

Abstract
Polycystin-1 (Pc1) cleavage at the G protein-coupled receptor (GPCR) proteolytic site (GPS) is required for normal kidney morphology in humans and mice. We found a complex pattern of endogenous Pc1 forms by GPS cleavage. GPS cleavage generates not only the heterodimeric cleaved full-length Pc1 (Pc1cFL) in which the N-terminal fragment (NTF) remains noncovalently associated with the C-terminal fragment (CTF) but also a novel (Pc1) form (Pc1deN) in which NTF becomes detached from CTF. Uncleaved Pc1 (Pc1U) resides primarily in the endoplasmic reticulum (ER), whereas both Pc1cFL and Pc1deN traffic through the secretory pathway in vivo. GPS cleavage is not a prerequisite, however, for Pc1 trafficking in vivo. Importantly, Pc1deN is predominantly found at the plasma membrane of renal epithelial cells. By functional genetic complementation with five Pkd1 mouse models, we discovered that CTF plays a crucial role in Pc1deN trafficking. Our studies support GPS cleavage as a critical regulatory mechanism of Pc1 biogenesis and trafficking for proper kidney development and homeostasis.
INTRODUCTION
Polycystin-1 (PC1) is encoded by the PKD1 gene that is mutated in autosomal dominant polycystic kidney disease (ADPKD), characterized by the development of numerous cysts in both kidneys and progressive renal failure (1). PC1 regulates terminal differentiation of tubular structures in kidney and liver (2–4), as well as maintaining the structural integrity of the kidney (5) and vasculature (6). Expression studies in human and mouse showed spatiotemporal regulated expression for PC1/Pc1 (7–10). In the fetal kidney, immunolocalization of endogenous PC1/Pc1 was observed at apical (most predominant) and basolateral plasma membranes of ureteric bud and collecting ducts (CDs) (11–13). During late renal morphogenesis, Pc1 expression increases significantly during planar cell polarity (PCP)-dependent convergent extension and late collecting duct branching elongation (9, 14). In adult kidney and other tissues or cell lines, PC1/Pc1 localization was reported in a range of subcellular compartments to apical and basal lateral plasma membranes (12), cell-cell junctions (15, 16), and primary cilia (17, 18). Based on its complex structure and expression patterns, native Pc1 is thought to act on the cell surface and may have multiple cellular functions in vivo.
PC1 is a 4,302-amino-acid (aa) glycoprotein with a huge N-terminal extracellular region containing protein-protein interaction motifs, an 11-transmembrane (TM) domain, and an ∼200-aa C-terminal cytoplasmic tail that can activate a number of signaling pathways (19, 20) (Fig. 1A). The N-terminal extracellular region is separated from the 11-TM domain by the G protein-coupled receptor (GPCR) proteolytic site (GPS) motif of ∼50 aa (19, 20). Initial studies with recombinant PC1 showed that PC1/Pc1 is cleaved within the GPS motif (21) at the tripeptide HL↓T3041 located ∼20 aa before the first TM domain, resulting in an ∼370-kDa N-terminal fragment (NTF) and an ∼150-kDa C-terminal fragment (CTF) (22). Following GPS cleavage, Pc1 NTF remains noncovalently associated with the CTF. In addition, a significant proportion of the overexpressed recombinant PC1/Pc1 remains uncleaved in various mammalian cells.
FIG 1.
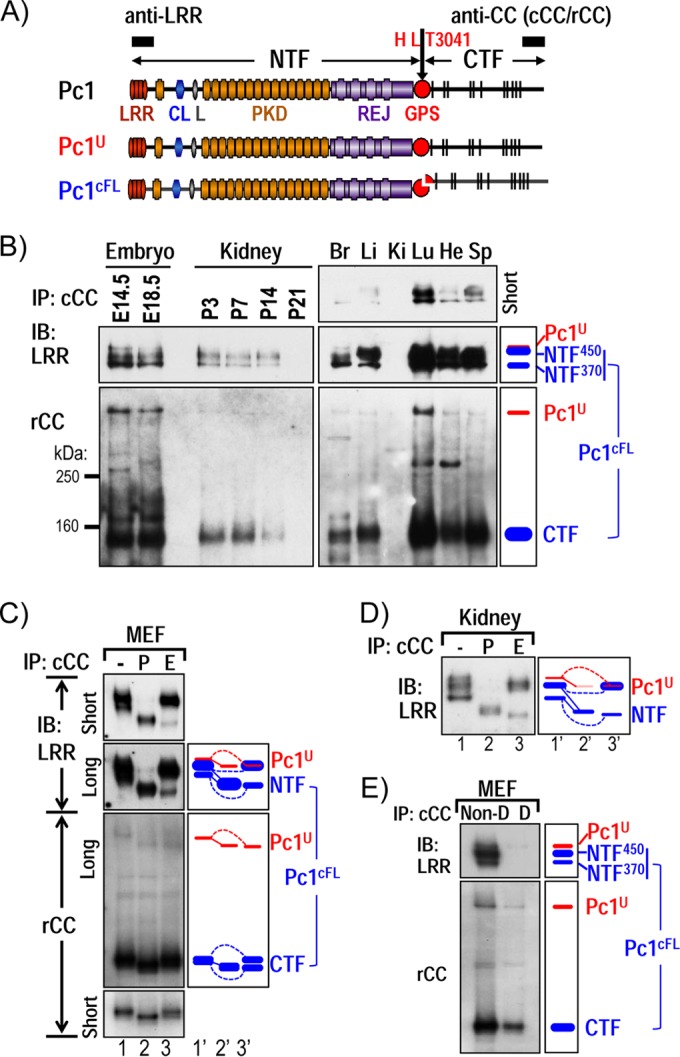
Characterization of endogenous Pc1U and Pc1cFL molecules in normal mouse tissues. (A) Schematic structure of mouse polycystin-1 (Pc1). LRR, leucine-rich repeat; CL, C-type lectin; L, LDL-A; PKD, polycystic kidney disease repeats; REJ, receptor for egg jelly; GPS, G-protein-coupled receptor proteolytic site. Pc1 cleavage occurs at HL↓T3041 site in the GPS motif, resulting in NTF and CTF fragments. Epitope positions of anti-LRR and anti-CC (chicken, cCC; rabbit, rCC) are shown by black boxes. The uncleaved full-length Pc1U (red) and the full-length cleaved Pc1cFL (blue) are schematized. The color code is maintained throughout the figures. (B) Endogenous Pc1 products were analyzed by immunoprecipitation (IP) with anti-cCC from wild-type (WT) mouse embryos (E14.5 and E18.5), kidneys (P3 to P21), and adult (2-month-old) tissues (Br, brain; Li, liver; Ki, kidney; Lu, lung; He, heart; Sp, spleen) and detected by immunoblotting (IB) with anti-LRR (upper panel) and anti-rCC (lower panel). The schematic diagram (right panel) provides an identification guide. (C) N-glycosylation modification of endogenous Pc1 from WT MEFs was monitored by IB on anti-cCC immunoprecipitates, either untreated (−) or treated with PNGase F (P) or endo-H (E). Pc1 products were detected with anti-LRR and anti-rCC from different exposures. The exclusive endo-H sensitivity of Pc1U contrasts with the partial endo-H sensitivity of Pc1cFL. Note that endo-H-deglycosylated Pc1U overlapped with the intense endo-H-resistant NTF450 band (lane 3). A schematic diagram provides an identification guide. (D) N-glycosylation modification of endogenous Pc1 from WT kidneys at P5 was analyzed as described for panel C and detected by IB with anti-LRR. Endogenous Pc1U is endo-H sensitive, whereas the Pc1 NTF subunit is both endo-H resistant and sensitive. (E) Noncovalent association of Pc1cFL subunits. MEF lysates were subjected to IP with anti-cCC under either nondenaturing conditions with 0.5% Triton X-100 (Non-D) or denaturing conditions with detergent SDS (0.1%; D), followed by IB with anti-LRR or anti-rCC. The NTF subunit was coprecipitated by the CTF subunit only under nondenaturing conditions. The schematic diagram provides an identification guide.
Missense mutations in the GPS or GPCR autoproteolysis-inducing (GAIN) domain of PKD1 disrupt cleavage of PC1 in recombinant systems (22–24) and prevent activation of the JAK-STAT pathway and induction of tubulogenesis in MDCK three-dimensional (3D) cultures (22). The first evidence for a functional and physiologic role of GPS cleavage for Pc1 came from analysis of mice homozygous for the knock-in missense change T3041V at the HL↓T3041 cleavage site (Pkd1V/V; the position of cleavage is indicated by the downward arrow), which produces a noncleavable Pc1 (Pc1V) (25, 26). In contrast to the embryonic-lethal Pkd1 null mice, which develop severely cystic kidneys starting at embryonic day 15.5 (E15.5) (2–4), Pkd1V/V mutant mice are viable with virtually normally appearing kidneys at birth. However, from postnatal day 3 (P3), Pkd1V/V mice develop massive cysts mainly in distal nephron segments, leading to death by ∼1 month (26). Therefore, GPS cleavage of Pc1 is not essential in embryonic kidneys but is fundamental in postnatal kidneys. Additionally, uncleaved full-length Pc1 (Pc1U) and GPS-cleaved Pc1 molecules appear to possess distinct biological functions. While Pc1U appears important for embryonic kidney development and postnatally for proximal tubule integrity, cleaved Pc1 is indispensable for intact structure of distal nephron segments after birth.
The GPS motif was first identified as an internal cleavage site in the neuronal GPCR protein latrophilin and was later found to be part of the ∼300-aa-long GAIN domain (23) present in ∼30 adhesion GPCRs (aGPCRs), the second largest GPCR subfamily characterized by an unusually large and complex ectodomain (27–29). A unique property of GPS cleavage is that the two resulting fragments remain associated noncovalently to form a stable but dissociable heterodimer via extensive networks of hydrophobic side chain interactions between the GPS/GAIN domain and the N-terminal stalk of the C-terminal fragment (23, 30–33). Previous studies on aGPCRs have suggested that GPS cleavage promotes efficient trafficking and signaling (34–36). It was also proposed that the NTF of the aGPCR heterodimers might inhibit receptor signaling (35, 37). Such inhibition can be relieved by conformational changes induced by ligand binding and activation of the G-protein-mediated signaling by the seven-TM CTF. Further, ligands that stabilize the heterodimer may antagonize aGPCR signaling while others may induce adhesion without dissociation of the NTF and CTF or inhibition of CTF signaling (38, 39). The NTF itself can serve additional functions independently of the CTF (40, 41). However, it remains unknown how cleavage affects the biochemical composition and the trafficking of these proteins in vivo to regulate the signaling pathways of the cleaved fragments.
In this study, we defined the molecular composition of endogenous Pc1 arising from GPS cleavage in the tissues and cells from postnatal and adult mice and describe its intracellular trafficking in vivo. We discovered novel complexity of endogenous Pc1 molecules arising from GPS cleavage and identified two distinct cleaved Pc1 molecules in vivo: (i) the heterodimeric cleaved full-length (Pc1cFL) form, in which the NTF subunit remains noncovalently associated to the CTF subunit, and (ii) the novel Pc1deN form, in which the NTF subunit is detached from the CTF. We show for the first time that GPS cleavage of endogenous Pc1 occurs in the endoplasmic reticulum (ER) and is not an absolute prerequisite for Pc1 trafficking to the Golgi compartment in vivo. Analysis using bacterial artificial chromosome (BAC) transgenesis establishes that Pc1deN does not traffic autonomously but is “carried” via Pc1cFL. This study on Pc1 biogenesis and trafficking leads to a model in which individual Pc1 forms play multiple roles in kidney development and homeostasis.
MATERIALS AND METHODS
Animals.
We previously produced the transgenic Pkd1extra and Pkd1TAG mouse lines, the knock-in Pkd1V/V and Pkd1MYC/MYC mice, and the knockout Pkd1ΔCMYC/ΔCMYC mice as well as obtained the N-ethyl-N-nitrosourea (ENU)-induced Pkd1m1Bei/m1Bei mice (26, 42–45). These mouse models were bred to a C57BL/6J genetic background. All animal experiments conformed to the standards of the Canadian Council of Animal Care of Institut de Recherches Cliniques de Montréal and of the Animal Care and Use Committees of Johns Hopkins School of Medicine and the University of Maryland School of Medicine.
Genotype analysis.
Mouse genotyping was accomplished on DNA extracted from tail biopsy specimens. The Pkd1TAG and Pkd1extra transgenes were genotyped by Southern blots using EcoRI (Pkd1 probe exon 7 to 15) and BamHI (Pkd1 probe exon 23 to 25), respectively (43, 46). To identify Pkd1V heterozygous mice, we used PCR amplification with the following oligonucleotides: forward, Pkd1 exon 23 (5′-CCA AAC AAC TCA GAC CAG G-3′), and reverse, Pkd1 intron 23 (5′-ACC AGG ACA GCA AGA AAA C-3′). These produce amplicons of 280 bp (wild-type [WT] Pkd1) and 320 bp (Pkd1V allele). The heterozygous or homozygous Pkd1V allele on the transgenic Pkd1extra or Pkd1TAG mouse background was distinguished by TaqMan gene copy number assay. Quantitative PCR (qPCR) was performed with the forward primer (Intron 23 Pkd1) 5′-TGC CTT TCT TCC CTC CTT GTC-3′, reverse primer (Flp recognition target [FRT] and linker from Pkd1V construct) 5′-GCC GAA GTT CCT ATT CTC TAG AAA GTA T-3′, and a TaqMan probe (FRT and linker from Pkd1V construct), 6FAM-CTC GAC GAA GTT CC-MGBNFQ (where FAM is 6-carboxyfluorescein and MGBNFQ is minor groove binder and nonfluorescent quencher). We used as a normalizer the Dolt gene with the forward primer (intron 1) 5′-GCC CCA GCA CGA CCA TT-3′, reverse primer (Intron 1) 5′-TAG TTG GCA TCC TTA TGC TTC ATC-3′, and a TaqMan probe with Dolt (VIC-CCA GCT CTC AAG TCG-MGBNFQ; Life Technologies). The PCRs were carried out with the PerfeCTa qPCR SuperMix (Quanta Biosciences) in an Mx4000, 3005P (Stratagene), or Viia7 apparatus (Life Technologies).
Histopathological analysis.
Transgenic Pkd1V/V; Pkd1extra mice, Pkd1V/V; Pkd1TAG mice, knock-in Pkd1V/V mice, and nontransgenic age-matched control Pkd1+/+ mice from different ages were sacrificed, and kidney tissues were readily removed. Kidneys were immediately placed in formalin or paraformaldehyde and then embedded in paraffin. Tissue sections (4 to 5 μm thick) were stained with hematoxylin and eosin (H&E) for morphological evaluation using an Axiophot Zeiss microscope (47). Cystic areas of kidneys at P10 from Pkd1V/V; Pkd1extra, Pkd1V/V; Pkd1TAG, and littermate Pkd1V/V and Pkd1+/+ controls were quantified at a magnification of ∼×1.5 to ×1.6 as a function of cyst percentage to total surface using a Leica MX12 microscope and Northern Eclipse software. In addition, cystic surface of the cortex versus medulla was evaluated for Pkd1V/V; Pkd1extra2 and Pkd1V/V controls using the same approach.
Protein analysis.
Immunoprecipitation (IP) studies of the endogenous polycystin-1 were accomplished on mouse tissue samples, embryos, or cells (murine embryonic fibroblasts [MEFs], collecting duct cells, and inner medullary collecting duct [IMCD] cells) homogenized in lysis buffer (20 mM sodium phosphate [pH 7.2], 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5 to 1% Triton X-100) and a cocktail of protease inhibitors (Sigma-Aldrich) (22). The homogenate was incubated for 1 h on ice and cleared of debris by centrifugation at 17,000 × g for 10 min at 4°C. Ten milligrams of protein lysates in 1 ml was typically used for IP with the chicken C-terminal Pc1 antibody (anti-cCC) and goat anti-chicken IgY–agarose beads (PrecipHen; Aves Labs) as described previously (26). The resulting IP products were loaded on 3 to 8% Tris-acetate-SDS-polyacrylamide precast gels or 4 to 12% Tris-glycine-SDS-polyacrylamide precast gels (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad). The membranes were incubated with rabbit polyclonal or rat monoclonal C-terminal anti-CC (anti-rCC) and a horseradish peroxidase (HRP)-conjugated secondary antibody as previously described (26). ECL Prime (GE Health Care Life Sciences) was used for detection on Kodak film or a ChemiDoc XRS+ Pharos imaging system (Bio-Rad). The membranes were then stripped using Restore Western blot buffer (Pierce, VWR) and reprobed with the anti-LRR (7e12) antibody directed to the LRR domain of Pc1 (Santa Cruz Biotechnology) (48). A similar protocol was performed for analysis of Pc1 in the Pkd1MYC/MYC and Pkd1ΔCMYC/ΔCMYC embryos, but the protein lysates were immunoprecipitated with a polyclonal anti-Myc (Cell Signaling Technology) and detected with a rabbit polyclonal anti-Myc (Cell Signaling Technology) or anti-LRR (7e12).
The immunodepletion studies were performed on kidneys, lungs, embryos, MEFs, and IMCD cells. According to the endogenous polycystin-1 expression levels, up to five rounds of immunoprecipitation were carried out to achieve complete depletion of both Pc1U and Pc1cFL. These IP products were monitored for intact Pc1cFL by coprecipitation of NTFs throughout the depletion procedure. The flowthrough fraction was immunoprecipitated with anti-CC (cCC) followed by Western blot analysis with anti-CC (rCC) and anti-LRR (7e12) to verify efficiency of immunodepletion.
Analysis of total protein lysates was carried out on kidneys, lungs, embryos, MEFs, and IMCD cells. Protein extracts were prepared as described previously (43, 44); usually, Pkd1extra (alone or with Pkd1V/V) was loaded at 1/10 of other samples for immunoblotting (IB). Membranes were incubated with anti-LRR (7e12) antibody and the internal control β-tubulin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Sigma-Aldrich and Abcam), followed by ECL Prime or ECL (GE Health Care Life Sciences) for detection. Total protein or IP samples were deglycosylated using peptide N-glycosidase F (PNGase F) or endoglycosidase H (endo-H; New England BioLabs) according to the manufacturer's instructions.
Cell and surface biotinylation studies.
Collecting duct (CD) cells were derived from Pkd1 WT postnatal kidneys for endogenous Pc1 analysis. Surface biotinylation experiments for CD monolayers were performed using a Pierce cell surface protein isolation kit (Thermo Scientific) according to the manufacturer's instructions. Anti-GM130 (Novus biologicals), a cis-Golgi marker, was used as a negative control for surface protein detection. Proteins on the blots were quantified using Quantity One software of the Pharos imaging system (Bio-Rad). The relative amounts of Pc1 NTF and CTF, detected by anti-LRR and CC, respectively, were determined by adjusting their signal intensities to those of noncleavable Pc1V loaded on the same blot.
Statistical analysis.
Values were expressed as means ± standard deviations. Statistical analysis was performed by one-way analysis of variance (ANOVA) with a Tukey correction test for multiple comparisons of the mean of each column to the mean of every other column and computed by Prism 6 software. A P value of 0.05 with a 95% confidence interval was considered significant.
RESULTS
Characterization of endogenous polycystin-1 (Pc1) products generated by GPS cleavage.
The cleaved polycystin-1 form consisting of the NTF associated with the CTF (Pc1cFL) (22) and the uncleaved full-length Pc1 (Pc1U) have previously been described in vitro and are illustrated in Fig. 1A. To identify the endogenous forms of Pc1 and their spatiotemporal expression patterns in vivo, we performed immunoprecipitation (IP) with a chicken antibody directed to the CTF (anti-cCC) from lysates of embryos, kidneys at different postnatal ages, and numerous tissues from adult mice, followed by immunoblot (IB) analysis with anti-rCC and anti-LRR separately. Embryo and adult tissues probed with anti-rCC showed a prominent ∼150-kDa band of Pc1, corresponding to the CTF subunit (Fig. 1B) as reported by Yu et al. (26). In addition, a weak but distinct ∼520-kDa Pc1 band corresponding to the Pc1U was observed. In kidneys, the Pc1U and the CTF subunit were detectable during postnatal development from P3 to P14, but their levels waned considerably thereafter, in contrast with the very high levels in adult lungs (Fig. 1B, bottom panel). Probing with anti-LRR detected a weak Pc1U band in the embryos and adult tissues, consistent with anti-rCC results (Fig. 1B, top panel), and, additionally, a strong Pc1 doublet (∼450 and ∼370 kDa) that is not recognized by anti-rCC (lower panel). Based on their molecular masses (MM) and coimmunoprecipitation (co-IP) with CTF, the doublet bands most likely represent the NTF subunits of Pc1cFL (a 450-kDa band [NTF450] and a 370-kDa band [NTF370], according to their MM) that is associated with the CTF (Fig. 1B, schematic right side). Decreased levels were observed for both coimmunoprecipitated NTF and CTF from the postnatal period to adulthood in the kidney samples. Together, these results indicate that Pc1cFL expression is developmentally regulated in the embryonic kidneys and in the adult tissues. Of interest, the stoichiometry of NTF450 and NTF370 varied among the samples examined; i.e., NTF370 was more abundant in the embryos and adult brain, while the NTF450 band was conversely more predominant in the others (Fig. 1B).
The consistent observation of a Pc1 NTF doublet raised the question of whether these bands represent two distinct Pc1cFL isoforms or result from differential N-glycosylation of Pc1cFL. To address this point, wild-type MEF cells, which express high endogenous levels of Pc1, were used to analyze the N-glycan modification of Pc1cFL with the N-deglycosylases PNGase F and endo-H (endoglycosidase H), which also serves to monitor protein trafficking along the secretory pathway (49–51). This approach is based on the characteristic nonuniform distribution of glycosylation enzymes along the intracellular secretory pathway, making the glycosylation pattern a useful marker indicating the localization of glycoproteins. The general rationale is that N-glycans of glycoproteins in the endoplasmic reticulum (ER) are all high mannose and are susceptible to removal by cleavage using PNGase F or endo-H, whereas complex N-glycans acquired in the medial/trans-Golgi compartment are resistant to removal by endo-H but remain sensitive to PNGase F. Sensitivity to endo-H is therefore indicative of proteins that are still in the ER, whereas proteins that acquire endo-H resistance have egressed the ER and transited through the Golgi compartment (49–51). Anti-cCC IP products from MEF protein extracts were treated with PNGase F or endo-H or left untreated (controls) and analyzed by IB with anti-rCC or -LRR antibodies (Fig. 1C). As seen with embryo and tissue samples (Fig. 1B, bottom panel), the Pc1 CTF in MEFs migrates as a pronounced band at ∼150 kDa (Fig. 1C, bottom panel). Treatment with PNGase F shifted the CTF to a slightly faster-migrating band at ∼140 kDa (the predicted MM of the CTF), whereas endo-H digestion resulted in appearance of two distinct bands at ∼150 and ∼140 kDa. These data indicate that the Pc1 CTF is composed of distinct species of very similar MW and different N-glycan types. Noticeably, the Pc1U was extensively N-glycosylated and exclusively sensitive to endo-H, as shown by its shift to ∼460 kDa (the predicted MM of full-length Pc1) upon treatment (Fig. 1C, bottom panel). The shift of Pc1U upon PNGase F treatment was also observed with anti-LRR (Fig. 1C, top panel). This result indicates that Pc1U is mainly localized to the ER. Importantly, both the NTF450 and NTF370 bands were reduced to a single one at ∼320 kDa, the predicted MM of NTF, by PNGase F treatment (Fig. 1C, top panel, lane 3). Analysis with endo-H indicated that NTF450 was endo-H resistant, while NTF370 was endo-H sensitive, as revealed by its shift to ∼320 kDa. Hence, the NTF doublet bands do not correspond to two distinct Pc1cFL isoforms but rather result from differential N-glycosylation modification in the NTF subunits of Pc1cFL. The NTF subunit therefore consists of both endo-H-resistant and -sensitive pools, as was found for the CTF subunits. Collectively, these data provide evidence for one single endogenous Pc1cFL form that could traffic from the Golgi compartment to the plasma membrane.
To determine whether differential N-glycosylation of endogenous Pc1cFL also occurs in the kidney, similar experiments were performed using P5 wild-type kidneys. Anti-LRR detected three bands from untreated kidney samples: the Pc1U and the more abundant doublet of NTF subunits from Pc1cFL (Fig. 1D). Pc1U was sensitive to both PNGase F and endo-H, as observed in MEFs, thereby pointing to ER localization. The NTF450 and NTF370 bands were reduced to a single band of ∼320 kDa by PNGase F treatment and exhibited endo-H resistance and sensitivity, respectively, supporting the presence of differential N-glycan modifications of Pc1cFL in the kidney. Analogous results were obtained with the lung (see Fig. 2C) and embryo (see Fig. 6). Collectively, these results show one single endogenous Pc1cFL form present in the ER and post-ER/Golgi compartments of kidneys and multiple tissues/cells and argue that Pc1 GPS cis-autoproteolytic cleavage occurs in the ER in vivo.
FIG 2.
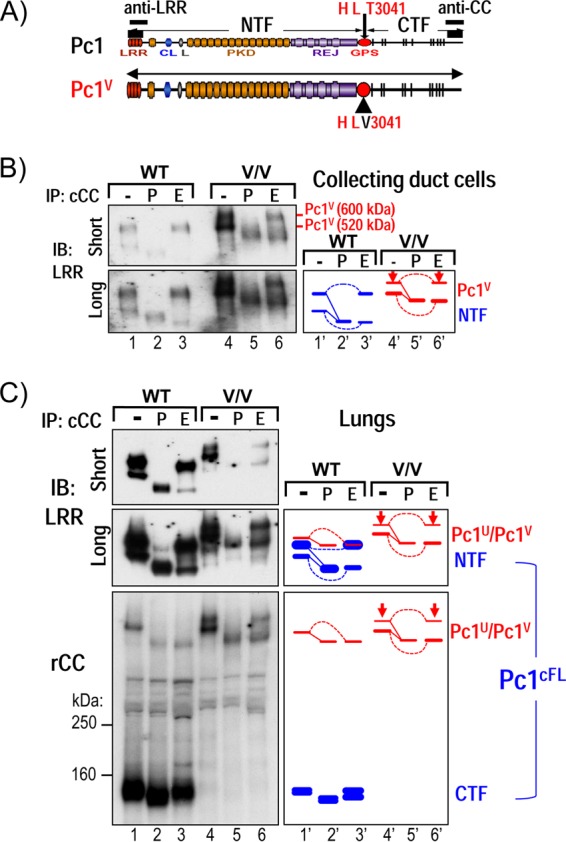
GPS cleavage is not a prerequisite for Pc1 intracellular trafficking. (A) Schematic structure of WT Pc1 and noncleavable Pc1V with a T3041V substitution at the HL↓T3041 cleavage consensus site, corresponding to the length of Pc1U. (B) N-glycosylation modification of Pc1 from collecting duct (CD) cells derived from WT and Pkd1V/V (V/V) postnatal kidneys was analyzed by IP with anti-cCC, either untreated (−) or treated with PNGase F (P) or endo-H (E), and then detected by IB with anti-LRR. In Pkd1V/V CD cells, the upper Pc1V band is endo-H resistant (arrow), and the lower Pc1V band is endo-H sensitive, as indicated in the schematic diagram. Note that Pc1U from WT CD cells was not detectable (lane 1). (C) N-glycosylation modification of Pc1 from WT and Pkd1V/V (V/V) postnatal lungs was analyzed for anti-cCC IP products with anti-LRR and anti-rCC, similarly as described for panel B. Of note, Pc1U is weakly detected in the WT lungs, indicated by a red line in the right diagram.
FIG 6.
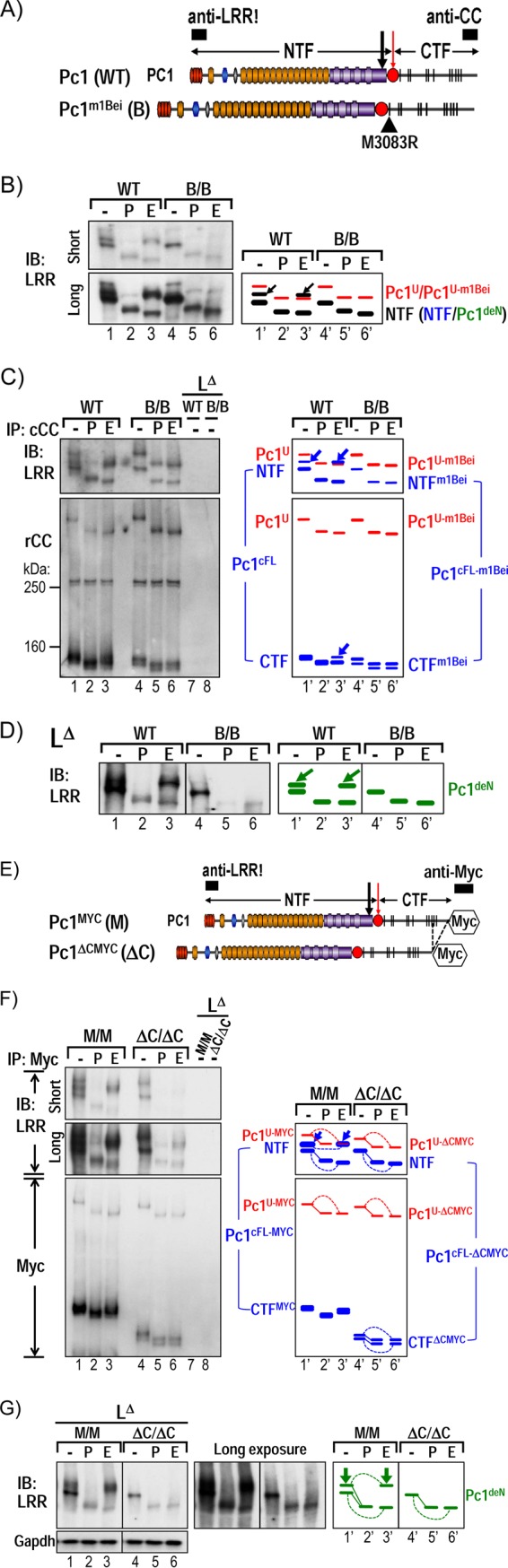
Intact CTF is required for intracellular trafficking of the Pc1deN form. (A) Schematic diagram of Pc1 from WT Pkd1 and Pkd1m1Bei alleles. The Pc1m1Bei contains a single substitution (M3083R) in the first TM domain of CTF (black triangle). Epitope positions of anti-LRR and anti-CC are indicated (black boxes). (B) Endogenous Pc1 forms from WT and homozygous Pkd1m1Bei/m1Bei (B/B) embryos (E12.5) were monitored by IB on total lysates either untreated (−) or deglycosylated with PNGase F (P) or endo-H (E) using anti-LRR. Pkd1m1Bei/m1Bei embryos express mutant full-length Pc1U-m1Bei, with exclusive endo-H sensitivity, similar to Pc1U in WT embryos (red). Pc1 NTF in Pkd1m1Bei/m1Bei embryos lacks endo-H resistance relative to WT embryos (arrows). Schematic diagram identifies the corresponding bands. (C) N-glycosylation status of endogenous Pc1U and Pc1cFL forms from WT and mutant Pkd1m1Bei/m1Bei embryos was monitored by IB on anti-cCC immunoprecipitates, either untreated (−) or treated with PNGase F (P) or endo-H (E). Pc1 products were detected with anti-LRR and anti-rCC as indicated. The absence of endo-H resistance of both Pc1 NTF (as observed for total NTF in panel B) and CTF subunits in Pkd1m1Bei/m1Bei embryos contrasts with endo-H resistance in WT embryos (blue, arrows). Re-IP of the flowthrough fractions with anti-cCC (LΔ) confirmed complete depletion of Pc1U and Pc1cFL from both WT and Pkd1m1Bei/m1Bei/ embryo lysates. The schematic diagram depicts corresponding bands. (D) N-glycosylation status of endogenous Pc1deN was analyzed by IB with anti-LRR from depleted lysates (LΔ) of WT and Pkd1m1Bei/m1Bei embryos following deglycosylation. Pc1deN of the Pkd1m1Bei/m1Bei embryos lacks endo-H resistance relative to WT embryos (arrows), as indicated by the schematic diagram at right. (E to G) Results of N-glycosylation analysis for endogenous Pc1 forms from E12.5 Pkd1MYC/MYC knock-in (M/M) and Pkd1ΔCMYC/ΔCMYC knockout (ΔC/ΔC) embryos using the same method as for the Pkd1m1Bei/m1Bei/ (B/B) embryos in panels A to C, except that anti-Myc was used to immunoprecipitate and detect endogenous Myc-tagged Pc1 molecules.
To define the nature of the association between CTF and NTF in the endogenous Pc1cFL form, lysates from MEFs were immunoprecipitated with anti-cCC antibody either under denaturing conditions (Fig. 1E, lane D, 0.1% SDS) to dissociate noncovalent protein interactions or nondenaturing conditions (Fig. 1E, lane Non-D, 0.5% Triton X-100). While Pc1 CTF and Pc1U were detected under both conditions (Fig. 1E, bottom panel), both NTF450 and NTF370 were detected only under nondenaturing conditions (Fig. 1E, top panel). Therefore, the endogenous Pc1cFL complex consists of NTF and CTF associated via noncovalent interactions.
Pc1 GPS cleavage is not a prerequisite for intracellular trafficking to the Golgi compartment.
Two possible mechanisms could be responsible for the lack of endo-H-resistant Pc1U. First, GPS cleavage is essential for Pc1 trafficking out of the ER. Second, Pc1U does exit the ER but becomes rapidly cleaved before or upon reaching the cis-Golgi network, consequently preventing detectable levels of endo-H-resistant Pc1U to be achieved at steady state. To differentiate between these two possibilities, we examined the N-glycosylation status of the noncleavable Pc1V in renal collecting duct cells from mutant Pkd1V/V mice (Table 1 and Fig. 2A). Two bands of ∼600 kDa and ∼520 kDa were detected with anti-LRR for Pc1V in untreated samples, whereby the lower band migrated to a similar position as the upper NTF band of wild-type collecting duct cells (Fig. 2B, lanes 1 and 4). Both Pc1V forms collapsed to a single band at the predicted MM of ∼460 kDa upon PNGase F treatment (lane 5). Importantly, the ∼600-kDa Pc1V was resistant to endo-H digestion, indicating that Pc1V localizes to a post-ER or -Golgi compartment. The ∼520-kDa Pc1V band, in contrast, was endo-H sensitive and shifted to ∼460 kDa upon endo-H treatment (Fig. 2B, lane 6). Similar results were obtained with the lungs of Pkd1V/V mice, whereby the lower Pc1V band comigrated with the Pc1U band of wild-type lung (Fig. 2C). Together, our data suggest that noncleavable Pc1V can traffic to the Golgi compartment as the endogenous cleaved form, Pc1cFL. These results, while divergent from models based on recombinant Pc1 and aGPCRs (52, 53), indicate that GPS cleavage is not a prerequisite for endogenous Pc1 intracellular trafficking to the Golgi compartment in vivo.
TABLE 1.
Pkd1 mouse lines
| Mouse line | Genetic modification | Description | Reference |
|---|---|---|---|
| Pkd1V/V | Knock-in | T3041V at GPS/GAIN domain produces noncleavable Pc1V | 26 |
| Pkd1extra2 | Transgenic (∼80 copies) | F3043X in Pkd1-BAC produces NTF-like protein | 44 |
| Pkd1extra39 | Transgenic (∼2 copies) | F3043X in Pkd1-BAC produces NTF-like protein | 44 |
| Pkd1TAG26 | Transgenic (∼15 copies) | Full-length Pkd1WT-BAC overexpresses endogenous Pc1 | 43 |
| Pkd1m1Bei/m1Bei | ENU mutagenesis | M3083R within the first transmembrane domain of CTF | 42 |
| Pkd1MYC/MYC | Knock-in | Produces fully functional Pc1 with a C-terminal 5×Myc tag | 45 |
| Pkd1ΔCMYC/ΔCMYC | Knockout | Produces 5×Myc-tagged Pc1 lacking C-terminal 257 aa | 45 |
Characterization of Pc1 products at the cell surface.
To identify the specific Pc1 forms localized to the cell surface, surface biotinylation experiments were performed in wild-type collecting duct cells. Intact monolayers were treated with or without a membrane-impermeant biotinylation reagent, and cell lysates were incubated with avidin-agarose and analyzed by IB (Fig. 3). The avidin-bound proteins from surface biotinylated cells contained the upper but not the lower Pc1 NTF product (Fig. 3, LRR blot, lane 4), consistent with their endo-H reactivity patterns (Fig. 2B, lane 3). CTF could also be detected in the biotinylated sample on the same blot after stripping (Fig. 3, CC blot, lane 4). The relative NTF or CTF quantity at the cell surface was determined by comparing the signal intensity of the NTF or CTF detected in the surface protein population relative to that in total lysates. We found that surface Pc1 NTF (NTF450) makes up about 50% of total cellular endo-H-resistant Pc1 NTF, whereas only about 5% of Pc1 CTF was located at the cell surface. This result indicates that while a small amount of Pc1 NTF can be associated with the CTF in the form of Pc1cFL at the cell surface, Pc1 NTF appeared predominantly as a stand-alone Pc1 molecule that is detached from the CTF.
FIG 3.
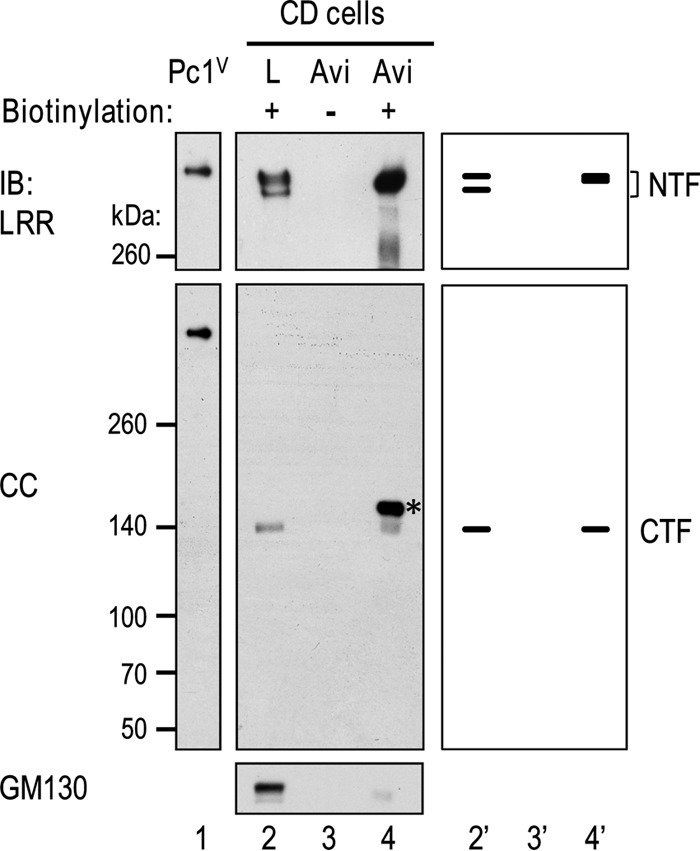
Identification and characterization of Pc1 products on cell surfaces of collecting duct (CD) cells. Confluent CD monolayers were untreated (lane 3) or treated with sulfo-NHS-SS-biotin [sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate] (lane 4). Protein lysates were prepared and incubated with NeutrAvidin-agarose (Avi, lanes 3 and 4). The bound proteins were eluted and analyzed by Western blotting using antibodies as indicated. The lack of detection of GM130 (a cis-Golgi protein) in the biotinylated protein population (lane 4) indicates that surface proteins were exclusively biotinylated. Total cell lysate (L) treated with biotin served as a positive control for NTF and CTF (lane 2), with an amount loaded that is equivalent to 1/20 of the amount used for NeutrAvidin-agarose binding. Recombinant Pc1V served to indicate the position of uncleaved Pc1 (lane 1). The schematic diagram at right provides an identification guide. The asterisk indicates a nonspecific band that is seen in the surface protein population (lane 4).
A novel endogenous detached form of Pc1 NTF: Pc1deN.
To determine whether a novel detached form of Pc1 NTF, here termed Pc1deN, exists in vivo, we devised an immunodepletion strategy that would specifically separate Pc1deN from other Pc1 forms (Fig. 4A). In this approach, Pc1cFL and Pc1U were quantitatively removed from total lysates by IP with anti-cCC under nondenaturing conditions, and the putative Pc1deN was assessed in flowthrough lysate (Fig. 4, LΔ) after depletion. Depletion efficiency was assessed by reprecipitation of the flowthrough lysate with anti-cCC and IB with anti-rCC or -LRR. We confirmed that the immunodepletion was complete by the absence of Pc1cFL and Pc1U signals in the unbound fraction via reprecipitation with anti-cCC (data not shown). Western blots of the depleted lysate (Fig. 4B, LΔ) with anti-LRR readily identified two Pc1deN products at ∼450 kDa (Pc1deN450) and ∼370 kDa (Pc1deN370) as in the original total lysate (Fig. 4B, L) in inner medullary collecting duct (IMCD) cells or kidney tissues (Fig. 4C). Pc1deN450 was endo-H resistant, Pc1deN370 was endo-H sensitive, and both had the same mobilities as the two NTF bands in the total lysate. Semiquantification of the signal intensities of Pc1 NTF bands in the flowthrough lysate and total lysate indicates that the relative ratio of Pc1deN to Pc1cFL is ∼10:1. Pc1deN was also found in the wild-type lung and MEF cells (data not shown), suggesting that this may be functional in vivo. These data show that GPS cleavage of Pc1 gives rise both to the Pc1cFL form and to a distinct and significant pool of Pc1deN that traffics from the ER to the Golgi compartment and to the cell surface (Fig. 3).
FIG 4.
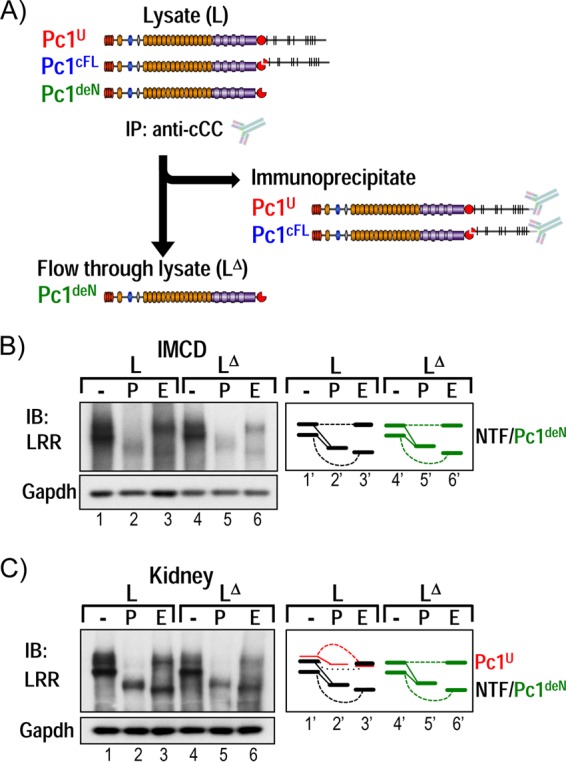
Identification of a novel endogenous Pc1 form: Pc1deN. (A) Immunodepletion strategy to identify the Pc1 NTF detached from the CTF subunit, Pc1deN. Pc1U and Pc1cFL are exhaustively immunoprecipitated from total lysates (L) with anti-cCC, and the putative Pc1deN is analyzed from immunodepleted lysates (LΔ) with anti-LRR. (B) N-glycosylation of endogenous Pc1deN in IMCD cells was analyzed from total lysate (L) and immunodepleted lysates (LΔ) by IB with anti-LRR, after deglycosylation with PNGase F (P) or endo-H (E) or no treatment (−). Note that the Pc1U form was not detectable. The depleted lysate (LΔ) is devoid of Pc1cFL (data not shown). Of interest, Pc1deN was detected in both endo-H-resistant (upper band) and -sensitive (lower band) forms, as schematically depicted at right. GAPDH was used as a loading control. (C) N-glycosylation of endogenous Pc1deN in P5 WT kidney was analyzed from total lysate (L) and immunodepleted lysates (LΔ) as described for IMCD cells in panel B. In total lysate, Pc1cFL overlapped with Pc1deN. The schematic diagram provides an identification guide.
Nonautonomous intracellular trafficking of the Pc1deN form.
The finding of Pc1deN as a significant form of endogenous GPS-cleaved Pc1 suggests that Pc1deN has a functional role in renal homeostasis. Consistent with this idea, the Pkd1V/V mouse mutant, which expresses a noncleavable Pc1 and therefore lacks the Pc1deN and Pc1cFL forms, gradually acquires cysts in the distal tubules and collecting ducts after birth (26). To determine if the cystic disease caused by the Pkd1V/V mutation could be rescued or ameliorated by coexpression of a Pc1 NTF-like protein, we crossed Pkd1V/V mice with two different lines of the Pkd1extra transgenic mouse model. The Pkd1extra transgenic mouse model expresses a Pc1 protein (Pc1extra) truncated at the GPS cleavage site by introduction of a stop codon at aa 3043 in a Pkd1-BAC vector (Table 1 and Fig. 5A) (44). The two different transgenic lines express Pc1extra with the correct temporal pattern of wild-type Pkd1 but at different levels in the kidney: the Pkd1extra39 line expresses Pc1extra at ∼15-fold over the endogenous Pc1 level, whereas the Pkd1extra2 line levels are ∼10 times that of the Pkd1extra39 line (Fig. 5B). Importantly, the transgenic Pkd1extra39 and Pkd1extra2 lines do not display any renal morphological abnormalities in the first few months of age.
FIG 5.

Analysis of Pc1deN functional role by a Pc1extra-BAC transgene in Pkd1V/V mice. (A) Schematic structure of endogenous Pc1 (Pkd1+/+), Pc1V (Pkd1V/V), and Pc1extra (Pkd1extra) proteins. Pc1extra protein was generated by insertion of a termination translation codon in exon 25 of Pkd1 at aa 3043 immediately following the GPS cleavage site. The epitope recognized by anti-LRR is indicated as a black box. (B) Pc1/Pc1V/Pc1extra protein expression levels in P10 kidneys were analyzed by IB with anti-LRR from mice with the genotypes indicated. Protein loading for Pkd1extra2 and Pkd1V/V; Pkd1extra2 mice was decreased by 10-fold (0.9 μg/lane) relative to all other kidney samples (9 μg). Pc1extra exhibits higher expression levels in line Pkd1extra2 than in line Pkd1extra39 and appears in both lines as a single band in comparison to the doublet detected in the wild-type Pc1. β-Tubulin was used as a loading control. (C) Histogram of the kidney weight-to-body weight ratio (KBW) for all genotypes as indicated. The ratios for the Pkd1V/V; Pkd1extra39, Pkd1V/V; Pkd1extra 2, and Pkd1V/V mice at P10 were significantly increased in comparison to the value for WT mice (*, P < 0.0001). n, number of mice. (D) Histopathological analysis (H&E staining) of Pkd1V/V; Pkd1extra kidneys at P10. Pkd1V/V; Pkd1extra39 and Pkd1V/V; Pkd1extra2 mice displayed numerous cysts throughout the kidney parenchyma comparable to Pkd1V/V mice. Scale bar, 100 μm. (E) Histogram of renal cystic index of Pkd1V/V; Pkd1extra kidneys at P10. Cystic involvement (percentage of cystic area) in the Pkd1V/V; Pkd1extra39 and Pkd1V/V; Pkd1extra2 lines shows no significant difference from that in the Pkd1V/V kidneys, but values were highly significant compare to control values (*, P < 0.0001). n, number of mice. (F) Renal cystic involvement in medulla versus cortex in Pkd1V/V and Pkd1V/V; Pkd1extra2 mouse lines at P10. For both Pkd1V/V and Pkd1V/V; Pkd1extra2 mouse lines, cyst surface area (%) is significantly higher in the medulla than in cortex (*, P < 0.0003). Values for the Pkd1V/V; Pkd1extra2 line are not significantly different from those of Pkd1V/V mice in cortex or medulla. n, number of mice. (G) Kaplan-Meier survival curves of the Pkd1V/V, Pkd1V/V; Pkd1extra39, and Pkd1V/V; Pkd1extra2 mice revealed similar life expectancies. (H) Pc1/Pc1V/Pc1extra N-glycosylation status at P10 kidneys was analyzed by IB with anti-LRR on kidney lysates from control Pkd1+/+, Pkd1V/V; Pkd1extra39, and Pkd1V/V; Pkd1extra2 mice, either untreated (−) or deglycosylated with PNGase F (P) or endo-H (E). Pc1 NTF in WT kidneys displayed both Pc1 endo-H-resistant and -sensitive forms, whereas Pc1extra in Pkd1V/V; Pkd1extra39 and Pkd1V/V; Pkd1extra2 kidneys is mainly endo-H sensitive. Protein loading for Pkd1V/V; Pkd1extra2 mice was decreased by 10-fold in comparison to other kidney samples. GAPDH served as a loading control.
Each Pkd1extra line was bred with the Pkd1V/+ mice to generate compound Pkd1V/V; Pkd1extra2 and Pkd1V/V; Pkd1extra39 animals. The compound Pkd1V/V; Pkd1extra mice expressed transgenic Pc1extra molecules in the kidney tissues similar to their respective Pkd1extra lines (Fig. 5B). Despite the high levels of Pc1extra, both lines of compound Pkd1V/V; Pkd1extra mice exhibited high kidney-to-body weight ratios similar to the Pkd1V/V mice, and these ratios were ∼7- to 8-fold increased over those of age-matched control wild-type mice (Fig. 5C). Further, these Pkd1V/V; Pkd1extra mice developed renal cystic expansion indistinguishable from the Pkd1V/V littermates at P10 (Fig. 5D). Analysis of the cystic index for these Pkd1V/V; Pkd1extra kidneys was comparable to that of the Pkd1V/V controls and significantly increased relative to age-matched wild-type controls (P < 0.0001) (Fig. 5E). Histomorphologic analysis of the cortex and medulla revealed significantly lower cystic involvement in the cortex than the medulla for both Pkd1V/V and Pkd1V/V; Pkd1extra kidneys (P < 0.0003), consistent with the preponderant distal nephron cystogenesis (Fig. 5F). Consequently, these compound Pkd1V/V; Pkd1extra mice had similar life expectancies as the Pkd1V/V littermates as determined from Kaplan-Meier curves (Fig. 5G). These results show that Pc1extra expression was not sufficient to prevent postnatal renal cystogenesis or affect the life span of the Pkd1V/V mice and suggest that Pc1deN acquires a critical property through GPS cleavage of nascent Pc1U.
To determine the reason for the absence of Pkd1V/V rescue by Pc1extra, the expression and posttranslational modification of Pc1extra were examined. In Pkd1V/V; Pkd1extra kidneys, the Pc1extra product was expressed at levels similar to those of Pkd1extra kidneys (Fig. 5B). However, it appeared as a single and intense band in both lines, whereas endogenous Pc1 from kidney tissues of nontransgenic mice migrated as a doublet. Moreover, the Pc1extra band in Pkd1V/V; Pkd1extra kidney lysates of both transgenic lines was N-glycosylated, similar to the NTF band in nontransgenic kidney, but was mainly endo-H sensitive (Fig. 5H). This result indicates that most Pc1extra molecules in the Pkd1V/V background do not exit the ER efficiently, unlike the endogenous wild-type Pc1deN derived via GPS cleavage, which moves throughout the secretory pathway. Our result suggests that the CTF of Pc1 is required for the Pc1deN to exit the ER.
Intact CTF is required for nonautonomous intracellular trafficking of the Pc1deN form.
To examine the dependence of Pc1deN trafficking on the Pc1 CTF, we undertook biochemical analysis of Pc1 forms in two different Pkd1 mouse models with mutations within the CTF, which we postulated impaired trafficking of the CTF. Importantly for this approach, the NTF of these Pkd1 mutants must have a wild-type GPS/GAIN domain that can undergo normal GPS cleavage.
The Pkd1m1Bei mouse was the first mutant CTF model examined. Pkd1m1Bei/m1Bei mice carry only a single amino acid substitution, M3083R, within the first TM domain of CTFm1Bei (Table 1 and Fig. 6A), which results in renal cyst formation starting at E15.5 (42, 54). Western blot analysis of Pkd1m1Bei/m1Bei embryo lysates with anti-LRR detected a single endo-H-sensitive NTF band (Fig. 6B, B/B) that comigrated with the Pc1 NTF370 in wild-type embryos. In addition, the Pc1U-m1Bei was exclusively endo-H sensitive (Fig. 6B). The cleavage pattern of the mutant Pc1 products was examined by immunodepletion (Fig. 4A). As shown in Fig. 6C, the Pc1cFL-m1Bei form is generated in the Pkd1m1Bei/m1Bei embryos, as evidenced by co-IP of NTF by the CTFm1Bei. The ability to undergo GPS cleavage suggests that Pc1m1Bei is able to fold properly within the GPS/GAIN domain. Notably, the CTFm1Bei appeared as a doublet, and both bands shifted similarly upon endo-H or PNGase F treatment (Fig. 6C, bottom panel). This doublet likely corresponds to the previously reported CTF isoforms resulting from alternative splicing of Pkd1 exon 31 (38 aa, 3.9 kDa) (55). Hence, both the NTF and CTF subunits of Pc1cFL-m1Bei appear exclusively endo-H sensitive, as was the Pc1U-m1Bei form (Fig. 6C), suggesting that the mutant Pc1cFL-m1Bei form, as well as the Pc1U-m1Bei form, is unable to egress from the ER despite proper cleavage. Most importantly, the Pc1deN form generated from Pc1cFL-m1Bei was essentially endo-H sensitive and appeared as a single band comigrating with the wild-type Pc1 NTF370 (Fig. 6D). Thus, despite having wild-type sequence, the Pc1deN of Pc1m1Bei was retained in the ER due to CTFm1Bei. Therefore, the wild-type CTF appears required for proper ER-Golgi compartment trafficking of both Pc1cFL and Pc1deN.
The second mutant mouse investigated, Pkd1ΔCMYC/ΔCMYC (Fig. 6, ΔC/ΔC), expresses cleavable Pc1 that is truncated by replacement of the C-terminal 257 aa of Pc1 with a Myc-epitope tag (45) (Table 1 and Fig. 6E). The phenotypically normal Pkd1MYC/MYC knock-in (Fig. 6, M/M) littermates, which express Myc-tagged full-length Pc1, served as controls. Similar to the Pkd1m1Bei mutation, this C-terminal truncation did not prevent formation of Pc1cFL but impaired its trafficking, as NTFΔCMYC and CTFΔCMYC were exclusively endo-H sensitive (Fig. 6F). Of note, it cannot be excluded that CTFΔCMYC may have a misfolded motif but with minor effect on the stability of the CTFΔCMYC doublet level. The mutant Pc1U-ΔCMYC form was also entirely endo-H sensitive, implying that the C-terminal domain of Pc1 is critical for intracellular trafficking of Pc1U as well as for Pc1cFL. The Pc1deN-ΔCMYC generated in Pkd1ΔCMYC/ΔCMYC embryos was detected as a single endo-H-sensitive band corresponding to the lower band of the Pc1deN-MYC doublet of the Pkd1MYC/MYC controls (Fig. 6G), as in the Pkd1m1Bei/m1Bei mutant. Together, the data from the two Pkd1 mutant mice exclude autonomous trafficking of Pc1deN and support the model that Pc1deN is carried to the Golgi compartment via Pc1cFL. Furthermore, these results highlight that the CTF is likely critical for proper trafficking of all Pc1 forms in vivo.
Functional rescue of Pkd1V/V phenotype by Pkd1-BAC transgenesis.
To provide evidence for the importance of an intact CTF in Pc1 trafficking, we determined whether the Pkd1V/V mouse phenotype could be rescued by the Pkd1TAG26-BAC transgene (Table 1 and Fig. 7A) that expresses wild-type Pc1 15-fold over endogenous levels in renal tissue (Fig. 7B). The Pkd1TAG26 mice were bred with Pkd1V/+ mice to generate the compound Pkd1V/V; Pkd1TAG animals. As shown in Fig. 7C, the Pkd1V/V; Pkd1TAG mice exhibited a normal kidney-to-body weight ratio (n = 8; 1.2 ± 0.1) that is similar to that of age-matched wild-type controls (n = 15; 1.3 ± 0.2) and is significantly decreased compared to that of Pkd1V/V controls (n = 10; 7.6 ± 2.4; P < 0.0001) at P10. Consistently, the Pkd1V/V; Pkd1TAG mice displayed normal kidney structure and function when analyzed at P10 and 3 months. The renal cystic area in Pkd1V/V; Pkd1TAG mice was significantly decreased (n = 7; 1.8 ± 0.9) compared to that of Pkd1V/V controls (n = 10; 32.2 ± 11.1; P < 0.0001) and was similar to that of age-matched normal controls (n = 6; 0.7 ± 0.3). Importantly, the Pkd1V/V; Pkd1TAG mice had a prolonged life expectancy compared to both the Pkd1V/V and the Pkd1TAG26 mice of up to 1 year. In addition, the Pkd1V/V; Pkd1TAG mice express the full complement of Pc1 cleaved products with N-glycosylation patterns for both the NTF and CTF identical to those of the endogenous Pc1 in nontransgenic kidneys (Fig. 7D and E). These data demonstrate that overexpression of wild-type Pc1 by the Pkd1TAG26 transgene can compensate for the mutant Pc1V and prevent Pkd1V/V renal cystogenesis. Our data provide evidence that the normal CTF is necessary for Pc1deN intracellular trafficking via Pc1cFL.
FIG 7.

Functional complementation of Pkd1V/V by Pkd1-BAC transgenic mice. (A) Schematic diagram of Pc1tg (Pkd1TAG) and Pc1V (Pkd1V/V). The epitopes recognized by anti-LRR and anti-CC are indicated as black boxes. (B) Protein expression of P10 kidneys from Pkd1+/+, Pkd1TAG, Pkd1V/V, and Pkd1V/V; Pkd1TAG mice were analyzed by IB using anti-LRR. Pc1 expression in Pkd1TAG and Pkd1V/V; Pkd1TAG mice was increased, and Pc1 migrated as a doublet, like endogenous Pc1. β-Tubulin served as a loading control. (C) Kidney histology (H&E staining) of Pkd1+/+, Pkd1V/V, and Pkd1V/V; Pkd1TAG mice. Pkd1V/V; Pkd1TAG mice showed complete rescue of the Pkd1V/V renal phenotype, similar to the WT controls at P10 and 3 months of age. Scale bar, 100 μm. (D) N-glycosylation status of Pc1 from Pkd1V/V; Pkd1TAG P10 kidneys was monitored by IB on anti-cCC immunoprecipitates, either untreated (−) or treated with PNGase F (P) or endo-H (E). Pc1 products were detected with anti-LRR and anti-rCC as indicated. Pc1cFL and Pc1U patterns in Pkd1V/V; Pkd1TAG kidneys are identical to those of the endogenous Pc1 in WT kidneys shown in Fig. 1D. The schematic diagram indicates different Pc1 forms. (E) Pc1 N-glycosylation status of wild-type Pkd1+/+ and Pkd1V/V; Pkd1TAG P10 kidneys was analyzed using total lysate (L) and immunodepleted lysate (LΔ) by IB with anti-LRR following deglycosylation. Pkd1V/V; Pkd1TAG kidneys produce both endo-H-resistant and -sensitive Pc1deN forms as in WT kidneys (left panel). The schematic diagram provides an identification guide.
DISCUSSION
Cis-autoproteolytic cleavage at the juxtamembrane GPS motif plays an essential role for the biological function of Pc1 (25, 26) and is disrupted by an increasing number of disease-associated PKD1 mutations (22–24). This study uncovered significant complexity of endogenous Pc1 biogenesis by GPS cleavage, with at least two distinct and coexisting cleaved Pc1 molecules in normal mouse tissues: (i) the heterodimeric Pc1cFL form that consists of the NTF noncovalently associated to the CTF and (ii) the novel Pc1deN form that represents the NTF detached from the CTF. Our results reveal that a small amount of uncleaved Pc1U resides primarily in the ER, whereas both Pc1cFL and Pc1deN molecules are generated early in the ER and progress through the secretory pathway. We found that Pc1deN is located at the cell surface. Moreover, the CTF plays a crucial and transient role for Pc1deN trafficking as determined by genetic and biochemical experiments in mice that express transgenic Pc1extra mimicking Pc1deN on a Pkd1V/V background or express two cleavable Pc1 proteins with different CTF mutations. The critical function of CTF for Pc1deN trafficking was shown by complementation analysis of the Pkd1V/V mouse mutant with the Pkd1TAG-BAC transgene.
The cleaved forms of Pc1 are predominant in whole embryos, in postnatal kidneys, and in various adult tissues. The finding of significant amounts of endo H-sensitive and -resistant populations of Pc1cFL indicate that GPS cleavage occurs early in the ER in vivo and that the resulting Pc1cFL then transits through the Golgi compartment. Pc1deN appears as abundant as, or in greater quantity than, Pc1cFL. Hence, Pc1cFL and Pc1deN together or independently are key contributors in renal development during postnatal periods and/or maintenance of homeostasis.
A surprising finding of the study is that GPS cleavage per se is not a prerequisite for endogenous Pc1 to exit the ER and transit through the Golgi compartment as determined by the identification of endo-H resistance of Pc1V. A dissociation of GPS cleavage from trafficking was previously shown for the native PKDREJ, a member of the polycystin-1 family, known to be naturally uncleaved and yet localized at the plasma membrane (56). Other reports, in contrast, suggested an essential role for GPS cleavage in progressing into the Golgi compartment based on impaired targeting of recombinant noncleavable GPS mutants in Pc1L3040H (52) and GPR56 (57), but the causal relationship was questioned due to possible protein misfolding. It is plausible that, for wild-type Pc1, Pc1U might also exit the ER as Pc1V but be efficiently converted to the cleaved forms by GPS cleavage before achieving endo-H resistance to detectable levels. The resulting Pc1cFL population is likely the predominant form that exits the ER. The trafficking and relative distribution of various Pc1 molecules in vivo are thus probably affected by the rate of GPS cleavage. Together, our results show that native Pc1 undergoes GPS cleavage prior to trafficking from the ER to the Golgi compartment but has the potential to transit independently of the GPS cleavage mechanism.
The identification of native Pc1deN as a major endogenous Pc1 molecule in tissues that are predominantly endo-H resistant and present at the plasma membrane of renal epithelial cells was striking. Pc1deN cannot be distinguished from the NTF subunits of Pc1cFL electrophoretically in total lysate and is only recognized using the immunodepletion strategy that specifically removes the other Pc1 forms. Pc1deN is more abundant than Pc1cFL at the plasma membrane of renal epithelial cells. This finding initially suggested that Pc1deN might traffic autonomously to reach the plasma membrane and play a critical functional role in renal homeostasis. However, BAC transgenic expression of Pc1extra, a Pc1 NTF-like protein, was unable to complement renal cystic progression and early postnatal death in the Pkd1V/V mice. While this finding precludes us from a functional evaluation of endogenous Pc1deN, it uncovered a novel trafficking mechanism for Pc1deN conferred by GPS cleavage that likely relies on a protein carrier or cofactor. Our biochemical analyses of mutant Pc1 with mutations in either the proximal or distal CTF region from the two Pkd1 mouse models, Pkd1m1Bei/m1Bei and Pkd1ΔCMYC/ΔCMYC, provided evidence that Pc1 CTF may be such a carrier for Pc1deN trafficking. Both Pc1deN and Pc1cFL were retained in the ER despite proper GPS cleavage in both mutants. This characterization not only demonstrates the molecular mechanism responsible for the null phenotype in these mouse mutants but also suggests the presence of at least two determinants within the proximal and distal regions of the CTF subunit. The requirement of the CTF for Pc1deN trafficking and function was demonstrated from biochemical and phenotypical complementation of the Pkd1V/V mouse mutant with the Pkd1TAG-BAC transgene. Together, our data thus show that early trafficking of Pc1deN does not occur autonomously but that Pc1deN is carried to intracellular compartments indirectly via Pc1cFL, followed by subsequent subunit dissociation.
Our finding of a small amount of Pc1cFL coexisting at the surface is consistent with the previous results in recombinant studies (16, 22, 52). One possible explanation for the observed Pc1deN excess (about 10-fold) is that Pc1cFL at the plasma membrane continuously undergoes subunit dissociation followed by internalization and degradation of the resulting dissociated CTF via its cytoplasmic PEST domain (58–60). An alternative explanation for the finding is the previously described cleavage events in the C-terminal tail of the CTF (59, 61, 62), which may result in C-terminal fragments that are translocated to the nucleus for signaling (59, 61). Since Pc1deN is predicted to contain no TM domain, it may be associated to the membrane via another cell surface receptor(s) and/or by lipid modifications, as previously proposed for CIRL/latrophilin and Sonic hedgehog (63, 64). Our result does not exclude the possibility that some of the CTF is dissociated from the NTF at the surface as described for CIRL/latrophilin (63, 64).
Based on these findings, we propose a GPS cleavage-based biogenesis and trafficking model for Pc1 with diverse functions (Fig. 8). Wild-type Pc1cFL dissociates to produce Pc1deN in the ER or traffics to the Golgi compartment (Fig. 8, step 1) and subsequently to the plasma membrane/cell-cell junctions (step 2), where it undergoes subunit (NTF and CTF) dissociation. The resulting Pc1deN may be associated to the membrane via another cell surface receptor(s) and/or by lipid modifications, but the released CTF from Pc1cFL may activate a signal pathway and then is quickly degraded. Pc1cFL may also traffic from the Golgi compartment to cilium for the GPS-dependent function (Fig. 8, step 3). The Pc1cFL-Bei/ΔCMYC mutants lacking the intact CTF cannot traffic from the ER to the Golgi compartment and to the plasma membrane and cilium.
FIG 8.
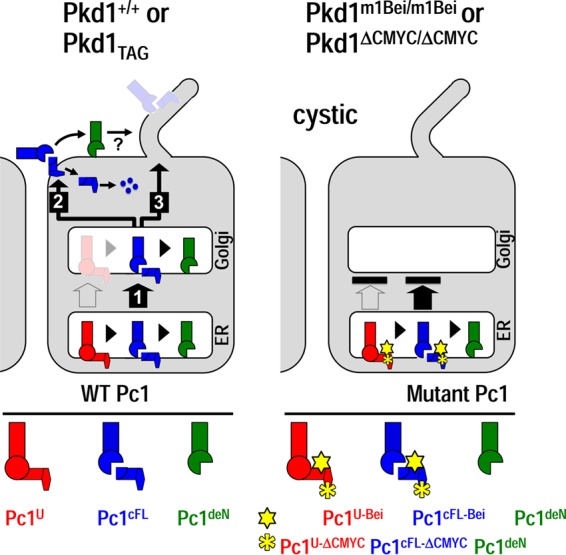
Model for the role of GPS cleavage in Pc1 biogenesis, trafficking, and functions. In wild-type kidneys (left panel), Pc1U is rapidly converted to Pc1cFL by GPS cleavage in the ER, resulting in small amounts that may exit the ER (open arrow). The resulting Pc1cFL is the main form that exits the ER (1) and traffics to the plasma membrane/cell-cell junctions (2) or other locations, possibly the primary cilium (3). Some of the Pc1cFL in the ER and Golgi compartment undergoes subunit dissociation, producing Pc1deN. The Pc1cFL on the plasma membrane/cell-cell junctions could also dissociate. The released CTF is likely rapidly degraded, as indicated by dots. The Pc1deN remains associated on the membrane, likely through the interaction with other membrane proteins or lipid modification, and accumulates over time (curved arrow). Pc1deN/Pc1cFL probably plays an important role at the cell membrane and/or at the cilium. In Pkd1m1Bei/m1Bei or Pkd1ΔCMYC/ΔCMYC pups (right panel) the mutant Pc1 is unable to exit the ER (black bar), leading to the development of massive cysts despite proper GPS cleavage. Schematized Pc1 forms in wild-type and mutant mice are illustrated below.
Our findings also shed light on human ADPKD pathogenic mechanisms triggered by various PKD1 mutations within the CTF subunit and show that these mutations can have as severe consequences as mutations in the NTF. This is consistent with results of a recent report showing that the type of PKD1 mutation, but not its protein location, correlated strongly with renal survival of the patients (65). Moreover, our data predict that a subset of PKD1 mutations affecting the CTF sequence would retain both Pc1cFL and Pc1deN in the ER without affecting GPS cleavage. Alleviation of such CTF carrier defects by providing a substitute could restore trafficking and function of both Pc1cFL and Pc1deN.
This study paves the way toward understanding the biochemical complexity and functions of the GPS-cleaved forms of endogenous Pc1. Crucial insights were devised for the functional role of the different Pc1 forms in renal development and homeostasis. Moreover, we identified for the first time that the CTF subunit can be a promising novel pharmacological target. Future studies will center on the development of innovative designs for therapeutic strategies that promote the trafficking and function of Pc1 forms affected by PKD1 mutations in ADPKD.
ACKNOWLEDGMENTS
We thank J. Calvet, Chiara Gamberi, and Owen Woodward for reading and commenting on the manuscript and B. Magenheimer and M. Chiaravalli for technical assistance.
This work was supported by grants from the Canadian Institutes of Health Research and the Polycystic Kidney Disease Foundation of Canada (to M.T.), by grants from the NIH (R01 DK062199 and P30 DK090868) and National Kidney Foundation of Maryland (to F.Q.), by a Frederick Banting and Charles Best of Canada Graduate Scholarship Award (to A.K.), and by a Korea Research Foundation grant by the Korean Government (KRF-2008-357-E00030, to H.K.).
Footnotes
Published ahead of print 23 June 2014
REFERENCES
- 1.Gabow PA. 1993. Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 329:332–342. 10.1056/NEJM199307293290508 [DOI] [PubMed] [Google Scholar]
- 2.Ahrabi AK, Jouret F, Marbaix E, Delporte C, Horie S, Mulroy S, Boulter C, Sandford R, Devuyst O. 2010. Glomerular and proximal tubule cysts as early manifestations of Pkd1 deletion. Nephrol. Dial. Transplant. 25:1067–1078. 10.1093/ndt/gfp611 [DOI] [PubMed] [Google Scholar]
- 3.Lu W, Shen X, Pavlova A, Lakkis M, Ward CJ, Pritchard L, Harris PC, Genest DR, Perez-Atayde AR, Zhou J. 2001. Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum. Mol. Genet. 10:2385–2396. 10.1093/hmg/10.21.2385 [DOI] [PubMed] [Google Scholar]
- 4.Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, Gabrielson K, Qian F, Mei C, Westphal H, Germino GG. 2004. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J. Am. Soc. Nephrol. 15:3035–3043. 10.1097/01.ASN.0000144204.01352.86 [DOI] [PubMed] [Google Scholar]
- 5.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. 2007. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat. Med. 13:1490–1495. 10.1038/nm1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K, Arnaout MA. 2000. Polycystin 1 is required for the structural integrity of blood vessels. Proc. Natl. Acad. Sci. U. S. A. 97:1731–1736. 10.1073/pnas.040550097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chauvet V, Qian F, Boute N, Cai Y, Phakdeekitacharoen B, Onuchic LF, Attie-Bitach T, Guicharnaud L, Devuyst O, Germino GG, Gubler MC. 2002. Expression of PKD1 and PKD2 transcripts and proteins in human embryo and during normal kidney development. Am. J. Pathol. 160:973–983. 10.1016/S0002-9440(10)64919-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foggensteiner L, Bevan AP, Thomas R, Coleman N, Boulter C, Bradley J, Ibraghimov-Beskrovnaya O, Klinger K, Sandford R. 2000. Cellular and subcellular distribution of polycystin-2, the protein product of the PKD2 gene. J. Am. Soc. Nephrol. 11:814–827 [DOI] [PubMed] [Google Scholar]
- 9.Guillaume R, D'Agati V, Daoust M, Trudel M. 1999. Murine Pkd1 is a developmentally regulated gene from morula to adulthood: role in tissue condensation and patterning. Dev. Dyn. 214:337–348. [DOI] [PubMed] [Google Scholar]
- 10.Guillaume R, Trudel M. 2000. Distinct and common developmental expression patterns of the murine Pkd2 and Pkd1 genes. Mech. Dev. 93:179–183. 10.1016/S0925-4773(00)00257-4 [DOI] [PubMed] [Google Scholar]
- 11.Van Adelsberg J, Chamberlain S, D'Agati V. 1997. Polycystin expression is temporally and spatially regulated during renal development. Am. J. Physiol. 272:F602–F609 [DOI] [PubMed] [Google Scholar]
- 12.Geng L, Segal Y, Peissel B, Deng N, Pei Y, Carone F, Rennke HG, Glucksmann-Kuis AM, Schneider MC, Ericsson M, Reeders ST, Zhou J. 1996. Identification and localization of polycystin, the PKD1 gene product. J. Clin. Invest. 98:2674–2682. 10.1172/JCI119090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palsson R, Sharma CP, Kim K, McLaughlin M, Brown D, Arnaout MA. 1996. Characterization and cell distribution of polycystin, the product of autosomal dominant polycystic kidney disease gene 1. Mol. Med. 2:702–711 [PMC free article] [PubMed] [Google Scholar]
- 14.Geng L, Segal Y, Pavlova A, Barros EJ, Lohning C, Lu W, Nigam SK, Frischauf AM, Reeders ST, Zhou J. 1997. Distribution and developmentally regulated expression of murine polycystin. Am. J. Physiol. 272:F451–F459 [DOI] [PubMed] [Google Scholar]
- 15.Kleymenova E, Ibraghimov-Beskrovnaya O, Kugoh H, Everitt J, Xu H, Kiguchi K, Landes G, Harris P, Walker C. 2001. Tuberin-dependent membrane localization of polycystin-1: a functional link between polycystic kidney disease and the TSC2 tumor suppressor gene. Mol. Cell 7:823–832. 10.1016/S1097-2765(01)00226-X [DOI] [PubMed] [Google Scholar]
- 16.Boletta A, Qian F, Onuchic LF, Bragonzi A, Cortese M, Deen PM, Courtoy PJ, Soria MR, Devuyst O, Monaco L, Germino GG. 2001. Biochemical characterization of bona fide polycystin-1 in vitro and in vivo. Am. J. Kidney Dis. 38:1421–1429. 10.1053/ajkd.2001.29282 [DOI] [PubMed] [Google Scholar]
- 17.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. 2003. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33:129–137. 10.1038/ng1076 [DOI] [PubMed] [Google Scholar]
- 18.Yoder BK, Hou X, Guay-Woodford LM. 2002. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 13:2508–2516. 10.1097/01.ASN.0000029587.47950.25 [DOI] [PubMed] [Google Scholar]
- 19.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. 1995. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 10:151–160. 10.1038/ng0695-151 [DOI] [PubMed] [Google Scholar]
- 20.Ponting CP, Hofmann K, Bork P. 1999. A latrophilin/CL-1-like GPS domain in polycystin-1. Curr. Biol. 9:R585–R588. 10.1016/S0960-9822(99)80379-0 [DOI] [PubMed] [Google Scholar]
- 21.Wei W, Hackmann K, Xu H, Germino G, Qian F. 2007. Characterization of cis-autoproteolysis of polycystin-1, the product of human polycystic kidney disease 1 gene. J. Biol. Chem. 282:21729–21737. 10.1074/jbc.M703218200 [DOI] [PubMed] [Google Scholar]
- 22.Qian F, Boletta A, Bhunia AK, Xu H, Liu L, Ahrabi AK, Watnick TJ, Zhou F, Germino GG. 2002. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. U. S. A. 99:16981–16986. 10.1073/pnas.252484899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arac D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Sudhof TC, Brunger AT. 2012. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 31:1364–1378. 10.1038/emboj.2012.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Gonzalez MA, Jones JG, Allen SK, Palatucci CM, Batish SD, Seltzer WK, Lan Z, Allen E, Qian F, Lens XM, Pei Y, Germino GG, Watnick TJ. 2007. Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease. Mol. Genet. Metab. 92:160–167. 10.1016/j.ymgme.2007.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian F. 2012. Polycystin-1, p 3728–3736 In Rawlings ND, Salvesen G. (ed), The handbook of proteolytic enzymes, 3rd ed. Academic Press, San Diego, CA [Google Scholar]
- 26.Yu S, Hackmann K, Gao J, He X, Piontek K, Garcia-Gonzalez MA, Menezes LF, Xu H, Germino GG, Zuo J, Qian F. 2007. Essential role of cleavage of polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc. Natl. Acad. Sci. U. S. A. 104:18688–18693. 10.1073/pnas.0708217104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fredriksson R, Lagerstrom MC, Hoglund PJ, Schioth HB. 2002. Novel human G protein-coupled receptors with long N-terminals containing GPS domains and Ser/Thr-rich regions. FEBS Lett. 531:407–414. 10.1016/S0014-5793(02)03574-3 [DOI] [PubMed] [Google Scholar]
- 28.Lin HH, Stacey M, Yona S, Chang GW. 2010. GPS proteolytic cleavage of adhesion-GPCRs. Adv. Exp. Med. Biol. 706:49–58. 10.1007/978-1-4419-7913-1_4 [DOI] [PubMed] [Google Scholar]
- 29.Sugita S, Ichtchenko K, Khvotchev M, Sudhof TC. 1998. α-Latrotoxin receptor CIRL/latrophilin 1 (CL1) defines an unusual family of ubiquitous G-protein-linked receptors. G-protein coupling not required for triggering exocytosis. J. Biol. Chem. 273:32715–32724 [DOI] [PubMed] [Google Scholar]
- 30.Abe J, Fukuzawa T, Hirose S. 2002. Cleavage of Ig-Hepta at a “SEA” module and at a conserved G protein-coupled receptor proteolytic site. J. Biol. Chem. 277:23391–23398. 10.1074/jbc.M110877200 [DOI] [PubMed] [Google Scholar]
- 31.Gray JX, Haino M, Roth MJ, Maguire JE, Jensen PN, Yarme A, Stetler-Stevenson MA, Siebenlist U, Kelly K. 1996. CD97 is a processed, seven-transmembrane, heterodimeric receptor associated with inflammation. J. Immunol. 157:5438–5447 [PubMed] [Google Scholar]
- 32.Krasnoperov VG, Bittner MA, Beavis R, Kuang Y, Salnikow KV, Chepurny OG, Little AR, Plotnikov AN, Wu D, Holz RW, Petrenko AG. 1997. alpha-Latrotoxin stimulates exocytosis by the interaction with a neuronal G-protein-coupled receptor. Neuron 18:925–937. 10.1016/S0896-6273(00)80332-3 [DOI] [PubMed] [Google Scholar]
- 33.Lin HH, Chang GW, Davies JQ, Stacey M, Harris J, Gordon S. 2004. Autocatalytic cleavage of the EMR2 receptor occurs at a conserved G protein-coupled receptor proteolytic site motif. J. Biol. Chem. 279:31823–31832. 10.1074/jbc.M402974200 [DOI] [PubMed] [Google Scholar]
- 34.Hsiao CC, Chen HY, Chang GW, Lin HH. 2011. GPS autoproteolysis is required for CD97 to up-regulate the expression of N-cadherin that promotes homotypic cell-cell aggregation. FEBS Lett. 585:313–318. 10.1016/j.febslet.2010.12.005 [DOI] [PubMed] [Google Scholar]
- 35.Paavola KJ, Stephenson JR, Ritter SL, Alter SP, Hall RA. 2011. The N terminus of the adhesion G protein-coupled receptor GPR56 controls receptor signaling activity. J. Biol. Chem. 286:28914–28921. 10.1074/jbc.M111.247973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Promel S, Frickenhaus M, Hughes S, Mestek L, Staunton D, Woollard A, Vakonakis I, Schoneberg T, Schnabel R, Russ AP, Langenhan T. 2012. The GPS motif is a molecular switch for bimodal activities of adhesion class G protein-coupled receptors. Cell Rep. 2:321–331. 10.1016/j.celrep.2012.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward Y, Lake R, Yin JJ, Heger CD, Raffeld M, Goldsmith PK, Merino M, Kelly K. 2011. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res. 71:7301–7311. 10.1158/0008-5472.CAN-11-2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paavola KJ, Hall RA. 2012. Adhesion G protein-coupled receptors: signaling, pharmacology, and mechanisms of activation. Mol. Pharmacol. 82:777–783. 10.1124/mol.112.080309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Promel S, Langenhan T, Arac D. 2013. Matching structure with function: the GAIN domain of Adhesion-GPCR and PKD1-like proteins. Trends Pharmacol. Sci. 34:470–478. 10.1016/j.tips.2013.06.002 [DOI] [PubMed] [Google Scholar]
- 40.Kaur B, Brat DJ, Devi NS, Van Meir EG. 2005. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene 24:3632–3642. 10.1038/sj.onc.1208317 [DOI] [PubMed] [Google Scholar]
- 41.Kaur B, Cork SM, Sandberg EM, Devi NS, Zhang Z, Klenotic PA, Febbraio M, Shim H, Mao H, Tucker-Burden C, Silverstein RL, Brat DJ, Olson JJ, Van Meir EG. 2009. Vasculostatin inhibits intracranial glioma growth and negatively regulates in vivo angiogenesis through a CD36-dependent mechanism. Cancer Res. 69:1212–1220. 10.1158/0008-5472.CAN-08-1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herron BJ, Lu W, Rao C, Liu S, Peters H, Bronson RT, Justice MJ, McDonald JD, Beier DR. 2002. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat. Genet. 30:185–189. 10.1038/ng812 [DOI] [PubMed] [Google Scholar]
- 43.Kurbegovic A, Cote O, Couillard M, Ward CJ, Harris PC, Trudel M. 2010. Pkd1 transgenic mice: adult model of polycystic kidney disease with extrarenal and renal phenotypes. Hum. Mol. Genet. 19:1174–1189. 10.1093/hmg/ddp588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurbegovic A, Trudel M. 2013. Progressive development of polycystic kidney disease in the mouse model expressing Pkd1 extracellular domain. Hum. Mol. Genet. 22:2361–2375. 10.1093/hmg/ddt081 [DOI] [PubMed] [Google Scholar]
- 45.Wodarczyk C, Rowe I, Chiaravalli M, Pema M, Qian F, Boletta A. 2009. A novel mouse model reveals that polycystin-1 deficiency in ependyma and choroid plexus results in dysfunctional cilia and hydrocephalus. PLoS One 4:e7137. 10.1371/journal.pone.0007137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thivierge C, Kurbegovic A, Couillard M, Guillaume R, Cote O, Trudel M. 2006. Overexpression of PKD1 causes polycystic kidney disease. Mol. Cell. Biol. 26:1538–1548. 10.1128/MCB.26.4.1538-1548.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Couillard M, Trudel M. 2009. C-myc as a modulator of renal stem/progenitor cell population. Dev. Dyn. 238:405–414. 10.1002/dvdy.21841 [DOI] [PubMed] [Google Scholar]
- 48.Ong AC, Harris PC, Davies DR, Pritchard L, Rossetti S, Biddolph S, Vaux DJ, Migone N, Ward CJ. 1999. Polycystin-1 expression in PKD1, early-onset PKD1, and TSC2/PKD1 cystic tissue. Kidney Int. 56:1324–1333. 10.1046/j.1523-1755.1999.00659.x [DOI] [PubMed] [Google Scholar]
- 49.Freeze HH. 2001. Use of glycosidases to study protein trafficking. Curr. Protoc. Cell Biol. Chapter 15:Unit 15.2. 10.1002/0471143030.cb1502s03 [DOI] [PubMed] [Google Scholar]
- 50.Kornfeld R, Kornfeld S. 1985. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54:631–664. 10.1146/annurev.bi.54.070185.003215 [DOI] [PubMed] [Google Scholar]
- 51.Stanley P. 2011. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 3:a005199. 10.1101/cshperspect.a005199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapin HC, Rajendran V, Caplan MJ. 2010. Polycystin-1 surface localization is stimulated by polycystin-2 and cleavage at the G protein-coupled receptor proteolytic site. Mol. Biol. Cell 21:4338–4348. 10.1091/mbc.E10-05-0407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krasnoperov V, Lu Y, Buryanovsky L, Neubert TA, Ichtchenko K, Petrenko AG. 2002. Post-translational proteolytic processing of the calcium-independent receptor of alpha-latrotoxin (CIRL), a natural chimera of the cell adhesion protein and the G protein-coupled receptor. Role of the G protein-coupled receptor proteolysis site (GPS) motif. J. Biol. Chem. 277:46518–46526. 10.1074/jbc.M206415200 [DOI] [PubMed] [Google Scholar]
- 54.Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, Maser RL, Grantham JJ, Calvet JP. 2006. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na+, K+, 2Cl− Co-transporter-dependent cystic dilation. J. Am. Soc. Nephrol. 17:3424–3437. 10.1681/ASN.2006030295 [DOI] [PubMed] [Google Scholar]
- 55.Xu H, Shen J, Walker CL, Kleymenova E. 2001. Tissue-specific expression and splicing of the rat polycystic kidney disease 1 gene. DNA Seq. 12:361–366 [DOI] [PubMed] [Google Scholar]
- 56.Butscheid Y, Chubanov V, Steger K, Meyer D, Dietrich A, Gudermann T. 2006. Polycystic kidney disease and receptor for egg jelly is a plasma membrane protein of mouse sperm head. Mol. Reprod. Dev. 73:350–360. 10.1002/mrd.20410 [DOI] [PubMed] [Google Scholar]
- 57.Jin Z, Tietjen I, Bu L, Liu-Yesucevitz L, Gaur SK, Walsh CA, Piao X. 2007. Disease-associated mutations affect GPR56 protein trafficking and cell surface expression. Hum. Mol. Genet. 16:1972–1985. 10.1093/hmg/ddm144 [DOI] [PubMed] [Google Scholar]
- 58.Kim H, Jeong W, Ahn K, Ahn C, Kang S. 2004. Siah-1 interacts with the intracellular region of polycystin-1 and affects its stability via the ubiquitin-proteasome pathway. J. Am. Soc. Nephrol. 15:2042–2049. 10.1097/01.ASN.0000133490.00348.59 [DOI] [PubMed] [Google Scholar]
- 59.Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T. 2006. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell 10:57–69. 10.1016/j.devcel.2005.12.005 [DOI] [PubMed] [Google Scholar]
- 60.Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. 1997. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc. Natl. Acad. Sci. U. S. A. 94:6965–6970. 10.1073/pnas.94.13.6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, Igarashi P, Bennett AM, Ibraghimov-Beskrovnaya O, Somlo S, Caplan MJ. 2004. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J. Clin. Invest. 114:1433–1443. 10.1172/JCI21753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woodward OM, Li Y, Yu S, Greenwell P, Wodarczyk C, Boletta A, Guggino WB, Qian F. 2010. Identification of a polycystin-1 cleavage product, P100, that regulates store operated Ca entry through interactions with STIM1. PLoS One 5:e12305. 10.1371/journal.pone.0012305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Porter JA, Young KE, Beachy PA. 1996. Cholesterol modification of hedgehog signaling proteins in animal development. Science 274:255–259. 10.1126/science.274.5285.255 [DOI] [PubMed] [Google Scholar]
- 64.Volynski KE, Silva JP, Lelianova VG, Atiqur Rahman M, Hopkins C, Ushkaryov YA. 2004. Latrophilin fragments behave as independent proteins that associate and signal on binding of LTX(N4C). EMBO J. 23:4423–4433. 10.1038/sj.emboj.7600443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cornec-Le Gall E, Audrezet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, Guillodo MP, Grall-Jezequel A, Saliou P, Ferec C, Le Meur Y. 2013. Type of PKD1 Mutation Influences Renal Outcome in ADPKD. J. Am. Soc. Nephrol. 24:1006–1013. 10.1681/ASN.2012070650 [DOI] [PMC free article] [PubMed] [Google Scholar]