

Abstract
Human mitochondria contain multiple copies of a circular genome made up of double-stranded DNA (mtDNA) that encodes proteins involved in cellular respiration. Transcript abundance of mtDNA-encoded genes varies between human individuals, yet the level of variation in the general population has not been systematically assessed. In the present study, we revisited large-scale RNA sequencing data generated from lymphoblastoid cell lines of HapMap samples of European and African ancestry to estimate transcript abundance and quantify expression variation for mtDNA-encoded genes. In both populations, we detected up to over 100-fold difference in mtDNA gene expression between individuals. The marked variation was not due to differences in mtDNA copy number between individuals, but was shaped by the transcription of hundreds of nuclear genes. Many of these nuclear genes were co-expressed with one another, resulting in a module-enriched co-expression network. Significant correlations in expression between genes of the mtDNA and nuclear genomes were used to identify factors involved with the regulation of mitochondrial functions. In conclusion, we determined the baseline amount of variability in mtDNA gene expression in general human populations and cataloged a complete set of nuclear genes whose expression levels are correlated with those of mtDNA-encoded genes. Our findings will enable the integration of information from both mtDNA and nuclear genetic systems, and facilitate the discovery of novel regulatory pathways involving mitochondrial functions.
Keywords: mitochondrial DNA-encoded genes, nuclear genes, gene expression variability, human populations, mitochondrial DNA copy number
The mitochondrion is the primary energy-generating organelle, harboring critical components of the electron transport chain for ATP synthesis through oxidative phosphorylation. In almost all eukaryotic cells, mitochondria have central roles in biosynthesis, homeostasis, and programmed cell death.1, 2, 3 Dysfunctional mitochondria have pleiotropic negative effects, giving rise to a large spectrum of defects that primarily affect tissues with high energy requirements such as the brain, heart, liver, skeletal muscles, kidney, and the endocrine and respiratory systems.4, 5, 6, 7 Despite the fundamental role of mitochondria in eukaryotic cell functions and human health, the transcriptional pattern of mtDNA has been overlooked by the majority of genomic investigations. Most transcriptome profiling studies do not take into account the transcripts of the mtDNA-encoded genes. Current procedures used for the analysis of RNA sequencing (RNA-seq) data typically filter out and discard short reads mapped onto mtDNA. As a result, none of the large-scale RNA-seq studies in humans have been focused on characterizing mtDNA transcription. Comprehensive transcriptomic data sets from large-scale RNA-seq studies have revealed significant expression variation for nuclear genes,8, 9 but to date virtually no progress has been made toward investigating expression variation in mtDNA genes. The lack of a precedent for evaluating the expression variability of mtDNA genes will ultimately hinder the understanding of mitochondrial biology as well as the advance of mitochondrial medicine.
To simultaneously estimate the expression of mtDNA and nuclear genes, in the present study we revisited published RNA-seq data8, 9 that included a large number of polyadenylated10, 11, 12, 13 mitochondrial transcripts. We quantified between-individual variation in the expression of mtDNA genes in HapMap samples of European (CEU) and African (YRI) ancestry to evaluate this variation at the population level. We detected co-varying expression between mtDNA and nuclear genes, and explored various potential mechanisms that may underlie the association.
Materials and methods
RNA sequencing data
Short sequence data produced for two RNA-seq studies8, 9 were obtained from GEO using accessions GSE19480 and GSE25030. The SRA (sequence read archives) files were downloaded and subsequently converted into FASTQ files using the NCBI SRA toolkit program, fastq-dump (v 2.1.16). Mitochondria and mitoplast RNA-seq data produced by Mercer et al14 were obtained from GEO using accession GSE30772.
CEU and YRI reference genomes
The human reference genome (hg19) was re-engineered for CEU and YRI by replacing the mtDNA sequence to produce two population-specific reference genomes. For CEU, the Revised Cambridge Reference Sequence (NC_012920) was obtained from the MITOMAP website (http://www.mitomap.org/MITOMAP). For YRI, the sequence of NC_001807 in GenBank's RefSeq database was used. CEU and YRI mtDNA sequences differ by 41 single-nucleotide variants and four indels. The GTF file of Gencode v11 with updated mtDNA annotation (for CEU and YRI, respectively) was used to guide the short-read mapping.
Estimation of transcript abundance
To estimate FPKM (fragments per kilobase of exon per million fragments), RNA-seq short reads were mapped to the corresponding population-specific reference genome using Tophat2 v2.0.1 and processed using Cufflinks v2.0.2. TopHat option – read-mismatches was set to 2 and 3 (ie, allowing up to 2 and 3 mismatches in final read alignments), for CEU and YRI, respectively, because the mtDNA nucleotide diversity in YRI is higher than that in CEU.15 By setting the TopHat option -g to 1, we deliberately allowed reads to be mapped on a single specific position on the genome. For each population, the expression values of log10(FPKM+1) for all genes were quantile-normalized across individuals.16 The gene expression level per mtDNA copy was computed according to 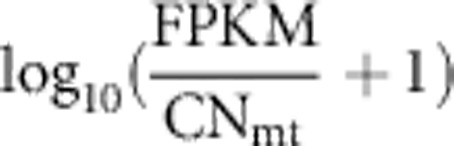 with CNmt being the mtDNA copy number. Thirteen representative housekeeping (HK) genes were randomly selected from the list of HK genes in a previous study.17
with CNmt being the mtDNA copy number. Thirteen representative housekeeping (HK) genes were randomly selected from the list of HK genes in a previous study.17
Weighted gene co-expression network analysis
The weighted gene co-expression network analysis (WGCNA)18 was used to identify nuclear gene modules in the co-expression network including mtDNA genes. For each module, we defined eigengene significance by measuring the correlation between the modules and the trait under consideration (ie, the average expression level of mtDNA genes). To ensure the results are robust to the WGCNA parameter settings,19 we allowed the key parameter, the thresholding power for network construction (or the power), to vary between 5, 6 (the optimal value determined by WGCNA), and 7, where the other parameters were kept fixed (ie, the smallest value of the scale independence=0.9, the minimum module size=30, and the maximum joining height=10). Thus, instead of producing WGCNA modules using one single value for the power, we ran WGCNA three times with three different power values and produced three sets of WGCNA modules. We then iterated all pairs of genes, and identified gene pairs in which two genes appeared in the same module in all three sets of WGCNA modules. The identified pairs of genes were represented with 1 in an adjacency matrix, while the rest pairs of genes were represented with 0. We used the MCL algorithm implemented in SBEToolbox20 to identify clusters of genes within the adjacency matrix. In this way, we identified genes in the same clusters that were highly connected and had been consistently grouped in all three sets of WGCNA modules.
Gene ontology analysis
For certain sets of genes, we computed enrichment scores for the gene ontology (GO) biological process and molecular function terms using DAVID.21 The program compares the annotation composition in a list of genes to that of background genes. The full set of genes expressed in LCLs (with average FPKM>1.0) was used as the background gene set.
Estimation of mtDNA copy number
The mtDNA copy number was estimated using the mtDNA/nDNA ratio.22, 23, 24, 25 For a given chromosomal region, short reads mapped to the region were retrieved from the BAM file at the FTP site of the 1000 Genomes Project26 using samtools.27 This was done for the whole mtDNA region and the autosomal regions; the numbers of short reads in the two regions were used to compute mtDNA/nDNA. If the read coverage along the genome is even, then the number of reads in different genomic regions of the same length should not vary substantially. That is to say, the autosomal regions could be selected arbitrarily as long as the total length of these regions is long enough (eg, comparable with the length of mtDNA). In this study, we chose to use the genic regions of the 13 HK genes. To confirm that mtDNA copy number estimation was indeed not affected by the selection of autosomal regions, we reestimated the mtDNA copy numbers for all samples 1000 times using randomly selected autosomal regions of the same total length. Each time, the result was compared with the result derived from the genic regions of the 13 HK genes. The correlations between estimates were consistently high (average Spearman correlation coefficient [SCC]=0.981; Supplementary Figure 1).
Analysis of eQTL
HapMap SNPs28 with minor allele frequency (MAF) of>5% were selected from CEU and YRI populations (∼2.2 million per population). We tested the eQTL associations between each SNP and gene expression with a linear regression model using Matrix eQTL.29 To establish the null distribution of P-values, randomly shuffled expression data were used to perform the regression analysis.
Distribution of source code and data
The associated source code and data are available at http://www.github.com/jamesjcai/mtRNA-seq.
Results
Expression levels of mtDNA genes
We used RNA-seq data sets generated from LCLs of 60 CEU8 and 69 YRI9 individuals to estimate expression levels for both mtDNA and nuclear genes. RNA-seq short reads were mapped onto population-specific reference genomes. Multiple-hit reads were discarded to avoid the influence of mapping artifacts caused by the improper assignment of mtDNA reads to nuclear genome sequences of mitochondrial origin (NUMTs), which include >500 sequences covering ∼627 kb or 0.021% of the human genome.30, 31 Our results showed that, overall, mtDNA genes were expressed significantly more abundantly than other genes (Kolmogorov–Smirnov (K–S) test: P=1.3e-12; Supplementary Figure 2). This is consistent with our expectation and indicates that the RNA-seq data sets are replete with mitochondrial transcripts. Additional analyses showed that the accuracy of this estimation was not affected by: (1) the number of mapped reads, (2) the ages of individuals from whom blood samples were collected, (3) the batch effect of cell line processing in the RNA-seq experiments, or (4) EBV copy number and/or cell doubling time for the LCLs (see Supplementary Note 1: analyses showing that the accuracy of mtDNA gene expression estimation is not affected by confounding factors).
Expression variability of mtDNA genes
There was substantial variation in mtDNA gene expression among CEU (Figure 1) and YRI (Supplementary Figure 3) individuals. The variation was more pronounced in mtDNA genes (Figure 1a) than HK genes (Figure 1b). To quantify this, we computed the coefficient of variation (CV) of expression for all genes with an average FPKM>2.0. Indeed, mtDNA genes tended to have a significantly larger CV than nuclear genes (K–S test: P=2.7e-6; see Supplementary Figure 4). We hypothesized that the marked variation in mtDNA gene expression was due to differences in mtDNA copy number between individuals. To test this, we estimated the mtDNA copy number for each sample using the ratio between the number of short reads mapped in mtDNA versus nuclear DNA (Materials and Methods). Our estimates showed a significant, positive correlation with the estimates obtained by Maranville et al32 using a PCR-based method with 46 YRI samples (Pearson correlation test: r=0.445, P=0.002). As before, we found considerable differences in mtDNA copy number among CEU and YRI individuals (Supplementary Figure 5), but this variation did not correlate with that of mtDNA gene expression (Spearman correlation test: ρ=0.067 and 0.112, P=0.61 and 0.42, for CEU and YRI, respectively). In fact, gene expression level per copy was found to be highly variable for mtDNA genes, ranging from 0.09 to 0.20 in CEU and 0.06 to 0.11 in YRI. These results suggest that differences in mtDNA copy number do not account for mtDNA gene expression variability.
Figure 1.

Between-individual expression variation and among-gene expression correlation for mtDNA genes and nuclear genes. The gene expression levels of (a) 13 mtDNA genes and (b) 13 housekeeping nuclear genes in 60 CEU samples are shown. Vertical axes represent the normalized expression level of genes; horizontal polylines across the display represent all individuals. The box plots depict the median, lower and upper quartiles of expression levels of each gene among samples. Pairwise correlations of expression between (c) mtDNA genes and (d) nuclear genes. SCCs are shown with font in the size proportional to their value. Indicators of significance: ***P<0.001, **P<0.01, and *P<0.05.
Coordinated expression of mtDNA genes
It is known that functionally related genes, such as the 13 mtDNA protein-coding genes, are likely to be expressed coordinately.33 Indeed, we observed remarkably strong, positive correlations between expression levels of mtDNA genes: SCCs between possible pairs ranged from 0.55 to 0.97 with an average of 0.86 (Figure 1c and Supplementary Figure 6). In contrast, SCCs between the HK genes were much smaller (Supplementary Figure 7a, b): only 7 out of 78 HK gene pairs showed a SCC>0.50 (Figure 1d). Additionally, we examined the SCCs between genes whose products form a single protein complex – the SNF2h/cohesion complex.34 For this particular example, the SCCs between genes of the same protein complex (Supplementary Figure 7c, d) were stronger than those between HK genes, but still much weaker than those between mtDNA genes.
Co-expression of mtDNA and nuclear genes
To examine co-expression of mtDNA and nuclear genes, we performed pairwise correlation tests and identified nuclear genes whose expressions were significantly correlated with one or more mtDNA genes (SCC with Bonferroni correction, adjusted P<0.05; Supplementary Table 1). These included 496 positive and 434 negative correlations between nuclear and mtDNA gene expression in CEU, plus 203 positive and 184 negative correlations in YRI. A total of 63 of these genes (29 positive and 34 negative) were found in common between CEU and YRI (Figure 2), including 15 ribosomal protein genes, ACIN1 (apoptotic chromatin condensation inducer protein activated by caspase-3, Figure 3a), and ZSWIM1 (zinc-finger SWIM domain-containing protein indirectly interacting with MT-ND2 through MAPK14,35 Figure 3b). GO analyses indicated that positively-correlated nuclear genes were often involved in the regulation of transcription; those negatively-correlated were more likely to be involved in translation (for details, see Supplementary Table 2).
Figure 2.

Co-varying expression network between mtDNA and nuclear genes. The consensus network shared between CEU and YRI is plotted. Connections are placed between genes that are correlated at |Spearman's ρ|>0.60 in CEU and YRI. The width of connection lines is proportional to the average absolute value of SCCs for the two populations. Positive and negative correlation relationships are presented as red and blue lines, respectively. Scatter plots between expression of MT-ATP6 and RSPO1 for CEU and YRI are shown to depict the degree of correlations.
Figure 3.

Links between mtDNA and nuclear genes whose expressions are co-varying. (a) ACIN1 mediates the apoptosis pathway, in which mitochondria are involved. (b) ZSWIM1 indirectly interacts with MT-ND2 through interaction with MAPK14. (c) Transcripts of AQR, CNOT4, MED28, PROSC and UQCR11 are detectable in mitochondrial preparation that contains both outer and inner membrane, but not in mitoplast preparation that contains only inner membrane.
We used several SCC cutoffs ranging from 0.5 to 0.9 to produce co-expression networks of different sizes. The size of the resulting co-expression networks decreased with the increase of the SCC cutoff value (Supplementary Table 3). The overlap between co-expression networks for CEU and YRI decreased rapidly with the increment of SCC cutoff. This low level of overlap might be due to that the overall expression distributions were different between CEU and YRI (Supplementary Figure 8) that may be attributed to the technical difference in RNA-seq procedures (ie, paired-end RNA-seq for CEU8 and single-end RNA-seq for YRI9) or differences in the number of passage from immortalization of LCLs between CEU and YRI samples.
Next, to examine the large-scale organization of co-expression networks, we used the WGCNA method18 to identify nuclear gene modules significantly correlated with mtDNA genes. WGCNA starts from the level of thousands of genes, with modules represented by their centroids, to assess relationships between modules and the trait under consideration.18 Using a conservative procedure that reduces the influence of WGCNA parameter choices (Materials and Methods), we identified a total of 98 gene clusters, each containing at least 30 genes. These clusters are available for download (Materials and Methods). All pairs of genes in one cluster had been consistently grouped into the same module, regardless of different values of the power used in WGCNA analysis.
A major concern when evaluating relationships between genes based on their expression is that transcriptional co-regulation among many genes can give rise to indirect interaction effects in expression data,36 and regular correlation networks cannot distinguish direct from indirect relationships.37, 38 To control for the indirect effects, we employed the approach developed by Schafer and Strimmer36 based on the graphical Gaussian model (GGM)39 to reconstruct a GGM network. In this network, each link indicated a partial correlation between two genes that remained after removing the effects of other genes. Our results showed that many nuclear genes were partially correlated with mtDNA genes (Supplementary Table 4), and that partial correlation of mtDNA genes with RSPO1, PRRC2C, EIF4E2, and TMEM101 appeared in Figure 2 showed significance.
Effects of subcellular co-localization
Transcript localization by mRNA trafficking is an important cellular process that controls subcellular distribution of mRNAs and the subsequent distribution of proteins.40 Through this process, transcripts of a number of nuclear genes preferentially localize to the vicinity of mitochondria.41, 42 We hypothesized that these nuclear genes are likely to be functionally related to mitochondria and that their expression is likely to be correlated with mtDNA genes. To test this, we obtained the RNA-seq data generated by Mercer et al14 using a subtractive approach. In that study, mRNAs were extracted and sequenced for a mitochondrial preparation as well as for a mitoplast preparation, obtained from the mitochondrial preparations stripped of their outer membrane. In the mitochondrial preparation, both outer and inner mitochondrial membranes were intact, whereas in the mitoplast preparation, only the inner membrane remained. We mapped short reads to estimate gene expression levels for the two preparations, and identified a total of 6472 genes that were expressed in the mitochondrial but not mitoplast preparations (FPKM cutoff=0.05). Only five of these genes were among the 29 nuclear genes positively correlated with mtDNA genes in both populations (Figure 3c). This ratio (17%) was significantly lower than expected (P<0.05, one-tailed χ2 test).
Nuclear SNPs associated with mtDNA gene expression?
Associations between gene expression and genotype (eQTLs) have been established for many nuclear genes.8, 9, 43, 44, 45, 46 We hypothesized that the expression variation of mtDNA genes may be associated with genotypes defined by autosomal SNPs, although the regulatory mechanisms through which these SNPs might affect mtDNA gene expression is not clear. Because all SNPs in mtDNA were of low frequency (MAF<10%), no cis-acting mitochondrial eQTLs (ie, the expression-controlling SNPs located in mtDNA) could be detected. We resorted to identify transacting mitochondrial eQTLs (ie, the mtDNA expression-controlling SNPs located in autosomes). However, using the established method,46 we detected no significant eQTL relationships between autosomal SNPs and mtDNA gene expression. Finally, we examined the existence of indirect links. For example, an autosomal SNP is associated with the expression of a nuclear gene (ie, the ‘regular' eQTL relationship), whereas the expression of this nuclear gene is correlated with the expression of mtDNA genes. In this way, the autosomal SNP is indirectly linked with mtDNA gene expression. We identified this kind of autosomal SNPs and tabulated these SNPs and corresponding nuclear genes whose expression was associated with mtDNA gene expression (Supplementary Table 5).
Discussion
We systematically examined the between-individual variation in the expression level of mtDNA genes, a subject that has been neglected by the overwhelming majority of previous studies. Using existing data sets, we quantified the population-level expression variability of mtDNA genes for European and African populations. Up to >100-fold between-individual difference in mtDNA gene expression was detected in both populations.
We further investigated whether the marked variation is due to differences in mtDNA copy numbers between-individual samples. Each mitochondrion contains between two and ten copies of mtDNA, cells have numerous mitochondria, and a cell may harbor several thousand mtDNA copies.47 It is generally believed that mtDNA gene expression is proportional to the number of mtDNA copies,48, 49 and the amount of mtDNA in a cell could provide a major regulatory point in mitochondrial activity.50, 51 Our results based on the population-level expression variability, however, do not appear to favor the idea.
We evaluated relationships between mtDNA and nuclear genes by constructing the co-expression network and using the WGCNA algorithm to detect the correlated genes and modules. Functional analyses of correlated genes and modules confirmed biological processes previously known to be associated with mitochondrial activities, such as apoptosis.52 These analyses also identified many genes involved in those biological processes previously unknown to be associated with mitochondrial activities.53 Furthermore, we evaluated whether functionally relevant pathways could be investigated by identifying associations between genetic variants (mainly, SNPs) and expression of mtDNA genes. However, we did not find evidence for the existence of any mitochondrial eQTL. We conclude that although genomic variants may make important contributions to the expression of mtDNA genes, the evidence to date is too weak to support such a conclusion. On the other hand, we know that evidence for mtDNA polymorphisms associated with susceptibility to complex disorders is also weak.6 Thus, establishing convincing relationships between phenotypes and mtDNA transcripts/variants remains highly challenging.
Several caveats and technical limitations are associated with our analysis. (1) Many factors that might alter the number of mtDNA copies could not be controlled. These factors include the stage of the cell cycle, the energetic requirements of the cells, the environmental effects on the redox balance of the cells, the stage of differentiation, and/or cell signaling mechanisms.54, 55 An accurate determination of the number of mtDNA copies per cell is technically difficult to achieve because both the number of mitochondria per cell and the number of mtDNA copies per mitochondrion vary.56, 57 Our analysis could only focus on the variability of mtDNA gene expression at the level of per sample, ignoring the details of expression variability at the level of per mtDNA copy. (2) Throughout the paper, the transcription of mtDNA genes was treated as occurring in a manner completely analogous to that of nuclear transcription. For mtDNA, its long polycistronic precursor transcripts are processed and released individually,58 and stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase.11 The differences in these detailed transcriptional and post-transcriptional processes between mtDNA genes7, 59 and nuclear genes were ignored in this study. (3) The two RNA-seq data sets8, 9 used in this study were derived from poly(A)-enriched RNA pools, and were not generated using strand-specific RNA-seq. These might influence the estimation of the levels of mtDNA gene expression because of the different polyadenylation statuses of mtDNA genes13, 60, 61 and the possible cross-mapping between L- and H-strand-derived mitochondrial transcripts.60 Nevertheless, because these technical limitations systematically influenced all samples in the same manner on the same order of magnitude, our main results in connection with the between-individual expression variability should not be affected.
In summary, we have taken the first step toward characterizing the population-level expression variability of mtDNA genes in humans, which is done through exploiting widely accessible yet completely untapped RNA-seq reads originating from the transcribed mitochondrial genome. In doing this, we established an analytical framework for future analyses of the interplay between the two human genomes. This study demonstrates the utility of publically available data for answering interesting questions in studies of natural human variation. Using this data, we have confirmed that there is a substantial amount of variation in mtDNA gene expression across individuals, rejecting the hypothesis that the transcript abundance of mtDNA genes is determined by the number of mtDNA copies. Next, the expression of mtDNA genes may be either positively or negatively associated with the expression of many known and unknown network modules. These modules contain many genes, many of whose functions were hitherto not known to be linked with mitochondrial function, underscoring the need to further study the underlying mechanisms of these associations to increase our understanding of the genetic basis of the expression regulation of mtDNA genes.
Acknowledgments
We thank all anonymous reviewers and section editor Peter Robinson for their valuable and constructive comments. We thank Hui Jiang, Yi Xing, Dongxiao Zhu, Stephen Montgomery, Maranville, and Di Rienzo for helpful discussions and/or sharing research data. EY is supported by CVM Postdoctoral Trainee Research Grant (02-144002-03504) and GW is supported by CVM Graduate Trainee Research Grant (02-291039-00002) at Texas A&M University. We acknowledge the Texas A&M Supercomputing Facility and the Whole Systems Genomics Initiative (WSGI) for providing computing resources and systems administration support.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Al Rawi S, Louvet-Vallee S, Djeddi A, et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–1144. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- Ni Chonghaile T, Sarosiek KA, Vo TT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mechanisms of disease: mitochondrial respiratory-chain diseases. New Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt TE, Shadel GS. A compendium of human mitochondrial gene expression machinery with links to disease. Environ Mol Mutagen. 2010;51:360–379. doi: 10.1002/em.20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Sammeth M, Gutierrez-Arcelus M, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JH, Tong L. Mitochondrial poly(A) polymerase and polyadenylation. Biochim Biophys Acta. 2012;1819:992–997. doi: 10.1016/j.bbagrm.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaike T, Suzuki T, Katoh T, Ueda T. Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J Biol Chem. 2005;280:19721–19727. doi: 10.1074/jbc.M500804200. [DOI] [PubMed] [Google Scholar]
- Gagliardi D, Stepien PP, Temperley RJ, Lightowlers RN, Chrzanowska-Lightowlers ZM. Messenger RNA stability in mitochondria: different means to an end. Trends Genet. 2004;20:260–267. doi: 10.1016/j.tig.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Slomovic S, Laufer D, Geiger D, Schuster G. Polyadenylation and degradation of human mitochondrial RNA: the prokaryotic past leaves its mark. Mol Cell Biol. 2005;25:6427–6435. doi: 10.1128/MCB.25.15.6427-6435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Neph S, Dinger ME, et al. The human mitochondrial transcriptome. Cell. 2011;146:645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MX, Sivakumar IK, Maragh S, et al. Next-generation sequencing of human mitochondrial reference genomes uncovers high heteroplasmy frequency. PLoS Comput Biol. 2012;8:e1002737. doi: 10.1371/journal.pcbi.1002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY. Human housekeeping genes are compact. Trends Genet. 2003;19:362–365. doi: 10.1016/S0168-9525(03)00140-9. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- Konganti K, Wang G, Yang E, Cai JJ. SBEToolbox: a Matlab toolbox for biological network analysis. Evol Bioinform Online. 2013;9:179–182. doi: 10.4137/EBO.S12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barazzoni R, Short KR, Nair KS. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J Biol Chem. 2000;275:3343–3347. doi: 10.1074/jbc.275.5.3343. [DOI] [PubMed] [Google Scholar]
- Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW, Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31:e61. doi: 10.1093/nar/gng060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evdokimovsky EV, Ushakova TE, Kudriavtcev AA, Gaziev AI. Alteration of mtDNA copy number, mitochondrial gene expression and extracellular DNA content in mice after irradiation at lethal dose. Radiat Environ Biophys. 2011;50:181–188. doi: 10.1007/s00411-010-0329-6. [DOI] [PubMed] [Google Scholar]
- Malik A, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction. Mitochondrion. 2012;13:481–492. doi: 10.1016/j.mito.2012.10.011. [DOI] [PubMed] [Google Scholar]
- The-1000-Genomes-Project-Consortium. Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The-International-HapMap-Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010;6:e1000834. doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone D, Calabrese FM, Lang M, Gasparre G, Attimonelli M. The reference human nuclear mitochondrial sequences compilation validated and implemented on the UCSC genome browser. BMC Genomics. 2011;12:517. doi: 10.1186/1471-2164-12-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranville JC, Luca F, Richards AL, et al. Interactions between glucocorticoid treatment and cis-regulatory polymorphisms contribute to cellular response phenotypes. PLoS Genet. 2011;7:e1002162. doi: 10.1371/journal.pgen.1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toung JM, Morley M, Li M, Cheung VG. RNA-sequence analysis of human B-cells. Genome Res. 2011;21:991–998. doi: 10.1101/gr.116335.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Schmiesing JA, et al. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature. 2002;418:994–998. doi: 10.1038/nature01024. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Chiang CY, Srivastava J, et al. A human MAP kinase interactome. Nat Methods. 2010;7:801–805. doi: 10.1038/nmeth.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer J, Strimmer K. An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics. 2005;21:754–764. doi: 10.1093/bioinformatics/bti062. [DOI] [PubMed] [Google Scholar]
- Lopez-Kleine L, Leal L, Lopez C. Biostatistical approaches for the reconstruction of gene co-expression networks based on transcriptomic data. Brief Funct Genomics. 2013;12:457–467. doi: 10.1093/bfgp/elt003. [DOI] [PubMed] [Google Scholar]
- Markowetz F, Spang R. Inferring cellular networks—a review. BMC Bioinformatics. 2007;8 (Suppl 6:S5. doi: 10.1186/1471-2105-8-S6-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werhli AV, Grzegorczyk M, Husmeier D. Comparative evaluation of reverse engineering gene regulatory networks with relevance networks, graphical gaussian models and bayesian networks. Bioinformatics. 2006;22:2523–2531. doi: 10.1093/bioinformatics/btl391. [DOI] [PubMed] [Google Scholar]
- Lecuyer E, Yoshida H, Parthasarathy N, et al. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell. 2007;131:174–187. doi: 10.1016/j.cell.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Sylvestre J, Vialette S, Corral Debrinski M, Jacq C. Long mRNAs coding for yeast mitochondrial proteins of prokaryotic origin preferentially localize to the vicinity of mitochondria. Genome Biol. 2003;4:R44. doi: 10.1186/gb-2003-4-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Uchiumi T, Saito T, et al. Localization of mRNAs encoding human mitochondrial oxidative phosphorylation proteins. Mitochondrion. 2012;12:391–398. doi: 10.1016/j.mito.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Montgomery SB, Dermitzakis ET. From expression QTLs to personalized transcriptomics. Nat Rev Genet. 2011;12:277–282. doi: 10.1038/nrg2969. [DOI] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy E, Yelensky R, Bonakdar S, et al. Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines. PLoS Genet. 2008;4:e1000287. doi: 10.1371/journal.pgen.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Clark AG, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sparman M, Sritanaudomchai H, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–372. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya J, Perez-Martos A, Garstka HL, Wiesner RJ. Regulation of mitochondrial transcription by mitochondrial transcription factor A. Mol Cell Biochem. 1997;174:227–230. [PubMed] [Google Scholar]
- Seidel-Rogol BL, Shadel GS. Modulation of mitochondrial transcription in response to mtDNA depletion and repletion in HeLa cells. Nucleic Acids Res. 2002;30:1929–1934. doi: 10.1093/nar/30.9.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- Williams RS. Mitochondrial gene expression in mammalian striated muscle. Evidence that variation in gene dosage is the major regulatory event. J Biol Chem. 1986;261:12390–12394. [PubMed] [Google Scholar]
- Eisenberg T, Buttner S, Kroemer G, Madeo F. The mitochondrial pathway in yeast apoptosis. Apoptosis. 2007;12:1011–1023. doi: 10.1007/s10495-007-0758-0. [DOI] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- Michel S, Wanet A, De Pauw A, Rommelaere G, Arnould T, Renard P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J Cell Physiol. 2012;227:2297–2310. doi: 10.1002/jcp.23021. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Kai Y, Maldonado E, Currin RT, Lemasters JJ. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy. 2009;5:1099–1106. doi: 10.4161/auto.5.8.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Kuroiwa T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp Cell Res. 1991;196:137–140. doi: 10.1016/0014-4827(91)90467-9. [DOI] [PubMed] [Google Scholar]
- Robin ED, Wong R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J Cell Physiol. 1988;136:507–513. doi: 10.1002/jcp.1041360316. [DOI] [PubMed] [Google Scholar]
- Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- Bestwick ML, Shadel GS. Accessorizing the human mitochondrial transcription machinery. Trends Biochem Sci. 2013;38:283–291. doi: 10.1016/j.tibs.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Neph S, Dinger ME, et al. The human mitochondrial transcriptome. Cell. 2011;146:645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperley RJ, Seneca SH, Tonska K, et al. Investigation of a pathogenic mtDNA microdeletion reveals a translation-dependent deadenylation decay pathway in human mitochondria. Hum Mol Genet. 2003;12:2341–2348. doi: 10.1093/hmg/ddg238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.