

Abstract
Here, we report the isolation and functional characterization of mAbs against two murine norovirus (MNV) strains, MNV-1 and WU20, which were isolated following oral infection of mice. The mAbs were screened for reactivity against the respective homologous and heterologous MNV strain by ELISA. Selected mAbs were of IgA, IgG1, IgG2a or IgG2b isotype and showed a range of Western blot reactivities from non-binding to strong binding, suggesting recognition of conformational and linear epitopes. Some of the anti-MNV-1 antibodies neutralized both MNV-1 and WU20 infections in culture and in mice, but none of the anti-WU20 mAbs neutralized either virus. The non-neutralizing anti-MNV-1 IgG2b antibody 5C4.10 was mapped to the S domain of the MNV-1 capsid, whilst the epitopes of the neutralizing anti-MNV-1 IgA antibodies 2D3.7 and 4F9.4 were mapped to the P domain. Generation of neutralization escape viruses showed that two mutations (V339I and D348E) in the C′D′ loop of the MNV-1 P domain mediated escape from mAb 2D3.7 and 4F9.4 neutralization. These findings broaden the known neutralizing epitopes of MNV to the main surface-exposed loops of the P domain. In addition, the current panel of antibodies provides valuable reagents for studying norovirus biology and development of diagnostic tools.
Introduction
Murine noroviruses (MNVs) are the most prevalent endemic pathogen in mice research facilities in the USA and Europe (Henderson, 2008). MNV has also been described in wild rodents, including house mice, large field Japanese mice and the European wood mouse (Ohsugi et al., 2013; Smith et al., 2012). MNV, like the related human noroviruses (HuNoVs), are non-enveloped, positive-sense, ssRNA viruses in the family Caliciviridae (Green, 2007). MNV and HuNoV are genetically similar, and both are gastrointestinal pathogens that are transmitted by the faecal–oral route (Wobus et al., 2006). HuNoVs are a leading cause of non-bacterial gastroenteritis (Glass et al., 2009). The development of HuNoV vaccines has been difficult due largely to the unavailability of tissue-culture systems and until very recently a small animal model (Taube et al., 2013). However, unlike HuNoV, MNV is cultivable and replicates well in mice. Thus, MNV provides a tool to study norovirus biology, host–microbe interactions and functional aspects of norovirus biology such as mechanisms of escape from antibody neutralization.
The human and murine NoV genome encodes three and four ORFs, respectively (Thackray et al., 2007). ORF2 encodes the major capsid protein (VP1), which is structurally divided into the N terminus (N), the shell (S) and the protruding (P) domains (Katpally et al., 2008; Prasad et al., 1994, 1996, 1999). The P domain is the exposed part of the virus capsid and is subdivided into P1 and P2 subdomains. The P2 subdomain is involved in binding cellular receptors (Choi et al., 2008; Tan et al., 2004) and interacts with neutralizing antibodies (Donaldson et al., 2010; Nilsson et al., 2003; Taube et al., 2010). We previously described the identification of neutralizing mAb A6.2 against MNV strain MNV-1 (Wobus et al., 2004), and mapped its epitope to the A′B′ and E′F′ loops of the MNV-1 P domain (Taube et al., 2010). Neutralization escape studies revealed that mutations in the E′F′ loop are necessary for virus escape from mAb A6.2 neutralization (Kolawole et al., 2014; Lochridge & Hardy, 2007). The identified mutations not only interfered with the antibody–antigen interface but some also stabilized a conformation of the A′B′ and E′F′ loops that could not be bound by mAb A6.2 (Kolawole et al., 2014), suggesting a novel mode of neutralization escape.
In the current study, we generated and characterized new anti-MNV mAbs from strain MNV-1- or WU20-infected mice. We identified mAbs binding to the MNV P domain that were of IgA isotype and were capable of neutralizing MNV in vitro and in vivo, as well as a non-neutralizing mAb of IgG isotype that mapped to the MNV S domain. Analysis of neutralization escape mutants indicated that, unlike mAb A6.2, amino acids in the C′D′ loop of the MNV P domain are necessary for escape from neutralization. These studies increase our understanding of the anti-MNV antibody response as well as creating useful reagents for future studies of norovirus biology.
Results and Discussion
Binding specificity of hybridoma supernatants
Mice were immunized twice with infectious MNV-1 or WU20 and boosted once with UV-inactivated homologous virus to generate anti-MNV-1 and anti-WU20 antibodies. Sera from infected mice produced significantly higher absorbance than pre-immune sera (data not shown). Following fusion and subcloning, nine hybridomas from the MNV-1-immunized animal and 10 hybridomas from the WU20-immunized animal with reactivities against the homologous virus by ELISA were selected for further analysis (Fig. 1, Table S1 available in the online Supplementary Material). IgG and IgA isotype controls were used as negative controls, whilst the previously isolated anti-MNV-1 mAb A6.2 (at 1 µg ml−1; Wobus et al., 2004) was used as a positive control. As expected, no reactivity was observed for the negative controls, whilst mAb A6.2 reacted not only with MNV-1 but also WU20. Hybridoma supernatants were also tested by ELISA for the ability to bind the heterologous virus (Fig. 1). Absorbances twice the negative control were considered positive (see dashed line in Fig. 1). Of the nine hybridomas isolated from the MNV-1-immunized animal, four were cross-reactive with WU20 (Fig. 1a), whilst all 10 hybridomas from the WU20-immunized animal cross-reacted with MNV-1, although with varying reactivities (Fig. 1b). In both cases, less reactivity was observed against the heterologous virus.
Fig. 1.
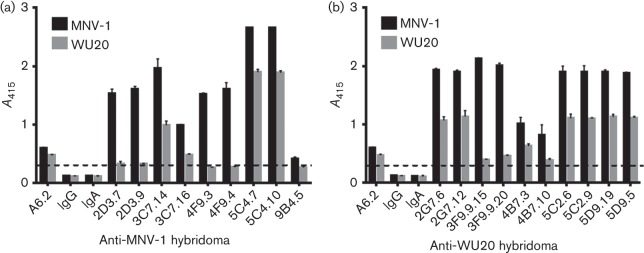
Reactivities of mAbs raised against MNV-1 and WU20. Plates were coated with equal amounts of MNV-1 or WU20 and hybridoma supernatants were tested for reactivity against MNV-1 (a) or WU20 (b) by ELISA. The mAb A6.2 (100 ng per well) was used as a positive control, whilst isotype controls of IgG2b (IgG) and IgA subclass were used as negative controls. Data are presented as means±sem of at least three independent experiments. The dashed line represents twice the absorbance values at 415 nm (A415) for the negative controls and indicates the threshold for a positive signal.
Next, the immunoglobulin isotype was determined for each hybridoma (Table S1). Four anti-MNV-1 hybridomas (2D3.7, 2D3.9, 4F9.3 and 4F9.4) were IgAs, whilst the remaining anti-MNV-1 and all anti-WU20 hybridomas were IgGs, IgG1, IgG2a or IgG2b. Hybridoma supernatants were further tested for their reactivity to MNV proteins by Western blotting using a 1 : 1 mix of MNV-1 and WU20 as antigen (Table S1 and Fig. S1). No reactivity with denatured MNV was observed for hybridomas 2D3.7, 2D3.9, 4F9.3, 9B4.5, 3F9.9.15 and 3F9.9.20, whilst all other hybridomas reacted against MNV-1 and WU20. To determine the domain of the viral capsid protein recognized by the antibodies, hybridoma supernatants were screened by ELISA for their ability to bind recombinantly expressed MNV-1 P or S domain (Table S1 and Fig. 2). ELISA was not performed with WU20 S and P domains due to unavailability of the reagents. Similar to the positive-control mAb A6.2, which is known to bind the MNV-1 P domain (Taube et al., 2010), MNV-1 hybridomas 2D3.7, 2D3.9 and 4F9.4 and WU20 hybridomas 3F9.9.15 and 3F9.9.20 reacted with the MNV-1 P domain. In contrast, the MNV-1 hybridomas 5C4.7 and 5C4.10 bound strongly to the MNV-1 S domain. No binding to either the P or S domain was observed for the remaining hybridoma supernatants.
Fig. 2.
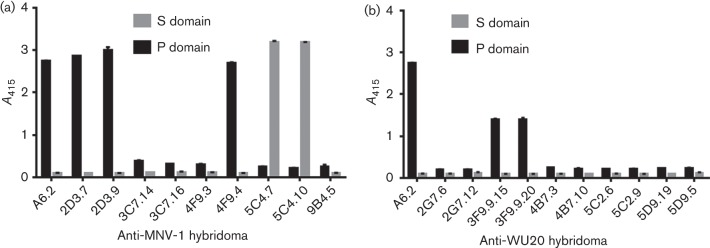
Epitope mapping of mAbs to the MNV-1 capsid protein P or S domain. Plates were coated with equal amounts of bacterially expressed MNV-1 P or S domain, and hybridoma supernatants were tested for reactivity against MNV-1 (a) or WU20 (b) by ELISA. Data are presented as means±sem of at least three independent experiments.
Taken together, these data suggested that 2D3.7, 2D3.9, 3F9.9.15 and 3F9.9.20 recognized a conformational epitope on the MNV-1 P domain because they reacted against the P domain by ELISA but not against denatured capsid protein by Western blotting. Hybridomas 5C4.7 and 5C4.10 bound the S domain by ELISA, whilst 4F9.4 bound the P domain, and all bound the capsid protein by Western blotting, suggesting these mAbs recognized a linear epitope on the S or P domain, respectively. Hybridomas 3C7.14, 3C7.16, 2G7.6, 2G7.12, 4B7.3, 4B7.10, 5C2.6, 5C2.9, 5D9.19 and 5D9.5, which reacted against the capsid protein by Western blotting but not against the P or S domain by ELISA, probably recognized linear epitopes that were inaccessible when the protein domains were folded. Hybridoma supernatant 9B4.5 reacted only weakly to MNV-1 virions and did not recognize linear epitopes by Western blotting or the individual capsid protein domains. This could be due to either low antibody yield from this hybridoma or a lower intrinsic affinity of this antibody. Lastly, hybridoma 4F9.3 specifically bound to MNV-1 virions by ELISA but not individual P or S domains or denatured capsid protein by Western blotting, suggesting that this hybridoma bound to a conformational epitope, but that slight differences in the folding of recombinantly expressed P and S domain compared with assembled virions might prevent binding to recombinant protein.
Neutralizing activity of hybridoma supernatants
To evaluate the neutralizing ability of each hybridoma, neutralization assays were carried out with 100 p.f.u. MNV-1 or WU20 (Fig. 3, Table S1). The neutralizing mAb A6.2 and immune sera from MNV-1- or WU20-infected mice were used as a positive control and neutralized both MNV-1 and WU20. The IgG isotype control did not neutralize either virus, and its titres were set to 100 % to normalize the data. MNV-1 hybridomas 2D3.7, 2D3.9, 4F9.3 and 4F9.4 effectively neutralized both MNV-1 and WU20, whilst MNV-1 hybridomas 3C7.14 and 3C7.16 partially neutralized both viruses (Fig. 3a). Hybridomas 3C7.16 and 9B4.5 were significantly less effective in neutralizing WU20 than MNV-1 (Fig. 3a). The S domain-specific hybridomas 5C4.7 and 5C4.10 (Fig. 3a) and all 10 WU20 hybridomas (Fig. 3b) did not decrease infectivity below 70 % for either MNV-1 or WU20, and thus were considered non-neutralizing.
Fig. 3.
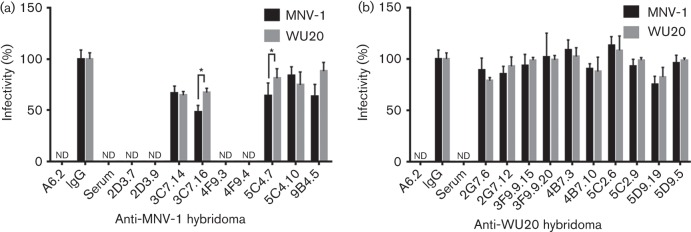
Analysis of mAb neutralization activity. MNV-1 or WU20 were incubated with hybridoma supernatants from an MNV-1-immunized (a) or WU20-immunized (b) mouse for 30 min at 37 °C before performing plaque neutralization assays. The percentage infectivity was determined by setting the virus titre for the isotype antibody as 100 %. Data are presented as means±sem of three independent experiments. Statistical analysis was performed using a t-test to compare the percentage infectivity of MNV-1 versus WU20. *P<0.05; nd, not detected (<1 % infectivity).
The ability of the P domain-binding mAbs 2D3.7, 2D3.9, 4F9.3 and 4F9.4 to neutralize MNV infection is consistent with previous observations that the P domain contains the binding sites for neutralizing antibodies (Donaldson et al., 2010; Nilsson et al., 2003; Taube et al., 2010). In contrast, the S domain-binding mAbs 5C4.7 and 5C4.10 are non-neutralizing but whether these mAbs are cross-reactive across genotypes and/or genogroups as described for other norovirus-specific S-domain mAbs (Almanza et al., 2008; Batten et al., 2006; Li et al., 2009; Parra et al., 2012) will need to be determined in future studies. The inability or partial ability of the other mAbs to neutralize MNV infection may be due to the use of hybridoma supernatants without adjusting for antibody concentration, because it is possible that the hybridomas contained a low level of antibody with little activity or a high level of antibody with no sensitivity.
In summary, four strongly neutralizing IgA hybridomas were isolated from the MNV-1-immunized animal that cross-neutralized WU20, whilst no neutralizing mAbs were isolated from the WU20-immunized animal. MNV-1 causes an acute infection and is cleared by 7 days post-infection, whilst WU20 is a persistent virus, which is shed for at least 35 days (Thackray et al., 2007). It is interesting to note that, whilst both immune sera neutralized either virus, suggesting that neutralizing antibodies are raised during infection with either virus, no WU20-neutralizing mAbs were isolated. Slight structural differences may occur in the immunodominant P2 domain, as two of seven amino acid differences in the P2 domain between MNV-1 and WU20 are located in the E′F′ loop, which constitutes the mAb A6.2-binding site. Thus, it is conceivable that the dominating immune response against WU20 is directed against non-neutralizing epitopes, providing a potential explanation for the inability of the host to clear WU20. However, future experiments will need to test this hypothesis.
Purification and analysis of select mAbs
Three of the MNV-1 hybridomas, 2D3.7, 4F9.4 and 5C4.10, were selected for purification and further characterization. mAbs 2D3.7 and 4F9.4 were selected for representing the first description of MNV-neutralizing IgAs, whilst 5C4.10 is, to the best of our knowledge, the first reported anti-MNV S-domain mAb. To verify the binding reactivities of the three purified mAbs, equal concentrations were tested for binding to MNV-1 and WU20 virions by ELISA and compared with the same concentrations of purified mAb A6.2 (Fig. 4). IgA and IgG isotype mAbs were also purified and used as negative controls as they did not react with either MNV-1 or WU20. The purified mAbs 2D3.7, 4F9.4 and 5C4.10 reacted more strongly with MNV-1 compared with the previously isolated mAb A6.2. Specifically, mAbs 2D3.7, 4F9.4 and 5C4.10 were two- to sevenfold more reactive than mAb A6.2 to MNV-1, with mAb 5C4.10 showing the strongest reactivity at 100 ng (Fig. 4a). All purified mAbs were less reactive to WU20 than MNV-1 at all concentrations tested, with mAb 4F9.4 being the least reactive to WU20 (Fig. 4b). In contrast, at 100 ng, mAb 5C4.10 reacted similarly to WU20 and MNV-1 (Fig. 4). The higher reactivities of 2D3.7 and 4F9.4 to MNV-1 compared with A6.2 suggested a higher affinity for this strain. Whilst binding of 2D3.7 and 4F9.4 to MNV-1 and WU20 at 10 and 100 ng was mostly saturated, 5C4.10 was fivefold more reactive than A6.2 at 100 ng, suggesting that binding to the S domain is not fully saturated. The high reactivity of 5C4.10 with both MNV-1 and WU20 makes this mAb a useful tool for the detection of viral capsid protein.
Fig. 4.
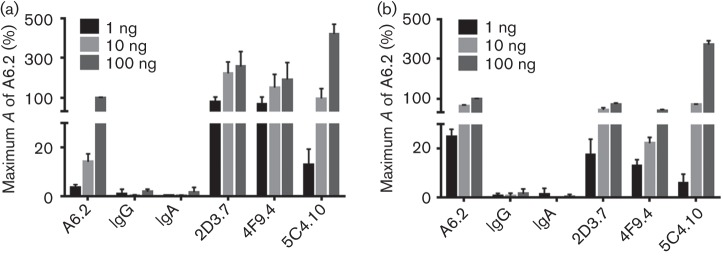
Binding activities of selected purified mAbs. Plates were coated with equal amounts of MNV-1 (a) or WU20 (b) and tested for reactivity against 1, 10 or 100 ng of the indicated purified mAbs by ELISA. Background absorbance (A) was subtracted from all absorbance values. The percentage maximum absorbance was calculated by setting the absorbance for 100 ng A6.2 antibody to 100 %. Data are presented as means±sem of at least three independent experiments.
Neutralization of MNV-1 P domain A′B′ and E′F′ loop mutants by mAbs 2D3.7 and 4F9.4
Previous work has demonstrated that the epitopes for the neutralizing mAb A6.2 are located on both the A′B′ and E′F′ loops (Katpally et al., 2008, 2010; Taube et al., 2010), whilst analysis of nine A′B′ loop and 12 E′F′ loop mutants demonstrated that amino acids in the E′F′ loop are critical for escape from neutralization (Kolawole et al., 2014). To determine whether residues in the A′B′ and E′F′ loops could also mediate escape from neutralization by mAbs 2D3.7 and 4F9.4, 21 mutants (A′B′ loop mutants: E296K, Q298E, Q298S, S299A, S299R, G300F, G300K, G300R and T301I, and E′F′ loop mutants: V378A, V378F, V378L, A381F, A381G, A382G, A382K, A382R, A382P, D385E, D385G, L386F) and WT MNV-1 were tested against mAbs 2D3.7 and 4F9.4 in plaque neutralization assays (Fig. 5 and data not shown). Similar data were obtained for all mutants; thus, only three mutants each for the A′B′ and E′F′ loops are shown for clarity. Antibody concentrations that reduced virus infectivity below 20 % were considered neutralizing, whilst those that reduced infectivities to 20–80 % were considered partially neutralizing. WT MNV-1 was partially neutralized at 60 ng and fully neutralized at 200 ng mAbs 2D3.7 and 4F9.4. All A′B′ and E′F′ loop mutants were fully neutralized by 200 ng mAbs 2D3.7 and 4F9.4, but susceptibility to neutralization varied greatly at 60 ng antibody (Fig. 5). A′B′ loop mutants E296K, S299R and G300K were fully or partially neutralized by mAbs 2D3.7 and 4F9.4, respectively, similarly to WT MNV-1 (Fig. 5a, b). E′F′ loop mutant V378F was neutralized at 60 ng with mAb 2D3.7 similarly to WT MNV-1, whilst A382K and L386F were partially neutralized at the same dose (Fig. 5c). Similarly, partial neutralization to WT MNV-1 was observed for the three mutants with mAb 4F9.4 (Fig. 5d). No A′B′ or E′F′ loop mutant escaped mAb 2D3.7 and 4F9.4 neutralization at 200 ng, suggesting that escape from antibody neutralization is primarily mediated by a different region of the MNV-1 P domain.
Fig. 5.

A′B′ and E′F′ loop MNV-1 P domain mutants do not mediate escape from mAbs 2D3.7 and 4F9.4 neutralization. WT MNV-1 and A′B′ loop mutants with IgA mAb 2D3.7 (a), A′B′ loop mutants with IgA mAb 4F9.4 (b), E′F′ loop mutants with IgA mAb 2D3.7 (c) and E′F′ loop mutants with IgA mAb 4F9.4 (d) were subjected to in vitro neutralization. Virus was incubated with the indicated concentrations of mAbs 2D3.7 and 4F9.4 for 30 min at 37 °C before performing plaque neutralization assays. The percentage infectivity was calculated by setting WT MNV-1 without mAb to 100 %. The dashed lines indicate 20 and 80 % infectivity. Data are presented as means±sem of three independent experiments.
Identification of mAb 2D3.7 and 4F9.4 neutralization escape mutants
Noroviruses, like all RNA viruses, have a high mutation rate (Bull et al., 2010). Therefore, mutations in the quasispecies occur during passaging that may allow the virus to adapt to environmental changes. We thus employed an in vitro passaging approach of MNV-1 in the presence of mAb stressor to generate escape viruses that would allow mapping of residues within the MNV-1 P domain that mediate escape from neutralization of mAbs 2D3.7 and 4F9.4. MNV-1 was serially passaged in two lineages through RAW 264.7 cells 20 times in the presence of increasing concentrations of the mAbs 2D3.7, 4F9.4 or a non-neutralizing IgA isotype control (Fig. 6a). Antibody concentrations were 40 ng for passage 1 (P1) and P2, 60 ng for P3, 100 ng for P4 and P5, 200 ng for P6–P8 and 600 ng for P9–P20. Escape from the IgA antibody-mediated selection occurred slower than has been reported for mAb A6.2 (Lochridge & Hardy, 2007). The starting virus stock (P0) was neutralized by 200 ng mAbs 2D3.7 (Fig. 6b) and 4F9.4 (Fig. 6c), reducing the virus infectivity to less than 1 %. The kinetics of neutralization escape were similar for both lineages with either mAb, and partial neutralization resistance occurred by P5 (Figs 6b and 6c). In the case of mAb 2D3.7, P20 viruses were resistant to 200 ng antibody (Fig. 6b), whilst for mAb 4F9.4, resistant viruses appeared by P15. As anticipated, viruses grown in the presence of non-neutralizing IgA isotype control were neutralized by both mAbs up to P20.
Fig. 6.
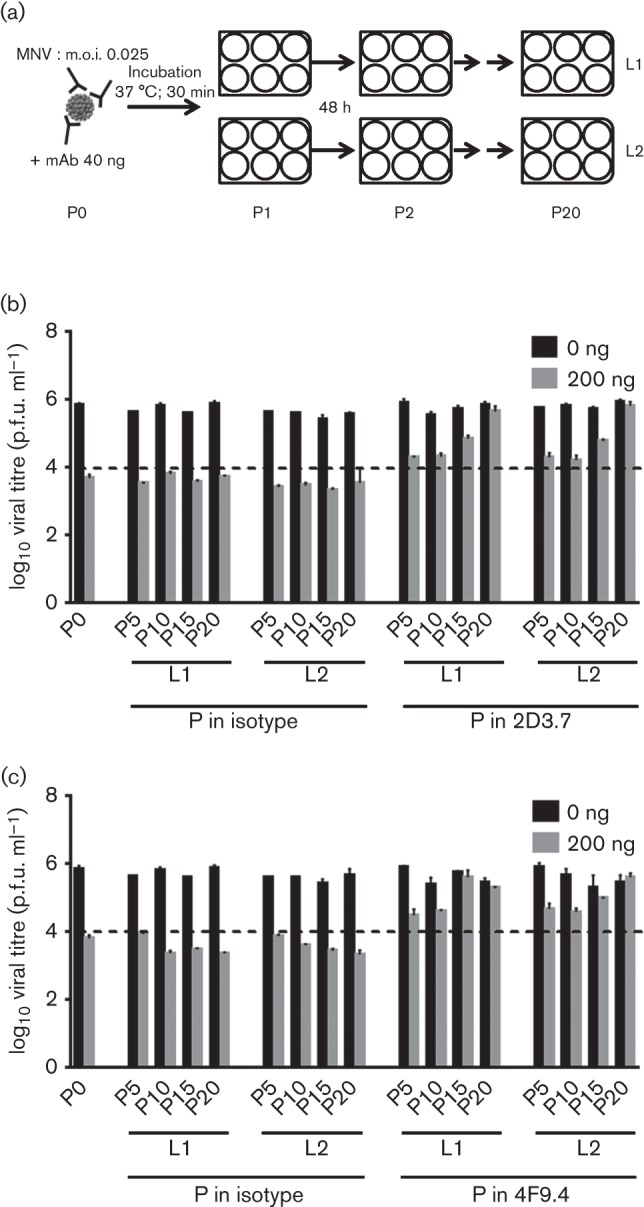
Isolation of 2D3.7 and 4F9.4 neutralizing escape mutants. (a) Schematic of the experimental set-up. MNV-1 was passaged through RAW 264.7 cells in the presence of increasing concentrations [40 ng for passage 1 (P1) and P2, 60 ng for P3, 100 ng for P4 and P5, 200 ng for P6–P8 and 600 ng for P9–P20] of neutralizing mAb 2D3.7 or 4F9.4 or a non-neutralizing IgA isotype control antibody. At each passage, MNV-1 (m.o.i. 0.025) was pre-incubated with the respective mAb for 30 min at 37 °C before infection for 48 h. For each antibody, two independent lineages (L1 and L2) were analysed. (b, c) Virus from the original stock (P0) and from P5, P10, P15 and P20 were incubated with 0 or 200 ng mAb 2D3.7 (b) or 4F9.4 (c) for 30 min at 37 °C and analysed by plaque neutralization assay. The dashed line indicates the level of neutralization at P0. Data are presented as means±sem of at least three independent experiments.
To identify the dominant mutations in the P domain of MNV grown under antibody selection, P0 and P20 viruses were Sanger sequenced. P0 and P20 viruses passaged with the IgA isotype control had the same sequence when compared with the WT MNV-1 genome (data not shown). In contrast, the mutations V339I and D348E, located in the C′D′ loop of the MNV-1 P domain (Taube et al., 2010), were identified after 20 passages in mAbs 2D3.7 and 4F9.4 (Fig. S2). These data suggested that mAbs 2D3.7 and 4F9.4 bind to similar regions of the viral capsid. Interestingly, whilst D348 is highly exposed on the surface of the P domain, V339 is buried between the E′F′ and the C′D′ loops and a portion of V339 is visible only from the side of the P domain in the B conformation (Fig. S2, right panel). Therefore, the V339I escape mutation is probably indirect, with the larger side chain affecting the conformation of these loops rather than directly blocking antibody binding.
Characterization of mAb 2D3.7 and 4F9.4 neutralization escape mutants
To verify that V339I and D348E mediated escape from neutralization by mAbs 2D3.7 and 4F9.4, single point mutations were introduced into the MNV-1 infectious clone and recombinant viruses were recovered. These mutations did not affect the growth of the recombinant viruses compared with the parental MNV-1 (Fig. 7a). Recombinant viruses were next subjected to in vitro neutralization by mAbs 2D3.7 and 4F9.4. WT MNV-1 was almost completely neutralized by 200 ng and partially neutralized by 60 ng mAb 2D3.7, whilst recombinant viruses V339I and D348E fully escaped neutralization of mAb 2D3.7 at all concentrations tested (Fig. 7b). In contrast, WT MNV-1 was almost completely neutralized by 60 and 200 ng and partially neutralized by 20 ng mAb 4F9.4, whilst recombinant viruses V339I and D348E escaped neutralization of 4F9.4 at lower doses of 20 and 60 ng but were neutralized at the higher dose of 200 ng (Fig. 7c). These data suggested differences in the reactivities of each mAb to the MNV-1 capsid antigen, with a stronger neutralizing activity for mAb 4F9.4, indicating that these mAbs may have arisen from two independent hybridomas.
Fig. 7.

Characterization of 2D3.7 and 4F9.4 neutralization escape mutants. (a) RAW 264.7 cells were infected with the indicated viruses at m.o.i. of 2 on ice for 1 h. The inoculum was removed and cells were infected until indicated time points. Virus titres were determined by plaque assay. (b, c) Indicated recombinant viruses were subjected to in vitro neutralization by mAbs 2D3.7 (b) and 4F9.4 (c). Virus was incubated with the indicated concentrations of mAbs 2D3.7 and 4F9.4 for 30 min at 37 °C before performing plaque neutralization assay. The percentage infectivity was calculated by setting WT MNV without mAb as 100 %. The dashed lines indicate 20 and 80 % infectivity. Data are presented as means±sem of three independent experiments. (d) Sequence alignment of the heavy and light chain sequences for mAbs 2D3.7 and 4F9.4. The amino acid sequence of 2D3.7 is shown. Identical amino acids of 4F9.4 are indicated by asterisks. The complementary determining region (CDR) sequences are underlined.
To clarify the independent origin of each mAb, the heavy and light chains for each mAb were sequenced. The sequences from both mAbs were very similar (Fig. 7d). Only two and one amino acid differences were observed in the heavy chain complementary determining regions (CDR) 2 and CDR3, respectively, whilst only one amino acid difference was observed in the light chain CDR3. Thus, the higher in vitro neutralization capacity of mAb 4F9.4 over 2D3.7 may be related to the four amino acid differences in the CDR regions of both mAbs. Furthermore, to verify the different reactivities, the Kd values for mAbs 2D3.7 and 4F9.4 were determined by plasmon surface resonance. Consistent with the ELISA and neutralization, Biacore data demonstrated that the Fabs from the IgA mAb had Kd values of 1.47×10−11 and 5.96×10−13 M, respectively (data not shown). In comparison, Fabs from the IgG mAb A6.2 had a Kd of 4×10−8 M (Kolawole et al., 2014). This may explain why it took 15 passages for MNV-1 to escape from mAbs 2D3.7 and 4F9.4 as opposed to five passages for mAb A6.2. There are at least two possible reasons why the IgAs, 2D3.7 and 4F9.4, had such a high affinity compared with the IgG A6.2. First, the viral load in the immunization yielding 2D3.7 and 4F9.4 may have been lower than that yielding A6.2 such that the high-affinity antibodies were preferentially selected. Secondly, it is also possible that the IgAs represent multiple rounds of exposure to antigen that resulted in more affinity maturation and isotypic switching (e.g. Scott et al., 1994).
Taken together, these data suggested that mAbs 2D3.7 and 4F9.4 arose independently, yet they shared the C′D′ loop of the MNV-1 P domain as their binding epitope and region for neutralization escape.
In vivo neutralization of MNV by mAbs
To determine whether the neutralizing mAbs 2D3.7 and 4F9.4 also neutralize MNV-1 in vivo, STAT1−/− mice, which support higher viral loads than WT mice, were used for neutralization studies as described previously (Kolawole et al., 2014). Mice were simultaneously infected with 106 p.f.u. WT MNV-1 orally and injected with 15, 100 or 500 µg mAb 2D3.7 or 4F9.4 or IgA isotype control intraperitoneally for 48 h. These concentrations of antibody were chosen based on our previous in vivo neutralization study with mAb A6.2, in which 15 µg A6.2 caused neutralization in systemic sites (lung, liver and spleen), and 100 µg A6.2 in addition neutralized MNV-1 in some regions in the intestine (stomach and duodenum) and mesenteric lymph nodes, whilst 500 µg A6.2 reduced viral titres in all sites except in faeces (Kolawole et al., 2014). No overt clinical symptoms are visible at this early time point. A good correlation was established between the in vitro and in vivo neutralization potency of the IgA mAbs. In vivo neutralization was dose dependent. After the administration of 15 or 100 µg mAb 2D3.7 or 4F9.4, viral titres were significantly reduced in some regions of the gastrointestinal tract (stomach, proximal ileum and colon) and systemic sites (liver, lung and spleen) compared with the IgA mAb isotype control samples (Fig. 8a, b). The highest mAb concentration of 500 µg per mouse completely neutralized MNV-1 infection in systemic sites and significantly decreased infection throughout the gastrointestinal tract and mesentery (Fig. 8c). However, no neutralization was observed in the faeces at any concentration (Fig. 8), similar to our previous in vivo neutralization experiments with mAb A6.2 (Kolawole et al., 2014). There was no significant difference between the neutralizing activities of mAbs 2D3.7 and 4F9.4 at the two lower concentrations of 15 and 100 µg, but 4F9.4 was more effective in neutralizing MNV-1 in the intestine at 500 µg compared with 2D3.7 (Fig. 8). These data demonstrated that mAbs 2D3.7 and 4F9.4 exhibit MNV-1 neutralization both in vitro and in vivo. Furthermore, the in vivo neutralization activity of mAbs 2D3.7 and 4F9.4 is similar to that of mAb A6.2 (Kolawole et al., 2014) when the mAbs are administered intraperitoneally, despite the difference in isotype. The inability of IgG (A6.2) (Kolawole et al., 2014) and IgA (2D3.7 and 4F9.4) neutralizing mAbs to reduce viral shedding in the faeces may reflect the inability of mAbs to reach specific sites in the intestine at sufficient doses following intraperitoneal inoculation. We think it is less likely that a large amount of input virus remained, as our previous study of MNV-1 replication kinetics demonstrated that input virus was no longer detectable at 24 h post-infection (Gonzalez-Hernandez et al., 2014). However, future studies will be required to investigate this further. Similarly, future studies will need to determine the extent of in vivo neutralization escape from mAbs 2D3.7 and 4F9.4 by mutants V339I and D348E.
Fig. 8.
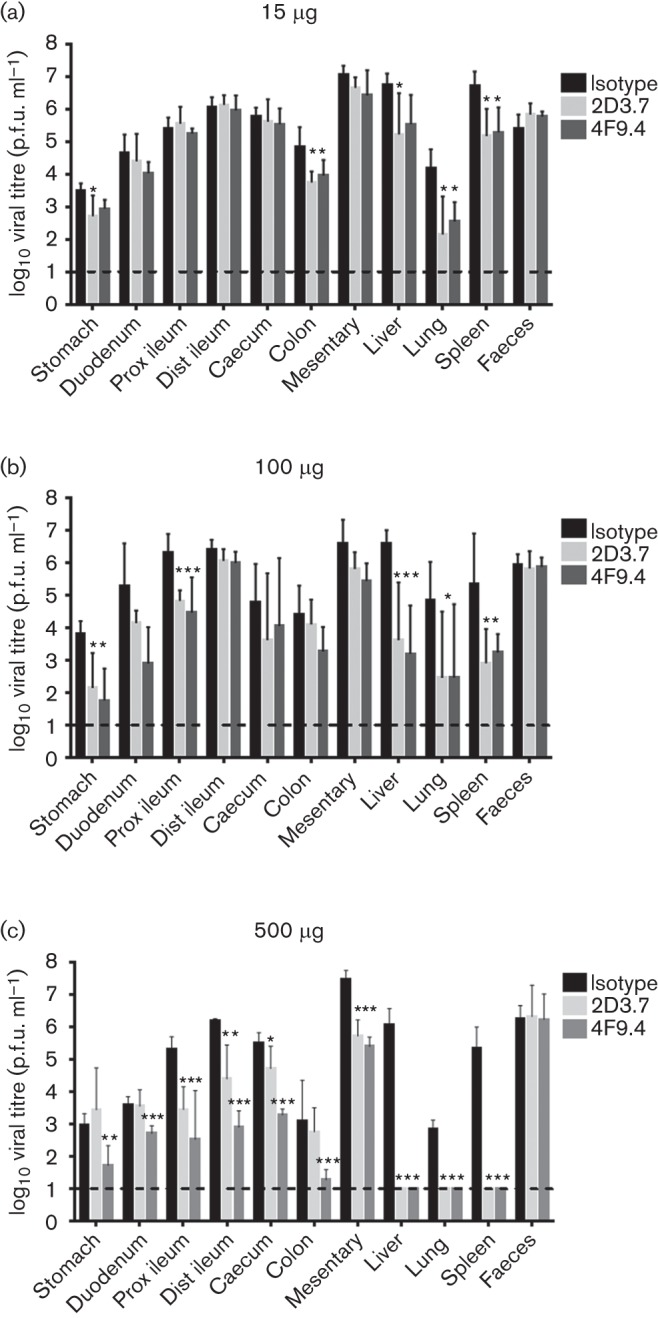
In vivo neutralization activity of mAbs 2D3.7 and 4F9.4. STAT1−/− mice were infected with 1×106 p.f.u. MNV-1 orally and injected intraperitoneally with 15 µg (a), 100 µg (b) or 500 µg (c) of mAb 2D3.7 or 4F9.4 or isotype control mAb. The indicated tissues were harvested 2 days post-infection and viral titres measured by plaque assay. Data are presented as means±sd of at least five mice per condition from at least two independent experiments. Statistical analysis was performed using a t-test to compare virus titre for each tissue between IgA mAb-infected mice and isotype control. *P<0.05; **P<0.01; **P<0.001. Prox ileum, proximal ileum; Dist ileum, distal ileum.
Conclusions
To date, only three MNV-1-neutralizing mAbs, A6.2, A6.1 and H6.1, have been described (Wobus et al., 2004). However, sequencing of the heavy and light chains of these three mAbs indicated they are identical (Kolawole et al., 2014, and data not shown). Therefore, our goal was to generate additional mAbs with specificities against MNV. To this end, BALB/c mice were infected with MNV-1 or WU20 and boosted with live and UV-inactivated homologous virus. Here, we have reported the generation of a panel of 19 mAbs with reactivities against MNV-1 and WU20 capsids. Two mAbs of IgA isotype were capable of neutralizing both MNV strains in vitro and MNV-1 in vivo. Their binding epitope was mapped to the C′D′ loop of the MNV-1 P domain. In addition, we isolated a non-neutralizing IgG isotype mAb with reactivity against the MNV-1 S domain. These mAbs increase the tools available for the study of MNV biology.
A wider collection of mAbs are available against human than murine NoVs, but collectively these studies highlight similarities regarding the epitopes of human and murine norovirus-neutralizing antibodies (Chen et al., 2013). Like the mAbs described in this report, mAbs raised against HuNoV virus-like particles (VLPs) locate epitopes in the variable P2 domain (Allen et al., 2009; Debbink et al., 2012) or in conserved regions of the S and P1 domains of HuNoV (Batten et al., 2006; Higo-Moriguchi et al., 2014; Li et al., 2010; Parker et al., 2005). Due to the lack of a HuNoV cell-culture system, surrogate neutralization assays have been developed for HuNoV VLP binding to histo-blood group antigens, the HuNoV attachment receptor, which locate the ‘neutralizing’ epitope to the P domain (Reeck et al., 2010). This is similar to findings from MNV, in which neutralizing epitopes and neutralizing escape mutations are located in the P2 domain (Katpally et al., 2008; Kolawole et al., 2014; Taube et al., 2010; this study). The similarities between MNV and HuNoV mAb-binding epitopes indicate that results from MNV mAb studies can provide information to better understand neutralizing antibody resistance, norovirus evolution and vaccine design for HuNoVs.
Methods
Cell culture.
Murine macrophage RAW 264.7 and 293T cells were purchased from the American Type Culture Collection (ATCC) and maintained as described previously (Taube et al., 2010; Wobus et al., 2004). The IgG isotype control was directed against coxsackievirus B4 (CV; clone 204-4) and was purchased from ATCC (HB 185). The IgA isotype control was directed against the F1 antigen from Yersinia pestis and was purchased from ATCC (HB-192).
Viruses.
The plaque-purified MNV-1 clone (GV/MNV1/2002/USA) MNV-1.CW3 (referred to as MNV-1) and the faecal isolate WU20 (GV/WU20/2005/USA) (Thackray et al., 2007) were amplified in RAW 264.7 cells and used at P6 for all studies. Concentrated virus stocks were generated as described previously (McCartney et al., 2008).
Ethics statement.
All mouse studies were performed in accordance with local and federal guidelines as outlined in the ‘Guide for the Care and Use of Laboratory Animals’ of the National Institutes of Health. Protocols were approved by the University of Michigan Committee on Use and Care of Animals (UCUCA No. 09710).
Generation of anti-MNV mAbs.
Specific-pathogen-free and MNV-seronegative BALB/c mice were purchased from Jackson Laboratories. One mouse each was infected orally with 4×106 p.f.u. MNV-1 or WU20 and reinfected at the same dose 42 days later. MNV-1 and WU20 were CsCl purified as described elsewhere (Karst et al., 2003) and 4×106 p.f.u. were UV inactivated for 30 min in a biosafety cabinet before boosting mice intraperitoneally with homologous virus either 14 (WU20) or 42 (MNV-1) days later. Spleens were harvested 4 days after the last booster in both cases. Splenocytes were fused with the P3×63-Ag8.653 murine myeloma cell line (ATCC CRL-1580; Kearney et al., 1979) using standard hybridoma technology (Gefter et al., 1977; Nishinarita et al., 1985). Briefly, splenocytes and myeloma cells were fused in 42 % PEG 4000/15 % DMSO at a ratio of 3–5 : 1 and plated in flat-bottomed, 96-well tissue-culture dishes (Corning) in Iscove’s modified DMEM (IMDM; Lonza) supplemented with 20 % heat-inactivated FBS, 2 mM l-glutamine, 100 U penicillin ml−1, 100 µg streptomycin ml−1 and 30 µM 2-mercaptoethanol with HAT (100 µM hypoxanthine, 0.4 µM aminopterin, 16 µM thymidine) as the hybridoma selection agent. BriClone Hybridoma Cloning Medium (QED Bioscience/Archport Ltd), an IL-6 growth factor-rich culture supplement, was added to the medium at a concentration of 5 % to support hybridoma cell growth. Cultures were fed on days 7, 9 and 12. Culture supernatants were screened for antibody production 14–20 days post-fusion by ELISA. Hybridomas that showed positive antibody production were subcloned in 96-well plates by limiting dilution at 0.5–2.5 cells per well in IMDM fusion medium containing mercaptoethanol, hypozanthine, thymidine and 5 % BriClone Hybridoma Cloning Medium without aminopterin.
MNV ELISA.
Hybridoma supernatants were screened for their ability to bind MNV by ELISA as described previously (Wobus et al., 2004) with some modifications. ELISA plates were coated with 0.059 mg ml−1 concentrated MNV-1 or WU20 in PBS. Supernatants were diluted 1 : 1, whilst purified mAbs were serially diluted from 1000 to 10 ng ml−1 in ELISA III buffer (0.15 M sodium chloride, 1 mM EDTA, 0.1 % BSA, 0.05 % Tween 20, in 50 mM Tris/HCl buffer, pH 7.4). A6.2 mAb (1 µg ml−1) was used as positive control and set to 100 % for purified mAbs. IgA and IgG isotype mAbs were used as negative controls.
Western blotting.
A 1 : 1 mix of WU20 and MNV-1 of concentrated virus or mock-infected lysate concentrated similarly to the virus was separated by 10 % SDS-PAGE. Proteins were transferred to a nitrocellulose membrane (Bio-Rad). The membrane was blocked with 5 % milk in phosphate buffered saline + 0.05 % Tween-20 (PBS-T) buffer overnight and probed with a 1 : 1000 dilution of hybridoma supernatant followed by a 1 : 5000 dilution of a goat anti-mouse HRP-conjugated secondary antibody, each for 1 h at room temperature. Proteins were visualized by chemiluminescence using Thermo Scientific Pierce ECL Western Blotting Substrate following the manufacturer’s instructions.
Plaque neutralization assay.
For neutralization assays using hybridoma supernatants, 100 p.f.u. MNV-1 or WU20 were diluted with 100 µl hybridoma supernatant and incubated for 30 min at 37 °C prior to performing a plaque assay as described previously (Gonzalez-Hernandez et al., 2012). Plaque neutralization assays with purified mAbs were performed as described previously (Kolawole et al., 2014). One hundred microliters of mAbs A6.2 or CV (2 ng ml−1) was used as positive and negative controls, respectively. The percentage infectivity was determined by setting the virus titre in the negative control to 100 %.
Sequencing of mAbs.
Total RNA was extracted from hybridoma cells expressing IgA mAbs 2D3.7 and 4F9.4 using the RNeasy system (Qiagen). The CDR was amplified by two rounds of PCR, sequenced and analysed at LakePharma.
Purification of mAbs.
The IgG mAbs 5C4.10, A6.2 and CV were grown in Bioreactor CELLine CL 1000 flasks (Sigma-Aldrich) as described previously (Lencioni et al., 2011) and purified with protein A HiTrap Sepharose columns (GE Healthcare) according to the manufacturer’s protocol. Purified mAbs were immediately dialysed into four changes of PBS overnight at 4 °C and stored at −20 °C. IgA mAbs 2D3.7, 4F9.4 and the isotype control were purified by first precipitating the IgA from the hybridoma cell-culture supernatant with a 50 % saturated (final concentration) solution of ammonium sulfate. The precipitate was collected by centrifugation and dialysed against 50 mM Tris/HCl buffer (pH 7.6). The IgA was then purified using a Superose 6 size-exclusion column equilibrated with 0.5 M NaCl, 50 mM Tris/HCl (pH 7.5) and 5 % glycerol (v/v). Protein elution was monitored via A280 and fractions containing IgA were identified by SDS-PAGE (with or without reducing agent).
Generation and purification of IgA Fab.
Purified IgA antibody solution was dialysed against 0.1 M sodium citrate buffer (pH 4.0). The antibody was then digested with pepsin (pepsin : IgA = 1 : 25, w/w) for 17 h at 37 °C. Digestion was stopped by neutralization of solution with 1 M Tris buffer, pH 9.0. The digested IgA solution was then dialysed against 50 mM Tris/HCl (pH 7.6) and applied to a Mono-Q column equilibrated with the same buffer. Bound IgA Fab was eluted with a NaCl gradient in the same Tris buffer.
Surface plasmon resonance analysis.
Experiments were performed as described previously (Kolawole et al., 2014). In brief, time biomolecular interaction analysis was performed by surface plasmon resonance using a BIAcore 2000 instrument equipped with an NTA sensor chip. Purified polyhistidine-tagged WT MNV-1 P domain was immobilized on the flow cell. IgA Fab fragments at a range of concentration of approximately 25–800 nM with duplicates were used as analytes along with several blanks using buffer only. The running buffer consisted of 100 mM sodium phosphate (pH 7.4), 400 mM NaCl, 40 µM EDTA and 0.005 % (v/v) Tween-20 surfactant. Data analysis was conducted with the BIAevaluation package. Curve fittings were done with the 1 : 1 Langmuir binding model. All fitting met the manufacturer’s prescribed requirements.
Generation of neutralization escape mutants.
Neutralization escape mutants were generated as described previously (Kolawole et al., 2014) with some modifications. MNV-1 was pre-incubated with 40 ng IgA mAbs 2D3.7, 4F9.4 or IgA isotype control at 37 °C for P1. The experiment was repeated for 20 passages, using the previous passage as inoculum for the subsequent round and increasing the mAb concentration from 40 ng for P2, 60 ng for P3, 100 ng for P4 and P5 and 200 ng for P6–P8 to 600 ng for P9 – P20. Plaque neutralization assays were performed for virus samples from P0, P5, P10, P15 and P20 to monitor the changes in mAb neutralization ability.
Mutagenesis of MNV-1.
Recombinant viruses were generated as described previously (Kolawole et al., 2014) using the primers 5′-CAACTGGAGATCGAGATCCAGACCGAGACCA-3′ (forward) and 5′-TGGTCTCGGTCTGGATCTCGATCTCCAGTTG-3′ (reverse) for MNV-1 with mutation V339I, and 5′-CACCAAGACTGGAGAGAAGCTCAAGGTCACC-3′ (forward) and 5′-GGTGACCTTGAGCTTCTCTCCAGTCTTGGTG-3′ (reverse) for D348E. Recovered viruses were plaque purified and mutations were confirmed by Sanger sequencing after amplifying the MNV-1 P domain (residues 225–541) with primers 5′-ATGAGGATGAGTGATGGCGCAG-3′ (forward) and 5′-TTATTGTTTGAGCATTCGGCCTG-3′ (reverse) specific to the viral sequence that harboured the C′D′ loop. Confirmed mutant viruses were amplified in RAW 264.7 cells. Viral growth curves were performed as described previously (Kolawole et al., 2014). Viruses at P2 were used for neutralization studies.
In vivo neutralization assays.
Six- to eight-week-old STAT1−/− mice (strain 2045) were purchased from Taconic Farms and housed at the University of Michigan animal care facility, where all experiments were conducted. Mice (seven to nine animals per group) were simultaneously infected orally with 106 p.f.u. virus and intraperitoneally with 15, 100 or 500 µg purified mAb 2D3.7 or 4F9.4 or the IgA isotype control in a volume of 100 µl PBS for 48 h. Mice were humanely euthanized according to approved protocols and the tissues processed as described elsewhere (Kolawole et al., 2014).
Statistical analyses.
Student’s t-test available in Prism v.6 (GraphPad) was used to perform statistical analyses. A P value less than 0.05 was considered significant.
Acknowledgements
This work was supported by Defense Advanced Research Projects Agency (DARPA) Contract HR0011-11-C-0093. The funders had no role in study design, data collection and analysis, the decision to publish or preparation of the manuscript. We thank Elizabeth Smith at the University of Michigan Hybridoma Core for performing the fusions and growing hybridoma cells, and members of the Wobus laboratory for helpful suggestions with manuscript figures.
Footnotes
One supplementary table and two figures are available with the online version of this paper.
References
- Allen D. J., Noad R., Samuel D., Gray J. J., Roy P., Iturriza-Gómara M. (2009). Characterisation of a GII-4 norovirus variant-specific surface-exposed site involved in antibody binding. Virol J 6, 150 10.1186/1743-422X-6-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almanza H., Cubillos C., Angulo I., Mateos F., Castón J. R., van der Poel W. H., Vinje J., Bárcena J., Mena I. (2008). Self-assembly of the recombinant capsid protein of a swine norovirus into virus-like particles and evaluation of monoclonal antibodies cross-reactive with a human strain from genogroup II. J Clin Microbiol 46, 3971–3979 10.1128/JCM.01204-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten C. A., Clarke I. N., Kempster S. L., Oliver S. L., Bridger J. C., Lambden P. R. (2006). Characterization of a cross-reactive linear epitope in human genogroup I and bovine genogroup III norovirus capsid proteins. Virology 356, 179–187 10.1016/j.virol.2006.07.034 [DOI] [PubMed] [Google Scholar]
- Bull R. A., Eden J. S., Rawlinson W. D., White P. A. (2010). Rapid evolution of pandemic noroviruses of the GII.4 lineage. PLoS Pathog 6, e1000831 10.1371/journal.ppat.1000831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Sosnovtsev S. V., Bok K., Parra G. I., Makiya M., Agulto L., Green K. Y., Purcell R. H. (2013). Development of Norwalk virus-specific monoclonal antibodies with therapeutic potential for the treatment of Norwalk virus gastroenteritis. J Virol 87, 9547–9557 10.1128/JVI.01376-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. M., Hutson A. M., Estes M. K., Prasad B. V. (2008). Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc Natl Acad Sci U S A 105, 9175–9180 10.1073/pnas.0803275105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debbink K., Donaldson E. F., Lindesmith L. C., Baric R. S. (2012). Genetic mapping of a highly variable norovirus GII.4 blockade epitope: potential role in escape from human herd immunity. J Virol 86, 1214–1226 10.1128/JVI.06189-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson E. F., Lindesmith L. C., Lobue A. D., Baric R. S. (2010). Viral shape-shifting: norovirus evasion of the human immune system. Nat Rev Microbiol 8, 231–241 10.1038/nrmicro2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gefter M. L., Margulies D. H., Scharff M. D. (1977). A simple method for polyethylene glycol-promoted hybridization of mouse myeloma cells. Somatic Cell Genet 3, 231–236 10.1007/BF01551818 [DOI] [PubMed] [Google Scholar]
- Glass R. I., Parashar U. D., Estes M. K. (2009). Norovirus gastroenteritis. N Engl J Med 361, 1776–1785 10.1056/NEJMra0804575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hernandez M. B., Bragazzi Cunha J., Wobus C. E. (2012). Plaque assay for murine norovirus. J Vis Exp 2012, e4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hernandez M. B., Liu T., Payne H. C., Stencel-Baerenwald J. E., Ikizler M., Yagita H., Dermody T. S., Williams I. R., Wobus C. E. (2014). Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J Virol 88, 6934–6943 10.1128/JVI.00204-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K. Y. (2007). Caliciviridae. In Fields Virology, 5th edn, pp. 949–980 Edited by Knipe D. M., Howley P. M. Philadelphia: Lippincott Williams & Wilkins [Google Scholar]
- Henderson K. S. (2008). Murine norovirus, a recently discovered and highly prevalent viral agent of mice. Lab Anim (NY) 37, 314–320 10.1038/laban0708-314 [DOI] [PubMed] [Google Scholar]
- Higo-Moriguchi K., Shirato H., Someya Y., Kurosawa Y., Takeda N., Taniguchi K. (2014). Isolation of cross-reactive human monoclonal antibodies that prevent binding of human noroviruses to histo-blood group antigens. J Med Virol 86, 558–567 10.1002/jmv.23734 [DOI] [PubMed] [Google Scholar]
- Karst S. M., Wobus C. E., Lay M., Davidson J., Virgin H. W., IV (2003). STAT1-dependent innate immunity to a Norwalk-like virus. Science 299, 1575–1578 [DOI] [PubMed] [Google Scholar]
- Katpally U., Wobus C. E., Dryden K., Virgin H. W., IV, Smith T. J. (2008). Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. J Virol 82, 2079–2088 10.1128/JVI.02200-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katpally U., Voss N. R., Cavazza T., Taube S., Rubin J. R., Young V. L., Stuckey J., Ward V. K., Virgin H. W., IV & other authors (2010). High-resolution cryo-electron microscopy structures of murine norovirus 1 and rabbit hemorrhagic disease virus reveal marked flexibility in the receptor binding domains. J Virol 84, 5836–5841 10.1128/JVI.00314-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney J. F., Radbruch A., Liesegang B., Rajewsky K. (1979). A new mouse myeloma cell line that has lost immunoglobulin expression but permits the construction of antibody-secreting hybrid cell lines. J Immunol 123, 1548–1550 [PubMed] [Google Scholar]
- Kolawole A. O., Li M., Xia C., Fischer A. E., Giacobbi N. S., Rippinger C. M., Proescher J. B., Wu S. K., Bessling S. L. & other authors (2014). Flexibility in surface-exposed loops in a virus capsid mediates escape from antibody neutralization. J Virol 88, 4543–4557 10.1128/JVI.03685-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencioni K. C., Drivdahl R., Seamons A., Treuting P. M., Brabb T., Maggio-Price L. (2011). Lack of effect of murine norovirus infection on a mouse model of bacteria-induced colon cancer. Comp Med 61, 219–226 [PMC free article] [PubMed] [Google Scholar]
- Li X., Zhou R., Wang Y., Sheng H., Tian X., Li H., Qiu H. (2009). Identification and characterization of a native epitope common to norovirus strains GII/4, GII/7 and GII/8. Virus Res 140, 188–193 10.1016/j.virusres.2008.12.004 [DOI] [PubMed] [Google Scholar]
- Li X., Zhou R., Tian X., Li H., Zhou Z. (2010). Characterization of a cross-reactive monoclonal antibody against Norovirus genogroups I, II, III and V. Virus Res 151, 142–147 10.1016/j.virusres.2010.04.005 [DOI] [PubMed] [Google Scholar]
- Lochridge V. P., Hardy M. E. (2007). A single-amino-acid substitution in the P2 domain of VP1 of murine norovirus is sufficient for escape from antibody neutralization. J Virol 81, 12316–12322 10.1128/JVI.01254-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney S. A., Thackray L. B., Gitlin L., Gilfillan S., Virgin H. W., IV, Colonna M. (2008). MDA-5 recognition of a murine norovirus. PLoS Pathog 4, e1000108 10.1371/journal.ppat.1000108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M., Hedlund K. O., Thorhagen M., Larson G., Johansen K., Ekspong A., Svensson L. (2003). Evolution of human calicivirus RNA in vivo: accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J Virol 77, 13117–13124 10.1128/JVI.77.24.13117-13124.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishinarita S., Claflin J. L., Lieberman R. (1985). T15 D region germ line amino acid sequences distinguished by monoclonal anti-idiotope antibody. J Immunol 134, 436–442 [PubMed] [Google Scholar]
- Ohsugi T., Matsuura K., Kawabe S., Nakamura N., Kumar J. M., Wakamiya M., Morikawa S., Urano T. (2013). Natural infection of murine norovirus in conventional and specific pathogen-free laboratory mice. Front Microbiol 4, 12 10.3389/fmicb.2013.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker T. D., Kitamoto N., Tanaka T., Hutson A. M., Estes M. K. (2005). Identification of Genogroup I and Genogroup II broadly reactive epitopes on the norovirus capsid. J Virol 79, 7402–7409 10.1128/JVI.79.12.7402-7409.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G. I., Abente E. J., Sandoval-Jaime C., Sosnovtsev S. V., Bok K., Green K. Y. (2012). Multiple antigenic sites are involved in blocking the interaction of GII.4 norovirus capsid with ABH histo-blood group antigens. J Virol 86, 7414–7426 10.1128/JVI.06729-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B. V., Rothnagel R., Jiang X., Estes M. K. (1994). Three-dimensional structure of baculovirus-expressed Norwalk virus capsids. J Virol 68, 5117–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B. V., Hardy M. E., Jiang X., Estes M. K. (1996). Structure of Norwalk virus. Arch Virol Suppl 12, 237–242 [DOI] [PubMed] [Google Scholar]
- Prasad B. V., Hardy M. E., Dokland T., Bella J., Rossmann M. G., Estes M. K. (1999). X-ray crystallographic structure of the Norwalk virus capsid. Science 286, 287–290 [DOI] [PubMed] [Google Scholar]
- Reeck A., Kavanagh O., Estes M. K., Opekun A. R., Gilger M. A., Graham D. Y., Atmar R. L. (2010). Serological correlate of protection against norovirus-induced gastroenteritis. J Infect Dis 202, 1212–1218 10.1086/656364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott B. B., Sadigh S., Andrew E. M., Maini R. N., Mageed R. A. (1994). Affinity maturation and isotype switch in clonally related anti-erythrocyte autoantibodies. Scand J Immunol 40, 16–21 10.1111/j.1365-3083.1994.tb03427.x [DOI] [PubMed] [Google Scholar]
- Smith D. B., McFadden N., Blundell R. J., Meredith A., Simmonds P. (2012). Diversity of murine norovirus in wild-rodent populations: species-specific associations suggest an ancient divergence. J Gen Virol 93, 259–266 10.1099/vir.0.036392-0 [DOI] [PubMed] [Google Scholar]
- Tan M., Hegde R. S., Jiang X. (2004). The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J Virol 78, 6233–6242 10.1128/JVI.78.12.6233-6242.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube S., Rubin J. R., Katpally U., Smith T. J., Kendall A., Stuckey J. A., Wobus C. E. (2010). High-resolution x-ray structure and functional analysis of the murine norovirus 1 capsid protein protruding domain. J Virol 84, 5695–5705 10.1128/JVI.00316-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube S., Kolawole A. O., Höhne M., Wilkinson J. E., Handley S. A., Perry J. W., Thackray L. B., Akkina R., Wobus C. E. (2013). A mouse model for human norovirus. MBio 4, e00450-13 10.1128/mBio.00450-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray L. B., Wobus C. E., Chachu K. A., Liu B., Alegre E. R., Henderson K. S., Kelley S. T., Virgin H. W., IV (2007). Murine noroviruses comprising a single genogroup exhibit biological diversity despite limited sequence divergence. J Virol 81, 10460–10473 10.1128/JVI.00783-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus C. E., Karst S. M., Thackray L. B., Chang K. O., Sosnovtsev S. V., Belliot G., Krug A., Mackenzie J. M., Green K. Y., Virgin H. W. (2004). Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol 2, e432 10.1371/journal.pbio.0020432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus C. E., Thackray L. B., Virgin H. W., IV (2006). Murine norovirus: a model system to study norovirus biology and pathogenesis. J Virol 80, 5104–5112 10.1128/JVI.02346-05 [DOI] [PMC free article] [PubMed] [Google Scholar]