PubMed] [Google Scholar]
- 21.Siemann EH, Creasy LL. Concentration of the phytoalexin resveratrol in wine. Am Soc Enol Vitic. 1992;43:49–52. [Google Scholar]
- 22.Gronbaek M, Deis A, Sorensen TI, Becker U, et al. Mortality associated with moderate intakes of wine, beer, or spirits. BMJ. 1995;310:1165–1169. doi: 10.1136/bmj.310.6988.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richard JL. Coronary risk factors. The French paradox. Arch Mal Coeur Vaiss. 1987;80:17–21. [PubMed] [Google Scholar]
- 24.Stewart JR, Artime MC, O’Brian CA. Resveratrol: A candidate nutritional substance for prostate cancer prevention. J Nutr. 2003;133:2440S–2443S. doi: 10.1093/jn/133.7.2440S. [DOI] [PubMed] [Google Scholar]
- 25.Goldberg DM, Yan J, Ng E, Diamandis EP, et al. A global survey of trans-resveratrol concentrations in commercial wines. Am J Enol Vitic. 1995;46:159–165. [Google Scholar]
- 26.Zern TL, West KL, Fernandez ML. Grape polyphenols decrease plasma triglycerides and cholesterol accumulation in the aorta of ovariectomized guinea pigs. J Nutr. 2003;133:2268–2272. doi: 10.1093/jn/133.7.2268. [DOI] [PubMed] [Google Scholar]
- 27.Fitzpatrick DF, Hirschfield SL, Coffey RG. Endothelium dependent vasorelaxing activity of wine and other grape products. Am J Physiol. 1993;265:H774–778. doi: 10.1152/ajpheart.1993.265.2.H774. [DOI] [PubMed] [Google Scholar]
- 28.Lekakis J, Rallidis LS, Andreadou I, Vamvakou G, et al. Polyphenolic compounds from red grapes acutely improve endothelial function in patients with coronary heart disease. Eur J Cardiovasc Prev Rehabil. 2005;12:596–600. doi: 10.1097/00149831-200512000-00013. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Zou J, Cao K, Hsieh TC, et al. Dealcoholized red wine containing known amounts of resveratrol suppresses atherosclerosis in hypercholesterolemic rabbits without affecting plasma lipid levels. Int J Mol Med. 2005;16:533–540. [PubMed] [Google Scholar]
- 30.Shen MY, Hsiao G, Liu CL, Fong TH, et al. Inhibitory mechanisms of resveratrol in platelet activation: Pivotal roles of p38 MAPK and NO/cyclic GMP. Br J Haematol. 2007;139:475–485. doi: 10.1111/j.1365-2141.2007.06788.x. [DOI] [PubMed] [Google Scholar]
- 31.Meeran SM, Katiyar SK. Cell cycle control as a basis for cancer chemoprevention through dietary agents. Front Biosci. 2008;13:2191–2202. doi: 10.2741/2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clément MV, Hirpara JL, Chawdhury SH, Pervaiz S. Chemopreventive agent resveratrol, a natural product derived from grapes, triggers CD95 signaling-dependent apoptosis in human tumor cells. Blood. 1998;92:996–1002. [PubMed] [Google Scholar]
- 33.Hadi SM, Asad SF, Singh S, Ahmad A. Putative mechanism for anticancer and apoptosis-inducing properties of plant-derived polyphenolic compounds. IUBMB Life. 2000;50:167–171. doi: 10.1080/152165400300001471. [DOI] [PubMed] [Google Scholar]
- 34.Belleri M, Ribatti D, Nicoli S, Cotelli F, et al. Antiangiogenic and vascular-targeting activity of the microtubule-destabilizing trans-resveratrol derivative 3,5,4′-trimethoxystilbene. Mol Pharmacol. 2005;67:1451–1459. doi: 10.1124/mol.104.009043. [DOI] [PubMed] [Google Scholar]
- 35.Surh YJ, Chun KS, Cha HH, Han SS, et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat Res. 2001;480–481:243–268. doi: 10.1016/s0027-5107(01)00183-x. [DOI] [PubMed] [Google Scholar]
- 36.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leonard SS, Xia C, Jiang BH, Stinefelt B, et al. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem Biophys Res Commun. 2003;309:1017–1026. doi: 10.1016/j.bbrc.2003.08.105. [DOI] [PubMed] [Google Scholar]
- 38.Manna SK, Mukhopadhyay A, Aggarwal BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: Potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol. 2000;164:6509–6519. doi: 10.4049/jimmunol.164.12.6509. [DOI] [PubMed] [Google Scholar]
- 39.Athar M, Back JH, Tang X, Kim KH, et al. Resveratrol: A review of preclinical studies for human cancer prevention. Toxicol Appl Pharmacol. 2007;224:274–283. doi: 10.1016/j.taap.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu X, Xuan Y. Bypassing cancer drug resistance by activating multiple death pathways--A proposal from the study of circumventing cancer drug resistance by induction of necroptosis. Cancer Lett. 2008;259:127–137. doi: 10.1016/j.canlet.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 41.Raptis S, Bapat B. Genetic instability in human tumors. EXS. 2006:303–320. doi: 10.1007/3-7643-7378-4_13. [DOI] [PubMed] [Google Scholar]
- 42.Bhat KPL, Kosmeder JW, 2nd, Pezzuto JM. Biological effects of resveratrol. Antioxid Redox Signal. 2001;3:1041–1064. doi: 10.1089/152308601317203567. [DOI] [PubMed] [Google Scholar]
- 43.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 44.Wei S, Kulp SK, Chen CS. Energy restriction as an antitumor target of thiazolidinediones. J Biol Chem. 2010;285:9780–9791. doi: 10.1074/jbc.M109.065466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cottart CH, Nivet-Antoine V, Laguillier-Morizot C, Beaudeux JL. Resveratrol bioavailability and toxicity in humans. Mol Nutr Food Res. 2010;54:7–16. doi: 10.1002/mnfr.200900437. [DOI] [PubMed] [Google Scholar]
- 46.Kraft TE, Parisotto D, Schempp C, Efferth T. Fighting cancer with red wine? Molecular mechanisms of resveratrol. Crit Rev Food Science Nutr. 2009;49:782–799. doi: 10.1080/10408390802248627. [DOI] [PubMed] [Google Scholar]
- 47.Trela BC, Waterhouse AL. Resveratrol: Isomeric molar absorptivities and stability. J Agric Food Chem. 1996;44:1253–1257. [Google Scholar]
- 48.Leiro J, Alvarez E, Arranz JA, Laguna R, et al. Effects of cis-resveratrol on inflammatory murine macrophages: Antioxidant activity and down-regulation of inflammatory genes. J Leukoc Biol. 2004;75:1156–1165. doi: 10.1189/jlb.1103561. [DOI] [PubMed] [Google Scholar]
- 49.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 50.Kelloff GJ, Sigman CC, Johnson KM, Boone CW, et al. Perspectives on surrogate end points in the development of drugs that reduce the risk of cancer. Cancer Epidemiol Biomarkers Prev. 2000;9:127–137. [PubMed] [Google Scholar]
- 51.Curtin J, Donovan M, Cotter T. Regulation and measurement of oxidative stress in apoptosis. J Immunol Meth. 2002;265:49–72. doi: 10.1016/s0022-1759(02)00070-4. [DOI] [PubMed] [Google Scholar]
- 52.Lee SK, Mbwambo ZH, Chung H, Luyengi L, et al. Evaluation of the antioxidant potential of natural products. Comb Chem High Throughput Screen. 1998;1:35–46. [PubMed] [Google Scholar]
- 53.Bhat KP, Pezzuto JM. Cancer chemopreventive activity of resveratrol. Ann N Y Acad Sci. 2002;957:210–229. doi: 10.1111/j.1749-6632.2002.tb02918.x. [DOI] [PubMed] [Google Scholar]
- 54.Martinez J, Moreno JJ. Effect of resveratrol, a natural polyphenolic compound, on reactive oxygen species and prostaglandin production. Biochem Pharmacol. 2000;59:865–870. doi: 10.1016/s0006-2952(99)00380-9. [DOI] [PubMed] [Google Scholar]
- 55.Yen GC, Duh PD, Lin CW. Effects of resveratrol and 4-hexylresorcinol on hydrogen peroxide-induced oxidative DNA damage in human lymphocytes. Free Radic Res. 2003;37:509–514. doi: 10.1080/1071576031000083099. [DOI] [PubMed] [Google Scholar]
- 56.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Subbaramaiah K, Chung WJ, Michaluart P, Telang N, et al. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J Biol Chem. 1998;273:21875–21882. doi: 10.1074/jbc.273.34.21875. [DOI] [PubMed] [Google Scholar]
- 58.Tsai SH, Lin-Shiau SY, Lin JK. Suppression of nitric oxide synthase and the down-regulation of the activation of NFkappaB in macrophages by resveratrol. Br J Pharmacol. 1999;126:673–680. doi: 10.1038/sj.bjp.0702357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Youn HS, Lee JY, Fitzgerald KA, Young HA, et al. Specific inhibition of MyD88-independent signaling pathways of TLR3 and TLR4 by resveratrol: molecular targets are TBK1 and RIP1 in TRIF complex. J Immunol. 2005;175:3339–3346. doi: 10.4049/jimmunol.175.5.3339. [DOI] [PubMed] [Google Scholar]
- 60.Kundu JK, Shin YK, Kim SH, Surh YJ. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-kappaB in mouse skin by blocking IkappaB kinase activity. Carcinogenesis. 2006;27:1465–1474. doi: 10.1093/carcin/bgi349. [DOI] [PubMed] [Google Scholar]
- 61.Korzeniewski N, Wheeler S, Chatterjee P, Duensing A, Duensing S. A novel role of the aryl hydrocarbon receptor (AhR) in centrosome amplification-implications for chemoprevention. Mol Cancer. 2010;9:153–163. doi: 10.1186/1476-4598-9-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ciolino HP, Daschner PJ, Yeh GC. Resveratrol inhibits transcription of CYP1A1 in vitro by preventing activation of the aryl hydrocarbon receptor. Cancer Res. 1998;58:5707–5712. [PubMed] [Google Scholar]
- 63.Hayes CL, Spink DC, Spink BC, Cao JQ, et al. 17 beta-estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc Natl Acad Sci USA. 1996;93:9776–9781. doi: 10.1073/pnas.93.18.9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peter Guengerich F, Chun YJ, Kim D, Gillam EMTS. Cytochrome P450 1B1: A target for inhibition in anticarcinogenesis strategies. Mutat Res. 2003;523–524:173–182. doi: 10.1016/s0027-5107(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 65.Neves MA, Dinis TC, Colombo G, Sáe Melo ML. Combining computational and biochemical studies for a rationale on the anti-aromatase activity of natural polyphenols. ChemMedChem. 2007;2:1750–1762. doi: 10.1002/cmdc.200700149. [DOI] [PubMed] [Google Scholar]
- 66.Gonelli A, Mischiati C, Guerrini R, Voltan R, et al. Perspectives of protein kinase C (PKC) inhibitors as anti-cancer agents. Mini Rev Med Chem. 2009;9:498–509. doi: 10.2174/138955709787847967. [DOI] [PubMed] [Google Scholar]
- 67.Jayatilake GS, Jayasuriya H, Lee ES, Koonchanok NM, et al. Kinase inhibitors from Polygonum cuspidatum. J Nat Prod. 1993;56:1805–1810. doi: 10.1021/np50100a021. [DOI] [PubMed] [Google Scholar]
- 68.Schneider Y, Vincent F, Duranton B, Badolo L, et al. Anti-proliferative effect of resveratrol, a natural component of grapes and wine, on human colonic cancer cells. Cancer Lett. 2000;158:85–91. doi: 10.1016/s0304-3835(00)00511-5. [DOI] [PubMed] [Google Scholar]
- 69.Khanduja KL, Bhardwaj A, Kaushik G. Resveratrol inhibits N-nitrosodiethylamine-induced ornithine decarboxylase and cyclooxygenase in mice. J Nutr Sci Vitaminol (Tokyo) 2004;50:61–65. [PubMed] [Google Scholar]
- 70.Chen C, Kong AN. Dietary chemopreventive compounds and ARE/EpRE signaling. Free Radic Biol Med. 2004;36:1505–1516. doi: 10.1016/j.freeradbiomed.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 71.Prochaska HJABS. Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: A screening assay for anticarcinogenic enzyme inducers. Anal Biochem. 1988;169:328–336. doi: 10.1016/0003-2697(88)90292-8. [DOI] [PubMed] [Google Scholar]
- 72.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 73.Athar M, Back JH, Kopelovich L, Bickers DR, Kim AL. Multiple molecular targets of resveratrol: anti-carcinogenic mechanisms. Arch Biochem Biophys. 2009;486:95–102. doi: 10.1016/j.abb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fulda S, Debatin KM. Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res. 2004;64:337–346. doi: 10.1158/0008-5472.can-03-1656. [DOI] [PubMed] [Google Scholar]
- 75.Corder R, Crozier A, Kroon PA. Drinking your health? It’s too early to say. Nature. 2003;426:119. doi: 10.1038/426119d. [DOI] [PubMed] [Google Scholar]
- 76.Yu C, Shin YG, Kosmeder JW, Pezzuto JM, van Breemen RB. Liquid chromatography/tandem mass spectrometric determination of inhibition of human cytochrome P450 isozymes by resveratrol and resveratrol-3-sulfate. Rapid Commun Mass Spectrom. 2003;17:307–313. doi: 10.1002/rcm.918. [DOI] [PubMed] [Google Scholar]
- 77.Walle T, Hsieh F, DeLegge MH, Oatis JJE, Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 78.Boocock DJ, Faust GE, Patel KR, Schinas AM, et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev. 2007;16:1246–1252. doi: 10.1158/1055-9965.EPI-07-0022. [DOI] [PubMed] [Google Scholar]
- 79.Kaldas MI, Walle UK, Walle T. Resveratrol transport and metabolism by human intestinal Caco-2 cells. J Pharm Pharmacol. 2003;55:307–312. doi: 10.1211/002235702612. [DOI] [PubMed] [Google Scholar]
- 80.Kang SS, Cuendet M, Endringer DC, Croy VL, et al. Synthesis and biological evaluation of a library of resveratrol analogues as inhibitors of COX-1, COX-2 and NF-kappaB. Bioorg Med Chem. 2009;17:1044–1054. doi: 10.1016/j.bmc.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 81.Maiti A, Cuendet M, Croy VL, Endringer DC, et al. Synthesis and biological evaluation of (+/−)-abyssinone II and its analogues as aromatase inhibitors for chemoprevention of breast cancer. J Med Chem. 2007;50:2799–2806. doi: 10.1021/jm070109i. [DOI] [PubMed] [Google Scholar]
- 82.Hoshino J, Park EJ, Kondratyuk TP, Marler L, et al. Selective synthesis and biological evaluation of sulfate-conjugated resveratrol metabolites. J Med Chem. 2010;53:5033–5043. doi: 10.1021/jm100274c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Song LL, Kosmeder JW, Lee SK, Gerhauser C, et al. Cancer chemopreventive activity mediated by 4′-bromoflavone, a potent inducer of phase II detoxification enzymes. Cancer Res. 1999;59:578–585. [PubMed] [Google Scholar]
- 84.Kim Y, Min HY, Park HJ, Lee EJ, et al. Suppressive effects of nitric oxide production and inducible nitric oxide synthase (iNOS) gene expression by Calystegia soldanella methanol extract on lipopolysaccharide-activated RAW 264.7 cells. Eur J Cancer Prev. 2004;13:419–424. doi: 10.1097/00008469-200410000-00010. [DOI] [PubMed] [Google Scholar]
- 85.Maiti A, Cuendet M, Kondratyuk T, Croy VL, et al. Synthesis and cancer chemopreventive activity of zapotin, a natural product from Casimiroa edulis. J Med Chem. 2007;50:350–355. doi: 10.1021/jm060915+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cao H, Yu R, Tao Y, Nikolic D, van Breemen RB. Measurement of cyclooxygenase inhibition using liquid chromatography–tandem mass spectrometry. J Pharm Biomed Anal. 2011;54:230–235. doi: 10.1016/j.jpba.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Y, Shin YG, Yu C, Kosmeder JW, Hirschelman WH, Pezzuto JM, van Breemen RB. Increasing the throughput and productivity of Caco-2 cell permeability assays using liquid chromatography-mass spectrometry: application to resveratrol absorption and metabolism. Comb Chem High Throughput Screen. 2003;6:757–767. doi: 10.2174/138620703771826865. [DOI] [PubMed] [Google Scholar]
- 88.Cuendet M, Guo J, Luo Y, Chen S, et al. Cancer chemopreventive activity and metabolism of isoliquiritigenin, a compound found in licorice. Cancer Prev Res. 2010;3:221–232. doi: 10.1158/1940-6207.CAPR-09-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marler L, Maiti A, Conda-Sheridan M, Cushman M, et al. Cancer chemopreventive potential of aromathecins and phenazines, novel natural product derivatives. Anticancer Res. 2010;30:4873–4882. [PubMed] [Google Scholar]
- 90.Kang Y-H, Pezzuto J. Quinones and Quinone Enzymes. In: Sies H, Packer L, editors. Methods in Enzymology. Elsevier Science; San Diego: 2004. pp. 380–415. [Google Scholar]
- 91.Buryanovsky L, Fu Y, Boyd M, et al. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry 2004. 2004;43:11417–11426. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Choi Y, Jermihov K, Nam SJ, Sturdy M, Maloney K, et al. Screening natural products for inhibitors of quinone reductase-2 using ultrafiltration LC-MS. Ana Chem. 2011;83:1048–1052. doi: 10.1021/ac1028424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wenzel E, Soldo T, Erbersdobler H, Somoza V. Bioactivity and metabolism of trans-resveratrol orally administered to Wistar rats. Mol Nut Food Res. 2005;49:482–494. doi: 10.1002/mnfr.200500003. [DOI] [PubMed] [Google Scholar]
- 94.Gehm BD, McAndrews JM, Chien P-Y, Jameson JL. Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc Nat Acad Sc USA. 1997;94:14138–14143. doi: 10.1073/pnas.94.25.14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bowers JL, Tyulmenkov VV, Jernigan SC, Klinge CM. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology. 2000;141:3657–3667. doi: 10.1210/endo.141.10.7721. [DOI] [PubMed] [Google Scholar]