

Abstract
Alzheimer's disease (AD) is the most common form of dementia, characterized by progressive loss of memory and cognitive dysfunctions. A central pathological event of AD is accumulation and deposition of cytotoxic amyloid-β peptide (Aβ) in the brain parenchyma. Heparan sulfate proteoglycans (HSPGs) and the side chains heparan sulfate (HS) are found associated with Aβ deposits in the brains of AD patients and transgenic animal models of AD. A growing body of evidence from in vitro and in vivo studies suggests functional roles of HSPG/HS in Aβ pathogenesis. Although the question of “how and why HSPG/HS is codeposited with Aβ?” still remains, it is within reach to understand the mechanisms of the events. Recent progress by immunohistochemical examination with advanced antibodies shed light on molecular structures of HS codeposited with Aβ. Several recent reports have provided important new insights into the roles of HSPG in Aβ pathogenesis. Particularly, experiments on mouse models revealed indispensible functions of HSPG in modulating Aβ-associated neuroinflammation and clearance of Aβ from the brain. Application of molecules to interfere with the interaction between HS and Aβ peptides has demonstrated beneficial effects on AD mouse models. Elucidating the functions of HSPG/HS in Aβ deposition and toxicity is leading to further understanding of the complex pathology of AD. The progress is encouraging development of new treatments for AD by targeting HS-Aβ interactions.
1. Introduction
Structure of Heparan Sulfate Proteoglycans. Heparan sulfate proteoglycans (HSPGs) are heavily glycosylated proteins, in which several heparan sulfate (HS) glycosaminoglycan (GAG) chains are covalently attached to a core protein. HSPGs are expressed on the cell surface and in the extracellular matrix (ECM) in all tissues. Cell surface HSPGs are membrane-spanning syndecans (SDCs) and lipid-anchored glypicans (GPCs). There are four members in SDC family (SDC 1–4) and six in GPC family (GPC 1–6). Secreted HSPGs are agrin, collagen type XVIII, and perlecan [1]. HS polysaccharide chains are characterized by highly structural heterogeneity with respect to the chain length and sulfation pattern, generated by a complex biosynthetic process within the Golgi apparatus [2, 3]. Functions of HSPGs are mainly attributed to the HS side chains that interact with a spectrum of protein ligands including growth factors, cytokines, enzymes, lipase, apolipoproteins, and protein components of the ECM, exerting biological activities in development, homeostasis, and diseases [3, 4].
The diverse functions of HS in different biological settings have been extensively studied, and substantial information is obtained. One of the most studied molecular mechanisms of HS is in signal transduction process, particularly growth factor medicated signaling. For example, HS mediates high affinity binding of fibroblast growth factor-2 (FGF-2) to its receptor promoting the formation of a stable tertiary signal complex of FGF-2-HS-FGF-2 receptor [5]. Apart from mediating growth factor activities, HS also functions as coreceptors in other biological activities, for example, modulating the interaction of neuropeptide agouti-related protein with melanocortin receptors 3 and 4 (MC3R and MC4R) in the hypothalamus and regulating food consumption [6–8]. Moreover, membrane HSPGs also act as endocytic receptors for diverse macromolecules such as lipid, growth factors, receptor ligands, and morphogens [1, 9].
Secreted HSPGs, agrin [10] and perlecan [11], constitute major structural molecules in the ECM and basement membrane (BM) along with collagens and other proteins (for review, see [12]). In the ECM, HSPG serves as storage for a number of molecules, such as growth factors and chemokines. In addition, HSPG also plays important roles in maintaining the integrity of ECM and BM [13, 14] and modulating cell mobility [15–17] (also for review, see [4]). In the BM, HSPG, along with collagen IV and laminin-entactin/nidogen complex, controls blood vessel permeability and takes a part in transportation of solutes between vessels and ECM [18, 19]. The ultrastructure of BM can be changed in disease conditions [20] and aging [21], probably due to abnormal production and breakdown of BM components including HSPGs [20].
Heparanase. Heparanase is an endo-β-glucuronidase that specifically cleaves HS side chains of HSPG, releasing oligosaccharide products at the size of 4–7 kDa (10–20 sugar units) [22]. Heparanase is normally expressed at a low level in majority of tissues including the brain [23]. Surprisingly, this unique HS-specific glycosidase is not essential for animal development and homeostasis, as demonstrated by targeted interruption of the heparanase gene in mouse [24]. The heparanase null mice produce longer HS chains in comparison to wildtype mice; however, there is no accumulation of the polysaccharide in organs, indicating that heparanase is not an indispensible enzyme for HS catabolism. In contrast, overexpression of heparanase in mice resulted in extensive modification of HS chains, producing short fragments with increased sulfation that exert higher potency for FGF-2-HS-FGF-2 receptor resembling [25]. This makes the heparanase transgenic mouse (Hpa-tg) a valuable tool for study of HS functions in different diseases [26–29]. Changes in expression of heparanase in tissues, mainly upregulation, have been reported in several diseases, particularly in cancers [30]. Increased expression of heparanase is detected in brain tumor glioma tissues from human and animal models, where heparanase is suggested to play an important role in the control of tumor cell proliferation and invasion [31]. Cerebral ischemia markedly increased heparanase levels in endothelial cells and astrocytes of mouse [32] and rat [33] brains. Available information suggests that heparanase may function as a regulatory factor in different pathological conditions, including tumor and inflammation, exerting its functions through modification of HS structure [34]. Moreover, heparanase has been shown to have nonenzymatic activities, most likely through direct interaction with cell surface receptors, which needs further investigations [35].
Aβ Pathology of Alzheimer's Disease. Alzheimer's disease (AD) is a major central nervous system disease characterized by a progressive neurodegeneration with a clinical phenotype of cognitive impairment. A histopathological hallmark of AD is extracellular Aβ deposition in brain parenchyma manifested as senile Aβ plaques [36]. The pathological Aβ peptides of 40 or 42 amino acids are products of sequential cleavage of the amyloid β precursor protein (AβPP), a transmembrane glycoprotein, by β-secretase (β-site APP cleaving enzyme 1: BACE1) [37] and γ-secretase, a multisubunit protease complex composed of at least 4 proteins including presenilin 1 and 2 [38]. Deposition of Aβ in the brain is attributed to excessive accumulation and aggregation of Aβ in the brain. Accumulation and deposition of Aβ most probably resulted from overproduction in the brain or/and impaired removal of Aβ from the brain [39]. Autosomal dominant mutations in three genes, that is, AβPP gene (APP) and presenilin 1 and 2 genes (PSEN1 and PSEN2), can cause early onset familial AD, accounting for <10% of AD cases [40–42]. All these mutations can result in overproduction of the Aβ peptides, leading to their accumulation and aggregation in the brain [43–45]. In clinic, the most common form of AD is late-onset sporadic AD accounting for about 90% of AD cases. Sporadic AD is not associated with genetic mutations, and no overproduction of Aβ was found. In these cases, it is generally believed that overall Aβ clearance is impaired, resulting in accumulation of Aβ peptides [46, 47]. In the brains of AD patients and some aging individuals with no clear diagnosis of dementia, Aβ is found to accumulate and deposit in blood vessel walls, named cerebral amyloid angiopathy (CAA), which has been interpreted as a sign of impaired Aβ clearance from the brain [48].
There are several ways for Aβ clearance, including degradation by proteolytic enzymes [49], receptor mediated Aβ transport across the blood-brain barrier (BBB) in which the main receptor is low-density lipoprotein receptor related protein-1 (LRP-1) [50], phagocytosis by innate immune cells (macrophages) [51], and perivascular drainage along the BM of blood vessels [52].
2. Interaction of HS with Aβ
Several in vitro studies demonstrate interaction of Aβ with GAGs including HS and heparin (a HS analogue with higher sulfation degree) [53–56]. It has been found that the HHQK domain at the N-terminus of Aβ is a HS binding motif and this sequence has also been shown to bind microglial cells, suggesting that microglia interact with Aβ through membrane associated HS [57]. Concurrently, a HS sequence of N-sulfated hexasaccharide domain containing critical 2-O-sulfated iduronic acid residues binds fibrillar Aβ and was identified in human cerebral cortex. Interestingly, this HS domain also serves as a binding site for the neuroprotective growth factor FGF-2. This evidence suggests that, in AD brain, neurotoxic Aβ may compete with neuroprotective FGF-2 for a common HS binding site [58]. Affinity of HS binding to Aβ is associated with its sulfation pattern, as heparin shows a higher affinity to Aβ, while desulfated HS essentially lost binding capacity to Aβ. This interaction is also dependent on chain length of the GAGs, as heparin fragments shorter than 6-sugar units do not bind to Aβ [58]. Furthermore, it has been proposed that the Aβ-HS interaction is mutually protective, such that HS is protected from heparanase degradation [53] and Aβ is protected from protease degradation [59].
3. Codeposition of HS with Aβ in AD Brain—Updated Findings
The presence of glycosaminoglycans (GAGs) in Aβ plaques in AD brain was first identified using Congo red staining for Aβ fibrils and Alcian blue dye for sulfated GAGs in brain sections of autopsy specimens of AD patients about 30 years ago [60]. The presence of HSPGs in Aβ plaques and CAA was later revealed by immunostaining with specific antibodies recognizing the core proteins of HSPGs [61–63]. With these antibodies, subtypes of HSPGs including SDC 1–3, GPC 1, and agrin have been immunolocalized in Aβ plaques and CAA of AD brains [64, 65]. Development of antibodies recognizing different Aβ fragments further promoted characterization of interaction between Aβ and HS.
Recent studies employed advanced type of anti-HS antibodies that differentially recognizes certain structures of HS polysaccharide chains [66, 67]. For example, phage display antibodies EV3C3 and HS4C3 recognize fully N-sulfated motifs in HS chain, while RB4EA12 and HS4E4 recognize partially N-sulfated and N-acetylated HS motifs [66, 68, 69]. Availability of these unique antibodies allowed us to analyze the molecular structure of HS codeposited with Aβ in the brain. By costaining the AD brain sections with an anti-HS phage display antibody HS4E4 and antibodies specific for Aβ species, we found that HS is differentially deposited with Aβ40 or Aβ42 in neuritic and diffuse plaques [70]. In sporadic AD cases, HS4E4 immunosignals are preferentially colocalized with Aβ40 in the cores of senile plaques; however, the HS4E4 signals are absent from Aβ42-rich diffuse deposits. In a recent study, antibodies (EV3C3 and HS4C3) recognizing highly N-sulfated HS detected strongest immunosignals in both fibrillar and nonfibrillar Aβ plaques, while antibodies (RB4EA12 and HS4E4) recognizing HS regions with lower degree of N-sulfation only stained fibrillar Aβ plaques [68], indicating a distinct property of HS structures in interaction with different Aβ aggregates in vivo. These reports are in agreement with our findings, confirming that only fibrillar Aβ plaques of Aβ40 deposits are colocalized with lower sulfated HS motifs. We have identified the membrane bound HSPGs, GPC 1, and SDC 3 in glial cells associated with Aβ deposits in dense core plaques, proximal to sites of HS accumulation, and suggested that HS codeposited with Aβ40 in neuritic plaques is mainly derived from glial cells [70]. RB4CD12 is another phage display antibody that recognizes highly sulfated domains of HS [71]. This antibody strongly stained both diffuse and neuritic Aβ plaques in the brains of AD and several transgenic AD mouse models. Interestingly, the RB4CD12 epitope accumulated in Aβ plaques can be demolished by extracellular sulfatases (Sulf-1 and Sulf-2) ex vivo [72], suggesting that 6-O-sulfated glucosamine residues are within the HS sequence interacting with Aβ.
These recent findings of selective deposition of HS with different species and forms of Aβ strongly suggest distinct roles of HS in Aβ aggregation and deposition. These studies point that HS/HSPG constitutes a part of Aβ plaques and the findings support the notion that HS plays a role in Aβ plaque formation and persistence.
4. HS Mediated Aβ Uptake—Implications in Aβ Cytotoxicity and Clearance
In the brain, Aβ are present in both extracellular and intracellular pools and extracellular Aβ contributes to intracellular Aβ through internalization mechanisms [73]. Cell types in the brain are known to engulf Aβ including neurons, endothelial cells [74], smooth muscle cells [75], and glial cells (microglia and astrocytes) [76, 77]. Internalization of Aβ into cells has been shown to be associated with Aβ cytotoxicity [78, 79]. Several cell surface macromolecules of microglia/macrophages are reported to play roles in Aβ uptake, including toll-like receptor [80], complement receptors [81], scavenger receptors [76, 82], LRP-1 [83], and transmembrane protein CD33, a member of the sialic acid-binding immunoglobulin-like lectins [84] (also for review, see [85]). HSPG functions as a cell surface receptor for entry of diverse macromolecules into cells; in this context, both the core protein and the HS side chains of HSPG are attributed to regulation of endocytosis (for review, see [9]). Having this in mind, we studied Aβ40 uptake and associated toxicity in Chinese hamster ovary (CHO) cell lines. After exposure to Aβ40, the CHO wildtype cells (CHO-WT) survived poorly, whereas the HS-deficient CHO pgsD-677 cells were resistant to the treatment. In correlation with Aβ cytotoxicity, the added Aβ40 was substantially uptaken by CHO-WT but barely by CHO pgsD-677 cells [86]. Likewise, Aβ40 cytotoxicity was attenuated in human embryonic kidney cells (HEK293) overexpressing heparanase due to extensive degradation of HS chains [86]. These findings suggest that cell surface HS mediates Aβ internalization and toxicity.
According to “amyloid hypothesis,” the cause of the majority form of AD, that is, late-onset sporadic, is due to impaired clearance of Aβ from the brain [47, 87]. Transport of Aβ across the BBB from brain to blood is an important route for Aβ clearance, where transcytosis requires Aβ to attach to cell surface after which it is internalized and subsequently released at the luminal side of the endothelium. LRP-1 at the surface of blood vessel endothelial and smooth muscle cells has been reported to function as Aβ cargo in this process [50, 75]. It has been recently reported that LRP-1 and HSPGs mediate Aβ internalization in a seemingly cooperative manner, in which HSPG is more important for Aβ binding to cell surface than LRP-1 [88]. Another important player in this context is apolipoprotein E (ApoE). ApoE and HS are consistently codetected in Aβ deposits and have been ascribed various roles in the pathogenesis of AD [89, 90]. ApoE can bind to HSPG forming functional complex of ApoE/HSPG; alternatively, it joins HSPG/LRP-1 uptake pathway in which ApoE first binds to HSPG and then presents to LRP-1 for uptake (for review, see [91]). The finding of codistribution of ApoE, HS, and LRP1 in Aβ40-positive microvasculature in the hippocampus of individuals with Down's syndrome (DS), diagnosed with AD, encouraged us to investigate correlation of these molecules in Aβ uptake and clearance [92]. We investigated the functional relationship between Aβ and ApoE and their interactions with cell surface HS and LRP-1 [92]. Coincubation of Aβ with CHO cells either deficient in HS (CHO pgsD677) or in LRP-1 (CHO 13-5-1) along with CHO-WT revealed that addition of ApoE in the cell culture increased Aβ association to the cells, which is dependent on presence of HSPG and LRP-1 on the cell surface. ApoE uptake by the cells does not require presence of both HSPG and LRP1; however, lack of HS in the CHO pgsD677 cells resulted in aberrant intracellular ApoE processing. These data propose that the complex interactions of ApoE, LRP-1, and HSPG facilitate Aβ internalization, which may represent one of major routes for Aβ clearance through transportation of ECM Aβ across BBB into the vessel lumen [92].
5. Heparanase in Aging and AD—Implications in Transmigration of Blood-Borne Monocytes
Heparanase expression in the brain is at marginally detectable level [23, 29], while, in several pathological conditions of the brain, expression of heparanase has been found elevated [31, 32]. Although limited information is available regarding the impact of heparanase on AD pathogenesis, Aβ40 has been shown to protect heparanase-catalyzed degradation of HSPGs in vitro with predicted effect contributing to the stability and persistence of Aβ plaques [53]. Our recent study has revealed increased vasculature expression of heparanase in the brains of AD patients and a mouse model that overexpresses human AβPP (Tg2576 mice) [29]. Since HS is involved in almost every step of Aβ pathogenesis found in AD (Figure 1), it is of great importance to study expression and activity of heparanase in the brain of aging subjects, both human and animal models.
Figure 1.
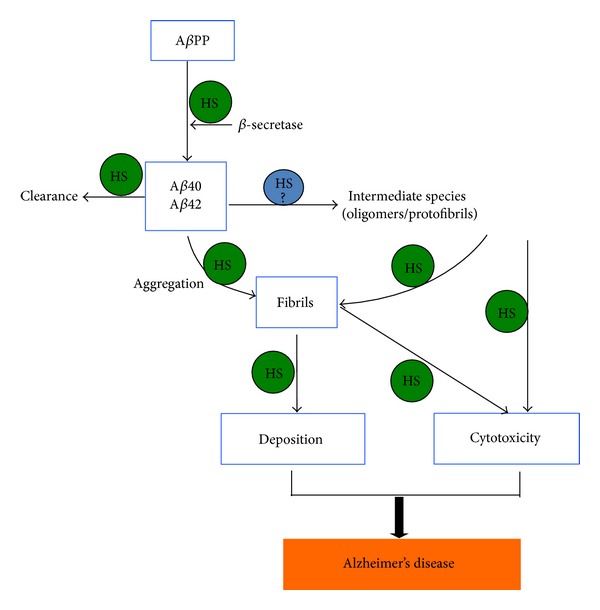
Heparan sulfate (HS) is involved in essentially each step of amyloid-β (Aβ) pathological development in Alzheimer's disease. HS modulates β-secretase (BACE) activity and accelerates Aβ aggregation and fibrillization. It is unclear whether HS is involved in formation of the toxic oligomers/protofibrils; however, HS mediates toxic effect of different types of Aβ fibrils. HS in the basement membrane participates in clearance of Aβ.
In the brain, perivascular macrophages derived from blood-borne mononuclear cells play an important role in Aβ clearance [51, 93, 94]; Aβ peptides are uptaken and subsequently degraded by proteases [95]. Several in vivo studies have demonstrated the multiple functions of HS and heparanase in inflammatory reactions with regard to infiltration of blood-borne immune cells into infected tissues [28, 96]. In this scenario, molecular structures of HS, for example, sulfation pattern and chain length, are pivotal in interaction between endothelial cells and leukocytes as well as with the soluble inflammatory cytokines. Accordingly, we have recently studied the potential roles of heparanase and HS in mediating blood-borne monocytes across blood vessel wall into the brain parenchyma on the transgenic mouse model overexpressing heparanase (Hpa-tg). Overexpression of heparanase resulted in shorter HS chains in the brain of Hpa-tg mouse [29]. In the study, we applied two experimental regimens, that is, localized cerebral microinjection of aggregated Aβ42 and systemic challenge by intraperitoneal injection of lipopolysaccharide (LPS), a bacterial endotoxin. Microinjection of aggregated Aβ42 into the brain elicited an inflammatory response restricted to the injection site of the wildtype mice, characterized by massive infiltration of microglia/macrophages. This inflammatory reaction clearly showed a beneficial effect for clearance of the injected Aβ. In comparison, recruitment and activation of immune cells (microglia and blood-borne monocytes) were significantly attenuated around the injection site of Hpa-tg mouse brain, which resulted in detainment of the injected Aβ42 [29]. The LPS-treated wildtype mice also showed massive activation of resident microglia as well as recruitment of monocyte-derived macrophages in the brain parenchyma, whereas Hpa-tg mice exhibited restricted inflammation with significantly fewer infiltrated macrophages. The mechanism for the reduced recruitment of inflammatory cells into the brain of Hpa-tg mice was verified with an in vitro BBB model constituted with primary endothelia cells and pericytes [29].
The integrity of ECM and the capillary vascular basement membrane (VBM) scaffold is often found severely damaged in association with Aβ deposition [97, 98], which may be responsible for perturbed elimination of solutes and Aβ from parenchyma, consequently leading to development of CAA [99]. As HSPGs are major components of the ECM and VBM and heparanase activity is strongly implicated in structural remodeling of the ECM and BM through degradation of HS, heparanase expression may markedly contribute to pathological changes in the ECM and VBM in AD brain, accordingly affecting Aβ clearance. There is essentially no information with this regard and studies are needed to explore the implications of HS in Aβ transportation and clearance.
6. Conclusion and Perspectives
Principle treatments for AD with regard to Aβ pathology are to reduce production, improve clearance, and prevent aggregation of the pathological peptides. Considering that HS-Aβ interaction contributes to every stage of the Aβ pathogenesis in AD, including production, clearance and accumulation, aggregation, and toxicity of Aβ (Figure 1), it is rational to hypothesize that interfering HS-Aβ interaction may have multiple beneficial effects. Earlier studies show that treatment with low molecular weight heparin (LMWH) reduced Aβ burden in the brain of an AD mouse model overexpressing human AβPP [100]; the effect is probably that the LMWH competes with endogenous HS, blocking the HS-Aβ interaction. This assumption is supported by our findings that the fragmentation of HS by overexpressed heparanase in mouse attenuated deposition of serum A amyloid (SAA; another amyloid protein) [27]. Though it is improper to use LMWH for treatment of AD, it is possible to apply non-anticoagulant LMWH or HS mimetics for the purpose. With the progress in characterization of HS molecular structures dissected from Aβ plaques, it should be possible to design compounds mimic to the HS structures that interact with Aβ to block its aggregation as well as to neutralize its toxicity. Moreover, targeting Aβ producing enzymes, that is, BACE1 and γ-secretase, constitutes one of the potential treatments for AD. Interestingly, HSPG has been found to modulate BACE activity [101, 102], and efforts are being made to synthesize HS-oligosaccharides as inhibitors of BACE [103]. In light of experimental and clinical evidences addressing the role of HS in Aβ pathology, it is plausible to expect that novel treatments by targeting HS-Aβ interaction may contribute to AD treatment and to improve effects of other treatments. Apart from designed synthesis of HS mimetics, natural anionic oligosaccharides, such as glycosaminoglycans isolated from marine animals and natural herbs, should also be explored for the potential to be developed as drug candidates for this particular application.
Acknowledgments
This work was supported by Grants from the Swedish Heart and Lung Foundation (20110131), Swedish Research Council (K2012-67X-21128-01-4), Stint (IB2012-4524), Polysackaridforskning Foundation (Uppsala), and National Natural Science Foundation of China (81373815 and 81202840).
Conflict of Interests
The authors declare that they have no conflict of interests regarding the publication of this paper.
References
- 1.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harbor Perspectives in Biology. 2011;3(7) doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kreuger J, Kjellén L. Heparan sulfate biosynthesis: regulation and variability. Journal of Histochemistry and Cytochemistry. 2012;60(12):898–907. doi: 10.1369/0022155412464972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindahl U, Li J. Interactions between heparan sulfate and proteins-design and functional implications. International Review of Cell and Molecular Biology. 2009;276:105–159. doi: 10.1016/S1937-6448(09)76003-4. [DOI] [PubMed] [Google Scholar]
- 4.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446(7139):1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 5.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64(4):841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- 6.Karlsson-Lindahl L, Schmidt L, Haage D, et al. Heparanase affects food intake and regulates energy balance in mice. PLoS ONE. 2012;7(3) doi: 10.1371/journal.pone.0034313.e34313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reizes O, Benoit SC, Strader AD, Clegg DJ, Akunuru S, Seeley RJ. Syndecan-3 modulates food intake by interacting with the melanocortin/AgRP pathway. Annals of the New York Academy of Sciences. 2003;994:66–73. doi: 10.1111/j.1749-6632.2003.tb03163.x. [DOI] [PubMed] [Google Scholar]
- 8.Strader AD, Reizes O, Woods SC, Benoit SC, Seeley RJ. Mice lacking the syndecan-3 gene are resistant to diet-induced obesity. Journal of Clinical Investigation. 2004;114(9):1354–1360. doi: 10.1172/JCI20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christianson HC, Belting M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biology. 2014;35:51–55. doi: 10.1016/j.matbio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Tsen G, Halfter W, Kroger S, Cole GJ. Agrin is a heparan sulfate proteoglycan. The Journal of Biological Chemistry. 1995;270(7):3392–3399. doi: 10.1074/jbc.270.7.3392. [DOI] [PubMed] [Google Scholar]
- 11.Murdoch AD, Dodge GR, Cohen I, Tuan RS, Iozzo RV. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. Journal of Biological Chemistry. 1992;267(12):8544–8557. [PubMed] [Google Scholar]
- 12.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nature Reviews Cancer. 2003;3(6):422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 13.Guretzki HJ, Schleicher E, Gerbitz KD, Olgemoller B. Heparin induces endothelial extracellular matrix alterations and barrier dysfunction. American Journal of Physiology. 1994;267(4, part 1):C946–C954. doi: 10.1152/ajpcell.1994.267.4.C946. [DOI] [PubMed] [Google Scholar]
- 14.Costell M, Gustafsson E, Aszódi A, et al. Perlecan maintains the integrity of cartilage and some basement membranes. Journal of Cell Biology. 1999;147(5):1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asplund A, Östergren-Lundén G, Camejo G, Stillemark-Billton P, Bondjers G. Hypoxia increases macrophage motility, possibly by decreasing the heparan sulfate proteoglycan biosynthesis. Journal of Leukocyte Biology. 2009;86(2):381–388. doi: 10.1189/jlb.0908536. [DOI] [PubMed] [Google Scholar]
- 16.Moon JJ, Matsumoto M, Patel S, Lee L, Guan J, Li S. Role of cell surface heparan sulfate proteoglycans in endothelial cell migration and mechanotransduction. Journal of Cellular Physiology. 2005;203(1):166–176. doi: 10.1002/jcp.20220. [DOI] [PubMed] [Google Scholar]
- 17.Floris S, van den Born J, van der Pol SMA, Dijkstra CD, de Vries HE. Heparan sulfate proteoglycans modulate monocyte migration across cerebral endothelium. Journal of Neuropathology and Experimental Neurology. 2003;62(7):780–790. doi: 10.1093/jnen/62.7.780. [DOI] [PubMed] [Google Scholar]
- 18.Yurchenco PD, Schittny JC. Molecular architecture of basement membranes. The FASEB Journal. 1990;4(6):1577–1590. doi: 10.1096/fasebj.4.6.2180767. [DOI] [PubMed] [Google Scholar]
- 19.Paulsson M. Basement membrane proteins: structure, assembly, and cellular interactions. Critical Reviews in Biochemistry and Molecular Biology. 1992;27(1-2):93–127. doi: 10.3109/10409239209082560. [DOI] [PubMed] [Google Scholar]
- 20.Farkas E, Luiten PGM. Cerebral microvascular pathology in aging and Alzheimer's disease. Progress in Neurobiology. 2001;64(6):575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 21.Hawkes CA, Gatherer M, Sharp MM, et al. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-β from the mouse brain. Aging Cell. 2013;12(2):224–236. doi: 10.1111/acel.12045. [DOI] [PubMed] [Google Scholar]
- 22.Pikas DS, Li J, Vlodavsky I, Lindahl U. Substrate specificity of heparanases from human hepatoma and platelets. Journal of Biological Chemistry. 1998;273(30):18770–18777. doi: 10.1074/jbc.273.30.18770. [DOI] [PubMed] [Google Scholar]
- 23.Zcharia E, Metzger S, Chajek-Shaul T, et al. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. The FASEB Journal. 2004;18(2):252–263. doi: 10.1096/fj.03-0572com. [DOI] [PubMed] [Google Scholar]
- 24.Zcharia E, Jia J, Zhang X, et al. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS ONE. 2009;4(4) doi: 10.1371/journal.pone.0005181.e5181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Escobar Galvis ML, Jia J, Zhang X, et al. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nature Chemical Biology. 2007;3(12):773–778. doi: 10.1038/nchembio.2007.41. [DOI] [PubMed] [Google Scholar]
- 26.Baker AB, Gibson WJ, Kolachalama VB, et al. Heparanase regulates thrombosis in vascular injury and stent-induced flow disturbance. Journal of the American College of Cardiology. 2012;59(17):1551–1560. doi: 10.1016/j.jacc.2011.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Escobar Galvis ML, Gong F, et al. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein a amyloidosis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(18):6473–6477. doi: 10.1073/pnas.0502287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Massena S, Christoffersson G, Hjertström E, et al. Achemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2010;116(11):1924–1931. doi: 10.1182/blood-2010-01-266072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, Wang B, O'Callaghan P, et al. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-β in murine brain. Acta Neuropathologica. 2012;124(4):465–478. doi: 10.1007/s00401-012-0997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. International Journal of Biochemistry and Cell Biology. 2006;38(12):2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Hong X, Nelson KK, deCarvalho AC, Kalkanis SN. Heparanase expression of glioma in human and animal models: laboratory investigation. Journal of Neurosurgery. 2010;113(2):261–269. doi: 10.3171/2009.9.JNS09682. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Zhang X, Lu Z, Yu SP, Wei L. Expression of heparanase in vascular cells and astrocytes of the mouse brain after focal cerebral ischemia. Brain Research. 2012;1433:137–144. doi: 10.1016/j.brainres.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi H, Matsumoto H, Kumon Y, et al. Expression of heparanase in nestin-positive reactive astrocytes in ischemic lesions of rat brain after transient middle cerebral artery occlusion. Neuroscience Letters. 2007;417(3):250–254. doi: 10.1016/j.neulet.2007.02.075. [DOI] [PubMed] [Google Scholar]
- 34.Vlodavsky I, Beckhove P, Lerner I, et al. Significance of heparanase in cancer and inflammation. Cancer Microenvironment. 2012;5(2):115–132. doi: 10.1007/s12307-011-0082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riaz A, Ilan N, Vlodavsky I, Li J, Johansson S. Characterization of heparanase-induced phosphatidylinositol 3-kinase-AKT activation and its integrin dependence. The Journal of Biological Chemistry. 2013;288(17):12366–12375. doi: 10.1074/jbc.M112.435172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 37.Cai H, Wang Y, McCarthy D, et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nature Neuroscience. 2001;4(3):233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 38.Bergmans BA, de Strooper B. γ-secretases: from cell biology to therapeutic strategies. The Lancet Neurology. 2010;9(2):215–226. doi: 10.1016/S1474-4422(09)70332-1. [DOI] [PubMed] [Google Scholar]
- 39.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 40.Janssen JC, Beck JA, Campbell TA, et al. Early onset familial Alzheimer's disease: mutation frequency in 31 families. Neurology. 2003;60(2):235–239. doi: 10.1212/01.wnl.0000042088.22694.e3. [DOI] [PubMed] [Google Scholar]
- 41.Raux G, Guyant-Maréchal L, Martin C, et al. Molecular diagnosis of autosomal dominant early onset Alzheimer's disease: an update. Journal of Medical Genetics. 2005;42(10):793–795. doi: 10.1136/jmg.2005.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. American Journal of Human Genetics. 1999;65(3):664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haass C, Hung AY, Selkoe DJ, Teplow DB. Mutations associated with a locus for familial Alzheimer's disease result in alternative processing of amyloid β-protein precursor. Journal of Biological Chemistry. 1994;269(26):17741–17748. [PubMed] [Google Scholar]
- 44.Citron M, Oltersdorf T, Haass C, et al. Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature. 1992;360(6405):672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 45.Cai X-D, Golde TE, Younkin SG. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259(5094):514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 46.Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science. 2010;330(6012):p. 1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer's Aβ peptide: the many roads to perdition. Neuron. 2004;43(5):605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 48.Weller RO, Massey A, Newman TA, Hutchings M, Kuo Y, Roher AE. Cerebral amyloid angiopathy: amyloid β accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. American Journal of Pathology. 1998;153(3):725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Aβ-degrading enzymes in Alzheimer's disease. Brain Pathology. 2008;18(2):240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-β peptide across the blood-brain barrier: implication for therapies in Alzheimer's disease. CNS and Neurological Disorders, Drug Targets. 2009;8(1):16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T. Macrophages in Alzheimer's disease: the blood-borne identity. Journal of Neural Transmission. 2010;117(8):961–970. doi: 10.1007/s00702-010-0422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-β peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathology. 2008;18(2):253–266. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bame KJ, Danda J, Hassall A, Tumova S. Aβ(1-40) prevents heparanase-catalyzed degradation of heparan sulfate glycosaminoglycans and proteoglycans in vitro. A role for heparan sulfate proteoglycan turnover in Alzheimer's disease. Journal of Biological Chemistry. 1997;272(27):17005–17011. doi: 10.1074/jbc.272.27.17005. [DOI] [PubMed] [Google Scholar]
- 54.Buée L, Ding W, Anderson JP, et al. Binding of vascular heparan sulfate proteoglycan to Alzheimer’s amyloid precursor protein is mediated in part by the N-terminal region of A4 peptide. Brain Research. 1993;627(2):199–204. doi: 10.1016/0006-8993(93)90321-d. [DOI] [PubMed] [Google Scholar]
- 55.Leveugle B, Scanameo A, Ding W, Fillit H. Binding of heparan sulfate glycosaminoglycan to β-amyloid peptide: inhibition by potentially therapeutic polysulfated compounds. NeuroReport. 1994;5(11):1389–1392. [PubMed] [Google Scholar]
- 56.Watson DJ, Lander AD, Selkoe DJ. Heparin-binding properties of the amyloidogenic peptides Aβ and amylin: dependence on aggregation state and inhibition by Congo red. The Journal of Biological Chemistry. 1997;272(50):31617–31624. doi: 10.1074/jbc.272.50.31617. [DOI] [PubMed] [Google Scholar]
- 57.Giulian D, Haverkamp LJ, Yu J, et al. The HHQK domain of β-amyloid provides a structural basis for the immunopathology of Alzheimer's disease. The Journal of Biological Chemistry. 1998;273(45):29719–29726. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 58.Lindahl B, Westling C, Giménez-Gallego G, Lindahl U, Salmivirta M. Common binding sites for β-amyloid fibrils and fibroblast growth factor-2 in heparan sulfate from human cerebral cortex. Journal of Biological Chemistry. 1999;274(43):30631–30635. doi: 10.1074/jbc.274.43.30631. [DOI] [PubMed] [Google Scholar]
- 59.Gupta-Bansal R, Frederickson RC, Brunden KR. Proteoglycan-mediated inhibition of Aβ proteolysis: a potential cause of senile plaque accumulation. The Journal of Biological Chemistry. 1995;270(31):18666–18671. doi: 10.1074/jbc.270.31.18666. [DOI] [PubMed] [Google Scholar]
- 60.Snow AD, Willmer J, Kisilevsky R. Sulfated glycosaminoglycans: a common constituent of all amyloids? Laboratory Investigation. 1987;56(1):120–123. [PubMed] [Google Scholar]
- 61.Perlmutter LS, Chui HC, Saperia D, Athanikar J. Microangiography and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer's disease. Brain Research. 1990;508(1):13–19. doi: 10.1016/0006-8993(90)91111-s. [DOI] [PubMed] [Google Scholar]
- 62.Snow AD, Mar H, Nochlin D, et al. The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer's disease. American Journal of Pathology. 1988;133(3):456–463. [PMC free article] [PubMed] [Google Scholar]
- 63.Su JH, Cummings BJ, Cotman CW. Localization of heparan sulfate glycosaminoglycan and proteoglycan core protein in aged brain and Alzheimer's disease. Neuroscience. 1992;51(4):801–813. doi: 10.1016/0306-4522(92)90521-3. [DOI] [PubMed] [Google Scholar]
- 64.Donahue JE, Berzin TM, Rafii MS, et al. Agrin in Alzheimer's disease: altered solubility and abnormal distribution within microvasculature and brain parenchyma. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(11):6468–6472. doi: 10.1073/pnas.96.11.6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Horssen J, Kleinnijenhuis J, Maass CN, et al. Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiology of Aging. 2002;23(4):537–545. doi: 10.1016/s0197-4580(02)00010-6. [DOI] [PubMed] [Google Scholar]
- 66.Kurup S, Wijnhoven TJM, Jenniskens GJ, et al. Characterization of anti-heparan sulfate phage display antibodies AO4B08 and HS4E4. The Journal of Biological Chemistry. 2007;282(29):21032–21042. doi: 10.1074/jbc.M702073200. [DOI] [PubMed] [Google Scholar]
- 67.van Kuppevelt TH, Dennisseni MA, van Venrooij WJ, Hoet RMA, Veerkamp JH. Generation and application of type-specific anti-heparan sulfate antibodies using phage display technology: further evidence for heparan sulfate heterogeneity in the kidney. The Journal of Biological Chemistry. 1998;273(21):12960–12966. doi: 10.1074/jbc.273.21.12960. [DOI] [PubMed] [Google Scholar]
- 68.Bruinsma IB, te Riet L, Gevers T, et al. Sulfation of heparan sulfate associated with amyloid-β plaques in patients with Alzheimer's disease. Acta Neuropathologica. 2010;119(2):211–220. doi: 10.1007/s00401-009-0577-1. [DOI] [PubMed] [Google Scholar]
- 69.Dam GBT, Kurup S, Van De Westerlo EMA, et al. 3-O-sulfated oligosaccharide structures are recognized by anti-heparan sulfate antibody HS4C3 . The Journal of Biological Chemistry. 2006;281(8):4654–4662. doi: 10.1074/jbc.M506357200. [DOI] [PubMed] [Google Scholar]
- 70.O’Callaghan P, Sandwall E, Li J, et al. Heparan sulfate accumulation with Aβ deposits in Alzheimer’s disease and Tg2576 mice is contributed by glial cells. Brain Pathology. 2008;18(4):548–561. doi: 10.1111/j.1750-3639.2008.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dennissen MABA, Jenniskens GJ, Pieffers M, et al. Large, tissue-regulated domain diversity of heparan sulfates demonstrated by phage display antibodies. The Journal of Biological Chemistry. 2002;277(13):10982–10986. doi: 10.1074/jbc.M104852200. [DOI] [PubMed] [Google Scholar]
- 72.Hosono-Fukao T, Ohtake-Niimi S, Hoshino H, et al. Heparan sulfate subdomains that are degraded by sulf accumulate in cerebral amyloid ß plaques of alzheimer's disease: evidence from mouse models and patients. The American Journal of Pathology. 2012;180(5):2056–2067. doi: 10.1016/j.ajpath.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 73.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nature Reviews Neuroscience. 2007;8(7):499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 74.Kandimalla KK, Scott OG, Fulzele S, Davidson MW, Poduslo JF. Mechanism of neuronal versus endothelial cell uptake of Alzheimer's disease amyloid β protein. PLoS ONE. 2009;4(2) doi: 10.1371/journal.pone.0004627.e4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kanekiyo T, Liu C, Shinohara M, Li J, Bu G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer's amyloid-β . Journal of Neuroscience. 2012;32(46):16458–16465. doi: 10.1523/JNEUROSCI.3987-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid β-protein via a scavenger receptor. Neuron. 1996;17(3):553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 77.Nielsen HM, Mulder SD, Beliën JAM, Musters RJP, Eikelenboom P, Veerhuis R. Astrocytic Aβ1-42 uptake is determined by Aβ-aggregation state and the presence of amyloid-associated proteins. GLIA. 2010;58(10):1235–1246. doi: 10.1002/glia.21004. [DOI] [PubMed] [Google Scholar]
- 78.Lai AY, McLaurin J. Mechanisms of amyloid-β peptide uptake by neurons: the role of lipid rafts and lipid raft-associated proteins. International Journal of Alzheimer's Disease. 2011;2011:11 pages. doi: 10.4061/2011/548380.548380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS Journal. 2010;277(6):1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 80.Tahara K, Kim H, Jin J, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain. 2006;129(11):3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fu H, Liu B, Frost JL, et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. GLIA. 2012;60(6):993–1003. doi: 10.1002/glia.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang CN, Shiao YJ, Shie FS, et al. Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiology of Disease. 2011;42(3):221–230. doi: 10.1016/j.nbd.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 83.N’Songo A, Kanekiyo T, Bu G. LRP1 plays a major role in the amyloid-β clearance in microglia. Molecular Neurodegeneration. 2013;8(supplement 1):p. P33. [Google Scholar]
- 84.Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer's disease risk gene cd33 inhibits microglial uptake of amyloid β . Neuron. 2013;78(4):631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Doens D, Fernandez PL. Microglia receptors and their implications in the response to amyloid β for Alzheimer's disease pathogenesis. Journal of Neuroinflammation. 2014;11:p. 48. doi: 10.1186/1742-2094-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sandwall E, O'Callaghan P, Zhang X, Lindahl U, Lannfelt L, Li J. Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology. 2010;20(5):533–541. doi: 10.1093/glycob/cwp205. [DOI] [PubMed] [Google Scholar]
- 87.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 88.Kanekiyo T, Zhang J, Liu Q, Liu C, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-β uptake. The Journal of Neuroscience. 2011;31(5):1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Holtzman DM, Bales KR, Tenkova T, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(6):2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang Q, Lee CYD, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Aβ . Neuron. 2008;58(5):681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer's disease: accidental encounters or partners? Neuron. 2014;81(4):740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O’Callaghan P, Noborn F, Sehlin D, et al. Apolipoprotein E increases cell association of amyloid-β 40 through heparan sulfate and LRP1 dependent pathways. Amyloid. 2014;21(2):76–87. doi: 10.3109/13506129.2013.879643. [DOI] [PubMed] [Google Scholar]
- 93.Simard AR, Soulet D, Gowing G, Julien J, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49(4):489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 94.Frenkel D, Wilkinson K, Zhao L, et al. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nature Communications. 2013;4, article 2030 doi: 10.1038/ncomms3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fiala M, Lin J, Ringman J, et al. Ineffective phagocytosis of amyloid-β by macrophages of Alzheimer's disease patients. Journal of Alzheimer's Disease. 2005;7(3):221–232. doi: 10.3233/jad-2005-7304. [DOI] [PubMed] [Google Scholar]
- 96.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nature Immunology. 2005;6(9):902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 97.Zarow C, Barron E, Chui HC, Perlmutter LS. Vascular basement membrane pathology and Alzheimer's disease. Annals of the New York Academy of Sciences. 1997;826:147–160. doi: 10.1111/j.1749-6632.1997.tb48467.x. [DOI] [PubMed] [Google Scholar]
- 98.Bonneh-Barkay D, Wiley CA. Brain extracellular matrix in neurodegeneration. Brain Pathology. 2009;19(4):573–585. doi: 10.1111/j.1750-3639.2008.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hawkes CA, Härtig W, Kacza J, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathologica. 2011;121(4):431–443. doi: 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- 100.Bergamaschini L, Rossi E, Storini C, et al. Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces pl aques and β-amyloid accumulation in a mouse model of Alzheimer’s disease. Journal of Neuroscience. 2004;24(17):4181–4186. doi: 10.1523/JNEUROSCI.0550-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Scholefield Z, Yates EA, Wayne G, Amour A, McDowell W, Turnbull JE. Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer's β-secretase. Journal of Cell Biology. 2003;163(1):97–107. doi: 10.1083/jcb.200303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Small DH, Klaver DW, Beckman M. Regulation of proBACE1 by glycosaminoglycans. Neurodegenerative Diseases. 2008;5(3-4):206–208. doi: 10.1159/000113703. [DOI] [PubMed] [Google Scholar]
- 103.Schwörer R, Zubkova OV, Turnbull JE, Tyler PC. Synthesis of a targeted library of heparan sulfate hexa- to dodecasaccharides as inhibitors of β-secretase: potential therapeutics for Alzheimer's disease. Chemistry A. 2013;19(21):6817–6823. doi: 10.1002/chem.201204519. [DOI] [PubMed] [Google Scholar]