

Abstract
The genus Negevirus consists of insect-only viruses isolated from mosquitoes and sandflies. Here, we report the successful construction of a full-length infectious cDNA clone of Negev virus (NEGV) strain M30957. Viral RNA was transcribed in vitro and virus was readily rescued with or without the use of a cap analogue. These results strongly suggest that NEGV, and likely other members within the genus, is a non-segmented, single-stranded, positive-sense RNA virus.
The newly proposed genus Negevirus consists of RNA viruses with genomes ~9–10 kb in length (Vasilakis et al., 2013). These viruses have been isolated from mosquito species encompassing three genera as well as from phlebotomine sandflies. Genus Negevirus genomes appear to contain three ORFs: (1) the largest, ~7 kb, contains putative non-structural proteins, (2) the second ORF consists of putative structural proteins and (3) the third encodes a protein of no known function. The 5′ and 3′ UTRs vary in length from 72 to 730 and 121 to 442 nt, respectively. All of the reported viruses within the genus contain a polyadenylate tail varying in length from 13 to 52 nt (Vasilakis et al., 2013). These viruses are distantly related to the genus Cilevirus, which comprises mite-transmitted plant viruses (Locali-Fabris et al., 2006).
The first member of the proposed genus Negevirus, Negev virus (NEGV), was initially isolated from Anopheles coustani mosquitoes collected in the Negev desert, Israel, in 1983. NEGV has subsequently been isolated from Culex species collected at various localities in Texas (Vasilakis et al., 2013). NEGV is unable to infect mammalian cells whereas it readily infects insect cells with peak titres of ~1010 p.f.u. ml−1. It causes extensive cytopathic effects (CPEs) within 12–24 h of infection in the C6/36 and C7/10 lines of Aedes albopictus mosquito cells. Additionally, Aedes aegypti mosquitoes are susceptible to oral infection (Vasilakis et al., 2013).
Reverse genetics is a powerful molecular tool that has facilitated the understanding of many aspects of genomic organization, viral replication and pathogenesis, and has revolutionized vaccine development (Wimmer et al., 2009; Wimmer & Paul, 2011). The construction of infectious cDNA clones and recombinant viral progeny has been reported for many positive- and negative-strand RNA viruses. Here, we report the successful construction of an infectious cDNA clone of NEGV to elucidate the details of genus Negevirus genomic organization.
NEGV strain M30957 was obtained from World Reference Center for Emerging Viruses and Arboviruses at the University of Texas Medical Branch (Galveston, Texas, USA). It was originally isolated from a pool of Culex coronator mosquitoes collected in Harris County, Texas in 2008. Virus was amplified on C7/10 cells for RNA extraction and an infectious cDNA clone was constructed using standard recombinant DNA techniques (Sambrook & Russell, 2001). Briefly, full-length viral genome was assembled in three fragments (Fig. 1). NEGV RNA was isolated from cell culture supernatant using the QIAamp Viral RNA Mini kit (Qiagen). cDNA was produced via reverse transcription by using random hexamers and SuperScript III (Invitrogen). cDNA fragments were amplified via PCR with NEGV-specific primers containing endonuclease restriction sites and Phusion DNA polymerase (New England BioLabs) (Fig. 1). Amplified PCR products were purified using the QIAquick Gel Extraction kit (Qiagen), digested with restriction endonucleases and cloned into pRS2 plasmid (a derivative of pUC19) (Fig. 1). Viral genome was placed under the control of an SP6 RNA polymerase promoter at the 5′ end and included a 3′ poly(A) tail with a downstream NotI linearization site for in vitro transcription (Fig. 1). Full-length plasmid cDNA was confirmed via Sanger sequencing.
Fig. 1.
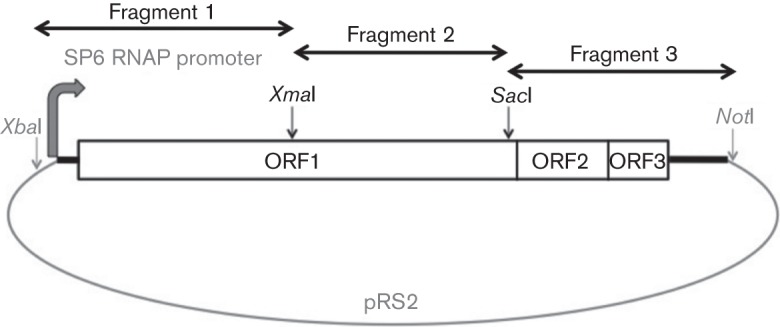
Schematic representation of the cDNA cloning strategy of NEGV. RNAP, RNA polymerase.
To rescue recombinant NEGV progeny, linearized plasmid was purified via phenol/chloroform extraction and 1 µg was utilized for the transcription reaction: 0.5 mM rNTP, 0.5 mM m7G(5′)ppp(5′)G RNA cap analogue (New England Biolabs), 0.5 µl RNase inhibitor and 1.25 µl SP6 polymerase (Ambion) in 25 µl total volume. Viral RNA transcripts were prepared in vitro with and without the m7G(5′)ppp(5′)G cap analogue (NEGV +cap and NEGV –cap, respectively). Transcription was performed for 2 h at 42 °C and then the transcription mixture placed on ice. Freshly confluent C7/10 cells seeded overnight were gently scraped into single-cell suspensions, washed five times with 1× PBS and resuspended in 450 µl 1× PBS. Cells were mixed with each transcription mixture (10 µg RNA), placed in 2 mm electroporation cuvettes and immediately electroporated (BTX ECM-830 Square Wave Electroporator; Harvard Apparatus) using the following conditions: 680 V, pulse length 99 μs, interval between pulses 200 ms and five pulses. Following electroporation, C7/10 cells were placed at 28 °C with 5 % CO2 and observed for CPEs. Recombinant NEGV progeny were readily produced with substantial CPEs by 12 h post-electroporation (h.p.e.) (Fig. 2a). Virus progeny produced plaques on ~80 % confluent C7/10 cell monolayers seeded overnight in six-well plates. Briefly, duplicate wells were infected with 0.1 ml aliquots from serial 10-fold dilutions in growth medium, then 0.4 ml of growth medium was added to each well to prevent cell desiccation and virus was adsorbed for 1 h. Following incubation, the virus inoculum was removed, and cell monolayers were overlaid with 3 ml of a 1 : 1 mixture of 2 % tragacanth and 2× modified Eagle’s medium with 5 % FBS, 2 % tryptose phosphate broth solution and 2 % penicillin/streptomycin. Cells were incubated at 28 °C with 5 % CO2 for 36 h for plaque development, the overlay was removed and monolayers were fixed with 3 ml 10 % formaldehyde in PBS for 30 min. Cells were stained with 2 % crystal violet in 30 % methanol for 5 min at room temperature; excess stain was removed under running water and plaques were counted. The successful recovery of recombinant NEGV was confirmed via full-length Sanger sequencing of plaque-purified virus subsequently amplified on C7/10 cells.
Fig. 2.

In vitro characterization of recombinant NEGV. Phase-contrast micrographs of C7/10 cells 12 h.p.e. with genomic NEGV RNA with or without addition of the cap analogue (a). Plaque phenotypes of the parental and recombinant viruses on monolayers of C7/10 cells (b). mfold-generated prediction of the RNA secondary structure of the NEGV 5′ UTR (c).
Surprisingly, NEGV could also be rescued without the use of the cap analogue, with a peak titre of 1.0×109 p.f.u. ml−1, identical to that derived using RNA transcripts generated with the cap analogue. In addition, both rescued viruses and the parental NEGV had the same plaque morphology on C7/10 cells (Fig. 2b). For comparison, similar virus rescue experiments with other positive-strand RNA viruses with capped genomes, in the absence of the cap analogue, yielded either no infectious virus or led to a 100-fold decrease of specific infectivity of the in vitro transcribed RNA (Ahlquist et al., 1984; Dasmahapatra et al., 1986; Polo et al., 1997; Pugachev et al., 1997; Rice et al., 1987). Therefore, our results strongly suggested the presence of an internal ribosome entry site (IRES) at the 5′ end of the NEGV genome, which would mediate cap-independent translation of the viral ORF to initiate replication. Indeed, analysis of 234 nt in the 5′ UTR of the NEGV genome using mfold (Zuker, 2003) predicted a highly organized RNA secondary structure (Fig. 2c) typical of IRES elements used by other positive-strand RNA viruses (Liu et al., 2009).
To assess potential phenotypic differences between the parental and clone-derived viruses (rescued with or without the use of the cap), one-step replication curves were performed with the parental and recombinant viruses in 50 % confluent C7/10 monolayers, in triplicate, with a m.o.i. of 10 p.f.u. per cell. Regardless of the rescue conditions, the replication kinetics of recombinant viruses were nearly identical to those of the WT parent with peak titres of 1.0×109 p.f.u. ml−1 (Fig. 3a).
Fig. 3.
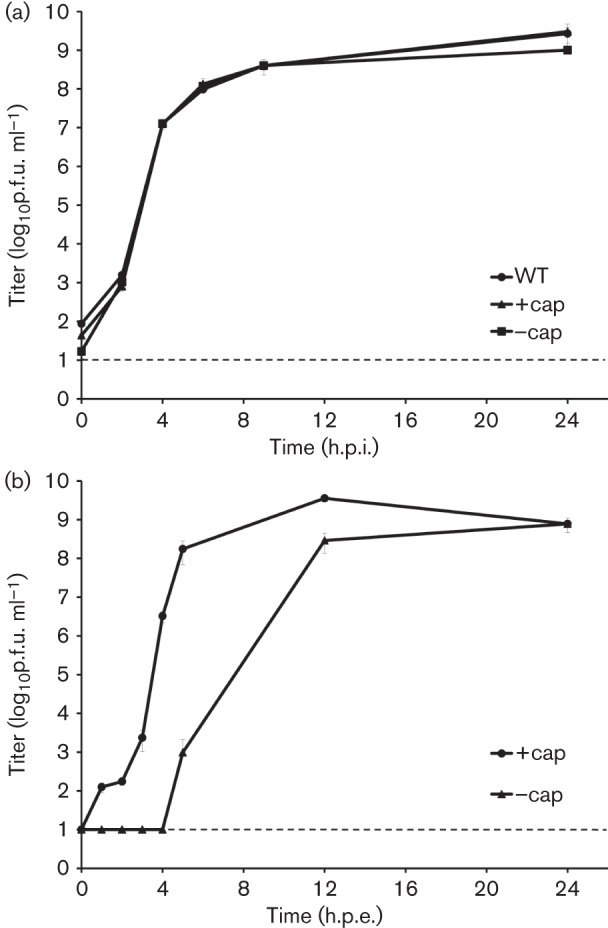
Replication kinetics of the parental and recombinant NEGV. (a) One-step replication curves after infection of C7/10 cells with NEGV variants. (b) Replication curves after electroporation of C7/10 cells with NEGV RNA in vitro transcribed with or without addition of the cap analogue. Each time point represents the mean titre from triplicates (infection) or duplicates (electroporation). Bar, sd; dashed line, limit of detection; h.p.i., hours post-infection.
Next, we compared replication of the recombinant viruses at the initial steps after cell transfection with transcribed RNA with and without the use of the cap. C7/10 cells were electroporated with 8 µg RNA in duplicate, transcribed with or without the cap as described above and samples of culture supernatants were collected every hour for the first 5 h, and then at 12 and 24 h.p.e. Interestingly, infectious virus was first detected at 1 h.p.e. for NEGV +cap, but only at 5 h.p.e. for NEGV –cap (Fig. 3b). Also, there was an estimated 2–3 h delay for the burst of virus production in NEGV –cap compared with NEGV +cap. However, both viruses reached the same peak titre of 109 p.f.u. ml−1 at 24 h.p.e. A possible explanation of this outcome is that IRES-mediated translation of viral proteins is less efficient than cap-mediated translation, which would result in a delay in initial accumulation of viral proteins that form replicative complexes. However, when those complexes begin to produce normally capped viral genomes, the rate of viral protein production increases, which in turn increases the rate of viral replication to the level observed after electroporation of capped RNA.
In summary, we describe the generation of an infectious cDNA clone of NEGV, the first isolated member of the newly proposed genus Negevirus. NEGV rescued in vitro in mosquito cells produced CPEs, plaque morphology and replication kinetics similar to the original virus. Remarkably, viable virus was readily obtained without the use of a cap analogue for in vitro genomic RNA synthesis, although its initial replication after electroporation was delayed compared with the capped RNA counterpart. Considering the highly organized predicted RNA secondary structure of the 5′ UTR, these data suggest the presence of an IRES at the 5′ end of the NEGV genome, which mediates low-efficiency translation. Additional experiments are needed to confirm the putative IRES and evaluate its role in NEGV replication. Our data suggest that genomes in the genus Negevirus likely consist of non-segmented, single-stranded, positive-sense RNA. The infectious NEGV cDNA clone we developed can be utilized to further characterize this newly discovered genus to elucidate many aspects of its biology, particularly its distinctive host specificity, and its potential for use in vector control or as a vehicle for vaccine production.
Acknowledgements
This work was supported by National Institutes of Health contract HHSN272201000040I/HHSN27200004/D04 (to R. B. T.).
References
- Ahlquist P., French R., Janda M., Loesch-Fries L. S. (1984). Multicomponent RNA plant virus infection derived from cloned viral cDNA. Proc Natl Acad Sci U S A 81, 7066–7070 10.1073/pnas.81.22.7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasmahapatra B., Dasgupta R., Saunders K., Selling B., Gallagher T., Kaesberg P. (1986). Infectious RNA derived by transcription from cloned cDNA copies of the genomic RNA of an insect virus. Proc Natl Acad Sci U S A 83, 63–66 10.1073/pnas.83.1.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wimmer E., Paul A. V. (2009). Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim Biophys Acta 1789, 495–517 10.1016/j.bbagrm.2009.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locali-Fabris E. C., Freitas-Astúa J., Souza A. A., Takita M. A., Astúa-Monge G., Antonioli-Luizon R., Rodrigues V., Targon M. L., Machado M. A. (2006). Complete nucleotide sequence, genomic organization and phylogenetic analysis of Citrus leprosis virus cytoplasmic type. J Gen Virol 87, 2721–2729 10.1099/vir.0.82038-0 [DOI] [PubMed] [Google Scholar]
- Polo S., Ketner G., Levis R., Falgout B. (1997). Infectious RNA transcripts from full-length dengue virus type 2 cDNA clones made in yeast. J Virol 71, 5366–5374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugachev K. V., Abernathy E. S., Frey T. K. (1997). Improvement of the specific infectivity of the rubella virus (RUB) infectious clone: determinants of cytopathogenicity induced by RUB map to the nonstructural proteins. J Virol 71, 562–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice C. M., Levis R., Strauss J. H., Huang H. V. (1987). Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J Virol 61, 3809–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Russell D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd edn Cold Spring Harbor, NY: Cold Spring Harbor Laboratory [Google Scholar]
- Vasilakis N., Forrester N. L., Palacios G., Nasar F., Savji N., Rossi S. L., Guzman H., Wood T. G., Popov V. & other authors (2013). Negevirus: a proposed new taxon of insect-specific viruses with wide geographic distribution. J Virol 87, 2475–2488 10.1128/JVI.00776-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer E., Paul A. V. (2011). Synthetic poliovirus and other designer viruses: what have we learned from them? Annu Rev Microbiol 65, 583–609 10.1146/annurev-micro-090110-102957 [DOI] [PubMed] [Google Scholar]
- Wimmer E., Mueller S., Tumpey T. M., Taubenberger J. K. (2009). Synthetic viruses: a new opportunity to understand and prevent viral disease. Nat Biotechnol 27, 1163–1172 10.1038/nbt.1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]