

Abstract
Chaperone proteins are critical for protein folding and stability, and hence are necessary for normal cellular organization and function. Recent studies have begun to interrogate the role of this specialized class of proteins in muscle biology. During development, chaperone-mediated folding of client proteins enables their integration into nascent sarcomeres. In addition to assisting with muscle differentiation, chaperones play a key role in maintenance of muscle tissues. Further, disruption of the chaperone network can result in neuromuscular disease. In this review, we discuss how chaperones are involved in myofibrillogenesis, sarcomere maintenance and muscle disorders. We also consider the possibilities of therapeutically targeting chaperones to treat muscle disease.
Keywords: Chaperones, sarcomere, myofibril, protein folding, contractile proteins
INTRODUCTION
Proper folding is a crucial step in protein synthesis that is mediated by a specialized class of proteins called chaperones. Chaperones are largely grouped into two categories, those that utilize ATP to mediate folding and those that are independent of ATP hydrolysis (Hartl and Hayer-Hartl 2009). Chaperones that require ATP utilize energy released from hydrolysis to drive conformational changes in the chaperone that assist in client protein folding. Chaperones that are independent of ATP often bind to nascent proteins and protect against non-specific hydrophobic interactions driving improper folding. General chaperones such as heat shock protein 90 (Hsp90) and Hsp70 are ubiquitously expressed and exhibit a wide range of client proteins (Kampinga and Craig 2010; Taipale et al. 2010). Specific chaperones such as αB-crystallin and uncoordinated-45 (UNC-45) are robustly expressed in fewer tissues and exhibit specialized functions with a more limited set of client proteins (Yu and Bernstein 2003; Wistow 2012). The activity of many chaperones, both general and specific, can also be modulated by co-chaperones that allow additional control with respect to client specificity and chaperone activity (Caplan 2003). In this review, we will discuss the role of both general and specific chaperones in the development and maintenance of the sarcomere, the fundamental contractile unit of muscle tissue (Figure 1). We will conclude with a discussion of chaperones in muscle disease and efforts to utilize chaperones as therapeutic targets.
Figure 1.
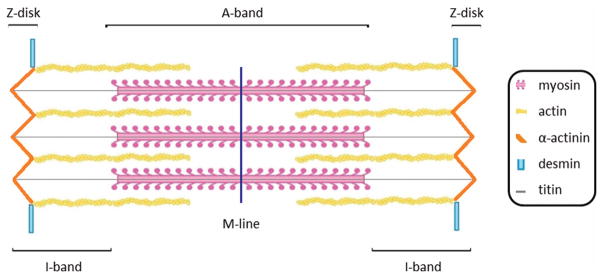
Representation of a simplified sarcomeric unit. Muscle fibers contain myofibrils with repeating sarcomeric units, the boundaries of which are defined by the Z-disk. The actin thin filaments are bound to the Z-disk and extend toward the middle of the sarcomere where they interact with the myosin motor domains of the thick filament. The I-bands are the region composed only of thin filaments whereas the A-band includes the region where the thick filaments interact with the thin filaments.
CHAPERONES IN MYOFIBRILLOGENESIS
Proper muscle development and function require a complex system of structural, motor, and chaperone proteins. Despite the great importance of muscle protein complexes, the details of sarcomere assembly or myofibrillogenesis remain unresolved (Sanger et al. 2005; Hawkins et al. 2008). One of the earliest steps in this process is actin nucleation followed by polymerization to form the thin filaments of the sarcomere (Ehler et al. 1999; Sparrow and Schöck 2009). These emerging filaments anchor their barbed ends to Z-bodies to form the I-Z-I bodies, precursors to the I-Z-I bands in mature muscle (Schultheiss et al. 1990; Young et al. 1998; Ojima et al. 1999). The Z-bodies incorporate α-actinin and titin (also known as connectin) early in development, eventually resulting in the α-actinin-rich Z-discs and the actin-containing I-bands (Fürst et al. 1989; Ehler et al. 1999). Thick myosin filaments are then integrated into this scaffold where they interact with titin, though how this process occurs is debated. One model says that thick filaments are formed separately from I-Z-I bodies but that both structures work together in assembling striated myofibrils (Holtzer et al. 1997). Another model states that initially, non-muscle myosin assembles with other components to form pre-myofibrils, but that in later stages muscle myosin takes over to finish the process of sarcomere assembly (Sanger et al. 2002). A third model proposes that titin serves as a scaffold onto which all of the sarcomeric components join to finish the process of myofibrillogenesis (Ehler et al. 1999; Van der Ven et al. 1999).
During myofibrillogenesis, protein chaperones are responsible for ensuring that each protein within the developing sarcomere is correctly folded. The importance of chaperones to this process is underscored by studies indicating that loss of chaperone activity can result in failure to properly develop sarcomeric structures and often leads to embryonic lethality (Venolia and Waterston 1990; Du et al. 2008; Hawkins et al. 2008; Lee et al. 2011b; Chen et al. 2012). Recent studies have begun to elucidate the complex network of chaperones that guide their respective client proteins into their correctly folded conformation and the roles they play in sarcomere assembly and muscle development (Figure 2).
Figure 2.

Chaperones with their respective client proteins during development and in the mature sarcomere. The chaperones Hsp90 and UNC-45 are responsible for myosin motor domain folding; myosin molecules are then incorporated into the thick filament (pink arrow). After sarcomere assembly, Hsp90 and UNC-45 appear to migrate to the Z-disk (blue, green arrows). The proposed UNC-45 activity modulator SmyD1 has been shown to interact with UNC-45 and is required for normal sarcomere development. It moves to the M-line following sarcomere assembly (purple arrow). The CCT (chaperonin) and GimC (prefoldin) complex is responsible for efficient folding of actin prior to incorporation into the thin filaments (yellow arrow), though Hsp25/27 and αB-crystallin have also been shown to facilitate actin folding. αB-crystallin assists in folding of titin and several intermediate filament proteins including desmin. Finally, N-RAP has been shown to assist in folding of the α-actinin protein that is incorporated into the Z-disk (orange arrows).
Hsp90 and UCS proteins in myosin and thick filament assembly
The general chaperone Hsp90 is widely expressed and is involved in a variety of activities such as cellular proliferation, organism development and cancer. Hsp90 consists of three functional domains: the N-terminal domain is responsible for ATP binding and ATPase activity, while the middle and C-terminal domains mediate chaperone dimerization (Street et al. 2012). After binding the nascent protein, cycles of ATP hydrolysis drive conformational changes in Hsp90 that in turn assist in folding client proteins within the cell (Taipale et al. 2010). Hsp90 function and specificity are also regulated by co-chaperones such as Cdc37, p23, Fk506bp1, CHIP, Sgta, Sha1 and UNC-45 (Li et al. 2012). In addition to its well-established activities, recent evidence has begun to elucidate the role of Hsp90 in mediating sarcomere development.
Early studies identified Hsp90 expression in somitic muscle progenitors of zebrafish embryos and showed that inhibition of Hsp90 blocked somite formation, indicating a key role in muscle development (Lele et al. 1999; Sass et al. 1999). Later work by Srikakulam and Winkelman (2004) found that inhibition of Hsp90 disrupted myofibril formation in the C2C12 mouse myoblast cell line in vitro. Using fluorescent microscopy, the authors determined that Hsp90 co-localized with the nascent myosin protein, indicating a potential role in myosin processing. Additionally, myosin expressed in vitro exhibits poor folding behavior that can be improved by the addition of Hsp90, further supporting its critical role as a myosin chaperone (Chow et al. 2002; Liu et al. 2008).
The role of Hsp90 in muscle development has been confirmed in vivo using genetic manipulation of zebrafish. Knockdown of the Hsp90a1 paralogue in zebrafish resulted in a paralyzed phenotype and yielded severely disrupted sarcomere development (Etard et al. 2007; Du et al. 2008). Interestingly, knockdown of the zebrafish paralogues Hsp90a2 and Hsp90b did not affect zebrafish muscle development, indicating a specific role for Hsp90a1 in sarcomere assembly. Further, the ATPase-deficient Hsp90a1 mutant slothu45 exhibited a similar phenotype to Hsp90a1 knockdown, confirming that the ATP-dependent activity of Hsp90 is necessary for muscle development (Hawkins et al. 2008). Hsp90 mutants lacking functional ATPase activity have also been shown to disrupt muscle development in C. elegans (Gaiser et al. 2011). Interestingly, knockdown of myosin expression or inhibition of myosin ATPase activity did not phenocopy knockdown of Hsp90 in zebrafish (Codina et al. 2010). This indicates that Hsp90 might be involved in the folding and integration of additional sarcomeric components.
Studies have begun to investigate how Hsp90 directs muscle development outside of its role in myosin folding. Yun and Matts (2005) showed that Hsp90 inhibition disrupted the interaction between Ccd37 and MyoD, key transcription factors in muscle differentiation. Loss of this interaction could be related to the depletion of Hsp90-dependent protein kinases, resulting in reduced levels of phosphorylated MyoD and its downstream target myogenin (Pelpel et al. 2000; Pratt and Toft 2003; Yun and Matts 2005). Therefore Hsp90 appears to play an indirect role in determining muscle cell identity during development in addition to its role as a protein chaperone for sarcomeric proteins.
The UCS (UNC-45/CRO1/SHE4) family of proteins is conserved across eukaryotic organisms and is critical to the proper folding of myosins within the cell (Hutagalung et al. 2002). The UCS protein UNC-45 consists of three distinct domains: the N-terminal tetratricopeptide repeat (TPR) domain, the central domain, and the UCS domain. The TPR domain has been shown to bind Hsp90 and Hsp70 whereas the C-terminal UCS domain binds myosin (Barral et al. 2002; Toi et al. 2003; Etard et al. 2007; Gazda et al. 2013). UNC-45 was initially discovered as a result of cloning the gene that yields an “uncoordinated” phenotype in C. elegans upon its mutation (Epstein and Thomson 1974; Barral et al. 2002; Yu and Bernstein 2003). Subsequent studies in both zebrafish and Drosophila have shown that loss of UNC-45 expression severely disrupts skeletal muscle sarcomere assembly (Wohlgemuth et al. 2007; Lee et al. 2011b). While initially described as an Hsp90 co-chaperone, a study by Melkani and colleagues (2010) found that Drosophila UNC-45 was able to assist in re-folding denatured citrate synthase as well as reduce heat-induced myosin aggregation in vitro. Further, studies in C. elegans and zebrafish found that the myosin-binding UCS domain is critical to sarcomere development while the Hsp90-binding TPR domain is dispensable (Bernick et al. 2010;Ni et al. 2011). These studies indicate that UNC-45 appears to be a distinct protein chaperone rather than solely an Hsp90 co-chaperone.
Though the precise mechanism of action for UNC-45 remains unclear, a recent study by Gazda and colleagues (2013) provided some interesting insights in this regard. In silico modeling showed that the UNC-45 UCS domain forms a long groove that could effectively bind proteins in an extended conformation prior to folding. This groove was previously identified a highly conserved region in the UCS domain (Lee et al. 2011a). Gazda and colleagues (2013) showed that mutations in this groove inhibited sarcomere formation in C. elegans similarly to loss of UNC-45 expression, indicating that this structure is essential to UNC-45 function. Further, the investigators showed that UNC-45 is capable of integrating into oligomers, the formation of which is required for sarcomere development. The authors propose that this oligomeric structure acts as a scaffold for myosin chaperones such as Hsp70 and Hsp90 and assists in myosin globular head folding at discrete intervals along the nascent sarcomere.
In addition to its role in skeletal muscle, UNC-45 is important in cardiac muscle. Knockdown of UNC-45 specifically in the Drosophila heart leads to myofibrillar disarray, low number of myofibrils, reduced levels of myosin and shorter lifespan (Melkani et al. 2011). Knockdown in zebrafish also results in cardiac dysfunction (Wohlgemuth et al. 2007). Biochemical and molecular experiments have confirmed that mouse UNC-45b forms complexes with both the alpha- and beta-cardiac myosins and GATA4, a transcription factor important in cardiogenesis (Chen et al. 2012), emphasizing UNC-45’s key role in cardiac development.
Invertebrates only have a single UNC-45 isoform, compared to the two isoforms, UNC-45a and UNC-45b, found in vertebrates (Price et al. 2002). Both mammalian isoforms have been shown to activate Hsp90-dependent myosin folding in vitro; however, the general form UNC-45a is expressed in most cell types while UNC-45b is expressed only in striated muscle, indicating a possible evolutionary sub-functionalization (Liu et al. 2008; Myhre and Pilgrim 2012). Experiments in C2C12 skeletal muscle cells demonstrated that UNC-45a is involved in cell proliferation and maintenance of myofibrils once they have formed, whereas UNC-45b has a role in sarcomere development (Price et al. 2002). A study by Wohlgemuth and colleagues (2007) found that knockdown of UNC-45b in zebrafish results in severe defects in both trunk muscle and cardiac development. Later work by Comyn and colleagues (2012) found that the combined loss of UNC-45a and UNC-45b phenotypically resembles loss of UNC-45b alone in developing zebrafish embryos. The authors concluded that the absence of a compound phenotype in double knockout embryos indicates that UNC-45 isoforms do not share redundant activity. It therefore appears that while both isoforms retain myosin chaperone activity, they are each utilized for independent functions within the cell. How these unique functions are regulated in cells that co-express both isoforms such as the developing myoblast could yield interesting insight into possible interacting partners of UNC-45.
Recent reports have begun to investigate the SET- and MYND-domain-containing protein 1 (SmyD1) as a potential modulator of UNC-45 function. SmyD1 was originally identified as a histone methyltransferase that is strongly expressed in heart and muscle tissues (Gottlieb et al. 2002; Tan et al. 2006). Targeted deletion of the mammalian homologue Bop in mice resulted in heart formation defects affecting the right ventricle (Gottlieb et al. 2002). Morpholino knockdown of the zebrafish alternative splicing isoforms SmyD1a and SmyD1b resulted in myofibrillar disorganization with cardiac and skeletal muscle malfunction in morphant embryos, further supporting its critical role in muscle development (Tan et al. 2006). Further, they showed that mutation of the SET-domain responsible for methyltransferase activity did not permit rescue of SmyD1 morphant knockdown embryos, supporting a role for methyltransferase activity in zebrafish muscle development. The precise mechanism of SmyD1 function in muscle remains unclear however. A recent study in zebrafish by Just and colleagues (2011) found that SmyD1 localizes to the M-line where it physically interacts with myosin. Interestingly, they showed that loss of methyltransferase activity did not impede myofibrillogenesis, indicating alternative roles for SmyD1 in regulating sarcomere development in contrast to the methyltransferase activity requirement noted by Tan and colleagues (2006). A study by Li and colleagues (2013) found that knockdown of the smyd1b gene in zebrafish led to increased expression of both hsp90 and unc45b with decreased myosin expression specifically at the protein level. The authors propose that myofibrillar defects could therefore be related to improper regulation of UNC-45b expression or activity, leading to increased myosin degradation. Together, these studies point to the complex role of UNC-45 and its interacting partners in sarcomere assembly and muscle development.
Chaperone proteins in actin, thin filament and Z-disk assembly
Chaperones are also involved in the proper folding of α-actinin and actin during myofibrillogenesis. N-RAP, for example, is a chaperone shown to be involved in α-actinin folding and integration into the Z-disk during myofibril assembly (Dhume et al. 2006). Chaperonin [CCT, also called TCP-1 Ring Complex (TRiC)] and its cofactor GimC (prefoldin) appear to be necessary for actin folding and assembly (Siegers et al. 1999; Grantham et al. 2002). In vitro experiments have shown that ribosome-associated actin chains bind to GimC, which remains bound to the nascent actin protein and delivers it to cytosolic TRiC (Hansen et al. 1999). The interaction of GimC and TRiC with actin folding intermediates constitutes the “folding compartment” and serves to accelerate actin folding by at least 5 times (Siegers et al. 1999). Finally, a study by Brown and colleagues (2007) confirmed that Hsp25/27 is a regulator of actin polymerization in Xenopus laevis frogs. Depletion of this protein resulted in cardia bifida, actin filament disarray, and myofibrillar disorganization. Thus, the presence of Hsp25/27 may be a critical requirement during myogenesis and heart tube formation.
Chaperone proteins in titin and intermediate filament assembly
The small heat shock protein (sHSP) αB-crystallin is commonly localized to the Z-disk and I-band regions of the sarcomere and appears to be involved in assembly of titin and intermediate filaments during myofibrillogenesis (Bullard et al. 2004). This chaperone is abundant in cardiac muscle cells, where it is necessary for the folding of titin filaments (Bennardini et al. 1992; Inagaki et al. 2006). In the heart, αB-crystallin has been shown to bind to the N2B region of cardiac titins, located in their N-terminal halves, near to the Z-disk (Bullard et al. 2004). αB-crystallin is also required for the assembly and folding of desmin, which forms an intermediate filament found at the Z-disk of the sarcomere (Bennardini et al. 1992). The role of αB-crystallin is not limited to folding of intermediate filaments however. A study by Singh and colleagues (2007) found that actin directly interacts with αB-crystallin in co-immunoprecipitation experiments, suggesting a further role in regulating actin filament dynamics. Moreover, in vitro experiments using chicken muscle myosin have shown that αB-crystallin prevents myosin aggregation and preserves its enzymatic activity under heat-shock conditions (Melkani et al. 2006). This suggests that αB-crystallin may be involved in nascent myosin folding and myofibrillogenesis.
Concluding remarks regarding chaperones in myofibrillogenesis
Formation of the sarcomere requires a series of highly regulated events that must integrate a wide variety of protein components into a cohesive unit. Chaperone proteins lay at the heart of this process, assisting in providing correctly folded elements to the nascent sarcomere for incorporation (summarized in Figure 2). Recent efforts have made strides in elucidating the complex network of both general chaperones (such as Hsp90 and TRiC/GimC) and specific chaperones that are highly expressed in muscle (such as UNC-45), as well as their interacting partners (summarized in Table 1). Further studies to uncover chaperone-binding partners in muscle could lead to a better understanding of the process of sarcomere assembly and function.
Table 1.
Chaperone and co-chaperone proteins and their known client and interacting partners
| Chaperone/Co-chaperone | Client/Interacting Proteins | References |
|---|---|---|
| Hsp90 | myosin | (Ni et al. 2011) |
| UNC-45 | (Barral et al. 1998; Barral et al. 2002; Price et al. 2002;Liu et al. 2008) | |
| UNC-45 | myosin | (Barral et al. 1998; Barral et al. 2002; Price et al. 2002;Toi et al. 2003; Lord and Pollard 2004) |
| Hsp90 | (Barral et al. 1998; Barral et al. 2002; Price et al. 2002; Liu et al. 2008) | |
| SmyD1 | (Tan et al. 2006; Just et al. 2011; Li et al. 2013) | |
| SmyD1* | myosin | (Just et al. 2011) |
| Hsp90 | (Li et al. 2013) | |
| UNC-45 | (Li et al. 2013) | |
| CCT & GimC | actin | (Siegers et al. 1999; Grantham et al. 2002) |
| myosin | (Srikakulam and Winkelmann 1999) | |
| Hsp25/27 | actin | (Brown et al. 2007) |
| αB-crystallin | myosin | (Melkani et al. 2006) |
| actin | (Singh et al. 2007) | |
| Titin | (Bennardini et al. 1992; Bullard et al. 2004; Inagaki et al. 2006) | |
| desmin | (Bennardini et al. 1992; Wang et al. 2001) | |
| N-RAP | α-actinin | (Dhume et al. 2006) |
not confirmed to be a chaperone or co-chaperone
CHAPERONES IN THE MAINTENANCE OF MUSCLE CELL STRUCTURE AND FUNCTION
Following a highly ordered assembly process, muscle cells utilize a complex series of pathways to maintain structural integrity and therefore function. It remains unclear whether sarcomeres are maintained passively through stochastic protein exchange and turnover or through an active and directed response to muscle stress. For the latter proposal, damaged sarcomeric proteins would have to be efficiently recognized, removed and replaced to maintain the repetitively organized structure. Studies over the past decade have shown that the active regulation of chaperone proteins plays an essential role in muscle maintenance. Recent work has begun to elucidate the sub-cellular localization and dynamic movement of chaperone molecules within functional sarcomeres (Figure 3), providing a better understanding of maintenance of muscle cell structure and function.
Figure 3.

Localization and movement of chaperones in response to stress in vertebrate sarcomeric structures. Myosin chaperones Hsp90 and UNC-45 are held in reserve in the Z-disk until cellular stress induces rapid localization to the A-band. Similarly, the intermediate filament chaperones αB-crystallin and Hsp25/27 are located in the cytosol and localize to titin near the Z-disk upon stress to the sarcomere.
The chaperones Hsp90 and UNC-45 are expressed in muscle cells after development, supporting their continued role in sarcomere maintenance. DAF-21, the C. elegans homologue of Hsp90, is largely localized along the I-band with some M-line localization at high expression levels (Gaiser et al. 2011). In contrast, UNC-45 appears to be largely localized to the A-band in C. elegans, consistent with its role as a myosin chaperone (Ao and Pilgrim 2000). However, more recent studies indicate that UNC-45 can exist as two different populations, one that is freely diffusible within the I-band and another that is stably bound within the A-band (Gaiser et al. 2011). Interestingly, zebrafish models suggest more dynamic localization patterns. Etard and colleagues (2008) showed that both Hsp90 and UNC-45b are localized within the Z-disk. Upon cellular stress to the muscle, both Hsp90 and UNC-45 rapidly localize to the myosin containing A-band, presumably to monitor for damaged myosin and assist in repair (Figure 3). While the precise function of this dynamic regulation remains unclear, it is possible that binding of Hsp90 and UNC-45 could impair myosin function. By holding these proteins in reserve within the Z-disk, the cell can quickly respond to damage while retaining functionality under normal circumstances.
Hsp70 also contributes to the overall homeostasis of muscle cells, though no studies have identified any specific localization within the sarcomere. Hsp70 is therefore thought to be a more ubiquitous contributor to the protection against cellular damage and dysfunction. In skeletal muscle, several studies have shown that Hsp70 expression is increased following eccentric exercise and injury (Morton et al. 2009; Paulsen et al. 2009; Senf et al. 2013). In Hsp70 knockout mice, inflammatory response to muscle injury is significantly delayed (Senf et al. 2013). This slowed response is followed by inflammation of the muscle, and in severe cases, necrosis and reduced ability to repair muscle fibers. Further, ectopic expression of Hsp70 in muscle tissue was able to stimulate regeneration in rescue experiments. Collectively, this study suggests that Hsp70 could be involved in the initial inflammatory response and in mediating the transition to muscle repair and recovery.
There is evidence to suggest that the localization of members of the sHSP family is dynamically regulated in response to muscle cell stress. Early studies of αB-crystallin indicated that it is expressed in muscle cells as a soluble and freely diffusible protein within the cell, though the authors did note a small subset that was localized to the Z-disk (Atomi et al. 1991). Later studies by Koh and Escobedo (2004) found that αB-crystallin rapidly localized to the Z-disk following mechanical stress such as lengthening contractions. In this same study, the authors determined that Hsp25/27 also exhibited a similar Z-disk translocation. A recent study by Kötter and colleagues (2014) refined this model, showing that αB-crystallin, along with Hsp25/27, binds to the titin immunoglobulin-like domain in the I-band region proximal to the Z-disk, where they may protect titin from aggregation. Interestingly, αB-crystallin is involved in facilitating the proper folding of intermediate filament proteins present within the Z-disk during development (Perng et al. 1999; Goldfarb and Dalakas 2009). As both Hsp25/27 and αB-crystallin translocate near to Z-disks after mechanical stress, they may act to protect the Z-disk proteins from further injury or aid in their repair.
These sub-cellular localization patterns and translocations imply a dynamic picture of chaperone movement and coordination with regard to muscle maintenance. Understanding this network is a formidable challenge. Additional real-time subcellular experiments will undoubtedly help clarify how chaperones respond to cellular stress. Interrogating the mechanisms that regulate translocation of chaperones into damaged muscle could also identify therapeutic targets to promote muscle repair following injury.
Chaperones and autophagy
Autophagy is an important subcellular degradation mechanism that mediates cellular remodeling and removal of damaged proteins and organelles (Mizushima 2007; Kundu and Thompson 2008; Levine and Kroemer 2008). Separate from the ubiquitin-proteasome system, autophagic degradation is lysosome-based and occurs by three different delivery pathways: chaperone-mediated autophagy (CMA), macroautophagy, and microautophagy (Sandri et al. 2013). Both CMA and macroautophagy have been shown to utilize chaperones and therefore will be focused on here.
CMA is a proteolytic pathway that utilizes ubiquitin-independent shuttling of chaperone substrates through the lysosomal membrane for degradation. This highly selective process targets only proteins bearing the KFERQ pentapeptide recognition sequence, which is responsible for the direct translocation of target proteins into the lysosome (Dice et al. 1990). CMA involves the chaperones Hsp70 and Hsp90 and co-chaperones including Bcl-2-associated athanogene 1 (BAG-1) (Agarraberes and Dice 2001).The role of CMA has not been extensively explored in skeletal muscle to date. However, a recent study determined that CMA components LAMPA and Hsc70 are increased in sporadic inclusion body myositis (s-IBM) muscle (Cacciottolo et al. 2014), implying that some aspects of autophagy may be mediated by CMA in skeletal muscle, particularly in the context of disease.
Chaperone-assisted selective autophagy (CASA) is a macroautophagic delivery mechanism in which chaperones selectively identify and deliver damaged proteins directly to the autophagosome (Gamerdinger et al. 2011). Formation of the CASA complex at the Z-disk is coordinated by Starvin (Stv) in Drosophila and its homologue Bcl-2-associated athanogene 3 (BAG-3) in vertebrates (Arndt et al. 2010). These proteins then recruit the co-chaperones Hsc70 and HspB8, and the ubiquitin ligase CHIP (Doong et al. 2000; Carra et al. 2008; Arndt et al. 2010). The co-chaperone HspB8 appears to be responsible for recognizing the mis-folded proteins, while Hsc70 is involved in substrate processing (Haslbeck et al. 2005; Carra et al. 2008). Arndt and colleagues (2010) propose that BAG-3 mediates the release of damaged components from the Z-disk, which are then ubiquitinylated by CHIP and degraded following recruitment of the ubiquitin adapter p62. During this process, BAG-3 is also ubiquitinylated by CHIP, which may lead to the co-degradation of BAG-3 (Arndt et al. 2010). Degradation of BAG-3 could represent a regulatory mechanism for the CASA pathway and warrants further investigation.
Interestingly, CASA appears to be dispensable for sarcomere assembly but is required for its maintenance. Loss of Stv allows for normal sarcomere development with lethality observed in early larval stages due to progressive disruption of the Z-disk (Coulson et al. 2005; Arndt et al. 2010). Similarly, BAG-3 knockout mice exhibit normal development with early lethality following muscle failure (Homma et al. 2006; Arndt et al. 2010). Recent studies indicate that unfolding of the Z-disk protein filamin caused by mechanical stress induces CASA activity, consistent with CASA’s role in maintenance (Ulbricht et al. 2013).
Despite significant progress, there remain fundamental questions regarding the role of CASA in muscle maintenance. While several of the components appear to be localized to the Z-disk, additional studies could reveal dynamic regulation similar to other chaperone systems discussed above. Additionally, CASA is largely unaffected in CHIP-deficient fibroblasts and CHIP-knockout mice do not suffer from myopathy, indicating the existence of functionally redundant ubiquitin ligases (Xu et al. 2002; Dai et al. 2003). Finally, loss of Stv in Drosophila or BAG-3 in mice phenotypically resembles a recently identified subset of myofibrillar myopathy (MFM) known to be associated with mutation of BAG-3, supporting the use of these loss-of-function mutants as clinical models (Selcen et al. 2009). Further interrogation of how CASA is regulated could yield important information on therapeutic intervention in diseases caused by BAG-3 or filamin mutations.
Concluding remarks on chaperones in muscle maintenance
Muscles derive their function from the precise integration of structural and functional proteins that must be maintained throughout the life of the organism. Recent efforts have begun to identify how chaperones play a role in sarcomere turnover and long-term stability. In vertebrates, chaperone proteins appear to be held in local reservoirs and dynamically localized to areas of potential muscle stress (Figure 3). It is possible that muscle cells utilize these chaperone reserves to assist in refolding damaged proteins without having to express and incorporate new proteins into the sarcomere. In the event that the damage is too severe, chaperones can mediate protein disposal through coordination with autophagic processes. Understanding how the cell regulates chaperone activity with respect to repair and protein turnover will likely have a dramatic impact on our ability to target chaperone proteins therapeutically.
CHAPERONES IN MUSCLE DISEASE
Diseases caused by improper chaperone activity
The first disease-associated mutation identified in a protein chaperone was the R120G mutation in human αB-crystallin (CRYAB), which was associated with a subset of myofibrillar myopathies (MFMs) now called αB-crystallinopathies (Vicart et al. 1998). Additional αB-crystallin mutations have been identified that result in C-terminal truncations and loss of chaperone activity (Selcen and Engel 2003). While the detailed mechanism of αB-crystallin function is still poorly understood, the conserved C-terminal α-crystallin domain is critical to the regulation of αB-crystallin activity within the cell. As many of the identified mutations are located within or near the α-crystallin domain, it is widely believed that loss of function within this domain is responsible for the aberrant activity of αB-crystallin mutants (Andley et al. 1996; Smulders et al. 1996). Crystallographic studies of the α-crystallin domain from αB-crystallin and related proteins have identified a dimeric structure that forms a collapsible groove that is thought to interact with the adjacent N-terminal extension (Bagnéris et al. 2009). In a follow up study, the authors found that the R120G mutation shifts the protein-protein interface and closes this gap, altering the multimer dynamics in favor of oligomerization over dimerization (Clark et al. 2011). This dominant negative activity would effectively reduce the levels of αB-crystallin available to the cell and prevent normal chaperone function.
αB-crystallin forms one of the major structural proteins of the eye lens and acts as a sHSP in several other tissues throughout the body (Ecroyd and Carver 2009). In muscle tissues, it is largely responsible for preventing the aggregation of desmin and other intermediate filament proteins (Goldfarb and Dalakas 2009). Clinically, the R120G mutation is associated with the formation of cataracts, weakness in the skeletal muscles and heart failure, which is a predominant cause of death (Vicart et al. 1998). The causal relationship between the R120G mutation and the clinically observed phenotype was shown using a transgenic mouse model (Wang et al. 2001). Expression of CryABR120G in mouse and Drosophila cardiomyocytes results in significantly impaired heart function that phenocopies human clinical cases (Rajasekaran et al. 2007; Xie et al. 2013). Interestingly, the observed cardiomyopathies appeared to result from an increase in reductive stress caused by activation of the glucose-6-phosphate dehydrogenase (G6PD) enzyme. In both models, knockdown of G6PD alleviated the cardiomyopathy and reduced protein aggregation. This implies that the underlying disease mechanism of CryABR120G might be related to metabolic disruption rather than accumulation of aggregated proteins. Though the mechanism remains unclear, these studies provide the foundation for novel treatments that could ameliorate cardiomyopathies commonly associated with impaired chaperone function.
Other proteins involved in chaperone function have been implicated in muscle disease, possibly through disruption of the CASA complex. Recent studies have identified BAG-3 mutations in a subset of childhood muscular dystrophies (Selcen et al. 2009). Mutation of the co-chaperone DnaJ (Hsp40) homolog, subfamily B, member 6(DNAJB6) has been implicated in the development of limb girdle muscular dystrophy type 1D (LGMD 1D) (Harms et al. 2012; Sarparanta et al. 2012). Expression of mutant DNAJB6 in zebrafish resulted in significant loss of muscle integrity and appeared to act in a dominant fashion by hijacking the activity of wild-type DNAJB6, BAG-3 and other members of the CASA complex. This could also be related to recent studies indicating a role for DNAJB6 in preventing aggregation of polyglutamine stretches (Gillis et al. 2013; Månsson et al. 2014) .
Despite the few examples cited above and the recent development of next generation sequencing techniques in determining genetic causes for human disease, exceedingly few mutations have been identified in chaperone proteins. While it is likely that disruption of such critical systems could result in embryonic lethality and be rarely observed, it is also possible that there could be functional redundancies among related protein chaperones. Future studies might therefore benefit from investigating how networks of chaperone proteins interact to compensate for possible deficiencies and mask loss-of-function mutations. Understanding this network could inform therapeutic approaches targeting chaperone proteins.
Several groups have begun to investigate how chaperone activity can be altered by mutations in client or interacting proteins in a variety of muscle disease models. Idiopathic inflammatory myopathies exhibit dramatic changes in Hsp70 levels in muscle and Hsp90 in both the muscle and invading immune cells, with similar findings observed in Duchenne muscular dystrophy (De Paepe et al. 2009; De Paepe et al. 2012). The authors conclude that both Hsp70 and Hsp90 are likely involved in mediating attempted muscle recovery while Hsp90 could play a role in promoting the cytotoxicity induced by invading immune cells. These findings point to the complex network of chaperone proteins that mediate interactions between both the endogenous cells of a tissue and migratory immune cells.
Recent studies have also begun to elucidate how regulation of the ubiquitin ligase protein degradation system is linked to the myosin chaperone UNC-45. Heightened expression of UNC-45 in C. elegans affects muscle development, highlighting the importance of maintaining normal chaperone levels during development (Landsverk et al. 2007). UNC-45 levels appear to be regulated by the CHN-1/UFD-2 complex during development in C. elegans (Hoppe et al. 2004). This complex requires CDC-48, the C. elegans homolog of the mammalian p97, the mutation of which has been shown to induce inclusion body myopathy in several disease models (Custer et al. 2010; Ritson et al. 2010). Interestingly, mutation of p97 reduces UNC-45 turnover, resulting in poor myofibrillar assembly and degradation (Janiesch et al. 2007). This indicates that mutations in the protein turnover systems can affect chaperone levels, which might contribute to associated disease progression. Further description of how chaperone networks are influenced by disease will be crucial to the successful implementation of chaperone-targeted therapeutics.
Chaperones as therapeutic options in neuromuscular disease
Chaperone proteins are currently being investigated as therapeutic targets in diseases where protein aggregation is thought to be the underlying molecular mechanism, though largely in the context of neurodegenerative disease. Increased expression of protein chaperones promotes cell viability and assists in clearance of protein aggregates in several disease models (Sakahira et al. 2002). However, the limiting factor for applying chaperone-based therapy to muscle disease has been development of therapeutic small molecules that are able to achieve similar results to chaperone over-expression systems (Table 2). Current efforts to modulate chaperone activity largely revolve around the feedback mechanism between heat shock factor 1 (HSF1) and Hsp90. HSF1 acts as a master regulator for several members of the HSP and sHSP families (de Thonel et al. 2012). Activation of this transcription factor results in heightened expression of Hsp70, Hsp90 and various co-chaperones and has been shown to provide therapeutic benefit in a variety of model systems.
Table 2.
Small-molecule therapeutics targeting HSF1 and Hsp90 in disease models.
| Chaperone Target | Drug | Disease Models | References |
|---|---|---|---|
| Hsp90 | geldanamycin | Parkinson's, Huntington's | (Sittler et al. 2001; McLean et al. 2004) |
| NXD30001 | ALS/SOD1G93A,glioblastoma | (Zhu et al. 2010; Cha et al.2014) | |
| alvespimycin | Castration-resistant prostate cancer | (Pacey et al. 2011) | |
| 17-AAG/tanespimycin | Breast cancer | (Banerji et al. 2005; Modi et al. 2011) | |
| IPI-504/retaspimycin hydrochloride | Gastrointestinal stromal tumor, non-small cell lung cancer | (Sequist et al. 2008; Wagner et al. 2013) | |
| STA-1474 | Osteosarcoma | (McCleese et al. 2009) | |
| PU-H71 | Breast cancer | (Caldas-Lopes et al. 2009) | |
| NVP-AUY922 | Several cancer models | (Eccles et al. 2008) | |
| HSF1 | celastrol | ALS/SOD1G93A,Huntington's, Spinal bulbar muscular dystrophy | (Piccioni et al. 2004; Kiaei et al. 2005; Zhang and Sarge 2007) |
| HSF1A | Huntington's, Machado-Joseph | (Neef et al. 2010) | |
| CYT1; CYT2; CYT3,CYT4 | Huntington's | (Zhang et al. 2009) | |
| PR-A1, A3; PR-C1;PR-D1; PR-F1 | Huntington's, cystic fibrosis | (Calamini et al. 2012) |
The celastrol family of molecules act as HSF1 agonists that activate the human heat shock response and induce expression of Hsp70, Hsp90 and other chaperones (Westerheide et al. 2004). Treatment of amyotrophic lateral sclerosis (ALS) and Huntington’s disease models with celastrol has shown therapeutic efficacy in reducing aggregate formation in neural tissues (Kiaei et al. 2005; Zhang and Sarge 2007). Despite these results, potential toxicity could be a limiting factor for widespread use in neural tissues or in muscle (Kalmar and Greensmith 2009; Hansen and Bross 2010). More recent studies have begun screening small molecule libraries for novel HSF1 agonists. Zhang and colleagues (2009) identified three geduin derivatives and one Sappanone A related molecule as activators of HSF1. Each of these small molecules increases HSF1 transcriptional activity and Hsp70 expression in cell cultures, indicating their potential use as therapeutics where Hsp70 has shown protective benefit. Neef and colleagues (2010) utilized a yeast screening system to identify HSF1A, a novel small molecule that interacts directly with HSF1 and can protect against neurodegeneration in a poly-glutamine fly model. A recent study by Calamini and colleagues (2012) screened over 900,000 small molecules and identified seven classes that exhibited HSF1 activation. Utilizing in vivo studies, the authors found that several of these compounds inhibited protein aggregation in C. elegans models, supporting their potential clinical use.
Inhibition of Hsp90 may represent a second viable therapeutic direction. Early studies indicated that inhibition of Hsp90 using geldanamycin dramatically increased HSF1 activity, which in turn increased expression of the chaperone Hsp70 (Zou et al. 1998). This increase in Hsp70 expression has been shown to inhibit aggregation of α-synuclein and huntingtin in Parkinson’s and Huntington’s disease models, providing a possible therapeutic intervention that might be translated to protein aggregation diseases in muscle (Sittler et al. 2001; McLean et al. 2004). Of note, a recent study by Cha and colleagues (2014) interrogated the ability of NXD30001, a derivative of radicicol, to ameliorate ALS symptoms in a superoxide dismutase G92A (SODG92A) mouse model. Treatment with the drug led to significantly reduced aggregation and motor neuron death, supporting its use in ameliorating aggregation diseases. Interestingly, the authors identified heightened expression of Hsp70 in skeletal muscle tissue following treatment, supporting NXD30001’s potential therapeutic use in muscle disease as well. However, as many Hsp90 inhibitors currently in development are being selected for anti-tumor activity, translation into the muscle disease field will require careful interrogation of potential cytotoxic effects. Small-molecule therapeutics targeting HSF1 and Hsp90 in disease models are summarized in Table 2.
The ability to modulate the activity of protein chaperones in vivo has shown great promise in several clinical settings, though none appear to be targeting muscle disease models to date (Neckers and Workman 2012). There are emerging lines of evidence to suggest that modulation of chaperone activity could provide therapeutic benefit for patients suffering muscle disorders accompanied by protein aggregation. A recent study from Melkani and colleagues (2013) showed that over-expression of UNC-45 in the Drosophila heart reduces huntingtin poly-glutamine aggregation and cardiomyopathy. Similarly, a study of oculopharyngeal dystrophy showed that over-expression of Hsp70 specifically within the muscle reduces the toxicity of mutant PABPN1 in a Drosophila model (Chartier et al. 2006). These studies, along with encouraging results in neurodegenerative disease discussed above, support exploration of chaperone activity modulators in muscle diseases where aggregation or improper protein folding is thought to contribute to the molecular mechanism.
Concluding remarks on chaperone proteins in disease
Disruption of chaperone networks can have catastrophic effects on muscle development and function, as demonstrated through direct mutation of chaperone proteins themselves or loss of function in key regulatory elements. There are a variety of chaperone activity modulators currently under investigation in cancer and neurodegenerative diseases. It will be interesting to see how these therapeutics developed for disparate diseases can be incorporated into treating muscle diseases associated with chaperone dysfunction and/or protein aggregation.
CONCLUSIONS
A protein’s activity is intrinsically related to its physical conformation and the process of adopting that conformation is regulated by protein chaperones. In this review, we discussed recent efforts to interrogate the roles of both general and specific chaperones in mediating muscle development, maintenance and health. While rare, disruption of these carefully tuned networks severely impairs muscle development and repair, exemplifying the absolute necessity for proper chaperone function. As we come to understand how different chaperones are involved in myofibril assembly and maintenance, we can begin to identify targets for therapeutic intervention. Modulating activity of chaperones during embryogenesis could allow for more normal myofibrillogenesis in muscle cells containing aberrantly folded contractile proteins, while progressive myopathies could benefit from enhancing activity of chaperones specifically involved in muscle maintenance. Recent efforts to develop small molecule therapeutics that target chaperones in cancer and neurodegenerative disease could serve as the foundation for their use in muscle disease.
Acknowledgments
Financial support: Grant Sponsor: National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases; Grant numbers: 3R01AR055958; 3R01AR055958-04S1
We appreciate helpful comments on the manuscript from Dr. Girish C. Melkani, Dr. Chi Lee and other members of the Bernstein lab at San Diego State University.
Footnotes
Conflict of Interest: The authors have no financial or other conflicts of interest to disclose.
LITERATURE CITED
- 1.Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114:2491–2499. doi: 10.1242/jcs.114.13.2491. [DOI] [PubMed] [Google Scholar]
- 2.Andley UP, Mathur S, Griest TA, Petrash JM. Cloning, expression, and chaperone-like activity of human alphaA-crystallin. J Biol Chem. 1996;271:31973–31980. doi: 10.1074/jbc.271.50.31973. [DOI] [PubMed] [Google Scholar]
- 3.Ao W, Pilgrim D. Caenorhabditis elegans Unc-45 Is a component of muscle thick filaments and colocalizes with myosin heavy chain B, but not myosin heavy chain A. J Cell Biol. 2000;148:375–384. doi: 10.1083/jcb.148.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Fürst DO, Saftig P, Saint R, Fleischmann BK, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–148. doi: 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 5.Atomi Y, Yamada S, Strohman R, Nonomura Y. Alpha B-crystallin in skeletal muscle: purification and localization. J Biochem (Tokyo) 1991;110:812–822. doi: 10.1093/oxfordjournals.jbchem.a123665. [DOI] [PubMed] [Google Scholar]
- 6.Bagnéris C, Bateman OA, Naylor CE, Cronin N, Boelens WC, Keep NH, Slingsby C. Crystal structures of α-crystallin domain dimers of αB-crystallin and Hsp20. J Mol Biol. 2009;392:1242–1252. doi: 10.1016/j.jmb.2009.07.069. [DOI] [PubMed] [Google Scholar]
- 7.Banerji U, O’Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23:4152–4161. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 8.Barral JM, Bauer CC, Ortiz I, Epstein HF. Unc-45 mutations in Caenorhabditis elegans implicate a CRO1/She4p-like domain in myosin assembly. J Cell Biol. 1998;143:1215–1225. doi: 10.1083/jcb.143.5.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barral JM, Hutagalung AH, Brinker A, Hartl FU, Epstein HF. Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin. Science. 2002;295:669–671. doi: 10.1126/science.1066648. [DOI] [PubMed] [Google Scholar]
- 10.Bennardini F, Wrzosek A, Chiesi M. Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res. 1992;71:288–294. doi: 10.1161/01.res.71.2.288. [DOI] [PubMed] [Google Scholar]
- 11.Bernick EP, Zhang P-J, Du S. Knockdown and overexpression of Unc-45b result in defective myofibril organization in skeletal muscles of zebrafish embryos. BMC Cell Biol. 2010;11:70–83. doi: 10.1186/1471-2121-11-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown DD, Christine KS, Showell C, Conlon FL. Small heat shock protein Hsp27 is required for proper heart tube formation. Genesis. 2007;45:667–678. doi: 10.1002/dvg.20340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, Ding L, Labeit S, Horwitz J, Leonard KR, et al. Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem. 2004;279:7917–7924. doi: 10.1074/jbc.M307473200. [DOI] [PubMed] [Google Scholar]
- 14.Cacciottolo M, Nogalska A, D’Agostino C, Engel WK, Askanas V. Chaperone-mediated autophagy components are upregulated in sporadic inclusion-body myositis muscle fibres. Neuropathol Appl Neurobiol. 2013;39:750–761. doi: 10.1111/nan.12038. [DOI] [PubMed] [Google Scholar]
- 15.Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, Chalfant MA, Saldanha SA, Hodder P, Tait BD, Garza D, et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol. 2012;8:185–196. doi: 10.1038/nchembio.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A, Guo Y, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A. 2009;106:8368–8373. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caplan AJ. What is a co-chaperone? Cell Stress Chaperones. 2003;8:105–107. doi: 10.1379/1466-1268(2003)008<0105:wiac>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carra S, Seguin SJ, Lambert H, Landry J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem. 2008;283:1437–1444. doi: 10.1074/jbc.M706304200. [DOI] [PubMed] [Google Scholar]
- 19.Cha JRC, St Louis KJH, Tradewell ML, Gentil BJ, Minotti S, Jaffer ZM, Chen R, Rubenstein AE, Durham HD. A novel small molecule HSP90 inhibitor, NXD30001, differentially induces heat shock proteins in nervous tissue in culture and in vivo. Cell Stress Chaperones. 2014;19:421–435. doi: 10.1007/s12192-013-0467-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chartier A, Benoit B, Simonelig M. A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. EMBO J. 2006;25:2253–2262. doi: 10.1038/sj.emboj.7601117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen D, Li S, Singh R, Spinette S, Sedlmeier R, Epstein HF. Dual function of the UNC-45b chaperone with myosin and GATA4 in cardiac development. J Cell Sci. 2012;125:3893–3903. doi: 10.1242/jcs.106435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chow D, Srikakulam R, Chen Y, Winkelmann DA. Folding of the striated muscle myosin motor domain. J Biol Chem. 2002;277:36799–36807. doi: 10.1074/jbc.M204101200. [DOI] [PubMed] [Google Scholar]
- 23.Clark AR, Naylor CE, Bagnéris C, Keep NH, Slingsby C. Crystal structure of R120G disease mutant of human αB-crystallin domain dimer shows closure of a groove. J Mol Biol. 2011;408:118–134. doi: 10.1016/j.jmb.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Codina M, Li J, Gutiérrez J, Kao JPY, Du SJ. Loss of Smyhc1 or Hsp90alpha1 function results in different effects on myofibril organization in skeletal muscles of zebrafish embryos. PloS One. 2010;5:e8416. doi: 10.1371/journal.pone.0008416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Comyn SA, Pilgrim D. Lack of developmental redundancy between Unc45 proteins in zebrafish muscle development. PloS One. 2012;7:e48861. doi: 10.1371/journal.pone.0048861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coulson M, Robert S, Saint R. Drosophila starvin encodes a tissue-specific BAG-domain protein required for larval food uptake. Genetics. 2005;171:1799–1812. doi: 10.1534/genetics.105.043265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Custer SK, Neumann M, Lu H, Wright AC, Taylor JP. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet. 2010;19:1741–1755. doi: 10.1093/hmg/ddq050. [DOI] [PubMed] [Google Scholar]
- 28.Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li H-H, Madamanchi N, Xu W, Neckers L, et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22:5446–5458. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhume A, Lu S, Horowits R. Targeted disruption of N-RAP gene function by RNA interference: a role for N-RAP in myofibril organization. Cell Motil Cytoskeleton. 2006;63:493–511. doi: 10.1002/cm.20141. [DOI] [PubMed] [Google Scholar]
- 30.Dice JF, Terlecky SR, Chiang HL, Olson TS, Isenman LD, Short-Russell SR, Freundlieb S, Terlecky LJ. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin Cell Biol. 1990;1:449–455. [PubMed] [Google Scholar]
- 31.Doong H, Price J, Kim YS, Gasbarre C, Probst J, Liotta LA, Blanchette J, Rizzo K, Kohn E. CAIR-1/BAG-3 forms an EGF-regulated ternary complex with phospholipase C-gamma and Hsp70/Hsc70. Oncogene. 2000;19:4385–4395. doi: 10.1038/sj.onc.1203797. [DOI] [PubMed] [Google Scholar]
- 32.Du SJ, Li H, Bian Y, Zhong Y. Heat-shock protein 90α1 is required for organized myofibril assembly in skeletal muscles of zebrafish embryos. Proc Natl Acad Sci U S A. 2008;105:554–559. doi: 10.1073/pnas.0707330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, Brandon A, de H, Gowan S, Boxall F, et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68:2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 34.Ecroyd H, Carver JA. Crystallin proteins and amyloid fibrils. Cell Mol Life Sci. 2009;66:62–81. doi: 10.1007/s00018-008-8327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehler E, Rothen BM, Hämmerle SP, Komiyama M, Perriard JC. Myofibrillogenesis in the developing chicken heart: assembly of Z-disk, M-line and the thick filaments. J Cell Sci. 1999;112 ( Pt 10):1529–1539. doi: 10.1242/jcs.112.10.1529. [DOI] [PubMed] [Google Scholar]
- 36.Epstein HF, Thomson JN. Temperature-sensitive mutation affecting myofilament assembly in Caenorhabditis elegans. Nature. 1974;250:579–580. doi: 10.1038/250579a0. [DOI] [PubMed] [Google Scholar]
- 37.Etard C, Behra M, Fischer N, Hutcheson D, Geisler R, Strähle U. The UCS factor Steif/Unc-45b interacts with the heat shock protein Hsp90a during myofibrillogenesis. Dev Biol. 2007;308:133–143. doi: 10.1016/j.ydbio.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 38.Etard C, Roostalu U, Strähle U. Shuttling of the chaperones Unc45b and Hsp90a between the A band and the Z line of the myofibril. J Cell Biol. 2008;180:1163–1175. doi: 10.1083/jcb.200709128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fürst DO, Osborn M, Weber K. Myogenesis in the mouse embryo: differential onset of expression of myogenic proteins and the involvement of titin in myofibril assembly. J Cell Biol. 1989;109:517–527. doi: 10.1083/jcb.109.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaiser AM, Kaiser CJO, Haslbeck V, Richter K. Downregulation of the Hsp90 system causesdefects in muscle cells of Caenorhabditis elegans. PLoS ONE. 2011;6:e25485. doi: 10.1371/journal.pone.0025485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gamerdinger M, Carra S, Behl C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: focus on BAG proteins. J Mol Med. 2011;89:1175–1182. doi: 10.1007/s00109-011-0795-6. [DOI] [PubMed] [Google Scholar]
- 42.Gazda L, Pokrzywa W, Hellerschmied D, Löwe T, Forné I, Mueller-Planitz F, Hoppe T, Clausen T. The myosin chaperone UNC-45 is organized in tandem modules to support myofilament formation in C. elegans. Cell. 2013;152:183–195. doi: 10.1016/j.cell.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gillis J, Schipper-Krom S, Juenemann K, Gruber A, Coolen S, Nieuwendijk R, van den Veen H, van Overkleeft H, Goedhart J, Kampinga HH, et al. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J Biol Chem. 2013;288:17225–17237. doi: 10.1074/jbc.M112.421685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–1813. doi: 10.1172/JCI38027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gottlieb PD, Pierce SA, Sims RJ, Yamagishi H, Weihe EK, Harriss JV, Maika SD, Kuziel WA, King HL, Olson EN, et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat Genet. 2002;31:25–32. doi: 10.1038/ng866. [DOI] [PubMed] [Google Scholar]
- 46.Grantham J, Ruddock LW, Roobol A, Carden MJ. Eukaryotic chaperonin containing T-complex polypeptide 1 interacts with filamentous actin and reduces the initial rate of actin polymerization in vitro. Cell Stress Chaperones. 2002;7:235–242. doi: 10.1379/1466-1268(2002)007<0235:ecctcp>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansen J, Bross P. A cellular viability assay to monitor drug toxicity. In: Bross P, Gregersen N, editors. Protein Misfolding and Cellular Stress in Disease and Aging. Methods in Molecular Biology. Humana Press; 2010. pp. 303–311. [DOI] [PubMed] [Google Scholar]
- 48.Hansen WJ, Cowan NJ, Welch WJ. Prefoldin-nascent chain complexes in the folding of cytoskeletal proteins. J Cell Biol. 1999;145:265–277. doi: 10.1083/jcb.145.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harms MB, Sommerville RB, Allred P, Bell S, Ma D, Cooper P, Lopate G, Pestronk A, Weihl CC, Baloh RH. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann Neurol. 2012;71:407–416. doi: 10.1002/ana.22683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 51.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 52.Hawkins TA, Haramis A-P, Etard C, Prodromou C, Vaughan CK, Ashworth R, Ray S, Behra M, Holder N, Talbot WS, et al. The ATPase-dependent chaperoning activity of Hsp90a regulates thick filament formation and integration during skeletal muscle myofibrillogenesis. Development. 2008;135:1147–1156. doi: 10.1242/dev.018150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holtzer H, Hijikata T, Lin ZX, Zhang ZQ, Holtzer S, Protasi F, Franzini-Armstrong C, Sweeney HL. Independent assembly of 1.6 microns long bipolar MHC filaments and I-Z-I bodies. Cell Struct Funct. 1997;22:83–93. doi: 10.1247/csf.22.83. [DOI] [PubMed] [Google Scholar]
- 54.Homma S, Iwasaki M, Shelton GD, Engvall E, Reed JC, Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. 2006;169:761–773. doi: 10.2353/ajpath.2006.060250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoppe T, Cassata G, Barral JM, Springer W, Hutagalung AH, Epstein HF, Baumeister R. Regulation of the myosin-directed chaperone UNC-45 by a novel E3/E4-multiubiquitylation complex in C. elegans. Cell. 2004;118:337–349. doi: 10.1016/j.cell.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 56.Hutagalung AH, Landsverk ML, Price MG, Epstein HF. The UCS family of myosin chaperones. J Cell Sci. 2002;115:3983–3990. doi: 10.1242/jcs.00107. [DOI] [PubMed] [Google Scholar]
- 57.Inagaki N, Hayashi T, Arimura T, Koga Y, Takahashi M, Shibata H, Teraoka K, Chikamori T, Yamashina A, Kimura A. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun. 2006;342:379–386. doi: 10.1016/j.bbrc.2006.01.154. [DOI] [PubMed] [Google Scholar]
- 58.Janiesch PC, Kim J, Mouysset J, Barikbin R, Lochmüller H, Cassata G, Krause S, Hoppe T. The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat Cell Biol. 2007;9:379–390. doi: 10.1038/ncb1554. [DOI] [PubMed] [Google Scholar]
- 59.Just S, Meder B, Berger IM, Etard C, Trano N, Patzel E, Hassel D, Marquart S, Dahme T, Vogel B, et al. The myosin-interacting protein SMYD1 is essential for sarcomere organization. J Cell Sci. 2011;124:3127–3136. doi: 10.1242/jcs.084772. [DOI] [PubMed] [Google Scholar]
- 60.Kalmar B, Greensmith L. Activation of the heat shock response in a primary cellular model of motoneuron neurodegeneration-evidence for neuroprotective and neurotoxic effects. Cell Mol Biol Lett. 2009;14:319–335. doi: 10.2478/s11658-009-0002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kiaei M, Nil K, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis. 2005;2:246–254. doi: 10.1159/000090364. [DOI] [PubMed] [Google Scholar]
- 63.Koh TJ, Escobedo J. Cytoskeletal disruption and small heat shock protein translocation immediately after lengthening contractions. Am J Physiol Cell Physiol. 2004;286:C713–722. doi: 10.1152/ajpcell.00341.2003. [DOI] [PubMed] [Google Scholar]
- 64.Kötter S, Unger A, Hamdani N, Lang P, Vorgerd M, Nagel-Steger L, Linke WA. Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J Cell Biol. 2014;204:187–202. doi: 10.1083/jcb.201306077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- 66.Landsverk ML, Li S, Hutagalung AH, Najafov A, Hoppe T, Barral JM, Epstein HF. The UNC-45 chaperone mediates sarcomere assembly through myosin degradation in Caenorhabditis elegans. J Cell Biol. 2007;177:205–210. doi: 10.1083/jcb.200607084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee CF, Hauenstein AV, Fleming JK, Gasper WC, Engelke V, Sankaran B, Bernstein SI, Huxford T. X-ray crystal structure of the UCS domain-containing UNC-45 myosin chaperone from Drosophila melanogaster. Structure. 2011a;19:397–408. doi: 10.1016/j.str.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee CF, Melkani GC, Yu Q, Suggs JA, Kronert WA, Suzuki Y, Hipolito L, Price MG, Epstein HF, Bernstein SI. Drosophila UNC-45 accumulates in embryonic blastoderm and in muscles, and is essential for muscle myosin stability. J Cell Sci. 2011b;124:699–705. doi: 10.1242/jcs.078964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lele Z, Hartson SD, Martin CC, Whitesell L, Matts RL, Krone PH. Disruption of zebrafish somite development by pharmacologic inhibition of Hsp90. Dev Biol. 1999;210:56–70. doi: 10.1006/dbio.1999.9262. [DOI] [PubMed] [Google Scholar]
- 70.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Zhong Y, Wang Z, Gao J, Xu J, Chu W, Zhang J, Fang S, Du SJ. Smyd1b is required for skeletal and cardiac muscle function in zebrafish. Mol Biol Cell. 2013;24:3511–3521. doi: 10.1091/mbc.E13-06-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta. 2012;1823:624–635. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 73.Liu L, Srikakulam R, Winkelmann DA. Unc45 activates Hsp90-dependent folding of the myosin motor domain. J Biol Chem. 2008;283:13185–13193. doi: 10.1074/jbc.M800757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lord M, Pollard TD. UCS protein Rng3p activates actin filament gliding by fission yeast myosin-II. J Cell Biol. 2004;167:315–325. doi: 10.1083/jcb.200404045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Månsson C, Kakkar V, Monsellier E, Sourigues Y, Härmark J, Kampinga HH, Melki R, Emanuelsson C. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. 2014;19:227–239. doi: 10.1007/s12192-013-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCleese JK, Bear MD, Fossey SL, Mihalek RM, Foley KP, Ying W, Barsoum J, London CA. The novel HSP90 inhibitor STA-1474 exhibits biologic activity against osteosarcoma cell lines. Int J Cancer J Int Cancer. 2009;125:2792–2801. doi: 10.1002/ijc.24660. [DOI] [PubMed] [Google Scholar]
- 77.McLean PJ, Klucken J, Shin Y, Hyman BT. Geldanamycin induces Hsp70 and prevents α-synuclein aggregation and toxicity in vitro. Biochem Biophys Res Commun. 2004;321:665–669. doi: 10.1016/j.bbrc.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 78.Melkani GC, Bodmer R, Ocorr K, Bernstein SI. The UNC-45 chaperone is critical for establishing myosin-based myofibrillar organization and cardiac contractility in the Drosophila heart model. PLoS ONE. 2011;6:e22579. doi: 10.1371/journal.pone.0022579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Melkani GC, Cammarato A, Bernstein SI. alphaB-crystallin maintains skeletal muscle myosin enzymatic activity and prevents its aggregation under heat-shock stress. J Mol Biol. 2006;358:635–645. doi: 10.1016/j.jmb.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 80.Melkani GC, Lee CF, Cammarato A, Bernstein SI. Drosophila UNC-45 prevents heat-induced aggregation of skeletal muscle myosin and facilitates refolding of citrate synthase. Biochem Biophys Res Commun. 2010;396:317–322. doi: 10.1016/j.bbrc.2010.04.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Melkani GC, Trujillo AS, Ramos R, Bodmer R, Bernstein SI, Ocorr K. Huntington’s disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart. PLoS Genet. 2013;9:e1004024. doi: 10.1371/journal.pgen.1004024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 83.Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, D’Andrea G, Dickler M, Moynahan ME, Sugarman S, et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17:5132–5139. doi: 10.1158/1078-0432.CCR-11-0072. [DOI] [PubMed] [Google Scholar]
- 84.Morton JP, Kayani AC, McArdle A, Drust B. The exercise-induced stress response of skeletal muscle, with specific emphasis on humans. Sports Med Auckl NZ. 2009;39:643–662. doi: 10.2165/00007256-200939080-00003. [DOI] [PubMed] [Google Scholar]
- 85.Myhre JL, Pilgrim DB. At the start of the sarcomere: A previously unrecognized role for myosin chaperones and associated proteins during early myofibrillogenesis. Biochem Res Int. 2012;2012:712315. doi: 10.1155/2012/712315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010;8:e1000291. doi: 10.1371/journal.pbio.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ni W, Hutagalung AH, Li S, Epstein HF. The myosin-binding UCS domain but not the Hsp90-binding TPR domain of the UNC-45 chaperone is essential for function in Caenorhabditis elegans. J Cell Sci. 2011;124:3164–3173. doi: 10.1242/jcs.087320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ojima K, Lin ZX, Zhang ZQ, Hijikata T, Holtzer S, Labeit S, Sweeney HL, Holtzer H. Initiation and maturation of I-Z-I bodies in the growth tips of transfected myotubes. J Cell Sci. 1999;112:4101–4112. doi: 10.1242/jcs.112.22.4101. [DOI] [PubMed] [Google Scholar]
- 90.Pacey S, Wilson RH, Walton M, Eatock MM, Hardcastle A, Zetterlund A, Arkenau H-T, Moreno-Farre J, Banerji U, Roels B, et al. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin Cancer Res. 2011;17:1561–1570. doi: 10.1158/1078-0432.CCR-10-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Paepe B, Creus KK, Martin J-J, Weis J, De Bleecker JL. A dual role for HSP90 and HSP70 in the inflammatory myopathies. Ann N Y Acad Sci. 2009;1173:463–469. doi: 10.1111/j.1749-6632.2009.04812.x. [DOI] [PubMed] [Google Scholar]
- 92.De Paepe B, Creus KK, Weis J, De Bleecker JL. Heat shock protein families 70 and 90 in Duchenne muscular dystrophy and inflammatory myopathy: Balancing muscle protection and destruction. Neuromuscul Disord. 2012;22:26–33. doi: 10.1016/j.nmd.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 93.Paulsen G, Lauritzen F, Bayer ML, Kalhovde JM, Ugelstad I, Owe SG, Hallén J, Bergersen LH, Raastad T. Subcellular movement and expression of HSP27, αB-crystallin, and HSP70 after two bouts of eccentric exercise in humans. J Appl Physiol. 2009;107:570–582. doi: 10.1152/japplphysiol.00209.2009. [DOI] [PubMed] [Google Scholar]
- 94.Pelpel K, Leibovitch M-P, Fernandez A, Leibovitch SA. Mutation of MyoD-Ser237 abolishes its up-regulation by c-Mos. FEBS Lett. 2000;474:233–237. doi: 10.1016/s0014-5793(00)01610-0. [DOI] [PubMed] [Google Scholar]
- 95.Perng MD, Cairns L, IJssel P, van den Prescott A, Hutcheson AM, Quinlan RA. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci. 1999;112:2099–2112. doi: 10.1242/jcs.112.13.2099. [DOI] [PubMed] [Google Scholar]
- 96.Piccioni F, Roman BR, Fischbeck KH, Taylor JP. A screen for drugs that protect against the cytotoxicity of polyglutamine-expanded androgen receptor. Hum Mol Genet. 2004;13:437–446. doi: 10.1093/hmg/ddh045. [DOI] [PubMed] [Google Scholar]
- 97.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 98.Price MG, Landsverk ML, Barral JM, Epstein HF. Two mammalian UNC-45 isoforms are related to distinct cytoskeletal and muscle-specific functions. J Cell Sci. 2002;115:4013–4023. doi: 10.1242/jcs.00108. [DOI] [PubMed] [Google Scholar]
- 99.Rajasekaran NS, Connell P, Christians ES, Yan L-J, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, Tang W, Winton MJ, Neumann M, Trojanowski JQ, et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. 2010;30:7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc Natl Acad Sci U S A. 2002;99:16412–16418. doi: 10.1073/pnas.182426899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sandri M, Coletto L, Grumati P, Bonaldo P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci. 2013;126:5325–5333. doi: 10.1242/jcs.114041. [DOI] [PubMed] [Google Scholar]
- 103.Sanger JW, Chowrashi P, Shaner NC, Spalthoff S, Wang J, Freeman NL, Sanger JM. Myofibrillogenesis in skeletal muscle cells. Clin Orthop. 2002;(403 Suppl):S153–162. doi: 10.1097/00003086-200210001-00018. [PubMed] [DOI] [PubMed] [Google Scholar]
- 104.Sanger JW, Kang S, Siebrands CC, Freeman N, Du A, Wang J, Stout AL, Sanger JM. How to build a myofibril. J Muscle Res Cell Motil. 2005;26:343–354. doi: 10.1007/s10974-005-9016-7. [DOI] [PubMed] [Google Scholar]
- 105.Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M, McDonald K, Stajich JM, Mahjneh I, Vihola A, et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet. 2012;44:450–455. doi: 10.1038/ng.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sass JB, Martin CC, Krone PH. Restricted expression of the zebrafish hsp90alpha gene in slow and fast muscle fiber lineages. Int J Dev Biol. 1999;43:835–838. [PubMed] [Google Scholar]
- 107.Schultheiss T, Lin ZX, Lu MH, Murray J, Fischman DA, Weber K, Masaki T, Imamura M, Holtzer H. Differential distribution of subsets of myofibrillar proteins in cardiac nonstriated and striated myofibrils. J Cell Biol. 1990;110:1159–1172. doi: 10.1083/jcb.110.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative αB-crystallin mutations. Ann Neurol. 2003;54:804–810. doi: 10.1002/ana.10767. [DOI] [PubMed] [Google Scholar]
- 109.Selcen D, Muntoni F, Burton BK, Pegoraro E, Sewry C, Bite AV, Engel AG. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83–89. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Senf SM, Howard TM, Ahn B, Ferreira LF, Judge AR. Loss of the inducible Hsp70 delays the inflammatory response to skeletal muscle injury and severely impairs muscle regeneration. PLoS ONE. 2013;8:e62687. doi: 10.1371/journal.pone.0062687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Jänne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A, et al. First-line gefitinib in patients with advanced non–small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 112.Siegers K, Waldmann T, Leroux MR, Grein K, Shevchenko A, Schiebel E, Hartl FU. Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin-GimC system. EMBO J. 1999;18:75–84. doi: 10.1093/emboj/18.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Singh BN, Rao KS, Ramakrishna T, Rangaraj N, Rao CM. Association of alphaB-crystallin, a small heat shock protein, with actin: role in modulating actin filament dynamics in vivo. J Mol Biol. 2007;366:756–767. doi: 10.1016/j.jmb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 114.Sittler A, Lurz R, Lueder G, Priller J, Hayer-Hartl MK, Hartl FU, Lehrach H, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 115.Smulders RHPH, Carver JA, Lindner RA, Boekel MAM, van Bloemendal H, de Jong WW. Immobilization of the C-terminal extension of bovine αA-crystallin reduces chaperone-like activity. J Biol Chem. 1996;271:29060–29066. doi: 10.1074/jbc.271.46.29060. [DOI] [PubMed] [Google Scholar]
- 116.Sparrow JC, Schöck F. The initial steps of myofibril assembly: integrins pave the way. Nat Rev Mol Cell Biol. 2009;10:293–298. doi: 10.1038/nrm2634. [DOI] [PubMed] [Google Scholar]
- 117.Srikakulam R, Winkelmann DA. Myosin II folding is mediated by a molecular chaperonin. J Biol Chem. 1999;274:27265–27273. doi: 10.1074/jbc.274.38.27265. [DOI] [PubMed] [Google Scholar]
- 118.Srikakulam R, Winkelmann DA. Chaperone-mediated folding and assembly of myosin in striated muscle. J Cell Sci. 2004;117:641–652. doi: 10.1242/jcs.00899. [DOI] [PubMed] [Google Scholar]
- 119.Street TO, Lavery LA, Verba KA, Lee C-T, Mayer MP, Agard DA. Cross-monomer substrate contacts reposition the Hsp90 N-terminal domain and prime the chaperone activity. J Mol Biol. 2012;415:3–15. doi: 10.1016/j.jmb.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 121.Tan X, Rotllant J, Li H, DeDeyne P, Du SJ. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc Natl Acad Sci U S A. 2006;103:2713–2718. doi: 10.1073/pnas.0509503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.De Thonel A, Le Mouël A, Mezger V. Transcriptional regulation of small HSP—HSF1 and beyond. Int J Biochem Cell Biol. 2012;44:1593–1612. doi: 10.1016/j.biocel.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 123.Toi H, Fujimura-Kamada K, Irie K, Takai Y, Todo S, Tanaka K. She4p/Dim1p interacts with the motor domain of unconventional myosins in the budding yeast, Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:2237–2249. doi: 10.1091/mbc.E02-09-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ulbricht A, Eppler FJ, Tapia VE, van der Ven PFM, Hampe N, Hersch N, Vakeel P, Stadel D, Haas A, Saftig P, et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol. 2013;23:430–435. doi: 10.1016/j.cub.2013.01.064. [DOI] [PubMed] [Google Scholar]
- 125.Van der Ven PF, Ehler E, Perriard JC, Fürst DO. Thick filament assembly occurs after the formation of a cytoskeletal scaffold. J Muscle Res Cell Motil. 1999;20:569–579. doi: 10.1023/a:1005569225773. [DOI] [PubMed] [Google Scholar]
- 126.Venolia L, Waterston RH. The unc-45 gene of Caenorhabditis elegans is an essential muscle-affecting gene with maternal expression. Genetics. 1990;126:345–353. doi: 10.1093/genetics/126.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vicart P, Caron A, Guicheney P, Li Z, Prévost M-C, Faure A, Chateau D, Chapon F, Tomé F, Dupret J-M, et al. A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 128.Wagner AJ, Chugh R, Rosen LS, Morgan JA, George S, Gordon M, Dunbar J, Normant E, Grayzel D, Demetri GD. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin Cancer Res. 2013;19:6020–6029. doi: 10.1158/1078-0432.CCR-13-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 130.Westerheide SD, Bosman JD, Mbadugha BNA, Kawahara TLA, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 131.Wistow G. The human crystallin gene families. Hum Genomics. 2012;6:26. doi: 10.1186/1479-7364-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wohlgemuth SL, Crawford BD, Pilgrim DB. The myosin co-chaperone UNC-45 is required for skeletal and cardiac muscle function in zebrafish. Dev Biol. 2007;303:483–492. doi: 10.1016/j.ydbio.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 133.Xie HB, Cammarato A, Rajasekaran NS, Zhang H, Suggs JA, Lin H-C, Bernstein SI, Benjamin IJ, Golic KG. The NADPH metabolic network regulates human αB-crystallin cardiomyopathy and reductive stress in Drosophila melanogaster. PLoS Genet. 2013;9:e1003544. doi: 10.1371/journal.pgen.1003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99:12847–12852. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Young P, Ferguson C, Banuelos S, Gautel M. Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. EMBO J. 1998;17:1614–1624. doi: 10.1093/emboj/17.6.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yu Q, Bernstein SI. UCS proteins: managing the myosin motor. Curr Biol. 2003;13:R525–527. doi: 10.1016/s0960-9822(03)00447-0. [DOI] [PubMed] [Google Scholar]
- 137.Yun B-G, Matts RL. Differential effects of Hsp90 inhibition on protein kinases regulating signal transduction pathways required for myoblast differentiation. Exp Cell Res. 2005;307:212–223. doi: 10.1016/j.yexcr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 138.Zhang B, Au Q, Yoon IS, Tremblay M-H, Yip G, Zhou Y, Barber JR, Ng SC. Identification of small-molecule HSF1 amplifiers by high content screening in protection of cells from stress induced injury. Biochem Biophys Res Commun. 2009;390:925–930. doi: 10.1016/j.bbrc.2009.10.079. [DOI] [PubMed] [Google Scholar]
- 139.Zhang Y-Q, Sarge KD. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med. 2007;85:1421–1428. doi: 10.1007/s00109-007-0251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhu H, Woolfenden S, Bronson RT, Jaffer ZM, Barluenga S, Winssinger N, Rubenstein AE, Chen R, Charest A. The novel Hsp90 inhibitor NXD30001 induces tumor regression in a genetically engineered mouse model of glioblastoma multiforme. Mol Cancer Ther. 2010;9:2618–2626. doi: 10.1158/1535-7163.MCT-10-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]