

Abstract
Largazole is a potent class I selective histone deacetylase (HDAC) inhibitor. The majority of largazole analogues to date have modified the thiazole-thiazoline and the warhead moiety. In order to elucidate class I-specific structure–activity relationships, a series of analogues with modifications in the valine or the linker region were prepared and evaluated for their class I isoform selectivity. The inhibition profile showed that the C2 position of largazole has an optimal steric requirement for efficient HDAC inhibition and that substitution of the trans-alkene in the linker with an aromatic group results in complete loss of activity. This data will aid the design of class I isoform selective HDAC inhibitors.
Keywords: Largazole, HDAC inhibitor, Class I histone deacetylase, Isoform selectivity, Structure–activity relationship
Largazole (1, Fig. 1), isolated from a cyanobacterium of the Symploca genus, possesses highly differential growth inhibitory activity towards cancer cells.1 This selectivity is attributed to its very potent class I histone deacetylase (HDAC) inhibitory activity.2,3 Because of its potency, class I selectivity, and in vivo efficacy,4 largazole (1) possesses great potential as an anti-cancer agent.5 Additionally, largazole (1) has shown a variety of other biological activities, such as the in vitro and in vivo induction of osteoblast differentiation biomarkers,6 the sensitization of EBV+ tumor cells to the anti-herpes drug ganciclovir (GCV),7 the inhibition of ubiquitin activating enzyme, E1,8 and the in vitro and in vivo induction of apoptosis in hepatic stellate cells (HSC) in liver fibrosis models.9 These promising biological activities led to 11 total syntheses2,3,10–18 and a wide variety of analogues of largazole (1).2–4,10,12,13,16,18–27
Figure 1.
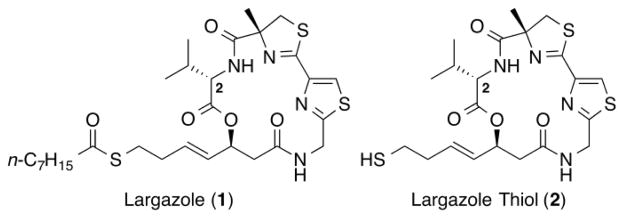
Structure of largazole (1) and largazole thiol (2).
To date, the majority of largazole analogues that have been synthesized and studied have altered the warhead or the thiazole-thiazoline moiety. For example, previous attempts to replace the thiol moiety of largazole thiol (2, Fig. 1) with other Zn2+ chelating groups have resulted in significant decreases in inhibitor potency.2,10,13,21,25 While the active site of HDACs is highly conserved, sequence variety in the cap region is relatively high.28 It is believed that the interactions between this hydrophobic cap region and the macrocycle of largazole (1) influence its class selectivity.5,29 However, changes in the thiazole-thiazoline unit of largazole (1) have not resulted in any significant improvement in its potency or isoform selectivity.18,21–23,26
In contrast, relatively little work has been done on the valine subunit at the C2 position or the nature of the linker as only aliphatic linkers have been studied. Furthermore, the limited work on these analogues has mostly focused on anti-proliferative activity rather than HDAC isoform selectivity. Here we report the class I isoform selectivity profile of several C2 and linker analogues of largazole (1) to provide valuable insights for future isoform selective analogue design.
Previous structure–activity relationships have shown that the valine residue at the C2 position of largazole (1) is able to withstand certain variations without significant loss in HDAC inhibitory activity.4,16,18,19,21 Therefore, compounds 3–6 were prepared by replacing the valine residue with aromatic (Phe, Tyr), basic (His), and acidic (Asp) amino acids to investigate the effect of different chemical functionalities on the class I HDAC isoform selectivity (Fig. 2).30 Compounds 3–6 have been shown to retain anti-proliferative activity in HCT116 colon cancer cells.4
Figure 2.
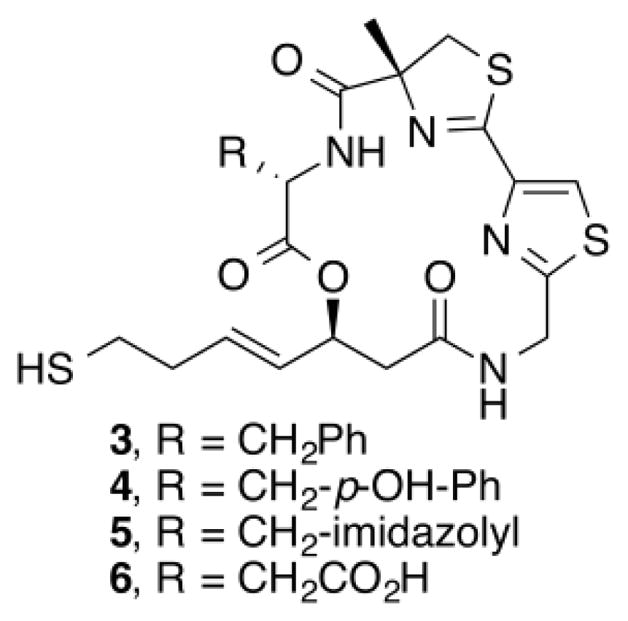
Structure of analogues 3–6.
The inhibition profile showed that 3–6 are very weak HDAC8 inhibitors, implying that the largazole scaffold has an intrinsic preference towards HDACs 1, 2, and 3 over HDAC8 (Table 1). Also, while 3–5 were comparable in potency to largazole thiol (2), aspartic acid analogue 6 experienced a significant reduction in activity. It is notable that there was an overall decrease in HDAC2 inhibition and that histidine analogue 5 showed minor selectivity towards HDAC1 over HDACs 2 and 3 (7- and 5.5-fold, respectively).
Table 1.
Class I HDAC isoform selectivity of 3–6
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | R | IC50 (nM)
|
|
|
|||||
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | ||||||
| 2a |
|
0.40 | 0.90 | 0.70 | 102 | 2.3 | 1.8 | ||
| 3 |
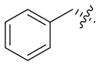
|
0.29 | 1.70 | 0.68 | NIb | 5.9 | 2.3 | ||
| 4 |
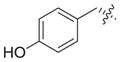
|
0.21 | 1.10 | 0.38 | NIb | 5.2 | 1.8 | ||
| 5 |
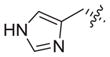
|
0.20 | 1.40 | 1.10 | NIb | 7.0 | 5.5 | ||
| 6 |
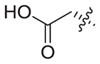
|
39 | 150 | 100 | NIb | 3.9 | 2.6 | ||
Data from ref. 5.
No inhibition up to 1 μM.
Based on these observations, a second set of analogues was designed to isolate possible interactions between HDAC1 and the imidazole ring of 5 (Fig. 3). Compounds 7–10 mimicked the position of the two different nitrogens present in histidine. We expected that the addition of an N-Boc group in 11 would block the hydrogen bond donating ability of the imidazole. The synthesis of 7–11 very closely followed that of 5 and 6.31
Figure 3.
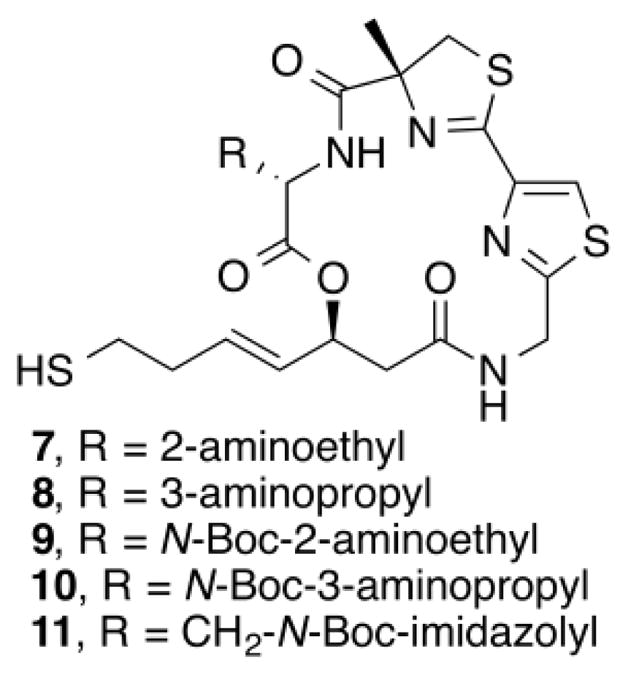
Structure of analogues 7–11.
As summarized in Table 2, compounds 7 and 8 showed decreased activity for all HDACs tested. However, the longer alkyl chain analogue 8 showed a higher potency than the shorter alkyl chain analogue 7 for HDACs 1, 2, and 3. The same trend was observed for the corresponding N-Boc protected analogues, as 10 was more potent than 9 for all HDACs tested. This suggests that there is an optimal steric requirement at the C2 position for efficient HDAC inhibitory activity. The N-Boc protected histidine analogue 11 showed very little change in potency or selectivity, demonstrating that the hydrogen bonding of the histidine might be a non-essential interaction in HDAC inhibition.
Table 2.
Class I HDAC isoform selectivity of 7–11
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | R | IC50 (nM)
|
|
|
|||||
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | ||||||
| 2a |
|
0.40 | 0.90 | 0.70 | 102 | 2.3 | 1.8 | ||
| 7 |
|
5.50 | 21.00 | 7.80 | NIb | 3.8 | 1.4 | ||
| 8 |
|
3.30 | 6.30 | 6.00 | NIb | 1.9 | 1.8 | ||
| 9 |
|
2.30 | 3.10 | 4.20 | NIb | 1.4 | 1.8 | ||
| 10 |
|
0.96 | 1.40 | 1.80 | 790 | 1.5 | 1.9 | ||
| 11 |
|
0.62 | 1.10 | 1.10 | 540 | 1.8 | 1.8 | ||
Data from ref. 5.
No inhibition up to 1 μM.
There are two phenylalanine residues (Phe150 and Phe205) in the active site of HDAC1.32 To utilize π–π stacking interactions for the improvement of HDAC1 selectivity, we designed linker analogues 12–14 via docking studies33 on a previously reported homology model of HDAC1 (Fig. 4).32 Compounds 12–14 were prepared34 and evaluated for their class I HDAC inhibition. Unfortunately, 12–14 showed no activity against class I HDACs. This complete lack of HDAC inhibition could be due to steric effects of bulky aromatic groups in the linker preventing the adoption of docking poses predicted in silico.
Figure 4.
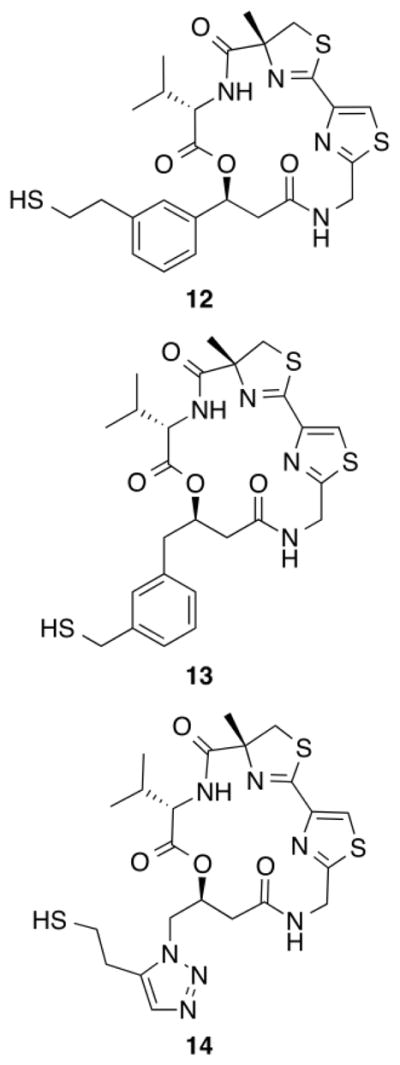
Structure of analogues 12–14.
In summary, we have prepared and tested a variety of C2 and linker analogues and probed them for their class I HDAC inhibition. Our results provide the following insights into the structure–activity relationship of largazole (1). First, the largazole scaffold has an innate preference for HDACs 1, 2, and 3, as every analogue prepared only weakly inhibits HDAC8. Second, the addition of aromatic groups to the linker region is detrimental to HDAC inhibition. Third, the hydrogen bonding of the imidazole ring in the histidine analogue 5 does not play an essential role in HDAC inhibition. Most importantly, the C2 position has an optimal steric requirement that needs to be met for efficient HDAC inhibition. Compounds 7–11 support this insight as the shorter alkyl chain showed weaker inhibition for all class I HDACs. This is further supported by the weak inhibitory activity of the aspartic acid analogue 6, and the relatively strong inhibition of 2–5 and 11. The bulkier groups in these analogues did not significantly reduce the inhibitory activity, suggesting that even larger groups could be installed at this position with minimal loss in HDAC inhibition activity. The trends presented here further extend the existing structure–activity relationship of largazole (1), and will aid in the design process of class I isoform selective HDAC inhibitors.
Supplementary Material
Acknowledgments
This work supported by the National Institutes of Health (National Cancer Institute, R01CA138544). We are grateful to the North Carolina Biotechnology Center (NCBC; Grant No. 2008-IDG-1010) for funding of the NMR instrumentation and to the National Science Foundation (NSF) MRI Program (Award ID No. 0923097) for funding mass spectrometry instrumentation. P.E.S. was supported by the Chemistry and Applications of Smart Molecules and Materials REU program funded by the NSF and DoD (NSF Award #1062607).
Footnotes
Supplementary data associated with this article can be found…
Notes
HL is a co-founder of Oceanyx Pharmaceuticals, Inc., which is negotiating licenses for largazole-related patents and patent applications.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Taori K, Paul VJ, Luesch H. J Am Chem Soc. 2008;130:1806. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 2.Ying Y, Taori K, Kim H, Hong J, Luesch H. J Am Chem Soc. 2008;130:8455. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 3.Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. J Am Chem Soc. 2008;130:11219. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu YX, Salvador LA, Byeon S, Ying Y, Kwan JC, Law BK, Hong J, Luesch H. J Pharmacol Exp Ther. 2010;335:351. doi: 10.1124/jpet.110.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong J, Luesch H. Nat Prod Rep. 2012;29:449. doi: 10.1039/c2np00066k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SU, Kwak HB, Pi SH, You HK, Byeon SR, Ying Y, Luesch H, Hong J, Kim SH. ACS Med Chem Lett. 2011;2:248. doi: 10.1021/ml1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh SK, Perrine SP, Williams RM, Faller DV. Blood. 2012;119:1008. doi: 10.1182/blood-2011-06-362434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ungermannova D, Parker SJ, Nasveschuk CG, Wang W, Quade B, Zhan G, Kuchta RD, Phillips AJ, Liu XD. PLoS ONE. 2012;7:e29208. doi: 10.1371/journal.pone.0029208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu YQ, Wang Z, Wang JN, Lam W, Kwong S, Li FR, Friedman SL, Zhou SY, Ren Q, Xu ZS, Wang XG, Ji L, Tang SB, Zhang H, Lui EL, Ye T. Liver Int. 2013;33:504. doi: 10.1111/liv.12034. [DOI] [PubMed] [Google Scholar]
- 10.Seiser T, Kamena F, Cramer N. Angew Chem Int Ed. 2008;47:6483. doi: 10.1002/anie.200802043. [DOI] [PubMed] [Google Scholar]
- 11.Ren Q, Dai L, Zhang H, Tan WF, Xu ZS, Ye T. Synlett. 2008:2379. [Google Scholar]
- 12.Numajiri Y, Takahashi T, Takagi M, Shin-Ya K, Doi T. Synlett. 2008:2483. [Google Scholar]
- 13.Nasveschuk CG, Ungermannova D, Liu XD, Phillips AJ. Org Lett. 2008;10:3595. doi: 10.1021/ol8013478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh AK, Kulkarni S. Org Lett. 2008;10:3907. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang B, Forsyth CJ. Synthesis. 2009:2873. [Google Scholar]
- 16.Zeng X, Yin BL, Hu Z, Liao CZ, Liu JL, Li S, Li Z, Nicklaus MC, Zhou GB, Jiang S. Org Lett. 2010;12:1368. doi: 10.1021/ol100308a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao Q, Wang LP, Jiao XZ, Liu XY, Wu QA, Xie PJ. Asian Nat Prod Res. 2010;12:940. doi: 10.1080/10286020.2010.510114. [DOI] [PubMed] [Google Scholar]
- 18.Benelkebir H, Marie S, Hayden AL, Lyle J, Loadman PM, Crabb SJ, Packham G, Ganesan A. Bioorg Med Chem. 2011;19:3650. doi: 10.1016/j.bmc.2011.02.024. [DOI] [PubMed] [Google Scholar]
- 19.Ying Y, Liu YX, Byeon SR, Kim H, Luesch H, Hong J. Org Lett. 2008;10:4021. doi: 10.1021/ol801532s. [DOI] [PubMed] [Google Scholar]
- 20.Bowers AA, Greshock TJ, West N, Estiu G, Schreiber SL, Wiest O, Williams RM, Bradner JE. J Am Chem Soc. 2009;131:2900. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowers AA, West N, Newkirk TL, Troutman-Youngman AE, Schreiber SL, Wiest O, Bradner JE, Williams RM. Org Lett. 2009;11:1301. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen F, Gao AH, Li J, Nan FJ. Chem Med Chem. 2009;4:1269. doi: 10.1002/cmdc.200900125. [DOI] [PubMed] [Google Scholar]
- 23.Souto JA, Vaz E, Lepore I, Poppler AC, Franci G, Alvarez R, Altucci L, de Lera AR. J Med Chem. 2010;53:4654. doi: 10.1021/jm100244y. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Huang PH, Chen CS, Forsyth CJ. J Org Chem. 2011;76:1140. doi: 10.1021/jo102478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhansali P, Hanigan CL, Casero RA, Tillekeratne LMV. J Med Chem. 2011;54:7453. doi: 10.1021/jm200432a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li XL, Tu ZC, Li H, Liu CP, Li Z, Sun Q, Yao YW, Liu JS, Jiang S. ACS Med Chem Lett. 2013;4:132. doi: 10.1021/ml300371t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schotes C, Ostrovskyi D, Senger J, Schmidtkunz K, Jung M, Breit B. Chem —Eur J. 2014;20:2164. doi: 10.1002/chem.201303300. [DOI] [PubMed] [Google Scholar]
- 28.Grozinger CM, Schreiber SL. Chem Biol. 2002;9:3. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 29.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. J Am Chem Soc. 2011;133:12474. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.For details on the synthesis of 3–6, see the Supporting Information.
- 31.For details on the synthesis of 7–11, see the Supporting Information.
- 32.Wang DF, Helquist P, Wiech NL, Wiest O. J Med Chem. 2005;48:6936. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- 33.For details on the docking study, see the Supporting Information.
- 34.For details on the synthesis of 12–14, see the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.