

Abstract
Retinoic-acid receptor-related orphan receptor-γt-positive (RORγt+) innate lymphoid cells (ILCs) produce interleukin (IL)-22 and IL-17, which are critical for protective immunity against enteric pathogens. The molecular mechanism underlying the development and survival of RORγt+ ILCs is not thoroughly understood. Here we show that Dedicator of cytokinesis 8 (DOCK8), a scaffolding protein involved in cytoskeletal rearrangement and cell migration, is essential for the protective immunity against Citrobacter rodentium. A comparative RNA sequencing-based analysis reveals an impaired induction of antimicrobial peptides in the colon of DOCK8-deficient mice, which correlates with high susceptibility to infection and a very low number of IL-22-producing RORγt+ ILCs in their GI tract. Furthermore, DOCK8-deficient RORγt+ ILCs are less responsive to IL-7 mediated signaling, more prone to apoptosis and produce less IL-22 due to a defect in IL-23-mediated STAT3 phosphorylation. Our studies reveal an unsuspected role of DOCK8 for the function, generation and survival of RORγt+ ILCs.
The mammalian gastrointestinal (GI) tract harbors a large number of microbiota 1. The mucosa of the GI tract is a critical interface for the host immune system and its associated microbial ecosystem 2. At the barrier surfaces, ILCs constitute a major fraction of the immune cell repertoire and serve a protective role in innate immune response to infectious microorganisms and in tissue remodeling after damage inflicted by injury or infection. ILCs are Lineage-negative, Id2-dependent cells with varying functions 3–5. Similar to T helper cell lineage, ILCs are classified into three groups 6, 7 based on their requirement of transcription factors and the effector cytokine production. Type 1 ILCs (ILC1) require transcription factor T-bet and produce interferon-γ, while type 2 ILCs (ILC2) depend on transcription factor GATA3 and produce Th2-associated cytokines IL-5 and IL-13 5, 6. Type 3 ILCs (ILC3) express RORγt transcription factor and produce IL-22 and/or IL-17 3, 5, 7. Type 3 ILCs include lymphoid tissue inducer cells (LTi) that mediate lymphoid tissue development in the fetus and its regeneration in the adult. Accordingly, RORγt-deficient mice lack lymphoid tissue inducer cells and lymph node formation 8. Although the transcription factors important for the generation of these cells have been identified, little is known about the factors important for their maintenance and regulation of cytokine production.
RORγt+ ILCs produce large quantities of IL-22 9–14. IL-22 has been shown to promote wound healing, proliferation, and anti-apoptotic pathways in the intestinal epithelial cells 15; it also up-regulates anti-microbial peptides and mucus production 9, 16. IL-22-mediated early induction of antimicrobial peptides, RegIIIγ and RegIIIβ in colonic epithelial cells is required for protection from C. rodentium infection 12, 16. Accordingly, IL-22-deficient mice show increased morbidity and mortality after C. rodentium infection16. Production of IL-22 by ILCs in response to C. rodentium infection is promoted by IL-23 12, 16, 17.
DOCK8 interacts with the Rho family small G protein Cdc42 18, 19. A very recent study has shown that in B cells, after ligation of TLR9, DOCK8 serves as an adaptor protein and links TLR9 to a STAT3 cascade, which is essential for TLR9-driven B cell proliferation and differentiation 20. DOCK8 mutation (null) is associated with combined immunodeficiency in humans 21, 22. DOCK8-deficient patients develop atopic dermatitis, Staphylococcus aureus skin abscesses or soft tissue infection, pneumonias, elevated serum IgE, eosinophilia, and are more susceptible to cancer 21, 23. The majority of these patients have low numbers of CD4+ and CD8+ T cells in the blood and spleen 21. However, it has not been investigated whether DOCK8-deficient patients also have defects in ILCs or altered microbial composition in their GI tracts, which would render them more susceptible to infections.
Herein, we present evidence that DOCK8 is essential for protective immunity against an enteric pathogen. Using DOCK8-deficient mice, we have shown that DOCK8 plays a crucial role in the survival of RORγt+ ILCs and their cytokine production. Indeed, we further determined that RORγt+ ILCs require DOCK8 for optimal STAT3 activation and IL-22 production upon IL-23 stimulation. Therefore, our studies suggest that besides having defective B-cell mediated responses, DOCK8-deficient patients may also have limited numbers of ILC3. Thus, our studies may help explain why the absence of DOCK8 contributes to patients’ susceptibility to a broad spectrum of pathogens.
Results
DOCK8 is required for the clearance of an enteric pathogen
To define the role of scaffolding protein DOCK8 in IL-23-mediated induction of IL-22, we utilized DOCK8pri/pri mice 24. These mice had defects in B cell immunological synapses, germinal center formation and long-lived antibody production. Along with B cell defects, these mice also had a defect in T cell survival and function 25.
Therefore, we first examined the total cell number as well as the cell composition in DOCK8pri/pri mice. As previously shown, these mice have significantly fewer cells in spleen and lymph nodes (LN) in comparison to age and sex-matched wild-type (WT) mice (Supplementary Fig. 1a, top). In contrast to the peripheral cell count, DOCK8pri/pri mice have a comparable number of lamina propria lymphocytes (LPL) in the GI tract, but significantly higher cells in the MLN in comparison to WT mice (Supplementary Fig. 1a, bottom). Consistent with previous findings, CD4+ and CD8+ T cells in the spleens of DOCK8pri/pri mice were reduced by 50–60% of WT levels 25, 26. Additionally, we did not find significant changes in the numbers of CD4+ and CD8+ T cells in the lamina propria (LP) of DOCK8pri/pri mice in comparison to WT (Supplementary Fig. 1b). Interestingly, DOCK8-deficiency did not affect the number of mature B cells in lymphoid organs except in the small intestine, where we found a significant reduction of the B cell percentage (Supplementary Fig. 1c). This reduction was also more pronounced when mice were infected with C. rodentium (Supplementary Fig. 1d and 2). The B cells that were found in the colonic LP expressed high levels of MHC Class II (Supplementary Fig. 1d). DOCK8 has been shown to be critical for interstitial dendritic cell (DC) migration during immune responses 26. Therefore, we assessed whether DOCK8 deficiency could affect the recruitment of myeloid cells in response to an infection. However, we did not find any reduction in the recruitment of CD11b+ or CD11c+ cells to the LP of DOCK8pri/pri mice after infection with C. rodentium (Supplementary Fig. 1e).
Since it has been shown that B cells play an important role in the clearance of enteric pathogens 27–29, we infected WT, DOCK8pri/pri mice, B cell-deficient (μMT), and IL-23R−/− mice with C. rodentium. Interestingly, similar to IL-23R−/− mice, all DOCK8pri/pri mice lost weight and succumbed to the infection (Fig. 1a and 1b). The spleens and livers of DOCK8pri/pri mice contained two to four log more bacteria than WT, indicating that DOCK8pri/pri mice were unable to control bacterial dissemination (Fig. 1c). Although we observed a defect in the recruitment of MHC class IIhigh B cells to the GI tract of DOCK8pri/pri mice (Supplementary Fig. 1c–d and 2), μMT mice survived, and were able to clear the infection (Fig. 1a and 1b). These results suggest that DOCK8 deficiency renders mice susceptible to C. rodentium infection independently of B cell function. IL-22−/− and IL-23p19−/− mice are highly susceptible to C. rodentium infection 16, 30, 31. Therefore, we used IL-23R−/− mice as a positive control for infection. Consistent with above, IL-23R−/− mice were highly susceptible to infection (Fig. 1a–b).
Figure 1. DOCK8 is required for clearance of enteric pathogen.<.

br>(a–c) WT, IL-23R−/−, DOCK8pri/pri and μMT mice were infected with C. rodentium (a) percent survival and (b) average body weight change of WT (n=5), IL-23R−/− (n=5), DOCK8pri/pri (n=6) and mMT (n=4) mice after infection. (c) C. rodentium CFU in the spleens and livers of infected mice at day 8 p.i. (d–e) DOCK8 is required in hematopoietic compartment for the survival. Bone marrow cells from mice of indicated genotype were transferred into irradiated Rag2−/−IL-2Rγc−/− mice. (d) Percent survival and (e) C. rodentium CFU in the livers of infected mice at day 8 p.i. (f) DOCK8-dependent gene expression in the colons of C. rodentium infected mice. (g) Colonic IL-22, RegIIIγ, and IL-23p19 expression in response to infection at day 8 p.i. Three independent experiments were performed with a minimum of four to six mice per group. The data shown are the mean ± SD. P-values were obtained by student’s t-test. **p< 0.01, ***p< 0.001. ns = not significant.
Next we determined whether DOCK8 was required within hematopoietic cells or non-hematopoietic cells. We generated chimeras by transferring bone marrow cells isolated from either DOCK8pri/pri or WT littermate mice into irradiated Rag2−/−IL-2Rγ−/− mice. Five weeks after transfer, these mice were infected with C. rodentium. As expected, similar to Rag2−/− IL-2Rγ−/− mice, all Rag2−/−IL-2Rγ−/− mice transferred with DOCK8pri/pri bone marrow succumbed to the infection, while WT bone marrow transferred mice survived the infection (Fig. 1d). The livers of Rag2−/−IL-2Rγ−/− mice and Rag2−/−IL-2Rγ−/− mice transferred with DOCK8pri/pri bone marrow contained significantly higher bacterial load than those transferred with WT bone marrow cells (Fig. 1e). These results suggest that DOCK8 is required within the hematopoietic compartment and plays an important role in the function of immune cells involved in protective immunity.
To understand the effect of DOCK8 on global transcription levels, we used RNA-sequencing to examine differential gene expression upon C. rodentium infection. RNA was purified from the infected colons of WT and DOCK8pri/pri mice. We evaluated the levels of the gene expression in the infected colons of WT and DOCK8pri/pri mice. Among the most highly expressed genes, RegIIIγ, RegIIIβ and IL-22 were highly defective in DOCK8pri/pri mice, in comparison to WT mice (Fig. 1f). Since it has been shown that early IL-22-dependent induction of RegIIIγ is critical for protective immunity against enteric pathogen 12, 16, we measured colonic IL-22 and RegIIIγ transcripts after infection with C. rodentium. Consistent with RNA-seq data, the levels of IL-22 and RegIIIγ expression were very low in the colon of DOCK8pri/pri mice in comparison to levels found in the colon of WT mice (Fig. 1g). This finding indicates that DOCK8 is required for the induction of IL-22 expression upon infection. IL-22 can be produced by many cell types, including ILCs and Th22 cells 3, 9, 11, 12, 31–33. Therefore, we addressed the cellular source of IL-22 that was critical for protection against enteric pathogen infection.
To assess whether DOCK8 is required for IL-22 production by ILCs or Th22 we crossed DOCK8pri/pri mice with Rag1−/− mice lacking T cells. These mice were also highly susceptible to enteric pathogen infection and failed to clear infection (Supplementary Fig. 3a–b). The spleens and livers of Rag1−/−DOCK8pri/pri mice contained significantly more bacteria than Rag1−/− mice, indicating that DOCK8 is required in innate immune cells for the control of enteric pathogen infection (Supplementary Fig. 3c). And similar to DOCK8pri/pri mice, Rag1−/−DOCK8pri/pri mice were also highly defective in IL-22-dependent RegIIIγ induction after enteric pathogen infection (Supplementary Fig. 3d). All together, these data indicate that DOCK8 is required for protective immunity against C. rodentium by regulating IL-22 production from innate immune cells.
Exogenous IL-22 protect DOCK8 KO mice during C. rodentium infection
Our data (Fig. 1 and Supplementary Fig. 3) clearly showed that DOCK8 was required for controlling C. rodentium infection. To test whether exogenous IL-22 could rescue the DOCK8pri/pri mice upon C. rodentium infection, we injected IL-22 cDNA via hydrodynamic delivery into DOCK8pri/pri mice. We had previously shown that a single injection of IL-22 encoding cDNA into IL-22-defective mice restored expression of IL-22 in the blood and colon, lasted as long as 10 days 34. We gave one injection of IL-22-encoding plasmid 2 days before infection and another at day 5 post-infection. DOCK8pri/pri mice that received the IL-22 plasmid survived an average of 5 days longer and had significantly less weight loss compared to empty plasmid recipients (Fig. 2a–b). In the IL-22 plasmid-treated group, splenic and liver bacteria counts were significantly lower compared to the empty plasmid-injected group (Fig. 2c). Thus, these sets of experiments collectively demonstrated that defective IL-22 production was an important cause of high bacteria load and death of DOCK8pri/pri mice during C. rodentium infection. Conversely, only 15% of DOCK8pri/pri mice that received the IL-22 plasmid survived the infection, suggesting that these mice might have other defects in innate immune cells, which were required for further survival. DOCK8 has been shown to be important for B cell immunological synapses, germinal centers and long-lived antibody production 24. It is also important for the survival and function of CD8+ T and NKT cells 25, 35. The DOCK8-deficiency also results in reduced CD4+ T cells and defective interstitial dendritic cell migration during immune responses 25, 26. These previous findings suggest that DOCK8 is also required for fully functional acquired immune response. It is important to mention that mice defective in the recombination activating gene become predisposed to C. rodentium infection at 3 to 4 weeks post-infection, suggesting that both innate and acquired immune response is required to clear C. rodentium infection (Supplementary Fig. 3a–b) 12, 16. Thus, partial restoration of survival in DOCK8pri/pri mice injected with IL-22 plasmid could be due to defective acquired immune responses.
Figure 2. Exogenous IL-22 protect DOCK8 KO mice during C. rodentium infection.<.
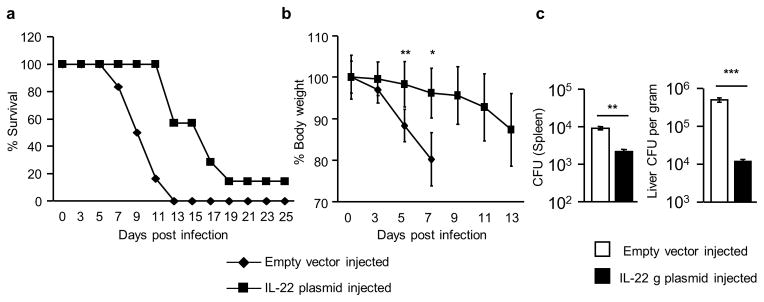
br>DOCK8pri/pri mice were administered with IL-22 expressing plasmid or empty vector via hydrodynamic injections at −2 and +5 days of C. rodentium infection. (a) Percent survival (b) average body weight changes after infection and (c) CFU in the spleens and livers of infected mice. All the data are representative of three independent experiments with five mice per group. The data shown are the mean ± SD. P-values were obtained by student’s t-test. *p< 0.05,**p< 0.01, ***p< 0.001.
RORγt+ ILCs require DOCK8 for optimal IL-22 production
Prior reports undoubtedly showed that ILC3 plays a decisive role in protection against C. rodentium infection by producing IL-22 early on 9, 10, 12, 13, 36–38. In this model, early during infection, classical DCs produce IL-23 that induces IL-22 production via ILC3 12, 16, 17. IL-22 enhances epithelial cell repair and prevents dissemination of bacteria. Therefore, we analyzed whether the defect in IL-22 production by DOCK8-deficient ILCs resulted from direct DOCK8-mediated signaling in ILCs or resulted from an indirect effect of DOCK8 on other cells important for its function. The defective IL-22 production in DOCK8pri/pri mice could be due to defective IL-23 expression in DCs. Therefore, we measured IL-23p19, a specific subunit of IL-23, in the colon of C. rodentium infected mice. Interestingly, IL-23 expression in the colon of C. rodentium infected DOCK8pri/pri mice was comparable to that of WT mice (Fig. 1g), suggesting that the IL-22 defect in DOCK8pri/pri mice is not due to impaired IL-23 expression in DCs. We also analyzed the expression of Dock8 in innate IL-23R+ cells and have found that Dock8 was highly expressed in these cells (Supplementary Fig. 4).
Next we investigated whether DOCK8 was also required for IL-22 production by RORγt+ ILCs upon IL-23 stimulation. Therefore, we examined IL-22 expression in the LPLs isolated from DOCK8pri/pri mice and their littermate control WT mice. As expected, in WT mice, ILC3 (CD3 negative) produced high amounts of IL-22 in response to IL-23. Compared to those in WT mice, both the percentage and absolute number of CD3−IL-22+ cells in the GI tract of DOCK8pri/pri mice were significantly reduced, indicating that DOCK8 was important for the production of IL-22 in the GI tract (Fig. 3a–b). However, this result could be misleading due to the paucity of RORγt+ ILC3 in DOCK8pri/pri mice that could have led to an overall reduction of IL-22 production. Thus, it remains uncertain whether DOCK8 directly regulates IL-22 production from each individual ILC3. To verify this, we purified CD90.2+IL-23R+ cells from the SI LPLs of Rag1−/−IL-23Rgfp/+ or Rag1−/−IL-23Rgfp/+DOCK8pri/pri mice and cultured them with or without IL-23. The IL-22 protein in the culture supernatant was measured by ELISA. The production of IL-22 by DOCK8-deficient RORγt+ ILCs was significantly reduced compared to those in WT mice (Fig. 3c). To further corroborate our findings, we measured IL-22 and IL-17 transcript levels in sorted ILC3s from Rag1−/−IL-23Rgfp/+ or Rag1−/−IL-23Rgfp/+DOCK8pri/pri mice (Supplementary Fig. 5). The IL-22 transcript level in DOCK8-deficient ILC3s was significantly reduced compared to those in WT mice. However, IL-17A transcript level was comparable in WT and DOCK8-deficient ILC3s. These results suggested that DOCK8 was required for RORγt+ ILCs optimal IL-22 production in response to IL-23 stimulation. We also investigated the function of DOCK8 on IL-22 production by CD4+T cells after C. rodentium infection. DOCK8-deficient CD4+ T cells are highly defective in IL-22 production after C. rodentium infection (Supplementary Fig. 6). However, IL-22 produced by CD4+T cells for protection against C. rodentium is not as crucial as IL22 produced by group 3 ILCs 39. Since IL-23 signals via STAT3-dependent mechanisms, we tested whether DOCK8 was also required in ILC3 for the activation of STAT3 in response to IL-23 stimulation. Thus, we stimulated freshly isolated ILC3 with IL-23 and assessed STAT3 phosphorylation (pY705-STAT3) by flow cytometry (Fig. 3d). Consistent with IL-23-mediated STAT3 phosphorylation in T cells 40, IL-23 induced STAT3 phosphorylation in WT ILCs, however, IL-23-mediated STAT3 activation was severely impaired in DOCK8-deficient ILCs (Fig. 3d). This data clearly show that DOCK8 is required for STAT3 activation and IL-22 production upon IL-23 stimulation.
Figure 3. DOCK8 is also required for the IL-23-mediated induction of IL-22.

(a) SI LPLs from WT and DOCK8pri/pri mice were stimulated with 20 ng/ml IL-23 for 4 hours in the presence of Golgi stop. RORγt and IL-22 expression in CD3− cells was analyzed by flow cytometry. (b) Percentage and absolute numbers of intestinal CD3−IL-22+ cells. (c) Sorted ILC3 (CD90.2+IL-23Rgfp/+) from intestinal LPLs of Rag1−/−IL-23Rgfp/+ and Rag1−/−IL-23Rgfp/+DOCK8pri/pri mice were cultured with or without IL-23 (20ng/ml). The IL-22 produced in culture supernatant was measured by ELISA. (d) FACS analysis for detection of pSTAT3 in ILC3 from Rag1−/−IL-23Rgfp/+ or Rag1−/−IL-23Rgfp/+DOCK8pri/pri mice. All the data are representative of three independent experiments with three mice per group. The data shown are the mean ± SD. P-values were obtained by student’s t-test. **p< 0.01, ***p< 0.001.
Profound reduction in the numbers of RORγt+ ILCs in the absence of DOCK8
The major innate cell population that expresses IL-23R in the intestine is Group 3 ILC, which is composed of LTi cells and ILC3s 41. Various ILC3s were described in humans and mice based on the effector cytokines they produce. These include but are not limited to (1) IL-22 producing NCR+ ILC39 which are also called ILC22, NK22, NKR-LTi or NCR2242 (2) NCR−IL-17A+IFN-γ+ double producing ILC343 and lastly (3) NCR−IL-17A+ ILC3s44. All of these subsets of ILC3s respond to IL-23 and produce the above mentioned cytokine. Thus, the group 3 of ILCs is very heterogeneous and is composed by numerous subsets of cells that can be distinguished by the expressions of various surface markers such a CD4 and NKP46. To determine whether DOCK8-deficiency affects the development of a particular subset of group 3 ILCs in the GI tract, we examined the RORγt+ ILC compartment of DOCK8pri/pri mice. Thus, we stained both the small intestine (SI) and colon lamina propria leukocytes (LPLs) with CD3, CD90.2, CD127, RORγt, CD4 and NKp46-specific antibodies. Both the percentage and number of RORγt+ ILCs (CD3−CD90.2+CD127+RORγt+) in the SI and colon were significantly reduced in DOCK8pri/pri mice (Fig. 4a–c and Supplementary Fig. 7a–c). The number of RORγt+ ILCs in the LPLs of SI of DOCK8pri/pri mice was reduced to 5.6 % of the number in WT mice. The percentage of CD4+ and NKp46+ ILCs among RORγt+ ILCs was comparable between DOCK8pri/pri and WT mice, despite the fact that their absolute numbers were significantly reduced in DOCK8pri/pri mice (Fig. 4a–c and Supplementary Fig. 7a–c). Next, we investigated whether the defect of RORγt+ ILCs in DOCK8pri/pri mice was only limited to the gut, or could be observed also in other organs. Both the percentage and absolute numbers of RORγt+ ILCs were significantly reduced in multiple organs of DOCK8-deficient mice, including spleens and lymph nodes (Supplementary Fig. 8a-c). Thus, DOCK8 is not only important for the survival of RORγt+ ILCs of the gut, but it is also required for their maintenance in other organs as well. We further explored whether DOCK8 requirement by RORγt+ ILCs was perinatal or postnatal. Therefore, we assessed the number of RORγt+ ILCs in the gut of WT and DOCK8pri/pri newborn mice. In contrast to the significant decrease of RORγt+ ILCs in the gut LPLs of adult DOCK8pri/pri mice, the frequency of total RORγt+ and perinatal CD4+ LTi cells in the gut lamina propria isolated from DOCK8pri/pri newborn mice was comparable to those from WT newborns, suggesting that DOCK8 is dispensable for the development of perinatal LTi cells in the gut (Supplementary Fig. 9 a–c). Recently, it was shown that ILC3s could be identified as CD45low and CD90.2high cells39. Therefore, in order to sort perinatal LTi cells, we stained newborn mice GI tract LPLs with CD45, CD3, CD90.2 and RORγt and assessed whether CD3− CD45low and CD90.2high cells could represent RORγt+ cells, as in adult mice (Supplementary Fig. 9d upper panel). Consistent with recently published work39, most of the RORγt+ cells found in LP of newborn mice could be identified as CD3− CD45low and CD90.2high cells. Then, we sorted CD4+ and CD4− perinatal LTi cells and assessed the expression of Dock8. Our result showed that CD4+ LTi cells do express Dock8 transcripts, but that expression level was significantly reduced compared to CD4− LTi and adult ILC3s (Supplementary Fig. 9d–e). All together these results suggest that DOCK8 is dispensable for the development of perinatal LTi cells in the gut.
Figure 4. Profound reduction of RORγt+ ILCs in the absence of DOCK8.

(a) RORγt+ ILCs were analyzed in small intestine LPLs by flow cytometry after gating on CD3−CD90.2+CD127+ cells. (b and c) Percentage and absolute numbers of CD3−CD90.2+, CD3−CD90.2+CD127+RORγt+, CD3−CD90.2+CD127+RORγt+CD4+ and CD3−CD90.2+CD127+RORγt+ NKp46+ cells of WT and DOCK8pri/pri mice. (d and e) Cell intrinsic functions of DOCK8 regulate RORγt+ ILCs. Mixed bone marrow chimeras generated by reconstituting 50% CD45.1 WT and 50% CD45.2 DOCK8pri/pri mice and transferred into lethally irradiated Rag2−/−IL-2Rγc−/− mice. (d) RORγt+ ILCs (CD3−CD90.2+CD127+) were analyzed four weeks post-transfer. (e) The percentage and absolute number of RORγt+ ILCs. Three independent experiments were performed with a minimum of three mice per group. The data shown are the mean ± SD. P-values were obtained by student’s t-test. **p< 0.01, ***p< 0.001. ns = not significant.
To further confirm our findings, we crossed DOCK8pri/pri mice with Rag1−/− IL-23Rgfp/+ and generated Rag1−/− IL-23gfp/+ DOCK8pri/pri mice in which IL-23R expression can be followed based on GFP expression 45. These mice have fewer CD90.2+ IL-23R+ cells compared to littermate control mice (Supplementary Fig. 10a). We also measured the level of IL-23R transcripts in the colons of Rag1−/− IL-23gfp/+ DOCK8pri/pri mice after C. rodentium infection. Consistent with DOCK8pri/pri mice, these mice also had significantly fewer IL-23R transcripts in comparison to Rag1−/− IL-23Rgfp/+ mice (Supplementary Fig. 10b–c). Altogether these results indicate that DOCK8 is involved in the development or survival of RORγt+ IL-23R+ ILCs. To determine whether DOCK8 was only required for the survival of RORγt+ ILCs, we compared the effect of DOCK8-deficiency on the generation of group 2 ILCs (ILC2) in the GI tract LPLs (Supplementary Fig. 11a–b). In contrast to ILC3 in the gut of DOCK8-deficient mice, ILC2 absolute numbers of DOCK8-deficient mice did not significant differ from the number of ILC2s found in DOCK8-sufficient mice. Interestingly, the percentage of ILC2 was inversely correlated with the percentage of ILC3 in DOCK8-deficient mice and was significantly elevated in DOCK8-deficient mice. These data suggest that DOCK8-deficiency specifically influences the generation/survival of RORγt+ ILCs, but not GATA3+ ILC2, in the gut.
To investigate whether the reduction in RORγt+ ILCs is due to a cell intrinsic defect, we generated mixed–bone marrow chimeric mice in which 50% of the reconstituting bone marrow was derived from WT CD45.1 donors and 50% was derived from DOCK8pri/pri CD45.2 congenic donors. Thus, WT and DOCK8pri/pri cells could be distinguished on the basis of CD45.1 and CD45.2 expressions respectively. We then evaluated the generation of RORγt+ ILCs in these chimeric mice. DOCK8-deficient hematopoietic progenitor cells failed to give rise to RORγt+ ILCs, whereas WT bone marrow progenitors developed normally into RORγt+ ILCs in irradiated Rag2−/−IL-2Rγ−/− mice (Fig. 4d–e and Supplementary Fig. 7d–e). These data suggests an intrinsic requirement of DOCK8 for the development of RORγt+ ILCs. Our data clearly show that DOCK8 is required for the generation/survival/function of ILC3. To test whether infusion of WT ILC3 could provide innate immune protection upon C. rodentium infection, 5 X 103 ILC3 sorted from Rag1−/−IL-23Rgfp/+ host was adaptively transferred in to Rag1−/−DOCK8pri/pri mice one hour after infection. DOCK8pri/pri mice that received the ILC3 survived an average of 3.5 days longer and had significantly less weight loss compared to untreated recipients (Supplementary Fig. 12a–b). In the ILC3-transfered group, splenic and liver bacteria counts were significantly lower compared to the untreated group (Supplementary Fig. 12c). Interestingly, the mean survival of DOCK8-deficient mice infused with WT ILC3s is comparable to the mean survival observed in mice that lack function ILC3s that were infused with WT ILC3s12, 39. It remains unclear why the adoptive transfer of WT ILCs could only provide partial protection, one possible explanation is that only limited number of transferred ILC3s can migrate to the colon or these cells have a short life span.
In a recent study, DOCK8 was shown to interact with Myd88 in B cells 20. However, we found that Myd88 was dispensable for the generation of RORγt+ ILCs (Supplementary Fig. 13). This result is consistent with prior findings, in which Myd88−/− mice did not show significant alteration in the development of RORγt+ ILCs 46. DOCK8 also interacts with WASp and regulates NK cell cytotoxicity 47. However, we did not find significant reductions of RORγt+ ILC3 in WASp−/− mice (Supplementary Fig. 13). Thus, DOCK8 is required for RORγt+ ILCs development in a cell-intrinsic manner and independent of Myd88 or WASp-mediated functions.
DOCK8 is required for the generation and survival of RORγt+ ILCs
To address why DOCK8pri/pri mice have drastic reductions in RORγt+ ILCs, we first tested whether these mice had an intact common lymphoid progenitors (CLPs) compartment. RORγt+ ILCs develop from CLPs (Lin−Sca-1lowc-KitIntIL-7Rα+) 48, 49, thus we evaluated the number of these cells in the bone marrow of WT and DOCK8pri/pri mice (Fig. 5a). Interestingly, we did not find any major difference in the percentage of CLPs among WT or DOCK8pri/pri mice. These data are consistent with the fact that DOCK8-deficient mice have normal lymph nodes and DOCK8 maybe required for RORγt+ ILCs survival only after birth. Next, we assessed whether DOCK8-deficient RORγt+ ILCs had a developmental defect due to defects in transcription factors implicated in their generation. These cells require transcription factors Id2 4, 50, RORγt 51 and Tox 52 for their development. Previous work in our laboratory has clearly shown all CD90.2+ IL-23R+ cells are RORγt+ ILCs 34. Therefore, we purified CD90.2+IL-23R+ and CD90.2+IL-23R− cells from Rag1−/− IL-23Rgfp/+ DOCK8pri/pri and Rag1−/− IL-23Rgfp/+ mice and measured transcripts for Id2, Tox and RORγt by quantitative PCR (Q-PCR). However, we observed comparable expression of these transcription factors in CD90.2+ IL-23R+ cells purified from the GI tract of Rag1−/− IL-23Rgfp/+ DOCK8pri/pri mice in comparison to controls (Supplementary Fig. 14). These data further support the idea that DOCK8 is not required for early development of RORγt+ ILCs, but may be required for their maintenance or survival. Recently, the aryl hydrocarbon receptor (Ahr), a ligand-dependent transcription factor, was shown to be important for the generation and maintenance of adult RORγt+ ILCs 10, 37, 38. Therefore, we also examined Ahr transcripts by Q-PCR, but its level of expression in CD90.2+ IL-23R+ cells was comparable to the levels found in WT cells (Supplementary Fig. 14). These data indicate that the effect of DOCK8 on RORγt+ ILCs occurs independently of Ahr expression. Recently it has been shown that Notch signaling is necessary for adult, but not fetal, development of RORγt+ ILCs 48. Thus, we examined the expression of Notch-1 and Notch-2 transcript in CD90.2+ IL-23R+ cells from Rag1−/− IL-23Rgfp/+ and Rag1−/− IL-23Rgfp/+ DOCK8pri/pri mice. CD90.2+ IL-23R+ cells from Rag1−/− IL-23Rgfp/+ DOCK8pri/pri mice had comparable expression of Notch-2, but slightly higher expression of Notch-1 (Supplementary Fig. 15). We also assessed the transcript of Hes1, a Notch transcriptional target, in CD90.2+ IL-23R+ cells but its expression was comparable. However, CD90.2+ IL-23R− cells from Rag1−/− IL-23Rgfp/+ DOCK8pri/pri mice had significantly higher expression of Hes1 transcript (Supplementary Fig. 15 top panel). More importantly we transduced bone morrow hematopoietic stem cells (HSCs) of DOCK8-deficient mice with retrovirus that contained constitutively active intracellular Notch and infused the transduced HSCs into Rag1IL2Rγ-deficient mice recipient. 3 weeks after HSCs reconstitution we analyzed the number of ILC3 in the gut of these transduced HSCs reconstituted mice. In these mice the number of ILC3 was vey limited. Thus overexpression of intracellular Notch could not rescue the ILC3 compartment in the absence of DOCK8 (Supplementary Fig. 15 bottom panel). Therefore, it is very unlikely that ILC3s defects observed in DOCK8-deficient mice are due to a defect in Notch mediated functions.
Figure 5. DOCK8 is required for the generation and survival of RORγt+ ILC.<.

br>(a) Dot plots showing the bone marrow common lymphoid progenitors (CLPs) from WT and DOCK8pri/pri mice. (b) DOCK8-deficient RORγt+ ILC3s have enhanced apoptosis. Rag1−/− and Rag1−/−DOCK8pri/pri SI LPLs were examined by flow cytometry for RORγt and apoptosis markers (Annexin V and 7AAD). All the data are representative of three independent experiments with three mice per group. (c) Histogram showing the IL-7Rα expression on ILC3 (left panel) and bar graph showing the MFI of IL-7Rα expression (right panel) (d) Histogram showing the phosphorylation of STAT5 in ILC3 upon IL-7 stimulation (left panel) and bar graph showing the MFI of pSTAT5 in ILC3 isolated from the SI LPLs of Rag1−/−IL-23Rgfp/+ or Rag1−/−IL-23Rgfp/+DOCK8pri/pri mice (right panel). All the data are representative of four independent experiments with two to four mice per group. The data shown are the mean ± SD. P-values were obtained by student’s t-test. **p< 0.01.
DOCK8 has been implicated in the survival and function of CD8+ T cells and NKT cells 25, 35. Therefore, we next investigated if DOCK8 also influences the survival of RORγt+ ILCs in the GI tract. We stained SI LPLs with 7-AAD and Annexin V to examine the apoptotic cell death. In contrast to CD90.2+RORγt+ cells in the GI tract of Rag1−/− mice, CD90.2+RORγt+ cells isolated from Rag1−/−DOCK8pri/pri mice consistently showed higher Annexin V and 7-AAD staining (Fig. 5b), indicating that intestinal RORγt+ ILCs without DOCK8 are more prone to apoptosis. However, the expression of anti-apoptotic gene Bcl2 was not affected in DOCK8pri/pri mice ILCs, as shown by FACS histogram (Supplementary Fig. 16a). Next we analyzed the expression of anti-apoptotic genes, Bcl-2, Bcl-xL and Mcl-1 in CD90.2+ IL-23R+ cells from Rag1−/− IL-23Rgfp/+ and Rag1−/− IL-23Rgfp/+ DOCK8pri/pri mice. CD90.2+ IL-23R+ cells from Rag1−/− IL-23Rgfp/+ DOCK8pri/pri mice had lower expression of Bcl-xL but no significant difference in expressions of Bcl-2 or Mcl-1 (Supplementary Fig. 16b). Thus DOCK8 may promote the survival of RORγt+ ILCs by inducing Bcl-xL expression.
Since the survival and the generation of RORγt+ ILCs are largely dependent on IL-7 53, 54 mediated signaling we analyzed IL-7Rα expression on RORγt+ ILCs (Fig. 5c). Boths RORγt+ ILCs isolated from DOCK8pri/pri or WT control mice had comparable level of expression of IL-7Rα. Thus, RORγt+ ILC defects in DOCK8-deficient mice are not due to defective IL-7 expression (Fig. 1f right panel) in the gut or defects in IL-7Rα expression on these cells. Next, we explored whether DOCK8-deficient ILC3 had normal response to IL-7 stimulation. Thus we isolated cells from SI LPLs of Rag1−/− IL-23Rgfp/+ and Rag1−/− IL-23Rgfp/+ DOCK8pri/pri mice and measured phosphorylation of STAT5 (pSTAT5) in response to IL-7. To our surprise, DOCK8-deficient ILC3s could not phosphorylate STAT5 as efficiently as WT ILC3s (Fig. 5d). DOCK8-deficient ILC3s have an impaired IL-7 mediated signaling, which may compromise their survival in the gut. All together, these data suggest that DOCK8 is critical for protective immunity against enteric pathogen by regulating the generation and survival of RORγt+ ILCs in the GI tract.
Discussion
In the present study, we have described how deficiency in DOCK8 leads to compromised protective immune response against an enteric pathogen. We have shown that DOCK8 expression in the hematopoietic compartment is required for protective immunity and its deficiency results in drastic reduction of RORγt+ ILCs in the GI tract. We have also presented evidence that DOCK8 positively regulates the survival of RORγt+ ILCs in a cell-intrinsic manner. The development of RORγt+ ILCs is dependent on several transcription factors including Id2 4, 50, RORγt 51,Tox 52 and Ikaros 55. Our studies show that DOCK8 is not required for the development of fetal LTi cells because DOCK8-deficient mice, in contrast to RORγt-deficient mice, develop normal lymph nodes and have normal LTi cells in their GI tract, so it is therefore likely that DOCK8 plays an important role in the maintenance of RORγt+ ILCs after birth or in adult mice. Our findings have uncovered a previously unsuspected role for DOCK8 in protective immunity at GI tract mucosa.
Recently, Ahr was shown to be important for the generation and maintenance of adult RORγt+ ILCs 10, 37, 38. More recently, the transcription factor T-bet (encoded by Tbx21) was also shown to be critical for the development of NKp46+ILC3 56–58. However, neither Ahr nor T-bet is required for the generation of fetal lymphoid tissue inducer (LTi) cells and as a result, these mice have normal lymph node formation. Unlike the above mentioned transcription factors, DOCK8 is unique in that it is not affecting a particular subset of ILC3, but required for the survival of all RORγt+ ILCs, including both CD4+ and NKp46+ subsets. The absence of DOCK8 may influence the microbial composition in the GI tract and that may influence the RORγt+ ILCs generation or survival. However, it is still under debate whether the development of IL-22-producing ILCs requires intestinal symbiotic microbes 11, 36, 42, 46, 54, 58. Our mixed bone marrow chimeric experiment has further demonstrated that RORγt+ ILCs from DOCK8-deficient mice are impaired in a cell-intrinsic manner. Although our findings indicate that DOCK8 is required for the generation and survival of RORγt+ ILCs, DOCK8 is also important for the survival and function of CD8+ T cells 25 and NKT cells 35. Therefore, DOCK8 in general may be required for the survival of many immune cells. However, more studies are required in order to determine by which mechanisms DOCK8 regulates cell survival.
DOCK8-deficient patients develop atopic dermatitis, Staphylococcus aureus skin abscesses or soft tissue infections, pneumonias, elevated serum IgE, eosinophilia and GI tract infections 21, 23, 59. The majority of these patients have low numbers of CD4+ and CD8+ T cells in the blood and spleen 21. However, it has not been investigated whether DOCK8-deficient patients also have defects in ILCs or altered microbial composition in their GI tracts. It is critical because dysbiosis in the microbial community can result in inflammatory diseases not only of the intestine, but also of organs at distal sites 60. It is widely accepted that RORγt+ ILCs are critical for protective immunity at barrier surfaces 11, 36. Therefore, our findings that DOCK8 is required for the generation of RORγt+ ILCs provides additional insight on the clinical features and the severe immunodeficiency observed in DOCK8-deficient patients. Recent data have provided evidence that commensal bacterial-derived signals limit steady-state serum IgE concentration and basophil-associated allergic inflammation 61. Since DOCK8-deficient patients have an autosomal recessive form of hyperimmunoglobulinemia E syndrome characterized by recurrent infections, increased susceptibility to atopic eczema and average serum IgE concentrations 10 times higher than those found in control subjects 21, 22, it is tempting to speculate that the increased susceptibility to bacterial infection and the elevated levels of IgE seen in DOCK8-deficient patients could be the result of dysbiosis in the microbial community, or inadequate control of pathogens due to a defect in the surveillance of the GI tract by ILCs. In addition to infection-susceptibility, DOCK8-deficient patients also have higher susceptibility to cancer 21, 23, 59. Using a mouse model, it has been shown that NKp46+ LTi cells are essential for melanoma rejection mediated by IL-12 62. In the above study, authors showed that IL-12-mediated repression of subcutaneous melanoma is independent of adaptive immunity or cNK cells, but requires innate linease-negative NKp46+ LTi cells. Tumor rejection by NKp46+ LTi was independent of IFN-γ, IL-22, lymphotoxin, perforin or IL-17, but required the expression of adhesion molecules in tumor vessels, which could facilitate the ability of leukocytes to evade the tumor 62. Thus, our findings that DOCK8-deficient mice have drastic reductions in RORγt+ ILCs could explain why DOCK8-deficient patients are more prone to cancer.
In summary, our data show that DOCK8 is critical for protective immunity against enteric pathogen infection, independent of its role in B cell-mediated functions. Our findings have revealed an unanticipated DOCK8 requirement for the generation and survival of RORγt+ ILCs. We also provide evidence that drastic reductions in the numbers of RORγt+ ILCs, as well as defective IL-22 production by these cells, are responsible for early mortality upon C. rodentium infection in DOCK8-deficient mice. To our knowledge, this is the first study that has identified the scaffolding protein DOCK8 as an important molecule for the generation, survival and optimal function of ILCs and protective immunity against enteric pathogen. Given that DOCK8-deficiency in human patients results in recurrent infections and increased vulnerability to cancer, these consequences may partly be due to defective RORγt+ ILCs in the GI tract.
Methods
Mice
Mice were bred and maintained under specific pathogen-free conditions in accordance with animal facilities at the Seattle Children’s Research Institute (SCRI). Experiments were conducted in accordance with the guidelines of the SCRI Animal Care and Use Committee. IL-23R−/− and DOCK8-deficient (DOCK8pri/pri) mice were generated as previously described 24, 45. WT (C57BL/6) and Rag1−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME) while Rag2−/−IL-2Rγ−/− mice were purchased from Taconic. Most of the animal used in the study were 6 to 8 weeks old male and female mice.
Citrobacter rodentium infection and assessment of CFU
C. rodentium was cultured overnight at 37° C in Luria broth with gentle agitation. After spinning down at 4500 rpm, they were suspended in PBS and the concentration was measured using a 600nm cell density meter (WPA Biowave CO8000). Mice were fasted for 8 hours before oral gavage of 2×109 CFU in 200μl PBS. Spleens and livers of infected mice were used to analyze the CFU. Spleens and livers were mechanically crushed in PBS and after serial dilution, plated on LB agar plates and incubated overnight at 37°C.
Bone marrow transfer
5×106 donor bone marrow cells were intravenously injected into recipient mice irradiated at 550 rads. Irradiated mice were treated with antibiotics (Baytril) for 2 weeks after injection and mice were infected 6 weeks post transfer.
Mixed bone marrow transfer
Recipient Rag2−/−IL-2Rγ−/− mice were irradiated 4 hours before transfer at 550 rad and treated with antibiotics (Baytril) for 2 weeks. Irradiated mice received 50/50 mixture of CD45.1+WT and CD45.2 DOCK8pri/pri bone marrow cells (5X106) intravenously and mice were analyzed for RORγt+ ILCs 5 weeks post-transfer.
Isolation of lamina propria cells
For GI tract lamina propria lymphocyte (LPL) isolation, colons and SI were isolated, flushed with ice cold PBS, the attached fat removed, and then cut open longitudinally. The samples were cleaned by shaking in ice cold PBS 4 times before tissue was cut into 1 cm pieces. The epithelial cells were removed by incubating the tissue in pre-digestion buffer (HBSS, 2mM EDTA and antibiotics) for 30 min at 37°C with shaking. The LPL cells were isolated by incubating the tissues in digestion buffer (HBSS, 10% FBS, 1mg/ml Collagenase (Worthington) and DNase (Sigma-Aldrich) for 20 min. The digested tissues were then filtered through 40μm filter. This step was repeated to get the complete digestion of the tissues. Cells were washed once in the ice cold medium (RPMI, 10% FBS and antibiotics), re-suspended in 10 ml of the 40% fraction of a 40:80 Percoll gradient, and overlaid on 5 ml of the 80% fraction in a 15 ml Falcon tube. Percoll gradient separation was performed by centrifugation for 20 min at 1800 rpm at room temperature. LPL cells were collected at the interphase of the Percoll gradient, washed, and re-suspended in medium.
Flow cytometric analysis
For flow cytometric analysis, cells were stained with antibodies to the following markers: CD3 {(FITC or PE, eBioscience, alexa fluor 647, BioLegend) (1:250 dilution)} CD90.2 {(PE-Cy5, PE-Cy7 Biolegend) (1:2000 dilution)}, CD4 {(PE, PE-Cy5, APC-efluor 780, or efluor 450, eBioscience) (1: 250 dilution)}, IL-22 {(APC, eBioscience) (1:125 dilution)}, RORγt {(PE, eBioscience) (1:200 dilution)}, CD127 {(PE or APC-efluor 780, eBioscience), NKp46 (eflour 660, eBiosciences), c-Kit (PE-Cy7, BD Biosciences) (1:125 dilution)}, SCA-1 {(PerCP-Cy5.5, eBiosciences) (1:500 dilution)} CD8 {(PE-Cy7, eBioscience) (1:2500 dilution)}, CD19 {(FITC, eBioscience), B220 (Alexafluor 647, Biolegend) (1:500 dilution)}, MHC class II {(PE, eBioscience), CD11c (APC, eBioscience) and CD11b (APC-efluor 780, eBioscience) (1:500 dilution)}. For cytokine production, cells were stimulated directly ex vivo by incubating for 4 hr with 20 ng/ml IL-23 (eBioscience) in the presence of Golgi plug (BD). Cells were fixed and permeabilized by using Foxp3 fix/perm buffer set Cat# 421403 (Biolegend), as described by manufacturers, and stained with IL-22 and RORγt. For pSTAT3, cells were stimulated by incubating for 20 min with 50 ng/ml rIL-23 (eBioscience) followed by fixation with 2% paraformaldehyde (PFA). PFA fixed cells were permeabilized with phosflow perm buffer III (BD biosciences) and stained with anti-STAT3 (pY705) antibody conjugated to Alexa fluor 647 (BD biosciences).
Gene expression
Total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA) and reverse transcribed with the iScript cDNA synthesis kit (BioRad, Hercules, CA). The real time PCR was conducted using Step One Plus Real Time PCR system (Applied Biosystems) with primer and probe listed in Supplementary Table 1. The reaction was set using Taq Man Fast Universal PCR Master Mix Cat# 43552042 (Applied Biosystems). The samples were normalized to the control housekeeping gene RPL-19 and reported according to the dCT method.
Measurement of IL-22 by ELISA
3000 Sorted SI LPLs were cultured in the absence or presence of 20 ng/ml rIL-23 (eBioscience CA, USA) for 24 hours. Supernatant was centrifuged and measured for IL-22 protein concentration by sandwich ELISA kit # 88-7522-86 (eBioscience CA, USA).
Hydrodynamic gene delivery of IL-22
For hydrodynamic injections, 15μg plasmid DNA was resuspended in 2ml of PBS and intravenously injected in 5–10 seconds using hydrodynamic tail vein injection. IL-22 protein production was validated from blood of mice that had received the hydrodynamic plasmid injections.
RNA sequencing
Total RNA was purified from C. rodentium infected colon with RNeasy mini kit (Qiagen). RNA sequencing was performed at Genomic Core Facility Southwestern Medical Center, University of Texas.
Statistical Analysis
For statistical analysis, p-values were calculated with a two-tailed unpaired student’s t test. p values of less than 0.05 were considered significant. Error bars denote ± SD.
Supplementary Material
Acknowledgments
This work was funded by US National Institutes of Health grant R21 (1 R21 AI101936), to MO. We thank Christopher C. Goodnow (John Curtin School of Medical Research and Australian Phenomics Facility, Australian National University, Australia) for providing DOCK8pri/pri mice, David Rawlings (Seattle Children’s Research Institute, Seattle, Washington, USA) for providing μMT, WASp−/− and Myd88−/− mice, Iaanis Aifantis (Dept. of pathology, New York University, NY, USA) for providing intracellular Notch retrovirus construct and Simon Glatigny (Benaroya Research Institute, Immunology Program, Seattle, WA 98101, USA) for helping with RNA sequencing. We would also like to thank Dawn Marie Pares for her outstanding administrative assistance.
Footnotes
Disclosure
The authors declared no conflict of interest.
Contributions
A.K.S. designed, performed and analyzed most experiments and wrote the manuscript, A.E. and M.F. performed and analyzed the IL-22 rescue experiments. E.B. designed and analyzed the experiments. M.O. supervised the study, analyzed the data and edited the manuscript.
Accession codes
RNA-sequencing data have been deposited in the gene expression omnibus (GEO) database under accession code GSE58765.
References
- 1.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 2.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 3.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 4.Yokota Y, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature. 1999;397:702–706. doi: 10.1038/17812. [DOI] [PubMed] [Google Scholar]
- 5.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 6.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells - how did we miss them? Nat Rev Immunol. 2013 doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 7.Tait Wojno ED, Artis D. Innate lymphoid cells: balancing immunity, inflammation, and tissue repair in the intestine. Cell Host Microbe. 2012;12:445–457. doi: 10.1016/j.chom.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eberl G. Inducible lymphoid tissues in the adult gut: recapitulation of a fetal developmental pathway? Nat Rev Immunol. 2005;5:413–420. doi: 10.1038/nri1600. [DOI] [PubMed] [Google Scholar]
- 9.Cella M, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiss EA, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- 11.Sawa S, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 12.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tumanov AV, et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe. 2011;10:44–53. doi: 10.1016/j.chom.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zenewicz LA, et al. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickert G, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 17.Satpathy AT, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14:937–948. doi: 10.1038/ni.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci. 2002;115:4901–4913. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 19.Ruusala A, Aspenstrom P. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS Lett. 2004;572:159–166. doi: 10.1016/j.febslet.2004.06.095. [DOI] [PubMed] [Google Scholar]
- 20.Jabara HH, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nat Immunol. 2012;13:612–620. doi: 10.1038/ni.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–2055. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelhardt KR, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–1302. e1284. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su HC. Dedicator of cytokinesis 8 (DOCK8) deficiency. Curr Opin Allergy Clin Immunol. 2010;10:515–520. doi: 10.1097/ACI.0b013e32833fd718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Randall KL, et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat Immunol. 2009;10:1283–1291. doi: 10.1038/ni.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Randall KL, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med. 2011;208:2305–2320. doi: 10.1084/jem.20110345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harada Y, et al. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood. 2012;119:4451–4461. doi: 10.1182/blood-2012-01-407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simmons CP, et al. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun. 2003;71:5077–5086. doi: 10.1128/IAI.71.9.5077-5086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maaser C, et al. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun. 2004;72:3315–3324. doi: 10.1128/IAI.72.6.3315-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bry L, Brenner MB. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol. 2004;172:433–441. doi: 10.4049/jimmunol.172.1.433. [DOI] [PubMed] [Google Scholar]
- 30.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 31.Basu R, et al. Th22 Cells Are an Important Source of IL-22 for Host Protection against Enteropathogenic Bacteria. Immunity. 2012;37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 33.Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity. 2009;31:15–23. doi: 10.1016/j.immuni.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 34.Eken A, Singh AK, Treuting PM, Oukka M. IL-23R innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 2013 doi: 10.1038/mi.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crawford G, et al. DOCK8 is critical for the survival and function of NKT cells. Blood. 2013;122:2052–2061. doi: 10.1182/blood-2013-02-482331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satoh-Takayama N, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Qiu J, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JS, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2012;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo X, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. 2014;40:25–39. doi: 10.1016/j.immuni.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parham C, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 41.Spits H, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 42.Sanos SL, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buonocore S, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powell N, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity. 2012;37:674–684. doi: 10.1016/j.immuni.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Awasthi A, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. 2009;182:5904–5908. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reynders A, et al. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat- lymphoid cells. EMBO J. 2011;30:2934–2947. doi: 10.1038/emboj.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ham H, et al. Dedicator of cytokinesis 8 interacts with talin and Wiskott-Aldrich syndrome protein to regulate NK cell cytotoxicity. J Immunol. 2013;190:3661–3669. doi: 10.4049/jimmunol.1202792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Possot C, et al. Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nat Immunol. 2011;12:949–958. doi: 10.1038/ni.2105. [DOI] [PubMed] [Google Scholar]
- 49.Mebius RE, et al. The fetal liver counterpart of adult common lymphoid progenitors gives rise to all lymphoid lineages, CD45+CD4+CD3- cells, as well as macrophages. J Immunol. 2001;166:6593–6601. doi: 10.4049/jimmunol.166.11.6593. [DOI] [PubMed] [Google Scholar]
- 50.Boos MD, Yokota Y, Eberl G, Kee BL. Mature natural killer cell and lymphoid tissue-inducing cell development requires Id2-mediated suppression of E protein activity. J Exp Med. 2007;204:1119–1130. doi: 10.1084/jem.20061959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eberl G, et al. An essential function for the nuclear receptor RORgamma;(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 52.Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat Immunol. 2010;11:945–952. doi: 10.1038/ni.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Satoh-Takayama N, et al. IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. J Exp Med. 2010;207:273–280. doi: 10.1084/jem.20092029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vonarbourg C, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 2010;33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang JH, et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity. 1996;5:537–549. doi: 10.1016/s1074-7613(00)80269-1. [DOI] [PubMed] [Google Scholar]
- 56.Sciume G, et al. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med. 2012;209:2331–2338. doi: 10.1084/jem.20122097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rankin LC, et al. The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat Immunol. 2013;14:389–395. doi: 10.1038/ni.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klose CS, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature. 2013;494:261–265. doi: 10.1038/nature11813. [DOI] [PubMed] [Google Scholar]
- 59.Sanal O, et al. Additional diverse findings expand the clinical presentation of DOCK8 deficiency. J Clin Immunol. 2012;32:698–708. doi: 10.1007/s10875-012-9664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10:311–323. doi: 10.1016/j.chom.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hill DA, et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med. 2012;18:538–546. doi: 10.1038/nm.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eisenring M, vom Berg J, Kristiansen G, Saller E, Becher B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol. 2010;11:1030–1038. doi: 10.1038/ni.1947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.