

Abstract
The use of and λ3- and λ5-iodanes in the oxidative dearomatization of phenols is a well-established and general procedure for the construction of cyclohexadienone structures. However, their use in asymmetric dearomatization reactions is quite underdeveloped and, despite work by several research groups over the past several years, a general chiral aryl iodide catalyst has yet to emerge. This article will serve to highlight the significant progress that has been made in this area and will reveal some of deficiencies in the literature that the author believes may be hindering further progress.
Keywords: hypervalent iodine, oxidation, phenols, asymmetric catalysis, catalyst design
The last three decades have seen tremendous growth in the chemistry of hypervalent organoiodine reagents (Figure 1).1 One of the main uses of these compounds is to serve as oxidants for a variety of organic functional groups. Despite the functional group selectivity often demonstrated by these reagents, their widespread adoption by the synthetic community has, in part, been slowed by concerns (perceived or otherwise2) about their stability and ease of preparation. As a result, the identification of conditions that employ catalytic amounts of an aryl iodide together with a stoichiometric oxidant3 (typically mCPBA) has generated renewed interest in hypervalent iodine intermediates and has stimulated the development of many new reactions.
Figure 1.
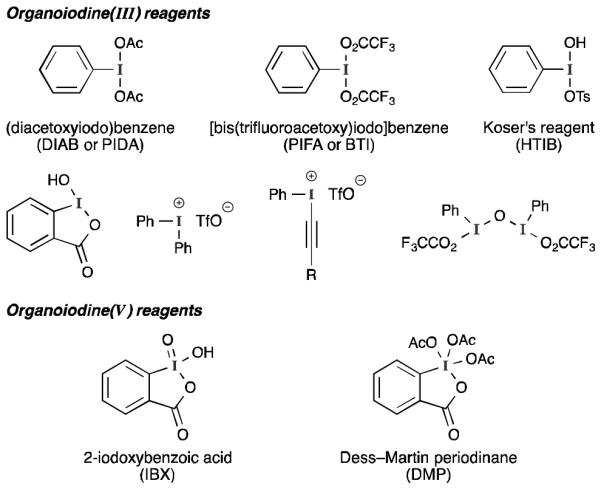
Some commonly encountered hypervalent iodine reagents.
This renewed interest in hypervalent iodine chemistry has also resulted in the development of several chiral hypervalent iodine reagents and catalysts.4 Although there are a few notable examples of asymmetric transformations that are promoted by stoichiometric amounts of a chiral iodine(III) reagent,5 this is a less-than-optimal approach. Unfortunately, developing catalytic variants of these reactions will be challenging owing to competitive oxidation of the substrate. However, this particular challenge does not appear to be a major issue with iodine(III)-mediated oxidative dearomatization of phenols.
The major problem associated with developing a general aryl iodide-based catalyst for asymmetric phenolic dearomatizations appears to be one of catalyst design. As will be seen, there are several factors that make this a particularly challenging task. However, it is a task that can be solved if certain considerations are made. The successful identification of an aryl iodide catalyst capable of performing asymmetric dearomatization reactions on a wide range of phenolic substrates will not only provide deeper insights into the structure and reactivity of hypervalent iodine intermediates, but will also have great potential in the areas of natural product synthesis and drug discovery.6,7
Mechanistic uncertainty
“… there are known knowns; there are things we know that we know. There are known unknowns; that is to say, there are things that we now know we don't know. But there are also unknown unknowns – there are things we do not know we don't know.”
–Donald Rumsfeld United States Secretary of Defense; February 12, 2002
The iodine(III) reagents PIDA and PIFA are the two most commonly used reagents for performing phenolic oxidative dearomatization reactions (eq 1).8 These two-electron oxidations involve the addition of a nucleophile to the aromatic ring of the phenol and result in the formation of either a 2,4-cyclohexadienone (1) or a 2,5-cyclohexadienone (2). Nucleophiles used in these conversions include: electron-rich arenes,9 heteroatom-based nucleophiles, (e.g. carboxylic acids, alcohols, sulfonamides),10 enol ethers,11 suitably activated olefins,12 enamides,13 and allyl silanes.11a,b,d,14 Both inter- and intramolecular reactions are possible.
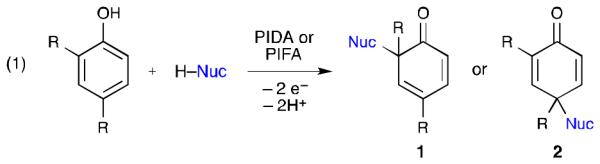
While the overall behavior of the reaction outlined in eq 1 is generally well understood, the mechanism of this process remains the subject of debate. Two mechanisms are commonly proposed for oxidative dearomatization reactions involving PhI(OAc)2.15,16 The first (Scheme 1, path A) involves ligand exchange between the phenol and iodine(III) carboxylate to give aryl-λ3-iodane 3.17 Nucleophilic attack as shown results in oxidation of the phenoxyl group, reduction of the iodine(III) center, and formation of dienone 2.18 Alternatively, the direct fragmentation of λ3-iodane 3 (Scheme 1, path B) through a unimolecular redox reaction would form phenoxenium ion 4,19 the subsequent trapping of which gives rise to dienone 2. Despite little experimental evidence for either of these two mechanisms,15 practitioners have adopted these pathways to rationalize the observed reactivity. For example, frontier molecular orbital analysis has been used to support the formation of a phenoxenium ion and to rationalize the observed regioselectivity (i.e., 1 vs. 2).20 However, while the positive charge of phenoxenium ion 4 is delocalized, two resonance forms (4b and 4c) place the positive charge α to a carbonyl, while another (4d) has a positively charged oxygen atom. Each of these would be expected to impart some degree of destabilization. Indeed, similar phenoxenium ions, generated by laser flash photolysis, have lifetimes between 3 and 170 ns in water.21,22
Scheme 1.
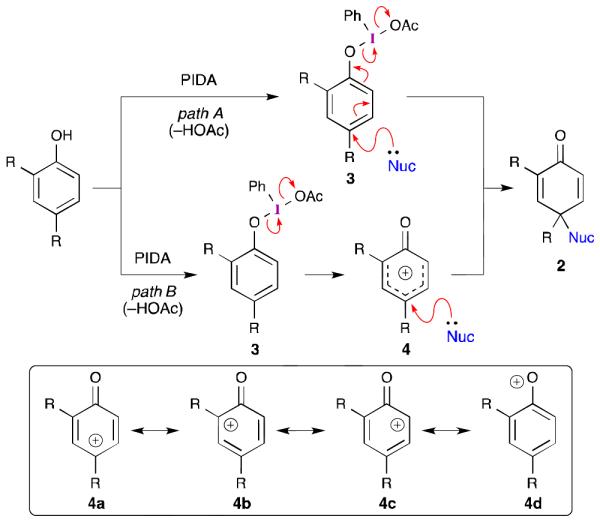
Commonly proposed mechanisms for the iodine(III)-mediated oxidation of phenols.
Another mechanistic possibility has been proposed by Felpin to rationalize the superiority of aryl trimethylsilyl ethers (5) in para-quinol formation.23 This proposal (Scheme 2) involves attack on the iodine by the carbon atom para to the silyl ether to give λ3-iodane 6. Nucleophilic displacement by water would then reduce the iodine and form dienone 7. While invoking an SN2 reaction at the tertiary carbon atom of 6 is unorthodox, there are several aspects of this intermediate that may facilitate this transformation. First, the C–I bond is expected to be quite long. This might make the tertiary carbon atom more planar than a typical sp3 carbon atom. Second, the sp2 centers attached to the tertiary carbon will reduce the steric crowding experienced by an approaching nucleophile. Third, and perhaps most importantly, the aryl-λ3-iodanyl group is considered to be a hypernucleofuge24 with a leaving group ability that is 106 times greater than triflate.25 This would certainly allow for facile nucleophilic substitution. Alternatively, such a potent leaving group might also open the door for an SN1-type reaction involving a phenoxenium ion reminiscent of 4. Although the formation of 6 does require the dearomatization of the starting phenol, this can be viewed as being nothing more than the first step of electrophilic aromatic substitution.
Scheme 2.
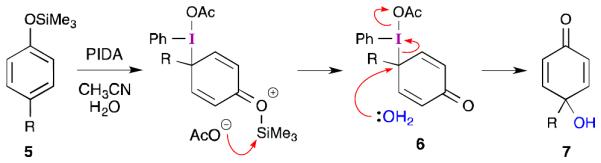
Alternative mechanism proposed by Felpin for the iodine(III)-mediated oxidation of aryl silyl ethers.23
The mechanism outlined in Scheme 2 is an atypical proposal for oxidative dearomatization reactions. However, Quideau has invoked intermediates similar to 6 in order to rationalize the regioselectivity observed during the arylation of phenols with Ph2ICl26 and to explain the stereoselectivity observed in PIDA-mediated diastereoselective spirocyclizations.27 This pathway is also consistent with other iodine(III)-mediated oxidations, which are thought to proceed by the initial formation of a C–I bond.28,29
Iodine(V) reagents can also perform oxidative dearomatization reactions on phenols.30 However, the postulated mechanism is significantly different, as illustrated in Scheme 3 for IBX-mediated ortho-quinol formation. Initial ligand exchange between IBX and the phenol likely generates intermediate 8. Subsequent 2,3-sigmatropic rearrangement results in oxygenation at the ortho-position and reduction of the iodine atom. This transformation is akin to the Mislow-Evans rearrangement31 of allylic sulfoxides. Once formed, λ3-iodane 9 can undergo either ligand exchange (producing ortho-iodosobenzoic acid (11) and quinol 10) or reductive cleavage (to give ortho-iodobenzoic acid (12) and the quinol).
Scheme 3.
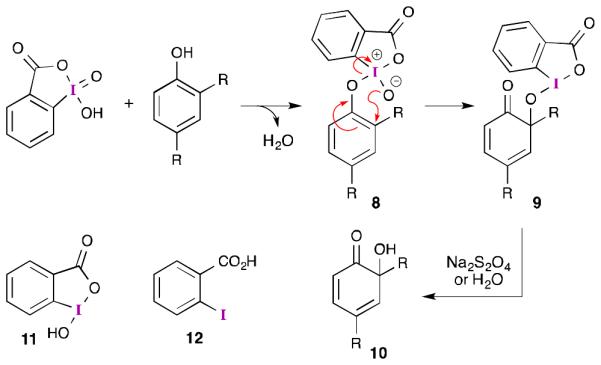
Proposed mechanism for the oxidation of ortho substituted phenols by iodine(V) reagents.
A consequence of this mechanism is that iodine(V)-mediated dearomatizations can only occur at the ortho position of the phenol. Furthermore, it is unlikely that external nucleophiles can be installed using iodine(V) oxidants. Because the goal of the present dialogue is to stimulate the development of a chiral aryl iodide catalyst capable of delivering a wide range of nucleophiles at either the ortho or para position (i.e., eqn 1), the remaining discussion will, for the most part, focus on iodine(III)-mediated reactions.
Catalytic Reactions
Others have reviewed the development of aryl iodide-catalyzed reactions,32 but a brief description of some key aspects and how they relate to phenolic dearomatizations is warranted. The presumed mechanism for aryl iodide-catalyzed dearomatization reactions is shown in Scheme 4. A not insignificant challenge that must be overcome is identifying a terminal oxidant that will react with the catalyst in preference to the substrate. This is especially important in asymmetric catalysis, where minimizing non-selective background reactions is important. It is also important to choose an oxidant that will not overoxidize the catalyst (i.e., form iodine(V) rather than iodine(III). mCPBA appears to offer the best balance as it does not react with most phenols at ambient temperature, but will easily oxidize the catalyst to the iodine(III) oxidation state with little competitive overoxidation.33 Other oxidants have been reported,34 but they have not achieved wide-spread use.
Scheme 4.
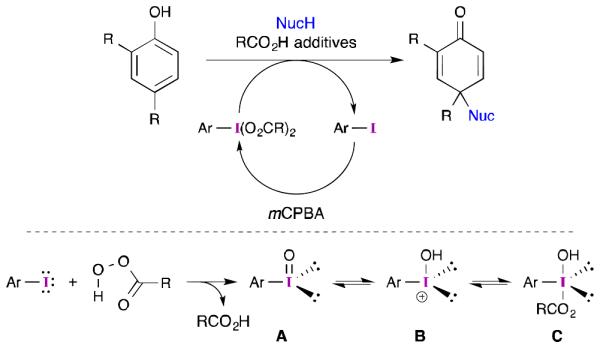
Presumed mechanism for catalytic phenolic dearomatizations.
A key piece of this picture that is not well understood is the identity of the iodine species that is formed upon reaction with the oxidant and how it relates to the iodine species that interacts with the phenol. It seems reasonable to assume that the reaction between an aryl iodide and mCPBA will initially form an iodosobenzene (A) and the corresponding carboxylic acid. Protonation of A will generate iodonium species B,35 which can then be trapped by the carboxylate and form C. Given a sufficient amount of carboxylic acid, iodine species C could be advanced to an iodine(III) dicarboxylate.33
In principle, A, B and C could all serve as functional oxidants for the phenolic substrate, but the stability and reactivity of the resulting iodine(III) phenolates may be quite different. Furthermore, as the oxidant (mCPBA) is consumed, the concentration of carboxylic acid will increase. This will almost certainly have some influence on the equilibrium concentrations of A, B and C. Indeed, others have found that acidic additives can have a beneficial influence on catalytic dearomatizations.3a,34a-b
Asymmetric Dearomatizations
We are unaware of a detailed mechanistic study of oxidative dearomatization reactions. This is understandable as it is difficult to imagine a set of experiments that would conclusively rule out one or more of the pathways discussed above. Furthermore, the highly unstable nature of the most relevant intermediates (3 and 4, Scheme 1) makes their spectroscopic detection and identification quite challenging.21,22 One potential test for the presence of phenoxenium ions is to perform enantioselective dearomatizations with asymmetric iodine(III) reagents, as the formation of a free phenoxenium ion would be expected to lead to diminished selectivity. Such reagents/catalysts are only now beginning to emerge and have given rise to conflicting mechanistic conclusions.
The first attempt at performing an enantioselective oxidative dearomatization was reported by Pelter and co-workers, who used a chiral λ3-iodane derived from PIDA and dibenzoyl tartaric acid to oxidize phenol 13 (Scheme 5).20 While reagent 1536 and its antipode successfully formed dienone 14, the product was isolated as a racemic mixture. These results were interpreted as evidence for phenoxenium ion formation (i.e., Scheme 1, path B). However, one could also conclude that this was simply a lessthan-ideal design for a chiral reagent. If this reaction proceeds according to the pathways outlined in Scheme 1, as suggested by the same authors,20 then the initial reaction between λ3-iodane 15 and the phenol should form an intermediate reminiscent of 16. Such an intermediate places all of the chiral information and the phenolate on opposite sides of the iodine(III) nucleus. Consequently, it is difficult to imagine how enantioinduction could be achieved even with a mechanism similar to that shown in Scheme 1, path A.
Scheme 5.
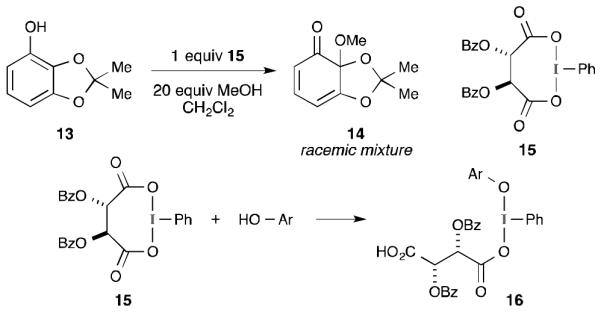
Initial attempt at asymmetric dearomatization with a tartrate-derived λ3-iodane.
Perhaps as a consequence of this negative result, and the associated difficulties with trying to develop an asymmetric reaction involving a phenoxenium ion, there were no further developments in the area of iodine(III)-mediated asymmetric dearomatization for about 10 years. Then, in a seminal report by Kita and co-workers, it was demonstrated that high enantioselectivity can be achieved in phenolic dearomatization reactions (Scheme 6).37 They found that by using conformationally rigid, chiral iodine(III) reagent 19, the asymmetric spirocyclization of napthols could proceed with high enantioselectivity (up to 86% ee). While μ-oxo-bridged compound 19 had to be used as a stoichiometric reagent, its reduced form (20a) could be recovered using silica gel chromatography. Furthermore, diiodide 20a could be used in catalytic quantities, along with mCPBA as a stoichiometric reoxidant, with some diminution in enantioselectivity (up to 69% ee). Interestingly, Kita observed a significant solvent dependence on enantioselectivity.37 The highest levels of enantio-control were observed in halogenated solvents: CCl4 (70% ee), CHCl3 (72% ee), CH2Cl2 (59% ee), and 1,2-DCE (60% ee). However, as the solvent polarity increased the enantioselectivity decreased: CH3CN (20% ee), 10:1 CH3CN/AcOH (16% ee), (CF3)2CHOH (0% ee). This was attributed to “discrete cationic intermediates”37a being formed in more polar solvents and suggested to the authors that in nonpolar solvents the reaction was proceeding through a mechanism akin to that shown in Scheme 1, path A.
Scheme 6.
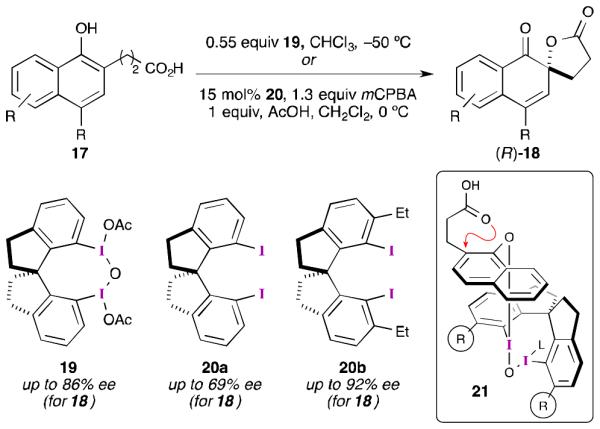
Kita’s asymmetric spirolactonization with conformationally rigid diiodide reagents and catalysts.
In subsequent work, Kita’s group proposed an empirically derived model (21) in order to rationalize the observed stereoselectivity.38 It was reasoned that incorporating substituents (R) ortho to each iodine atom would shield the bottom face of the bound substrate and lead to improved enantioselectivity. After screening several modified catalysts, it was determined that 20b afforded the best selectivity (up to 92% ee) for this transformation.
Ishihara and Uyanik have taken a different approach to aryl iodide catalyst design by appending stereogenic, flexible tethers to readily available 2-iodoresorcinol39 (22, Figure 2).40 They hypothesized that upon oxidation of the iodine center, the flexible arms would be able to achieve one of two possible “active” conformations. The first (23) would involve stabilizing n-σ* interactions41 between the iodine(III) center and Lewis basic groups (e.g., carbonyls) on the tether. This proposal is somewhat specious as these interactions (as drawn) occur in the same place where the non-bonding lone pairs on the iodine are located.42 Such interactions typically involve donation to the σ* orbital of the aryl–I bond.41b The second (24) would involve hydrogen-bonding interactions between the ligands attached to the iodine atom and a suitable H-bond donor (e.g., amides, carboxylic acids). In both cases, the additional non-covalent interactions would result in a reasonably rigid, C2-symmetric environment around the iodine(III) center. At the time, it was not clear which conformer would be favored, but this was answered by analysis of a related derivative (vida infra).
Figure 2.
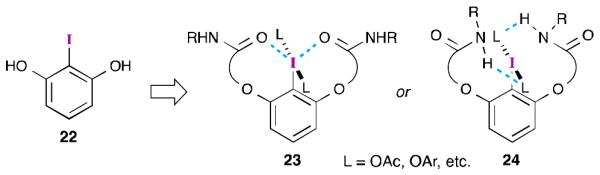
Ishihara and Uyanik’s design of flexible aryl iodide catalysts.
The initial dearomatization reaction investigated by Ishihara and Uyanik was the same asymmetric spirolactonization reported by Kita. After some experimentation with the substitution around the amide, it was determined that catalyst 25 provided the highest selectivity (up to 91% ee) with a range of substituted naphthol substrates (Scheme 7).40 The authors were also able to prepare iodine(III) derivative 26, and found that this reagent performed the spirocyclization with marginally better selectivity (95% ee). Interestingly, and in direct contrast to Kita’s work, little solvent dependence on enantioselectivity was observed with catalyst 2540b. For example, high enantioselectivity was observed even in CH3CN (83% ee), CH3NO2 (85% ee), and CF3CH2OH (70% ee). Often the best solvent choice was 2:1 CH2Cl2/CH3NO2. Significant diminution in selectivity was observed only with (CF3)2CHOH (41% ee). It should be noted that others have shown that chiral λ3-iodane 26, and some related compounds, can function as an oxidant in several asymmetric transformations,4,5 albeit in stoichiometric amounts.
Scheme 7.
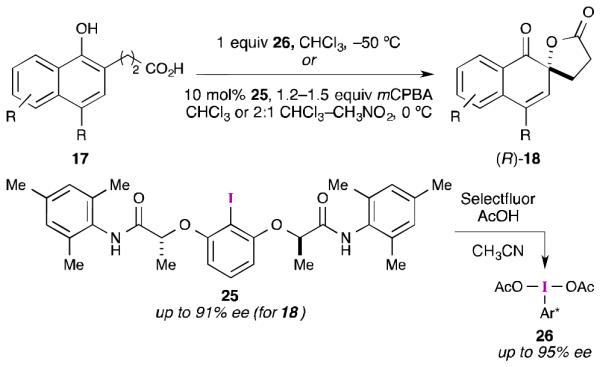
Ishihara and Uyanik’s asymmetric spirolactonization with flexible aryl iodide reagents and catalysts.
Following their success with the asymmetric spirocyclization of naphthols, Ishihara and Uyanik turned their attention to spirolactonization reactions of simple phenols (27, Scheme 8).43 This work was complicated by the propensity of the 2,4-cyclohexadienone products (28) to undergo dimerization reactions. Consequently, many of the initial products were trapped as Diels-Alder adducts (29). Aryl iodide 25 was a competent catalyst for these reactions, but the yields were poor. This could be rectified by using aryl iodide 30, which contains a modified tether. Interestingly, it was found that alcohol additives (MeOH or (CF3)2CHOH) were beneficial to the reactions and their use increased both yield and enantioselectivity.
Scheme 8.
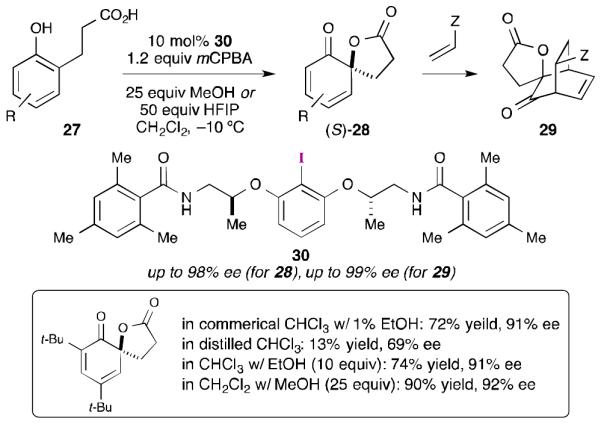
Ishihara and Uyanik’s asymmetric spirolactonization of simple phenols with flexible aryl iodide reagents and catalysts.
At this point, it is not entirely clear why the alcohol additives are so beneficial. Ishihara and Uyanik proposed that the added methanol serves to modify the structure of the active iodine(III) oxidant (Scheme 9). In doing so, the associative pathway for nucleophile addition is favored over the dissociative pathway. While this proposal still needs to be investigated, the authors were able to prepare dimethoxy substituted derivative 31 through solvolysis of the corresponding iodine(III) dicarboxylate. Thorough NMR analysis44 of 31 revealed that this complex likely contains hydrogen bonds between the amide and the methoxy groups. This structural feature was confirmed though X-ray diffraction analysis.
Scheme 9.

Ishihara and Uyanik’s proposal for the influence of methanol during asymmetric spirolactonizations with flexible aryl iodide catalysts. X-ray structure obtained from the Cambridge Crystallographic Database (CCDC 917160).
In addition to the spirolactonization reactions reported by Kita and Ishihara, Birman45 and Quideau46 have shown that asymmetric hydroxylation can also occur at the ortho position (Scheme 10). In both cases, the oxygen atom being incorporated into the product likely originates from the hypervalent iodine. However, the mechanism by which this occurs is quite dependent on the iodine oxidation state involved. Birman was able to construct several oxazoline-based iodine(V) reagents and found tert-leucine-derived reagent 34 afforded the highest selectivity (Scheme 10a).45 The iodine(V) oxidation state was established by several diagnostic IR stretches and NMR chemical shifts. Here, the hydroxyl group is likely being delivered to the substrate from one of the iodoxy groups in 34, as shown in Scheme 3.
Scheme 10.
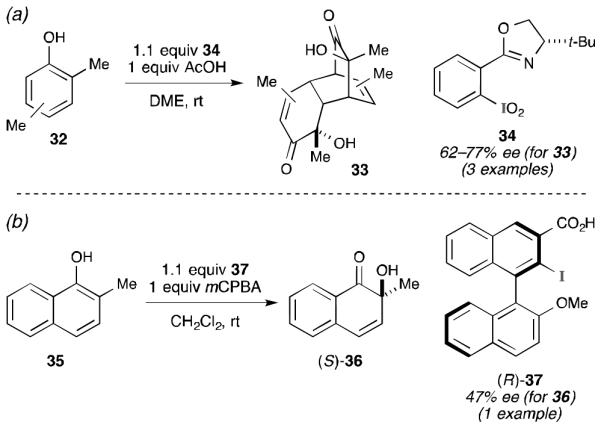
(a) Birman’s asymmetric ortho hydroxylation. (b) Quideau’s asymmetric ortho hydroxylation.
In Quideau’s work (Scheme 10b), the mechanism by which the oxygen atom is delivered is less clear. Unlike Birman’s oxazoline-derived aryl iodides, Quideau and co-workers experienced great difficulty in efficiently oxidizing and isolating their chiral aryl iodide reagents. Consequently, they resorted to an in situ oxidation using mCPBA. While this approach was successful in providing enantioenriched quinol 36,46 it complicates the mechanistic analysis. The authors proposed two models in order to explain the stereochemical outcome (Scheme 11). The first proposal involves complex 38, in which the iodine is in the +3 oxidation state.46 Because these reactions are run under anhydrous conditions, water cannot serve as an external nucleophile and oxygen atom source. Consequently, the authors invoked a mechanism involving internal delivery of the hydroxyl group with concomitant reduction of the iodine center. The internal delivery of a trans ligand to the 2-position of a coordinated naphtholate seems unlikely and likely requires a pseudorotation about the hypervalent nucleus before the transfer happens.47 While the iodine(III) oxidation state of this proposal holds with the known reactivity of mCPBA toward aryl iodides,32,33 it invokes a new mechanism by which a “nucleophile” can be delivered that is distinct from the proposals outlined in Schemes 1 and 2. The second proposal advanced by Quideau involves initial oxidation of the aryl iodide to the +5 oxidation state.46 Once the substrate is coordinated as indicated by complex 39, the oxygen atom can be delivered as shown. Finally, hydrolysis of iodine(III) intermediate 40 furnishes the observed product. Here, the oxygen delivery mechanism is in line with what has been previously proposed for iodine(V)-mediated ortho-hydroxylation reactions (i.e., Scheme 3),30 but the iodine(V) oxidation state is atypical for what is usually observed for mCPBA-mediated oxidations of aryl iodides.
Scheme 11.
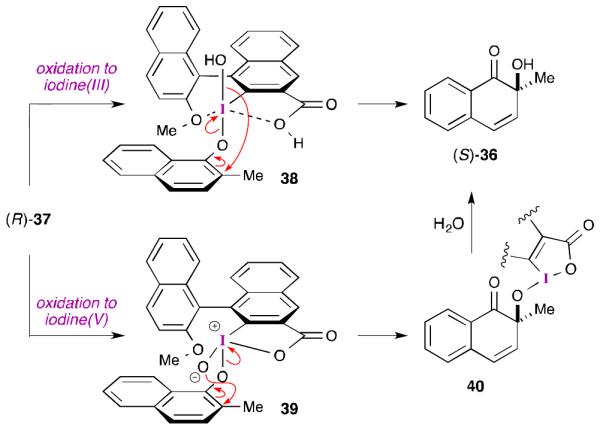
Proposed models for asymmetric induction in Quideau’s ortho hydroxylation.
Up to this point, the discussion of asymmetric dearomatization has centered on reactions in which the nucleophile becomes attached to the ortho position of the starting phenol (e.g., 1). As discussed before, iodine(III) oxidants are also able to promote reactions in which the nucleophile becomes attached to the para position of the phenol (e.g., 2). Designing an asymmetric version of such a reaction is more difficult as the developing stereocenter appears to be quite distant from the chiral environment presented by the catalyst/reagent. But is this really the case?
In order to better understand the structure of postulated iodine(III) phenoxide intermediate 3, we carried out a DFT study (M06-2X/6–31G(d) for C,H,O and SDD for iodine) with phenoxide 41.48 The calculations revealed that the aromatic ring of the phenoxide was partially blocked by the aromatic ring of the λ3-iodane (41a, Figure 3). We were intrigued by the possibility of using this feature as a means to shield one enantioface of the substrate and set out to install a chiral tether between the iodine(III) carboxylate and one of the ortho positions on the aryl iodide. Eventually, these designs led to catalyst 42, which can be prepared from L-(+)-dimethyl tartrate and 8-iodotetralone.
Figure 3.
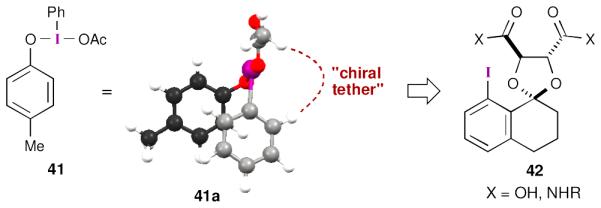
Harned’s computationally guided design of aryl iodide catalysts.
These catalysts were evaluated for their ability to produce enantioenriched para-quinols (44) from simple phenols (Scheme 12).48 In this regard, bisamide 45 provided the best balance of yield and enantioselectivity. Notably, the quinol products could be produced with up to 60% ee even in a polar reaction medium (9:1 acetonitrile/water).49 When 18O labeled water was used in the reaction, incorporation of the label was observed by mass spectrometric analysis of the product. While this does show that the hydroxyl group is originating from water and not mCPBA, it does not rule out a rapid exchange of water (as hydroxide) onto the iodine(III) center, followed by an internal delivery similar to that proposed by Quideau. The use of catalyst 45 could also be extended to other nucleophiles. For example, when methanol was used as the solvent, methyl ether 46 was formed in 30% ee. Similarly, spirocycle 47 was generated in 40% ee by using a tethered nucleophile.
Scheme 12.
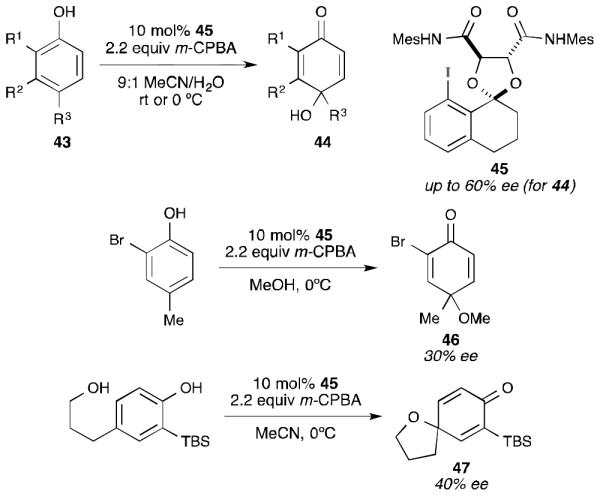
Harned’s asymmetric dearomatization with nucleophile incorporation at the para position.
Known knowns
The hypervalent iodine-mediated dearomatization of phenols is a well-known and highly useful reaction. At the same time, this reaction is not well understood. So what is known? In broad terms, this can be narrowed down to three main areas:
How substitution around the phenol substrate governs reactivity is fairly well understood. Electron-deficient phenols have higher oxidation potentials and tend to react slower than electron-rich phenols. A phenol that is too electron-deficient will not be a competent substrate for hypervalent iodine-mediated dearomatizations. In addition, the substituents have an influence on the charge distribution and LUMO coefficients of the corresponding phenoxenium ion, which can be used to predict the product distribution of the reaction.20 As mentioned above, the scope of the nucleophile has also been established.
The use of catalytic amounts of aryl iodides, along with a stoichiometric oxidant, is now well established and in many cases expected.
Asymmetric induction can be achieved for reactions occurring at both the ortho and para positions of the starting phenol.
Known unknowns
So what work still needs to be done? The following discussion outlines what I have identified as being key pieces of the puzzle that are missing and therefore hindering efforts at designing a highly selective aryl iodide catalyst. This is not meant to be an exhaustive list and there are surely some unknowns that have been overlooked.
Most glaring is the lack of mechanistic understanding of these reactions. This is understandable as many of the intermediates that would be the most interesting to study spectroscopically are too unstable to isolate or too fleeting to detect easily (e.g., phenoxenium ions). To our knowledge, even a “simple” kinetic study of iodine(III)- or iodine(V)-mediated oxidative dearomatization has not been performed.50 Further confusing the matter are the seemingly contradictory solvent effects on enantioselectivity observed by Kita and Ishihara/Uyanik. These results have been interpreted as evidence for a change in mechanism (i.e., Scheme 1, path A in non-polar solvents; Scheme 1, path B in polar solvents). However, the structure of the iodine(III) intermediates may also be changing, which would lead to different levels of enantiocontrol. For example, in the case of the Ishihara/Uyanik catalyst, polar solvents may disrupt the hydrogen bonds offered by the amide portions of the catalyst.
Surprisingly, the extreme reactivity of phenoxenium ions21,22 has not been considered in this context. Could it be possible that such an intermediate is trapped by the nucleophile before it has time to become separated from any chiral environment provided by the aryl iodide? If so, then the question of whether λ3-iodane 3 decomposes by an associative or dissociative mechanism does not really matter. In principle, both could lead to high enantioselectivity, provided an iodine(III)-bound phenoxide can be designed to favor exposure of one enantioface of the phenol over the other. Notably, both Kita and Ishihara/Uyanik saw little-to-no asymmetric induction with methoxy substituted naphthols.37,40 This might suggest that phenoxenium ion stability plays an important role: a more stable phenoxenium ion would have time to become solvent separated from the reduced aryl iodide.
Assuming for the moment that the preferred mechanism is the direct addition of the nucleophile (Scheme 1, path A), the three-dimensional shape of the iodine(III)-bound phenoxide will be critical for controlling which enantioface is exposed to the nucleophile. The problem in this case is that these complexes are too reactive to be adequately characterized by spectroscopic means. While Kita has proposed a model for explaining the observed selectivity, this is an empirical model. What happens after one of the iodine(III) centers is reduced? Is the other able to act independently (perhaps with a less organized model) or does the reoxidation of the first iodine atom happen too quickly? Similarly, Ishihara and Uyanik have shown some evidence for hydrogen bonding in their iodine(III) system (Scheme 9), but exactly how this translates to stereoselectivity once the substrate is bound is not clear.
The problem of structure also arises when one strips away the matter of enantioselectivity and simply considers running these reactions catalytically. What is the oxidation state of the iodine atom? Most likely, reactions run with mCPBA stop at iodine(III), but Quideau’s results46 suggest that iodine(V) may be possible in some cases. If iodine(III) is formed, then what are the other ligands around the iodine atom besides the aromatic group (e.g., Scheme 4). If the initially formed iodine(III) intermediate looks like complex C in Scheme 4, then which ligand undergoes substitution with the phenol? The ligand environment will almost certainly have a profound influence on both the reactivity and shape of the presumed iodine(III) phenoxide. Also, how does changing the electronics of the aryl iodide affect conversion and catalyst activity? Many simple aryl iodides can function as catalysts, with differing levels of efficiency, but these observations have really only been qualitative in nature.3a,34b,51 How these substituents influence the kinetics of the reaction are not well established.
Going forward, computational techniques will certainly play an increasingly important role in answering these questions. Indeed, computational studies of hypervalent iodine compounds have been performed, but have not addressed the questions posed here.26,27,52 The trouble with this approach is that without some experimental support (kinetics, transition state energies, etc.), computational results will not be entirely conclusive. Nevertheless, they will play an important role in designing new mechanistic experiments. For example, computational work by Goddard53,54 and Legault55 has shown that some iodine(V) and iodine(III) compounds must undergo a “hypervalent twist” before a bound substrate can undergo oxidation. This twisting was also incorporated into Quideau’s proposed mechanisms for ortho- hydroxylation.46
Similarly, computational investigation into the structure of certain key intermediates may prove beneficial for the rational design of novel catalyst architectures. The advantage being that the reactivity of intermediates that are too unstable for structural studies (NMR, X-ray) can still be probed. Once again, without some spectroscopic characterization of these intermediates it will be difficult to say how meaningful these theoretical results will be, but as these regents are synthesized and tested, the corresponding models and proposals can be further refined.
Unknown unknowns
By their very nature, unknown unknowns cannot be delineated, but they surely exist. Only by continued experimentation will new unknowns be revealed. Further experiments can then be designed and more knowns generated.
Concluding thoughts
Through this article I hope to have conveyed to the reader that asymmetric hypervalent iodine-mediated dearomatization of phenols is a tractable problem. In some ways, this problem can be likened to the field of asymmetric transition metal catalysis. In that field, a multitude of chiral ligands have been developed both to render a particular reaction asymmetric and to realize improved selectivity/reactivity in existing transformations. Often, the first ligand pulled from the shelf is effective at demonstrating that a particular transformation can be rendered asymmetric (say 20% ee or higher), but only through continued screening of different ligand architectures (e.g., BINAP, Trost ligand, PHOX, MOP, etc.) will synthetically useful levels of enantioselectivity be realized (>90% ee).56 At present, the primary problem of enantioselective aryl iodide catalysis is that only a handful of catalyst architectures have been described and few, if any, are commercially available. Only with the continued development (rational or otherwise) of new architectures57 will a truly general catalyst for asymmetric dearomatization be discovered.
Even though critical knowledge gaps still exist in this area, I find it remarkable how much rational catalyst design can still be accomplished. Furthermore, I think it is reasonable to conclude that as the mechanistic and structural questions raised above are answered, our ability to further evolve these designs will progress. The only way this can be accomplished is through continued experimentation, both at the bench and in silico. With this in mind I leave the reader with another quote or, perhaps, a rallying cry…
"Data! Data! Data!" he cried impatiently. "I can't make bricks without clay."
–Sir Arthur Conan Doyle,
The Adventure of the Copper Beeches
Acknowledgments
The University of Minnesota and the NIH (R21DA033556) are acknowledged for financial support. I am especially grateful to Patrick Lang, Alison Thorsness, William Wolf, Kelly Volp, and Nick Moon for their contributions, both published and unpublished, to this project in my own laboratory. I would also like to thank Prof. Christopher Douglas and Prof. Christopher Cramer for helpful discussions pertaining to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Moriarty RM, Prakash O. Acc. Chem. Res. 1986;19:244. Reviews and monographs: [Google Scholar]; (b) Zhdankin VV, Stang PJ. Chem. Rev. 2002;102:2523. doi: 10.1021/cr010003+. [DOI] [PubMed] [Google Scholar]; (c) Zhdankin VV, Stang PJ. Chem. Rev. 2008108:5299. doi: 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Varvoglis A. Hypervalent Iodine in Organic Synthesis. Academic Press; San Diego: 1997. [Google Scholar]; (e) Wirth T, editor. Hypvervalent Iodine Chemistry. Springer; Berlin: 2003. [Google Scholar]; (f) Zhdankin VV. Hypervalent iodine chemistry: preparation, structure and synthetic applications of polyvalent iodine compounds. Wiley; Chichester, U.K.: 2014. [Google Scholar]
- 2.(a) McQuaid KM, Pettus TRR. Synlett. 2004:2403–2405. doi: 10.1055/s-2004-832814. See note 4 in: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Plumb JB, Harper DJ. Chem. Eng. News. 1990 Jul 16;:3. [Google Scholar]; (c) Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277–7287. See note 3e in: [Google Scholar]
- 3.(a) Dohi T, Maruyama A, Yoshimura M, Morimoto K, Tohma H, Kita Y. Angew. Chem. Int. Ed. 2005;44:6193–6196. doi: 10.1002/anie.200501688. [DOI] [PubMed] [Google Scholar]; (b) Ochiai M, Takeuchi Y, Katayama T, Sueda T, Miyamoto K. J. Am. Chem. Soc. 2005;127:12244–12245. doi: 10.1021/ja0542800. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ngatimin M, Lupton DW. Aust. J. Chem. 2010;63:653–658. Reviews: [Google Scholar]; (b) Liang H, Ciufolini MA. Angew. Chem. Int. Ed. 2011;50:11849–11851. doi: 10.1002/anie.201106127. [DOI] [PubMed] [Google Scholar]
- 5.(a) Fujita M, Wakita M, Sugimura T. Chem. Commun. 2011;47:3983–3985. doi: 10.1039/c1cc10129c. [DOI] [PubMed] [Google Scholar]; (b) Röben C, Souto JA, González Y, Lishchynskyi A, Muñiz K. Angew. Chem. Int. Ed. 2011;50:9478–9482. doi: 10.1002/anie.201103077. [DOI] [PubMed] [Google Scholar]; (c) Farid U, Wirth T. Angew. Chem. Int. Ed. 2012;51:3462–3465. doi: 10.1002/anie.201107703. [DOI] [PubMed] [Google Scholar]; (d) Fujita M, Yoshida Y, Miyata K, Wakisaka A, Sugimura T. Angew. Chem. Int. Ed. 2010;49:7068–7071. doi: 10.1002/anie.201003503. [DOI] [PubMed] [Google Scholar]; (e) Farid U, Malmedy F, Claveau R, Albers L, Wirth T. Angew. Chem. Int. Ed. 2013;52:7018–7022. doi: 10.1002/anie.201302358. [DOI] [PubMed] [Google Scholar]
- 6.(a) Silva LF, Jr., Olofsson B. Nat. Prod. Rep. 2011;28:1722–1754. doi: 10.1039/c1np00028d. [DOI] [PubMed] [Google Scholar]; (b) Roche SP, Porco JA., Jr. Angew. Chem. Int. Ed. 2011;50:4068–4093. doi: 10.1002/anie.201006017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pouységu L, Deffieux D, Quideau S. Tetrahedron. 2010;66:2235–2261. [Google Scholar]
- 7.Zhuo C-X, Zhang W, You S-L. Angew. Chem. Int. Ed. 2012;51:12662–12686. doi: 10.1002/anie.201204822. [DOI] [PubMed] [Google Scholar]
- 8.(a) Pelter A, Ward RS. Tetrahedron. 2001;57:273–282. [Google Scholar]; (b) Moriarty RM, Prakash O. Org. React. 2001;57:327. [Google Scholar]
- 9.(a) Krishna KVR, Sujatha K, Kapil RS. Tetrahedron Lett. 1990;31:1351–1352. For example: [Google Scholar]; (b) Liang J, Chen J, Liu J, Li L, Zhang H. Chem. Commun. 2010;46:3666–3668. doi: 10.1039/c001465f. [DOI] [PubMed] [Google Scholar]; (c) Jacquemont G, Canesi S. J. Org. Chem. 2012;77:7588–7594. doi: 10.1021/jo301408j. [DOI] [PubMed] [Google Scholar]
- 10.Dohi T, Uchiyama T, Yamashita D, Washimi N, Kita Y. Tetrahedron Lett. 2011;52:2212–2215. [Google Scholar]
- 11.(a) Quideau S, Looney MA, Pouységu L. Org. Lett. 1999;1:1651–1654. [Google Scholar]; (b) Quideau S, Pouységu L, Oxoby M, Looney MA. Tetrahedron. 2001;57:319–329. [Google Scholar]; (c) Lebrasseur N, Fan G-J, Oxoby M, Looney MA, Quideau S. Tetrahedron. 2005;61:1551–1562. [Google Scholar]; (d) Zheng C, Wang L, Li J, Wang L, Wang DZ. Org. Lett. 2013;15:4046–4049. doi: 10.1021/ol401863k. [DOI] [PubMed] [Google Scholar]
- 12.(a) Wang X, Porco JA., Jr. Angew. Chem. Int. Ed. 2005;44:3067–3071. doi: 10.1002/anie.200500247. [DOI] [PubMed] [Google Scholar]; (b) Guérard KC, Guérinot A, Bouchard-Aubin C, Ménard M-A, Lepage M, Beaulieu MA, Canesi SJ. Org. Chem. 2012;77:2121–2133. doi: 10.1021/jo300169k. [DOI] [PubMed] [Google Scholar]
- 13.(a) Kita Y, Tohma H, Inagaki M, Hatanaka K, Yakura T. J. Am. Chem. Soc. 1992;114:2175–2180. [Google Scholar]; (b) Honda T, Shigehisa H. Org. Lett. 2006;8:657–659. doi: 10.1021/ol052841m. [DOI] [PubMed] [Google Scholar]; (c) Shigehisa H, Takayama J, Honda T. Tetrahedron Lett. 2006;47:7301–7306. [Google Scholar]
- 14.(a) Nicolaou KC, Edmonds DJ, Li A, Tria GS. Angew. Chem. Int. Ed. 2007;46:3942–3945. doi: 10.1002/anie.200700586. [DOI] [PubMed] [Google Scholar]; (b) Sabot C, Commare B, Duceppe M-A, Nahi S, Guérard KC, Canesi S. Synlett. 2008:3226–3230. [Google Scholar]
- 15.(a) Swenton JS, Callinan A, Chen Y, Rohde JJ, Kerns ML, Morrow GW. J. Org. Chem. 1996;61:1267–1274. [Google Scholar]; (b) Pelter A, Hussain A, Smith G, Ward RS. Tetrahedron. 1997;53:3879–3916. [Google Scholar]; (c) Kita Y, Arisawa M, Gyoten M, Nakajima M, Hamada R, Tohma H, Takada T. J. Org. Chem. 1998;63:6625–6633. [Google Scholar]
- 16.(a) Kita Y, Tohma H, Hatanaka K, Takada T, Fujita S, Mitoh S, Sakurai H, Oka S. J. Am. Chem. Soc. 1994;116:3684–3691. Kita has found that oxidative dearomatizations and biaryl couplings of phenol ethers proceed through radical cation intermediates. These reactions seem to require the use of PIFA in either trifluoroethanol or hexafluoroisopropanol. See: [Google Scholar]; (b) Tohma H, Morioka H, Takizawa S, Arisawa M, Kita Y. Tetrahedron. 2001;57:345–352. [Google Scholar]; (c) Dohi T, Ito M, Yamaoka N, Morimoto K, Fujioka H, Kita Y. Tetrahedron. 2009;65:10797–10815. [Google Scholar]
- 17.Powell WH. Pure Appl. Chem. 1984;56:769–778. IUPAC introduced the “lambda nomenclature” (λn) in 1983 as a means to describe the bonding of a heteroatom in a nonstandard valence state (n). Iodine’s standard valence state is 1. Thus, λ3-iodane and λ5-iodane refer to organoiodides in the +3 and +5 oxidation state, respectively. See: [Google Scholar]
- 18. For simplicity, the formation of 2,4-cyclohexadienone 1 is not shown in Scheme 1. The formation of this product likely involves a mechanism similar to that for 2, but requires nucleophilic attack at the ortho carbon of the phenol.
- 19.Hanway PJ, Winter AH. J. Am. Chem. Soc. 2011;133:5086–5093. doi: 10.1021/ja1114612. Although they are isoelectronic to phenylnitrenes, recent work has revealed phenoxenium ions to be closed-shell singlets in the ground state. See: [DOI] [PubMed] [Google Scholar]
- 20.Kürti L, Herczegh P, Visy J, Simonyi M, Antus S, Pelter A. J. Chem. Soc., Perkin Trans. 1999;1:379–380. [Google Scholar]
- 21.Wang Y-T, Jin KJ, Leopold SH, Wang J, Peng H-L, Platz MS, Xue J, Phillips DL, Glover SA, Novak M. J. Am. Chem. Soc. 2008;130:16021–16030. doi: 10.1021/ja805336d. and references therein. [DOI] [PubMed] [Google Scholar]
- 22.Hanway PJ, Xue J, Bhattacharjee U, Milot MJ, Ruixue Z, Phillips DL, Winter AH. J. Am. Chem. Soc. 2013;135:9078–9082. doi: 10.1021/ja403370k. [DOI] [PubMed] [Google Scholar]
- 23.Felpin F-X. Tetrahedron Lett. 2007;48:409–412. [Google Scholar]
- 24.Ochiai M. In: Chemistry of Hypervalent Compounds. Akiba K.-y., editor. Wiley-VCH; New York: 1999. pp. 359–387. [Google Scholar]
- 25.Okuyama T, Takino T, Sueda T, Ochiai M. J. Am. Chem. Soc. 1995;117:3360–3367. [Google Scholar]
- 26.Ozanne-Beaudenon A, Quideau S. Angew. Chem. Int. Ed. 2005;44:7065–7069. doi: 10.1002/anie.200501638. [DOI] [PubMed] [Google Scholar]
- 27.Pouységu L, Chassaing S, Dejugnac D, Lamidey A-M, Miqueu K, Sotiropoulos J-M, Quideau S. Angew. Chem. Int. Ed. 2008;47:3552–3555. doi: 10.1002/anie.200705816. [DOI] [PubMed] [Google Scholar]
- 28.(a) Koser GF, Relenyi AG, Kalos AN, Rebrovic L, Wettach RH. J. Org. Chem. 1982;47:2487–2489. For example: [Google Scholar]; (b) Moriarty RM, Khosrowshahi JS, Prakash O. Tetrahedron Lett. 1985;26:2961–2964. [Google Scholar]; (c) Koser GF, Lodaya JS, Ray DG, III, Kokil PB. J. Am. Chem. Soc. 1988;110:2987–2988. [Google Scholar]; (d) Moriarty RM, Vaid RK, Hopkins TE, Vaid BK, Prakash O. Tetrahedron Lett. 1990;31:201–204. [Google Scholar]; (e) Zhdankin VV, Mullikin M, Tykwinski R, Berglund B, Caple R, Zefirov NS, Koz’min AS. J. Org. Chem. 1989;54:2605–2608. [Google Scholar]; (f) Barton DHR, Jaszberenyi JC, Leßmann K, Timár T. Tetrahedron. 1992;48:8881–8890. [Google Scholar]; (g) Shi L, Kim Y-J, Gin DY. J. Am. Chem. Soc. 2001;123:6939–6940. doi: 10.1021/ja015991a. [DOI] [PubMed] [Google Scholar]; (h) Boye AC, Meyer D, Ingison CK, French AN, Wirth T. Org. Lett. 2003;5:2157–2159. doi: 10.1021/ol034616f. [DOI] [PubMed] [Google Scholar]; (i) Allen AE, MacMillan DWC. J. Am. Chem. Soc. 2010;132:4986–4987. doi: 10.1021/ja100748y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Fujita M, Yoshida Y, Miyata K, Wakisaka A, Sugimura T. Angew. Chem. Int. Ed. 2010;49:7068–7071. doi: 10.1002/anie.201003503. [DOI] [PubMed] [Google Scholar]; (k) Farid U, Wirth T. Angew. Chem. Int. Ed. 2012;51:3462–3465. doi: 10.1002/anie.201107703. [DOI] [PubMed] [Google Scholar]; (l) Farid U, Malmedy F, Claveau R, Albers L, Wirth T. Angew. Chem. Int. Ed. 2013;52:7018–7022. doi: 10.1002/anie.201302358. [DOI] [PubMed] [Google Scholar]; (m) Moon NG, Harned AM. Tetrahedron Lett. 2013;54:2960–2963. [Google Scholar]
- 29.Lubriks D, Sokolovs I, Suna E. Org. Lett. 2011;13:4324–4327. doi: 10.1021/ol201665c. [DOI] [PubMed] [Google Scholar]
- 30.(a) Magdziak D, Rodriguez AA, Van De Water RW, Pettus TRR. Org. Lett. 2002;4:285–288. doi: 10.1021/ol017068j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pezzella A, Lista L, Napolitano A, d’Ischia M. Tetrahedron Lett. 2005;46:3541–3544. [Google Scholar]; (c) Bernini R, Crisante F, Barontini M, Fabrizi G. Synthesis. 2009:3838–3842. [Google Scholar]
- 31.Evans DA, Andrews GC. Acc. Chem. Res. 1974;7:147–155. [Google Scholar]
- 32.(a) Richardson RD, Wirth T. Angew. Chem. Int. Ed. 2006;45:4402–4404. doi: 10.1002/anie.200601817. [DOI] [PubMed] [Google Scholar]; (b) Dohi T, Kita Y. Chem. Commun. 2009:2073–2085. doi: 10.1039/b821747e. [DOI] [PubMed] [Google Scholar]; (c) Chapter 4 in reference 1f.
- 33.(a) Iinuma M, Moriyama K, Togo H. Synlett. 2012:2663–2666. [Google Scholar]; (b) Tohma H, Maruyama A, Maeda A, Maegawa T, Dohi T, Shiro M, Morita T, Kita Y. Angew. Chem. Int. Ed. 2004;43:3595–3598. doi: 10.1002/anie.200454234. [DOI] [PubMed] [Google Scholar]
- 34.(a) Yu Z, Ju X, Wang J, Yu W. Synthesis. 2011:860–866. With urea·H2O2: [Google Scholar]; (b) Dohi T, Minamitsuji Y, Maruyama A, Hirose S, Kita Y. Org. Lett. 2008;10:3559–3562. doi: 10.1021/ol801321f. [DOI] [PubMed] [Google Scholar]; (c) Yakura T, Omoto M, Yamauchi Y, Tian Y, Ozono A. Tetrahedron. 2010;66:5833–5840. With Oxone: [Google Scholar]; (d) Minamitsuji Y, Kato D, Fujioka H, Dohi T, Kita Y. Aust. J. Chem. 2009;62:648–652. With peracetic acid: [Google Scholar]; Liu H, Tan C-H. Tetrahedron Lett. 2007;48:8220–8222. With sodium perborate: [Google Scholar]
- 35.Miyamoto K, Yokota Y, Suefuji T, Yamaguchi K, Ozawa T, Ochiai M. Chem.—Eur. J. 2014;20:5447–5453. doi: 10.1002/chem.201304961. Miyamoto and co-workers have found that hydroxyl(phenyl)-λ3-iodane can be stabilized by 18-crown-6 and is an efficient oxidant for hydroxyaromatics. See: and references therein. [DOI] [PubMed] [Google Scholar]
- 36.Ray DG, Koser GF. J. Org. Chem. 1992;57:1607–1610. The authors of reference 20 prepared oxidant 15 by ligand exchange of PIDA and dibenzoyl tartrate and represented its structure as shown in Scheme 5. However, in an earlier report Koser found that a similar ligand exchange reaction results in oligomeric material containing oxidized iodobenzene and tartarate subunits. See: [Google Scholar]
- 37.Dohi T, Maruyama A, Takenaga N, Senami K, Minamitsuji Y, Fujioka H, Caemmerer SB, Kita Y. Angew. Chem. Int. Ed. 2008;47:3787–3890. doi: 10.1002/anie.200800464. [DOI] [PubMed] [Google Scholar]
- 38.Dohi T, Takenaga N, Nakae T, Toyoda Y, Yamasaki M, Shiro M, Fujioka H, Maruyama A, Kita Y. J. Am. Chem. Soc. 2013;135:4558–4566. doi: 10.1021/ja401074u. [DOI] [PubMed] [Google Scholar]
- 39.Tsujiyama S, Suzuki K, Guthrie HM, Gibney HM, Curran DP. Org. Synth. 2007;84:272–284. [Google Scholar]
- 40.(a) Uyanik M, Yasui T, Ishihara K. Angew. Chem. Int. Ed. 2010;49:2175–2177. doi: 10.1002/anie.200907352. [DOI] [PubMed] [Google Scholar]; (b) Uyanik M, Yasui T, Ishihara K. Tetrahedron. 2010;66:5841–5851. [Google Scholar]
- 41.(a) Zhdankin VV, Koposov AE, Smart JT, Tykwinski RR, McDonald R, Morales- Izquierdo A. J. Am. Chem. Soc. 2001;123:4095–4096. doi: 10.1021/ja0155276. For examples of non-bonding secondary interactions in iodine(III) and iodine(V) compounds, see: [DOI] [PubMed] [Google Scholar]; (b) Ochiai M. Coord. Chem. Rev. 2006;250:2771–2781. [Google Scholar]
- 42.(a) Koser GF. In: The Chemistry of Halides, Pseudo-halides and Azides. Patai S, Rappoport Z, editors. Wiley; New York: 1983. pp. 721–740. As shown below, the preferred bonding mode for organoiodine(III) compounds consists of a T-shaped geometry with the most electronegative ligands being collinear and in apical positions. This arrangement provides greater stabilization of the 3c-4e hypervalent bond and places the nonbonding electrons of the iodine atom in equatorial positions. For further discussions on the structural properties of hypervalent iodine compounds, see: The Chemistry of the Functional Groups, Supplement D, Part 1. [Google Scholar]; (b) Ochai M. In: Hypvervalent Iodine Chemistry. Wirth T, editor. Springer; Berlin: 2003. pp. 5–68. [Google Scholar]; (c) Zhdankin VV. Hypervalent iodine chemistry: preparation, structure and synthetic applications of polyvalent iodine compounds. Wiley; Chichester, U.K.: 2014. pp. 4–11. [Google Scholar]
- 43.Uyanik M, Yasui T, Ishihara K. Angew. Chem. Int. Ed. 2013;52:9215–9218. doi: 10.1002/anie.201303559. [DOI] [PubMed] [Google Scholar]
- 44. Compound 31 was isolated as a 1:1 complex with methanol. The NMR analysis of this complex was performed in CDCl3 without additional methanol. See the supporting information of reference 43 for more information.
- 45.Boppisetti JK, Birman VB. Org. Lett. 2009;11:1221–1223. doi: 10.1021/ol8029092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quideau S, Lyvinec G, Marguerit M, Bathany K, Ozanne-Beaudenon A, Buffeteau T, Cavagnat D, Chénedé A. Angew. Chem. Int. Ed. 2009;48:4605–4609. doi: 10.1002/anie.200901039. [DOI] [PubMed] [Google Scholar]
- 47.Finet J-P. Ligand Coupling Reactions with Heteroatomic Compounds. Vol. 18. Pergamon; Oxford: 1998. The Ligand Coupling Mechanism; pp. 9–46. For an excellent discussion of these issues, see: Tetrahedron Organic Chemistry Series, Volume. [Google Scholar]
- 48.Volp KA, Harned AM. Chem. Commun. 2013;49:3001–3003. doi: 10.1039/c3cc00013c. [DOI] [PubMed] [Google Scholar]
- 49. Due to the limitations of using water as a nucleophile, we were unable to carry out an extensive solvent screen. For more information, see the supporting information for reference 48.
- 50.DeMunari S, Frigerio M, Santagostino M. Kinetic studies of alcohol oxidation using IBX and Dess-Martin periodinane have been performed. J. Org. Chem. 1996;61:9272–9279. [Google Scholar]
- 51.(a) Yakura T, Konishi T. Synlett. 2007:765–768. [Google Scholar]; (b) Dohi T, Takenaga N, Fukushima K.-i., Uchiyama T, Kato D, Shiro M, Fujioka H, Kita Y. Chem. Commun. 2010;46:7697–7699. doi: 10.1039/c0cc03213a. [DOI] [PubMed] [Google Scholar]
- 52.(a) Boucher M, Macikenas D, Ren T, Protasiewicz JD. J. Am. Chem. Soc. 1997;119:9366–9376. [Google Scholar]; (b) Landrun GA, Goldberg N, Hoffman R, Minyaev RM. New. J. Chem. 1998:883–890. [Google Scholar]; (c) Zhdankin VV, Arbit RM, Lynch BJ, Kiprof P. J. Org. Chem. 1998;63:6590–6596. [Google Scholar]; (d) Carroll MA, Martín-Santamaría S, Pike VW, Rzepa HS, Widdowson DA. J. Chem. Soc., Perkin Trans. 1999;2:2707–2714. [Google Scholar]; (e) Hirt UH, Schuster MFH, French AN, Wiest OG, Wirth T. Eur. J. Org. Chem. 2001:1569–1579. [Google Scholar]; (f) Kiprof P. ARKIVOC. 2005;iv:19–25. [Google Scholar]; (g) Mocci F, Uccheddu G, Frongia A, Cerioni G. J. Org. Chem. 2007;72:4163–4168. doi: 10.1021/jo070111h. [DOI] [PubMed] [Google Scholar]; (h) Uyanik M, Akakura M, Ishihara K. J. Am. Chem. Soc. 2009;131:251–262. doi: 10.1021/ja807110n. [DOI] [PubMed] [Google Scholar]; (i) Szabó KJ. J. Mol. Catal. A: Chem. 2010;324:56–63. [Google Scholar]; (j) Norrby P-O, Petersen TB, Bielawski M, Olofsson B. Chem.—Eur. J. 2010;16:8251–8254. doi: 10.1002/chem.201001110. [DOI] [PubMed] [Google Scholar]; (k) Moorthy JN, Senapati K, Parida KN, Jhulki S, Sooraj K, Nair NN. J. Org. Chem. 2011;76:9593–9601. doi: 10.1021/jo201491q. [DOI] [PubMed] [Google Scholar]; (l) Sajith PK, Suresh CH. Inorg. Chem. 2012;51:967–977. doi: 10.1021/ic202047g. [DOI] [PubMed] [Google Scholar]; (m) Guilbault A-A, Basdevant B, Wanie V, Legault CY. J. Org. Chem. 2012;77:11283–11295. doi: 10.1021/jo302393u. [DOI] [PubMed] [Google Scholar]
- 53.Su JT, Goddard WA., III J. Am. Chem. Soc. 2005;127:14146–14147. doi: 10.1021/ja054446x. [DOI] [PubMed] [Google Scholar]
- 54.(a) Moorthy JN, Singhal N, Senapati K. Tetrahedron Lett. 2008;49:80. For experimental confirmation of Goddard’s proposal, see: [Google Scholar]; (b) Moorthy JN, Senapati K, Parida KN, Jhulki S, Sooraj K, Nair NN. J. Org. Chem. 2011;76:9593–9601. doi: 10.1021/jo201491q. [DOI] [PubMed] [Google Scholar]
- 55.Guilbault A-A, Legault CY. ACS Catal. 2012;2:219–222. [Google Scholar]
- 56.(a) Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. For notable examples of this, see: [DOI] [PubMed] [Google Scholar]; (b) Phan DHT, Kim B, Dong VM. J. Am. Chem. Soc. 2009;131:15608–15609. doi: 10.1021/ja907711a. and supporting information. [DOI] [PubMed] [Google Scholar]; (c) Coulter MM, Kou KGM, Galligan B, Dong VM. J. Am. Chem. Soc. 2010;132:16330–16333. doi: 10.1021/ja107198e. and supporting information. [DOI] [PubMed] [Google Scholar]; (d) Linton EC, Kozlowski MC. J. Am. Chem. Soc. 2008;130:16162–16163. doi: 10.1021/ja807026z. and supporting information. [DOI] [PubMed] [Google Scholar]; (e) Crouch IT, Neff RK, Frantz DE. J. Am. Chem. Soc. 2013;135:4970–4973. doi: 10.1021/ja401606e. and supporting information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brenet S, Berthiol F, Einhorn J. Eur. J. Org. Chem. 2013:8094–8096. [Google Scholar]