

Abstract
Anticancer drugs are combined in an effort to treat a heterogeneous tumor or to maximize the pharmacodynamic effect. The development of combination regimens, while desirable, poses unique challenges. These include the selection of agents for combination therapy that may lead to improved efficacy while maintaining acceptable toxicity, the design of clinical trials that provide informative results for individual agents and combinations, and logistical and regulatory challenges. The phase 1 trial is often the initial step in the clinical evaluation of a combination regimen. In view of the importance of combination regimens and the challenges associated with developing them, the Clinical Trial Design (CTD) Task Force of the National Cancer Institute (NCI) Investigational Drug Steering Committee developed a set of recommendations for the phase 1 development of a combination regimen. The first two recommendations focus on the scientific rationale and development plans for the combination regimen; subsequent recommendations encompass clinical design aspects. The CTD Task Force recommends that selection of the proposed regimens be based on a biological or pharmacological rationale supported by clinical and/or robust and validated preclinical evidence, and accompanied by a plan for subsequent development of the combination. The design of the phase 1 clinical trial should take into consideration the potential pharmacokinetic and pharmacodynamic interactions as well as overlapping toxicity. Depending on the specific hypothesized interaction, the primary endpoint may be dose optimization, pharmacokinetics, and/or pharmacodynamic (i.e., biomarker).
Introduction
In most tumors, no single pathway has been identified that uniquely drives the malignant process. A more favorable therapeutic response may be obtained by combining drugs that target multiple pathways and/or inhibit resistance mechanisms (e.g., pharmacodynamic modulation). The past decade has seen the development of a vast array of new drugs focusing predominantly on specific molecular targets or pathways of interest. Perhaps the greatest clinical benefit from this approach has been demonstrated in malignancies driven predominantly by an identifiable molecular aberration(1-5). However, resistance usually develops(6-10). Conversely, in tumors that are genetically diverse (multiple “driver” mutations/alterations), focusing on a single target in an unselected population has had modest results(11-16). Combining molecularly targeted and/or cytotoxic drugs may be one strategy to overcome these limitations and improve efficacy(17).
The design and conduct of the phase 1 combination trial present specific challenges, such as the optimum selection of agents to combine among the range of possible combinations; the selection of the appropriate dose and schedule (including which drug or drugs to dose escalate); drug-drug interactions; overlapping toxicities; logistical and regulatory challenges. To address these challenges, the Investigational Drug Steering Committee (IDSC) of the National Cancer Institute (NCI) appointed a Clinical Trial Design (CTD) Task Force composed of academics, pharmaceutical industry representatives, and patient advocates, to develop recommendations (Table 1) similar to those developed previously for phase 1 and phase 2 clinical trials(18, 19). The CTD Task Force focused on development of combinations of systemic agents (marketed or investigational), with consideration of the proposed mechanism of action, pharmacokinetics, and expected toxicities. The recommendations provide pragmatic clinical guidelines rather than a rigid set of rules and do not encompass in depth details of study designs or regulatory or logistic challenges of combination regimens. The consensus recommendations were reviewed and approved by the IDSC on March 13, 2012(20) (Figure 1).
Table 1. Past and present members of the of the Investigational Drug Steering Committee Clinical Trial Design Task Force.
| Position | Name |
|---|---|
| Co-chairs | Mark J. Ratain, Michael LeBlanc |
| Members | Laurence H. Baker, Penelope Bradbury, Lee Ellis, Elizabeth Garrett-Mayer, Richard Gaynor, Gary Gordon, Susan Groshen, Robert Iannone, Patricia Keegan, Patricia M. LoRusso, Stuart Lutzker, Channing J. Paller, Gary Rosner, Larry Rubinstein, Daniel Sargent, Lalitha K. Shankar, Manish Sharma, Anthony Shields, David R. Spriggs |
| NCI liaison | S. Percy Ivy |
| Past chair and co-chair | Lesley Seymour, Donald Berry |
| Non-voting members | Rajeev Agarwal, Lori Minasian, Peter Ujhazy |
| IDSC patient advocate | Deborah Collyar |
| Past members | John Crowley, Afshin Dowlati, Jeffrey Humphrey, Mario Sznol, Miguel Villalona-Calero, Siu-Long Yao |
Figure 1.
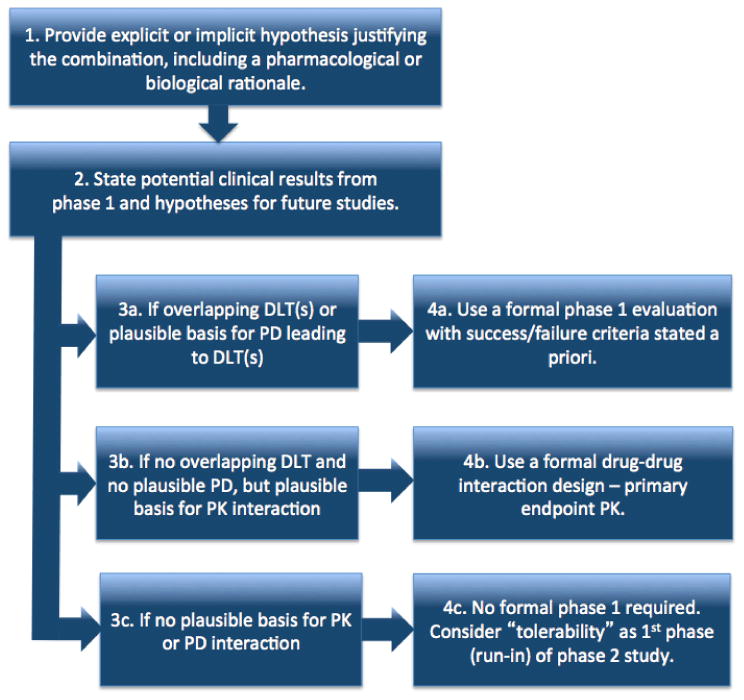
Process for determination of phase 1 combination trial design. DLT = dose limiting toxicity; PD = pharmacodynamic; PK = pharmacokinetic.
Consensus Recommendations
Recommendation 1
All phase 1 combination trials should state an explicit or implicit hypothesis justifying the combination, including a pharmacological or biological rationale that includes at least one of the following: in vitro data, in vivo data, or clinical data. The rationale may extrapolate from results with similar drugs and may be based on in silico analyses. The hypothesis supporting the combination should be clearly stated in the protocol.
Given the vast number of combinations of anticancer drugs that could be evaluated(21), priority should be given to those combinations that are based on the strongest rationale and are most likely to result in clinically significant therapeutic advances. The phase 1 study should, therefore, have a clearly referenced rationale justifying evaluation of the combination. The overarching hypothesis to combine anticancer drugs is to enhance antitum or effects (Table 2). The underlying hypothesis should include a pharmacologic and biologic rationale supported by at least one of the following: in vitro data, in vivo data, or clinical data.
Table 2. Hypotheses justifying combination trials.
| Target | Tumor type | Examples |
|---|---|---|
| Target multiple mechanisms of action | Gastric, colorectal, NSCLC | Addition of HER-2 (22), EGFR (23) or VEGF (46-48, 60) pathway inhibitors broadened anticancer activity of standard chemotherapy while minimizing cross-resistance. |
| Optimize the inhibition of a specific target or pathway | Melanoma | Combination of CTLA-4 and PD-1 receptor inhibitors resulted in tumor regression beyond that expected from monotherapy (61). |
| Target a potential resistance mechanism (bypass pathway) | Breast, melanoma, NSCLC | Addition of an mTOR inhibitor to antiestrogens (62), a MEK inhibitor to a BRAF kinase inhibitor (34), or an EGFR inhibitor to a MET inhibitor (49, 63) results in restoration of sensitivity and decreased proliferation in cell lines and, for NSCLC patients, to increased progression free survival. |
NSCLC = non-small cell lung cancer; HER-2 = human epidermal growth factor receptor 2; EGFR = epidermal growth factor receptor; VEGF = vascular endothelial growth factor; CTLA-4 = cytotoxic T-lymphocyte-associated antigen 4; PD-1 = programmed death 1; mTOR= mammalian target of rapamycin; MEK = mitogen-activated protein/extracellular signal-regulated kinase kinase.
The level of preclinical data required to predict a benefit in clinical trials is currently unknown, since preclinical studies do not generally predict success of clinical trials(22-24). Preclinical data may help in the prioritization of combinations to advance to clinical trials and in the design of the subsequent clinical trial (see recommendations 3 and 4)(25). Poor therapeutic indices may be associated with the high attrition rates found in oncology drug development, yet appear to be infrequently evaluated (26). Preclinical models have been shown to predict some non-hematologic toxicities in humans, including skin toxicity and gastrointestinal toxicities associated with EGFR inhibitors (27). Toxicities such as myalgias, arthralgias, and headaches are unable to be detected in preclinical studies.
The additive or synergistic effects of a drug combination can be evaluated in cell line assays, however, determining synergy in vivo is complex, standard definitions of synergy in vivo do not exist, and such standard definitions are used exclusively with in vitro models (28). Another consideration is the potential for antagonistic effects of agents when combined and the effect of sequence, rarely tested preclinically. The addition of gefitinib or erlotinib to chemotherapy has not conferred a demonstrable clinical benefit. Subsequent preclinical studies demonstrated the concurrent administration of an EGFR TKI with standard chemotherapy in NSCLC may be antagonistic, whereas sequential administration may have improved activity (29-31). This potential can be explored using preclinical models and may inform the design of schedules in the clinical trials. The IDSC has outlined some considerations regarding the selection of agents to take forward into clinical trials(18). However, negative preclinical results may not be reported due to publication bias, limiting relevant evidence and possibly contributing to high failure rates in oncology drug development. Publication of negative results through journals such as the Journal of Negative Results in BioMedicine(32, 33), may provide important information for combination trial design.
Recommendation 2
The potential results and next steps of the development plan of the combination should be clearly described. The description should include two parts: the rationale for why the biologic or pharmacologic interactions should translate into clinical effects, and one or more examples of phase 2 studies to test the hypothesis. The phase 2 example(s) should follow the guidelines of the Phase 2 Consensus Recommendations of the NCI's IDSC CTD Task Force.
In addition to a robust underlying hypothesis outlining why specific agents should be combined (recommendation 1), a clearly defined plan of how the combination will be evaluated in phase 1 and 2 clinical studies (recommendations 3 and 4), and the anticipated outcome of those trials, should be outlined. The trial design may aim to optimize a toxicity endpoint, pharmacodynamics biomarker or be descriptive and exploratory. As there are an infinite number of maximally tolerated doses for a drug combination, which may also be the case when optimizing a PD biomarker, the recommendation is not intended to be restrictive in nature. However, the rationale for the design should consider future trials. For example, a 3-part phase 1/2 trial explored the potential for a MEK inhibitor to delay the resistance to BRAF inhibition and the safety of the combination: (Part A) determine potential pharmacokinetic interactions, (Part B) evaluated toxicity, safety, and pharmacokinetics of escalating doses of both agents, (Part C) proof of principle in randomized phase 2 study evaluating progression-free survival(34). If the development plan for a combination is unclear or not feasible, it calls into question the rationale for undertaking the phase 1 trial in the first place. Furthermore, specific criteria including decision rules for success (e.g., a regimen that can be moved forward in development) and failure (e.g., a regimen that is too toxic for further evaluation) should be defined and fully developed in the clinical protocol (Table 3). Trametinib and dabrafenib were successfully combined with no significant incremental toxicity and fewer squamous cell carcinomas than for patients receiving monotherapy, and proof of principle was demonstrated with an improvement in progression-free survival with the combination(34). In contrast, when low-dose sorafenib and bevacizumab were combined, the tolerated doses of both agents was a quarter to half the single-agent dose used in other solid tumor studies because of unexpectedly severe toxicities, including hand-foot syndrome, hypertension, proteinuria, and thrombocytopenia(35).
Table 3. Selected pharmacokinetic interactions.
| Drug-drug or drug-food combination | PK interaction | Mechanism |
|---|---|---|
| Gefitinib with bexarotene | Plasma levels of gefitinib significantly reduced (64, 65). | Gefitinib is metabolized by multiple cytochrome P450 enzymes, including bexarotene. |
| Temsirolimus with lenalidomide | Administration of temsirolimus increased maximum concentration and area under the concentration-time curve of lenalidomide(43). | Lenalidomide is P-glycoprotein substrate. |
| Imatinib, dasatinib, and nilotinib with high fat meals | AUC increased by 82% when nilotinib was given 30 minutes after a high fat meal (66). | Oral TKIs have a high risk of PK interactions when administered in conjunction with high-fat meals |
| Imatinib, dasatinib, and nilotinib with ketoconazole, levothyroxine, and verapamil | Imatinib exposure increased following ketoconazole coadministration (67). | Oral TKIs such as have a high risk of drug interactions when administered with drugs affecting CYP3A4. |
AUC = area under the curve; TKI = tyrosine kinase inhibitors; PK = pharmacokinetic; CYP3A4 = cytochrome P450 3A4.
Recommendation 3
The design of combination phase 1 studies should address the following three factors: overlapping dose limiting toxicities (DLTs); a plausible mechanistic basis for a pharmacodynamic interaction leading to DLTs; and a plausible mechanistic basis for a pharmacokinetic interaction.
Due to the differences in pharmacology, mechanism of action, and toxicity of individual agents, the design of the phase 1 combination study should be tailored to the specific drugs to be combined. This may involve a formal phase 1 dose escalation trial (36), a pharmacokinetic endpoint (37), or a safety run-in to a phase 2 study. Three important considerations are (a) the potential for overlapping DLTs, (b) pharmacodynamic interactions, and (c) pharmacokinetic interactions.
Overlapping DLTs may limit escalation of doses to levels required for optimal activity or may affect dose intensity due to dose reductions when a regimen is administered chronically. Even when overlapping DLTs do not exist, pharmacodynamic interactions may result in toxicity and impact dose. Combining bevacizumab and sorafenib(35, 38) or sunitinib(39) resulted in proteinuria and thrombocytopenia, requiring modification of dosing and scheduling. The additive effects of mild overlapping adverse events may impair tolerance, particularly for drugs that are intended to be administered chronically(28).
Pharmacokinetic interactions may alter the absorption, distribution, metabolism, and excretion of one or both drugs. (Table 3) However, pharmacokinetic assessments of drug-drug interaction should be routinely included in phase 1 trial design only when scientific justification for such interactions, at pharmacologically achievable drug concentrations, has been identified(40). We recommend completing an initial pharmacokinetic analysis of the individual drug prior to initiating the next drug to increase the reliability of evidence for or against an interaction(37, 41, 42), rather than performing pharmacokinetic studies of all drugs in the combination and comparing the results to historical controls(43).
Combining agents with conflicting requirements with respect to the timing of meals adds challenges. Understanding these potential interactions will help generate hypotheses to be addressed in the phase 1 clinical trial, which will then influence the subsequent clinical trial design. Explicit instructions to trial participants are critical to ensure careful treatment selection for future trials.
Drug scheduling of combinations may affect additive or synergistic effects on efficacy, toxicity, or both. Combining drugs with on-off scheduling, such as sunitinib (4 weeks on, 2 weeks off is standard) and capecitabine (2 weeks on, 1 week off), will require the evaluation of sequences different from those used when the drugs are administered as single agents. If drug-drug interactions affect exposure to either drug or their metabolites, then applying common single-agent, on-off schedules may result in variable drug exposure(21). Preclinical studies could identify alternate schedules that result in more consistent drug exposures. Similar scheduling and sequencing issues may arise when intravenously administered drugs are combined with oral agents. Drug sequencing may also be a design issue when combining drugs with pharmacokinetic or pharmacodynamic interactions and short half-lives. Scheduling and dosing decisions also affect toxicities, and novel schedules such as alternating administration of the drugs may allow extended administration when concurrent administration is too toxic(44).
Recommendation 4
Selection of the clinical trial design should be based on the scientific rationale, underlying data and hypothesis for the combination, and the intended development plan for the combination (recommendations 1-3). Recommendation 4 is not intended to be prescriptive, but to provide pragmatic guidelines on the selection of the phase 1 trial design for a drug combination.
No single standard exists for phase 1 trial design for combinations, but the design should address the specific hypothesis (recommendation 3) and subsequent development plans for the combination (recommendation 2). The classic DLT-driven cohort expansion design (3+3) has most commonly been used(45). To date, most combinations have added one or more investigational agents to a standard backbone already in clinical practice(34, 46-49). One challenge is the need to distinguish the incremental toxicity of the combination. Hamberg et al. propose some solutions, including a 3+3+3 design, which may reduce the chance of falsely declaring the maximum tolerated dose has been reached (when in fact observed DLTs might be due to the standard therapy alone)(21). Another alternative is to include controls in phase 1 trials, either an intrapatient control (e.g., by introducing the novel agent after the standard backbone has been started) or the randomization of patients to commence the combination upfront or in a staggered approach(21). Mathematical modeling can also be used to refine dose escalation based on data that emerges during the trial. A Bayesian approach has been proposed to address background toxicity that arises when a new agent is added to standard treatment(21). In addition, if the combination is intended to be developed in several disease settings with different backbones, multi-arm studies may also be more efficient than a series of separate single-agent trials(50-52). Further, a phase 1 trial may fail to reflect optimal dose-relationships. Thus investigators may need to compare alternate doses and schedules in subsequent trials (21). While there may be multiple appropriate phase 1 design options, the proposed design should be fully specified in the protocol as an algorithm, including any stage-wise decision rules, so that the statistical properties of the proposed phase 1 design can be evaluated under hypothesized outcome probability models.
Recommendation 4A
Combination therapies with overlapping DLTs or a plausible basis for a pharmacodynamic interaction leading to DLTs require formal phase 1 evaluation. The selected doses to be studied should be justified based on the specific phase 2 plans.
Where overlapping DLTs exist or where pharmacodynamic interaction may be anticipated, a formal phase 1 design is required to evaluate toxicity, and to explore the recommended phase 2 doses and possibly the optimal schedule of the combination. The selection of doses to be evaluated should be justified based on preclinical data, nature of interaction of the study drugs, and dependence of the target for DLTs versus efficacy. In addition, the criteria for success and failure of a combination should be defined. An inability to escalate one or more drugs such that the combined dose would be expected to have greater efficacy than the single-agent counterparts may mean the combination is not suitable for progressing to a phase 2 study. In addition, DLTs occurring after the first cycle can prevent administration of subsequent cycles and should be factored into determination of recommended phase II dosing. Finally, toxicity may be unacceptable in the population for which the combination is planned to be developed.
When considering dose escalation for both agents, a model-based approach can be very helpful, particularly when it considers both toxicity and efficacy(53-55). When one of the agents has markedly less single-agent activity or is being added primarily as a modulator, the agent with greater single-agent activity should, in general, be maintained at or near its single-agent dose (MTD) while gradually titrating the second agent.
Recommendation 4B
Combinations without overlapping DLTs and without a plausible basis for pharmacodynamic interaction, but with a plausible pharmacokinetic interaction, should be studied using a formal drug-drug interaction design. The primary endpoint is pharmacokinetics. Crossover design is often optimal.
In phase 1 trials where pharmacokinetic interactions are anticipated (e.g., drugs metabolized by the same pathway(56)), a drug-drug interaction design should be considered. This would facilitate pharmacokinetic analyses for both single agents and the combination. A common method of assessing pharmacokinetic interaction uses crossover study designs that limit the number of patients required, and allow testing for different effects based upon different sequential ordering of the agents. Statistical analysis employing repeated measures over the same patient can account for inter-patient variability for some of the endpoints and thereby increases statistical power. Combinations involving drugs with long half-lives, such as vismodegib, require phase 1 designs with pharmacokinetic washout periods(57). Crossover studies may be randomized designs where drug 1 is followed by drug 1 and drug 2, or drug 1 and drug 2 are followed by drug 1, with a washout period, with extensive pharmacokinetic sampling during both the single-agent and combination phases. An alternative is the single sequence crossover design in which drug 1 plus drug 2 always follows drug 1, or the reverse(58). In both of these designs, both interpatient and intrapatient variability may need to be considered.
Where pharmacokinetic interactions could lead to drug accumulation, washout periods and/or low initial doses should be considered. The decision to escalate to the next dose would be based on interim pharmacokinetic results in which the predefined dose-escalation rule might be “If the drug A level at steady state increases less than x% over the previous level, and no DLT is present, then use dose B, otherwise use dose C.”In situations in which CYP3A4 interactions are expected, for example, this pharmacokinetic-informed dose escalation method is appropriate. The hypothesis-driven development plan performed in recommendation 2, in case of clinically significant pharmacokinetic interactions, must be based upon a reasonable assurance that any dose reductions in individual agents, necessitated by the need to maintain acceptable toxicity, preserves the expectation of superior efficacy for the combination(59).
Recommendation 4C
Combinations without overlapping toxicities, without a plausible basis for a pharmacodynamic interaction leading to a DLT, and without a plausible basis for a pharmacokinetic interaction do not require a formal phase 1 study. A pilot or safety run-in for tolerability can be conducted as an initial step of a phase 2 study.
If a combination regimen is not anticipated to have overlapping toxicity, pharmacodynamic or pharmacokinetic interaction, a formal phase 1 trial may not be required. Instead, a short pilot or safety run-in period may be undertaken as the initial part of a phase 2 trial to explore a limited number of dose levels (for example clinicaltrials.gov identifier NCT01839487 or NCT01708993), or to evaluate an anticipated recommended phase 2 dose directly prior to enrolling patients in the phase 2 portion of the trial. If this approach is chosen, the phase 2 design could include an early safety interim analysis to ensure the regimen is tolerable(21). However, prior to determining that a formal phase I trial is not required, consideration should be given to involving PK, PD and data safety experts.
Summary
The CTD Task Force formulated recommendations for the design of early-phase combination trials of anticancer agents based on consensus developed with members of the IDSC, the Task Force, and external experts. The selection of the proposed regimens is based on a biological or pharmacological rationale supported by clinical and preclinical data, accompanied by a plan for subsequent development of the combination trials. The potential pharmacokinetic and pharmacodynamic interactions, as well as overlapping toxicities, should be considered. Depending on the specific hypothesized interaction, the primary endpoint may be dose optimization, pharmacokinetics, and/or pharmacodynamics. The rationale and design of combination clinical trials should be carefully evaluated. Additional guidance may be obtained by consulting with regulatory authorities (e.g., the FDA and/or EMEA, the European Medicines Agency) prior to trial initiation. These recommendations complement consensus guidelines on the design of phase 1 and phase 2 clinical trials testing cancer therapeutics(18, 19), and were reviewed and formally approved by the IDSC.
Acknowledgments
Special thanks to Amy Gravell. P.A. Bradbury was supported by a Cancer Care Ontario Research Chair in Experimental Therapeutics.
Footnotes
Disclosure of Potential Conflicts of Interest: L. Baker is a consultant/advisory board member for Cytrx, Immune Design, and Morphotek. L.M. Ellis is a consultant/advisory board member for Amgen, Genentech, and ImClone Systems. G. Rosner reports receiving speakers bureau honoraria from Novartis. M.J. Ratain is a consultant/advisory board member for AbbVie, Daiichi Sankyo, and Genentech. No potential conflicts of interest were disclosed by the other authors.
Note: C.J. Paller and P.A. Bradbury contributed equally to this article.
References
- 1.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 3.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 4.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kantarjian HM, Talpaz M, O'Brien S, Giles F, Garcia-Manero G, Faderl S, et al. Dose escalation of imatinib mesylate can overcome resistance to standard-dose therapy in patients with chronic myelogenous leukemia. Blood. 2003;101:473–5. doi: 10.1182/blood-2002-05-1451. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 8.Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12:5764–9. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 9.Mehra R, Camidge DR, Sharma S, Felip E, Tan DS, Vansteenkiste JF, et al. First-in-human phase I study of the ALK inhibitor LDK378 in advanced solid tumors. J Clin Oncol. 2012;30(suppl;abstr3007) [Google Scholar]
- 10.Soverini S, Martinelli G, Rosti G, Bassi S, Amabile M, Poerio A, et al. ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol. 2005;23:4100–9. doi: 10.1200/JCO.2005.05.531. [DOI] [PubMed] [Google Scholar]
- 11.Chee KG, Longmate J, Quinn DI, Chatta G, Pinski J, Twardowski P, et al. The AKT inhibitor perifosine in biochemically recurrent prostate cancer: a phase II California/Pittsburgh cancer consortium trial. Clin Genitourin Cancer. 2007;5:433–7. doi: 10.3816/CGC.2007.n.031. [DOI] [PubMed] [Google Scholar]
- 12.Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 13.Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 14.Krug LM, et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have failed prior pemetrexed and either cisplatin or carboplatin therapy: A phase III, randomized, double-blind, placebo-controlled trial. ECCO-ESMO. 2011 Abstract 3BA. [Google Scholar]
- 15.Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13:528–38. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 16.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 17.Lorusso PM, Eder JP. Therapeutic potential of novel selective-spectrum kinase inhibitors in oncology. Expert Opin Investig Drugs. 2008;17:1013–28. doi: 10.1517/13543784.17.7.1013. [DOI] [PubMed] [Google Scholar]
- 18.LoRusso PM, Boerner SA, Seymour L. An overview of the optimal planning, design, and conduct of phase I studies of new therapeutics. Clin Cancer Res. 2010;16:1710–8. doi: 10.1158/1078-0432.CCR-09-1993. [DOI] [PubMed] [Google Scholar]
- 19.Seymour L, Ivy SP, Sargent D, Spriggs D, Baker L, Rubinstein L, et al. The design of phase II clinical trials testing cancer therapeutics: consensus recommendations from the clinical trial design task force of the national cancer institute investigational drug steering committee. Clin Cancer Res. 2010;16:1764–9. doi: 10.1158/1078-0432.CCR-09-3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. [Accessed 25th March 2012]; http://transformingtrials.cancer.gov/steering-committees/investigational-drug.
- 21.Hamberg P, Ratain MJ, Lesaffre E, Verweij J. Dose-escalation models for combination phase I trials in oncology. Eur J Cancer. 2010;46:2870–8. doi: 10.1016/j.ejca.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Dancey JE, Chen HX. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat Rev Drug Discov. 2006;5:649–59. doi: 10.1038/nrd2089. [DOI] [PubMed] [Google Scholar]
- 23.Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84:1424–31. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res. 2003;9:4227–39. [PubMed] [Google Scholar]
- 25.Holbeck S, Collins JM, Doroshow JH. NCI-60 combination screening matrix of approved anticancer drugs. Eur J Cancer. 2012;48(Suppl6,abstr27) [Google Scholar]
- 26.Ocana A, Amir E, Yeung C, Seruga B, Tannock IF. How valid are claims for synergy in published clinical studies? Ann Oncol. 2012;23:2161–6. doi: 10.1093/annonc/mdr608. [DOI] [PubMed] [Google Scholar]
- 27.Park SR, Davis M, Doroshow JH, Kummar S. Safety and feasibility of targeted agent combinations in solid tumours. Nat Rev Clin Oncol. 2013;10:154–68. doi: 10.1038/nrclinonc.2012.245. [DOI] [PubMed] [Google Scholar]
- 28.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 29.Gatzemeier U, Pluzanska A, Szczesna A, Kaukel E, Roubec J, De Rosa F, et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol. 2007;25:1545–52. doi: 10.1200/JCO.2005.05.1474. [DOI] [PubMed] [Google Scholar]
- 30.Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol. 2004;22:777–84. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Mok TW, Wu YL, Thongprasert S, Yu CJ, Zhang L, Ladreran GE, et al. A randomized placebo-controlled phase III study of intercalated erlotinib with gemcitabine/platinum in first-line advanced non-small cell lung cancer (NSCLC): FASTACT-II. J Clin Oncol. 2012;30(suppl;abstr7519) [Google Scholar]
- 32.Begley CG, Ellis LM. Drug development: Raise standards for preclinical cancer research. Nature. 2012;483:531–3. doi: 10.1038/483531a. [DOI] [PubMed] [Google Scholar]
- 33. [accessed 10Feb2014]; www.jnrbm.com.
- 34.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azad NS, Posadas EM, Kwitkowski VE, Steinberg SM, Jain L, Annunziata CM, et al. Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. J Clin Oncol. 2008;26:3709–14. doi: 10.1200/JCO.2007.10.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18:1726–34. doi: 10.1158/1078-0432.CCR-11-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gangadhar TC, Cohen EE, Wu K, Janisch L, Geary D, Kocherginsky M, et al. Two drug interaction studies of sirolimus in combination with sorafenib or sunitinib in patients with advanced malignancies. Clin Cancer Res. 2011;17:1956–63. doi: 10.1158/1078-0432.CCR-10-2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JM, Sarosy GA, Annunziata CM, Azad N, Minasian L, Kotz H, et al. Combination therapy: intermittent sorafenib with bevacizumab yields activity and decreased toxicity. Br J Cancer. 2010;102:495–9. doi: 10.1038/sj.bjc.6605514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rini BI, Garcia JA, Cooney MM, Elson P, Tyler A, Beatty K, et al. A phase I study of sunitinib plus bevacizumab in advanced solid tumors. Clin Cancer Res. 2009;15:6277–83. doi: 10.1158/1078-0432.CCR-09-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu K, House L, Ramirez J, Seminerio MJ, Ratain MJ. Evaluation of utility of pharmacokinetic studies in phase I trials of two oncology drugs. Clin Cancer Res. 2013;19:6039–4. doi: 10.1158/1078-0432.CCR-13-0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Connolly RM, Rudek MA, Garrett-Mayer E, Jeter SC, Donehower MG, Wright LA, et al. Docetaxel metabolism is not altered by imatinib: findings from an early phase study in metastatic breast cancer. Breast Cancer Res Treat. 2011;127:153–62. doi: 10.1007/s10549-011-1413-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Huang Y, Navarro MT, Hisoire G, Caulfield JP. A proof-of-concept and drug-drug interaction study of pamapimod, a novel p38 MAP kinase inhibitor, with methotrexate in patients with rheumatoid arthritis. J Clin Pharmacol. 2010;50:1031–8. doi: 10.1177/0091270009357433. [DOI] [PubMed] [Google Scholar]
- 43.Hofmeister CC, Yang X, Pichiorri F, Chen P, Rozewski DM, Johnson AJ, et al. Phase I trial of lenalidomide and CCI-779 in patients with relapsed multiple myeloma: evidence for lenalidomide-CCI-779 interaction via P-glycoprotein. J Clin Oncol. 2011;29:3427–34. doi: 10.1200/JCO.2010.32.4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 45.Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst. 2009;101:708–20. doi: 10.1093/jnci/djp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 47.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 48.Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009;373:1525–31. doi: 10.1016/S0140-6736(09)60569-9. [DOI] [PubMed] [Google Scholar]
- 49.Scagliotti GV, Novello S, Schiller JH, Hirsh V, Sequist LV, Soria JC, et al. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer. 2012;13:391–5. doi: 10.1016/j.cllc.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Mita AC, Takimoto CH, Mita M, Tolcher A, Sankhala K, Sarantopoulos J, et al. Phase 1 study of AMG 386, a selective angiopoietin 1/2-neutralizing peptibody, in combination with chemotherapy in adults with advanced solid tumors. Clin Cancer Res. 2010;16:3044–56. doi: 10.1158/1078-0432.CCR-09-3368. [DOI] [PubMed] [Google Scholar]
- 51.Goss G, Shepherd FA, Laurie S, Gauthier I, Leighl N, Chen E, et al. A phase I and pharmacokinetic study of daily oral cediranib, an inhibitor of vascular endothelial growth factor tyrosine kinases, in combination with cisplatin and gemcitabine in patients with advanced non-small cell lung cancer: a study of the National Cancer Institute of Canada Clinical Trials Group. Eur J Cancer. 2009;45:782–8. doi: 10.1016/j.ejca.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 52.Laurie SA, Gauthier I, Arnold A, Shepherd FA, Ellis PM, Chen E, et al. Phase I and pharmacokinetic study of daily oral AZD2171, an inhibitor of vascular endothelial growth factor tyrosine kinases, in combination with carboplatin and paclitaxel in patients with advanced non-small-cell lung cancer: the National Cancer Institute of Canada clinical trials group. J Clin Oncol. 2008;26:1871–8. doi: 10.1200/JCO.2007.14.4741. [DOI] [PubMed] [Google Scholar]
- 53.Harrington JA, Wheeler GM, Sweeting MJ, Mander AP, Jodrell DI. Adaptive designs for dual-agent phase I dose-escalation studies. Nat Rev Clin Oncol. 2013;10:277–88. doi: 10.1038/nrclinonc.2013.35. [DOI] [PubMed] [Google Scholar]
- 54.Mandrekar SJ, Cui Y, Sargent DJ. An adaptive phase I design for identifying a biologically optimal dose for dual agent drug combinations. Stat Med. 2007;26:2317–30. doi: 10.1002/sim.2707. [DOI] [PubMed] [Google Scholar]
- 55.Thall PF, Cook JD. Dose-finding based on efficacy-toxicity trade-offs. Biometrics. 2004;60:684–93. doi: 10.1111/j.0006-341X.2004.00218.x. [DOI] [PubMed] [Google Scholar]
- 56.Reardon DA, Vredenburgh JJ, Desjardins A, Peters KB, Sathornsumetee S, Threatt S, et al. Phase 1 trial of dasatinib plus erlotinib in adults with recurrent malignant glioma. J Neurooncol. 2012;108:499–506. doi: 10.1007/s11060-012-0848-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rudin CM. Vismodegib. Clin Cancer Res. 2012;18:3218–22. doi: 10.1158/1078-0432.CCR-12-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewis LD. Drug-drug interactions: is there an optimal way to study them? Br J Clin Pharmacol. 2010;70:781–3. doi: 10.1111/j.1365-2125.2010.03829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, et al. A Phase I/II Trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res. 2013;19:900–8. doi: 10.1158/1078-0432.CCR-12-1707. [DOI] [PubMed] [Google Scholar]
- 60.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 61.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O'Reilly T, Evans DB, et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005;11:5319–28. doi: 10.1158/1078-0432.CCR-04-2402. [DOI] [PubMed] [Google Scholar]
- 63.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 64.Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13:3731–7. doi: 10.1158/1078-0432.CCR-07-0088. [DOI] [PubMed] [Google Scholar]
- 65.Padda SK, Chhatwani L, Zhou L, Jacobs CD, Lopez-Anaya A, Wakelee HA. Phase I and pharmacokinetic study of bexarotene in combination with gefitinib in the third-line treatment of non-small-cell lung cancer: brief report. Anticancer Drugs. 2013;24:731–5. doi: 10.1097/CAD.0b013e32836100d7. [DOI] [PubMed] [Google Scholar]
- 66.Full Prescribing Information and Warning of QT Prolongation and Sudden Deaths for nilotinib http://dailymed.nlm.nih.gov/dailymed/archives/fdaDrugInfo.cfm?archiveid=6775 2008 [March 26, 2013]
- 67.Haouala A, Widmer N, Duchosal MA, Montemurro M, Buclin T, Decosterd LA. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. 2011;117:e75–87. doi: 10.1182/blood-2010-07-294330. [DOI] [PubMed] [Google Scholar]