

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Phosphomannose isomerase deficiency, mannosephosphate isomerase deficiency, MPI-CDG, CDG-Ib, Saguenay-Lac Saint-Jean syndrome, protein-losing enteropathy-hepatic fibrosis syndrome.
1.2 OMIM# of the disease
602579.
1.3 Name of the analysed genes or DNA/chromosome segments
MPI.
1.4 OMIM# of the gene(s)
154550.
1.5 Mutational spectrum
Eighteen mutations have been reported, including 15 missense mutations, 2 frame-shift-causing mutations and 1 splicing defect;1 (http://www.lovd.nl/MPI). One frame-shift mutation was caused by an insertion resulting in an unstable transcript that was hardly detectable at the mRNA level. This emphasizes the importance of mutation analysis at the genomic DNA level.2 The standard reference sequence indicating reported variants (ENSG00000178802) and a reference for exon numbering (ENST00000352410) can be found at http://www.ensemble.org.
1.6 Analytical methods
Sanger sequencing of the eight exons and flanking intronic sequences of the MPI gene (NCBI reference sequence: NM_002435.1).
1.7 Analytical validation
Sanger sequencing identifies mutations in >99% of patients with an enzymatically confirmed MPI deficiency. Deep intronic mutations, large deletions and duplications would not be detected using this approach.
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence. If known to be variable between ethnic groups, please report)
Some 25 patients have been reported (‘Asian', Austrian, Bulgarian, Canadian, Danish, Dutch, French, German, Italian, Lebanese, Polish, Spanish and Turkish). The frequency and the prevalence of the disease are not known.
1.9 Diagnostic setting

Comment: MPI-CDG is an autosomal recessive disorder with a hepatic-intestinal presentation (review in de Lonlay and Seta3). Digestive symptoms are vomiting, intractable diarrhoea and malnutrition, while hepatic disease is characterized by hepatomegaly and liver fibrosis. In addition, there is frequent thrombosis and hyperinsulinism. Untreated patients manifesting the disease before one year of age died in the first years of life.4, 5 Intrafamilial heterogeneity was shown in two siblings: one died at 5 years with hepatic fibrosis while the other survived into adulthood without treatment.6 The biochemical screening test ‘par excellence' is serum transferrin isoelectrofocusing. The next diagnostic step is enzymatic analysis of phosphomannose isomerase activity in leucocytes or fibroblasts. The diagnosis has to be confirmed by mutation analysis of MPI. This will permit heterozygote detection in the family and prenatal diagnosis.
2. TEST CHARACTERISTICS
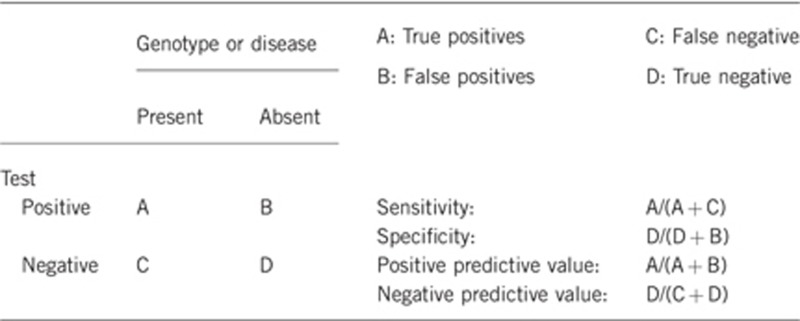
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Close to 100% when using the serum transferrin isoelectrofocusing test.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Close to 100% when using the serum transferrin isoelectrofocusing test. This test can be positive in secondary glycosylation disturbances such as galactosemia and hereditary fructose intolerance.7, 8
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Close to 100%.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Close to 100%.
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
100%, based on positive transferrin isoelectrofocusing screening and MPI mutation analysis.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
100%.
Index case in that family had not been tested:
100%.
3. CLINICAL UTILITY
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
Often the clinical picture is suggestive for this disorder. The blood sampling for the serum transferrin isoelectrofocusing screening test and that for the measurement of PMI enzymatic activity in leucocytes or fibroblasts and MPI analysis are a minor burden to the patient.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
The enzymatic test is only available in a limited number of laboratories (see orpha.net for the list).
3.1.4 Will disease management be influenced by the result of a genetic test?
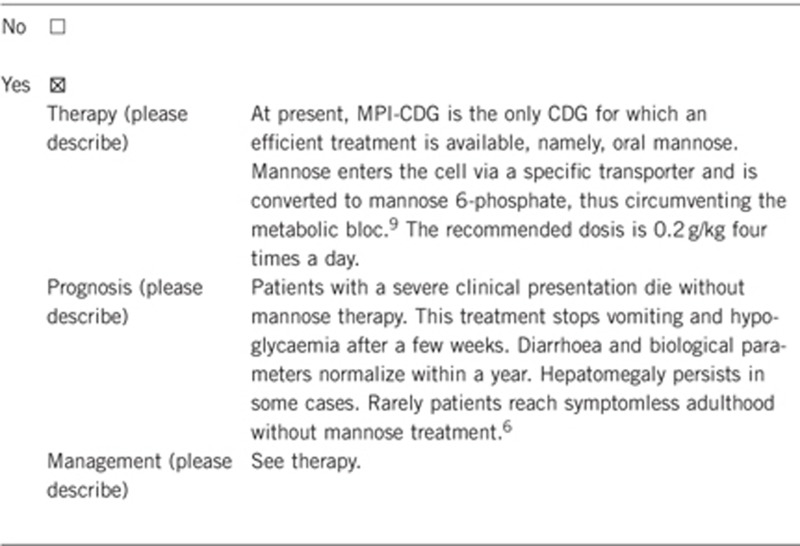
3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
In affected persons mannose should be started immediately after the diagnosis in order to prevent liver cirrhosis and liver insufficiency.
If the test result is negative (please describe):
No further follow-up is required in subjects from families with a known genetic defect.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
No lifestyle adaptations or other type of prevention can be applied, and no medical follow-up is required in persons with a negative genetic test within a family with identified mutations.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes. If the index case has known mutations, family members can be screened for disease or carriership.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes. A positive genetic test result permits prenatal diagnosis in siblings.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Not applicable as an efficient treatment is available.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics.
The authors declare no conflict of interest.
References
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30:1–14. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- Schollen E, Dorland L, de Koning TJ, et al. Genomic organization of the human phosphomannose isomerase (MPI) gene and mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG-Ib) Hum Mutat. 2000;16:247–252. doi: 10.1002/1098-1004(200009)16:3<247::AID-HUMU7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta. 2009;1792:841–843. doi: 10.1016/j.bbadis.2008.11.012. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet. 1998;62:1535–1539. doi: 10.1086/301873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillaumier-Barrot S, Le Bisec C, de Lonlay P, et al. Protein losing enteropathy-hepatic fibrosis syndrome in Saguenay-Lac St-Jean, Quebec is a congenital disorder of glycosylation type Ib. J Med Genet. 2002;39:849–851. doi: 10.1136/jmg.39.11.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal V, Kjaergaard S, Davis JA, et al. Genetic and metabolic analysis of the first adult with congenital disorder of glycosylation type Ib: longterm outcome and effects of mannose supplementation. Mol Genet Metab. 2001;73:77–85. doi: 10.1006/mgme.2001.3161. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Pirard M, Adamowicz M, Pronicka E, Van Schaftingen E. Inhibition of phosphomannose isomerise by fructose 1-phosphate: an explanation for defective N-glycosylation in hereditary fructose intolerance. Pediatr Res. 1996;40:764–766. doi: 10.1203/00006450-199611000-00017. [DOI] [PubMed] [Google Scholar]
- Sturiale L, Barone R, Fiumara A, et al. Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology. 2005;15:1268–1276. doi: 10.1093/glycob/cwj021. [DOI] [PubMed] [Google Scholar]
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998;101:1414–1420. doi: 10.1172/JCI2350. [DOI] [PMC free article] [PubMed] [Google Scholar]