

Abstract
Mice lacking the transcription factor NF-E2 p45-related factor 2 (Nrf2) develop more severe nonalcoholic steatohepatitis (NASH), with cirrhosis, than wild-type (Nrf2+/+) mice when fed a high-fat (HF) diet for 24 weeks. Although NASH is usually associated with insulin resistance, HF-fed Nrf2−/− mice exhibited better insulin sensitivity than HF-fed Nrf2+/+ mice. In livers of HF-fed mice, loss of Nrf2 resulted in greater induction of lipogenic genes, lower expression of β-oxidation genes, greater reduction in AMP-activated protein kinase (AMPK) levels, and diminished acetyl coenzyme A (CoA) carboxylase phosphorylation than in the wild-type livers, which is consistent with greater fatty acid (FA) synthesis in Nrf2−/− livers. Moreover, primary Nrf2−/− hepatocytes displayed lower glucose and FA oxidation than Nrf2+/+ hepatocytes, with FA oxidation partially rescued by treatment with AMPK activators. The unfolded protein response (UPR) was perturbed in control regular-chow (RC)-fed Nrf2−/− mouse livers, and this was associated with constitutive activation of NF-κB and JNK, along with upregulation of inflammatory genes. The HF diet elicited an antioxidant response in Nrf2+/+ livers, and as this was compromised in Nrf2−/− livers, they suffered oxidative stress. Therefore, Nrf2 protects against NASH by suppressing lipogenesis, supporting mitochondrial function, increasing the threshold for the UPR and inflammation, and enabling adaptation to HF-diet-induced oxidative stress.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a common condition that is associated with obesity, type 2 diabetes mellitus, and insulin resistance (1, 2). It represents a spectrum of phenotypes ranging from simple steatosis (fatty infiltration) through nonalcoholic steatohepatitis (NASH) to fibrosis and ultimately cirrhosis. However, the molecular events that dictate the evolution of NASH are not well defined (3).
Consumption of a high-fat (HF) diet, or excess lipogenesis, can lead to endoplasmic reticulum (ER) stress, which in turn stimulates the unfolded protein response (UPR) that attenuates transcriptional and translational programs to restore ER homeostasis (4–6). Initiation of the UPR is carried out by activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and protein kinase RNA (PKR)-like ER kinase (PERK). If ER stress is unresolved, ATF6, IRE1, and PERK stimulate lipogenesis by activating sterol regulatory element binding protein 1c (SREBP-1c), X-box binding protein 1 (XBP1), CCAAT/enhancer-binding protein β (C/EBPβ), and eukaryotic initiation factor 2α (eIF2α) (7–10). Steatosis can arise during unresolved ER stress as a consequence of upregulation of C/EBP homologous protein (CHOP) because it suppresses the expression of key metabolic gene regulators (11, 12).
Progression of liver steatosis to NASH requires the coexistence of inflammation. NASH may develop in individuals with metabolic syndrome because excessive delivery of fatty acids (FA) and triglycerides to the liver leads to increased lipid metabolism, causing oxidative stress through overproduction of reactive oxygen species (ROS) (13). Oxidative stress activates nuclear factor κB (NF-κB) and c-Jun N-terminal kinase (JNK), leading to induction of inflammatory genes, which in turn increase neutrophil recruitment to the liver and exacerbate both oxidative stress and inflammation (14). Besides oxidative stress, NF-κB and JNK can also be activated by ER stress (15, 16), and the fact that ER stress is accentuated by ROS (17) indicates that inflammation in NAFLD is probably inextricably connected with both ER and oxidative stress. Once established, chronic inflammation results in induction of apoptotic genes via JNK/c-Jun signaling (14), which leads to hepatocyte death and fibrosis (18).
The cap 'n' collar (CNC) basic-region leucine zipper (bZIP) transcription factor NF-E2 p45-related factor 2 (Nrf2, also called Nfe2l2) enables cells to adapt to oxidative stress by transactivating cytoprotective genes that contain antioxidant response element (ARE) sequences in their promoters (19–21). Under nonstressed conditions, Nrf2 activity is restricted by its constitutive proteasomal degradation, which is mediated by Kelch-like ECH-associated protein 1 (Keap1), a cullin-3 ubiquitin ligase substrate adaptor (22–24). However, the ability of Keap1 to direct Nrf2 for proteasomal degradation is blocked by thiol-reactive agents, which modify Cys residues in the substrate adaptor (25–27), resulting in induction of ARE-driven genes that provide protection against electrophiles and prooxidants (28).
Nrf2 may play a pivotal role in the development of NASH, because it represses the expression of genes involved in FA synthesis (29–31) and antagonizes inflammation (32). Global knockout of Nrf2 profoundly increases the susceptibility of mice to NASH when they are placed on a methionine- and choline-deficient (MCD) diet (33, 34), whereas genetic activation of Nrf2 by knockdown of Keap1 decreases their sensitivity to NASH caused by the MCD diet (35). The MCD diet, however, is of limited value as a model for human disease because it affects only the liver, produces rapid weight loss, and does not cause insulin resistance, and therefore, challenge with an HF diet provides a more relevant means of producing NAFLD experimentally. It is therefore notable that Nrf2 has been shown to influence hepatic lipid metabolism in HF-fed mice (36–44). Although the duration of HF diet feeding has varied enormously in such studies, it has generally been found that the livers of Nrf2−/− mice accumulate lipid to a greater extent than those of wild-type mice. Some researchers have reported greater expression of lipid metabolism-associated transcription factors and lipid metabolism-associated enzymes/proteins in Nrf2−/− mice than in the wild type, but consistent changes in gene expression have not been observed across all studies. Furthermore, in accordance with the view that Nrf2 inhibits hepatic lipid accumulation, pharmacological activation of Nrf2 has been found to downregulate lipogenesis genes (37, 38, 45). Surprisingly, however, genetic activation of Nrf2 has been reported to increase steatosis and inflammation in both leptin-deficient and HF-fed animals (46, 47), and it has been speculated that Keap1 may influence NASH by uncharacterized mechanisms that do not involve Nrf2. Consistent with this hypothesis, evidence has been provided that Keap1 suppresses activation of the JNK/c-Jun pathway by FA in an Nrf2-independent manner (48).
The primary emphasis of all the HF diet studies in Nrf2−/− mice outlined above has been on the expression of lipid metabolism genes. Little is therefore known about how loss of Nrf2 affects HF diet-stimulated ER stress and oxidative stress or how it influences the metabolic activity of hepatocytes. Moreover, the possible contribution of Nrf2 to insulin resistance during development of NASH is unknown. Thus, during the present study, we examined whether knockout of Nrf2 produces peripheral insulin resistance and whether this predicates increased sensitivity of the liver to NASH upon feeding an HF diet. Moreover, we tested the hypothesis that steatosis in livers of Nrf2−/− mice fed an HF diet disturbs the UPR and causes oxidative stress, both of which drive inflammation.
MATERIALS AND METHODS
Animals.
Throughout this study, male mice were examined. The Nrf2−/− and Nrf2+/+ animals, created by Itoh et al. (49) and provided kindly by Ken Itoh and Masayuki Yamamoto, were backcrossed over six generations onto a C57BL/6 background as described previously (50). All animal care protocols and procedures were performed in accordance to the Animal Scientific Procedures Act (1986) and with the approval of the University of Dundee Animal Ethics Committee. From 8 to 10 weeks of age, the mice were provided ad libitum either regular chow (RC), purchased from SDS Ltd. (Witham, Essex, United Kingdom), or an HF diet, obtained from Testdiets (International Product Supplies, London, United Kingdom). The RC contained 7.5% fat by energy; the HF diet contained 45% fat by energy. Fat mass, spontaneous locomotor activity, and food intake were determined (typically on mice between 24 and 30 weeks of age, which had been fed from the age of 8 to 10 weeks on either the HF diet or RC diet for 16 or 20 weeks) as described previously (51). For analysis of insulin signaling (ex vivo), mice (typically 28 weeks of age) were subjected to an overnight fast and injected with 2 units of insulin/kg body weight or an equal volume of saline intraperitoneally. The quadriceps muscle and liver were collected in liquid nitrogen 5 and 6 min after injection, respectively.
The vast majority of biochemical and molecular biology analyses were performed on livers of Nrf2−/− and Nrf2+/+ mice that had been placed on an RC or HF diet for 24 weeks (32 to 34 weeks of age). Upon sacrifice of these animals, plasma was collected, and the livers were removed. A lobe from each liver was preserved in formalin for histological analyses, and the remainder was snap-frozen in liquid nitrogen.
Physiological and clinical-chemistry measurements.
The EchoMRI-900 quantitative nuclear magnetic resonance (qMR) system (Echo Medical Systems, Houston, TX) was used to determine fat mass and lean mass in conscious mice. Blood samples were collected via tail vein or cardiac puncture performed on terminally anesthetized mice. Blood glucose, triglycerides, cholesterol and free fatty acid, and plasma leptin and insulin were measured as described previously (51). Plasma β-hydroxybutyrate was measured using a colorimetric assay (Cayman Chemical Company, Ann Arbor, MI). Plasma alanine aminotransferase (ALT) activity was measured using kits on a Daytona autoanalyzer (Randox). Glucose and insulin tolerance tests were carried out on mice as described elsewhere (51). The respiratory exchange ratio (RER) and O2 consumption were determined by open-circuit indirect calorimetry (Columbus Instruments).
Histology.
Formalin-fixed murine liver specimens were processed for hematoxylin and eosin staining as described previously (33). The severity of liver disease was evaluated histologically using the NAFLD activity score (NAS), which is the standard system for reporting the extent of damage (52); it represents the combined semiquantitated pathology score for steatosis, inflammation, and hepatocyte ballooning.
Reticulin and Van Gieson's staining of liver sections was undertaken by standard methods. Staining for nitrotyrosine protein adducts was performed as described previously (53), and a terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay was performed as described elsewhere (54). For electron microscopy analysis, livers were fixed in 2.5% glutaraldehyde-4% paraformaldehyde in 0.1 M sodium cacodylate buffer, postfixed in 1% aqueous osmium tetroxide, dehydrated in ethanol, transferred to propylene oxide, and then embedded in Durcupan resin (Sigma). Sections were cut on a Leica ultramicrotome and collected on Pioloform B (polyvinyl butyral)-coated copper grids, stained with uranyl acetate and lead citrate, and examined in a Jeol EX electron microscope. Images were collected on digital-imaging plates and processed in a Ditabis (Pforzeim, Germany) scanner.
Antibodies.
Antibodies against acetyl coenzyme A (CoA) carboxylase (ACC), phosphorylated ACC (p-ACC), phosphorylated AMP-activated protein kinase (p-AMPK), CHOP, cleaved caspase 3, cleaved caspase 9, eIFα, 78-kDa glucose-regulated protein (GRP78, also called BiP), high-mobility group protein B1 (HMGB1), IRE1α, PERK, JNK, and p-JNK were purchased from Cell Signaling (Invitrogen). The antibodies against activating transcription factor 4 (ATF4), pHistone H2AX, NF-κB, and procaspase 9 were obtained from Santa Cruz. The antibody against ATF6 was from Imgenex, and that against XBP1 was from Abcam. The antibody against actin was from Sigma-Aldrich, and that against total AMPK (α1 and α2 subunits) was from DSTT (University of Dundee). The antibody against nitrotyrosine was from Millipore. Antisera against mouse glutathione S-transferase a1 (Gsta1), mouse Gstm1, rat GSTA4, human glutamate-cysteine ligase catalytic (GCLC) and modifier (GCLM) subunits, and rat NAD(P)H:quinone oxidoreductase 1 (NQO1) were produced in house (50, 55).
Biochemical analyses.
Portions (approximately 100 mg) of frozen mouse liver were pulverized individually under liquid nitrogen, using a mortar and pestle. The ground material from each sample was resuspended in 1 ml of ice-cold 50 mM HEPES buffer, pH 7.5, that contained 150 mM NaCl, 1 mM dithiothreitol, and protease inhibitors before being homogenized. Thereafter, cytosol was prepared at 4°C from individual livers after two centrifugation steps (15,000 × g for 45 min and 100,000 × g for 90 min). For AMPK, ACC, and JNK Western blots, whole-cell soluble extracts were prepared from the resuspended material by a single centrifugation step (15,000 × g for 15 min at 4°C). For the NF-κB Western blots, nuclear extracts were prepared from frozen liver using the Pierce NE-PER kit (ThermoScientific Life Science Research Products, Rockford, IL). Protein concentrations in samples were measured as described previously, as were total glutathione, reduced glutathione (GSH), and oxidized glutathione (GSSG) (33). Liver malondialdehyde (MDA) and protein oxidation levels were measured using a TBARS assay kit (Cayman Chemical Company) and an OxyBlot detection kit (Millipore), respectively.
Gene expression profiling.
The expression of hepatic genes in the Nrf2−/− and Nrf2+/+ mice fed an RC or HF diet was measured by TaqMan real-time PCR using an Applied Biosystems Prism model 7700 sequence detector instrument (50). mRNAs for the following classes of proteins were monitored using commercial primer and probe sets, all of which were purchased from Life Technologies: lipid-associated transcription factors (farnesoid X receptor [FXR; Mm00436425_m1], liver X receptor alpha [LXRα; Mm00443451_m1], LXRβ [Mm00437265_g1], MLX-interacting protein-like [Mlxipl, also called carbohydrate-responsive element binding protein {ChERBP}; Mm00498811_m1], peroxisome proliferator-activated receptor alpha [PPARα; Mm00440939_m1], PPARγ [Mm01184322_m1], retinoid X receptor alpha [RXRα; Mm00441185_m1], small heterodimer partner [Shp; Mm00442278_m1], Srebf1 [whose mRNA encodes Srebp-1c; Mm00550338_m1], and Srebf2 [whose mRNA encodes Srebp-2; Mm01306293_m1]), FA synthesis proteins (acetyl-CoA carboxylase alpha [Acaca; Mm01304277_m1], ATP-citrate lyase [Acly; Mm01302282_m1], elongation of very long-chain fatty acids protein 5 [Elovl5; Mm00506717_m1], Elovl6 [Mm00851223_s1], and fatty acid synthase [Fasn; Mm00662319_m1]), the FA desaturation enzyme stearoyl-CoA desaturase (Scd1; Mm00772290_m1), triglyceride assembly proteins (fatty acid-binding protein 1 [Fabp1; Mm00444340_m1] and Fabp5 [Mm00783731_s1]), enzymes involved in FA oxidation (acetyl-CoA acyltransferase 1 [Acaa1; Mm00728460_s1], acetyl-CoA carboxylase beta [Acacb; Mm01204683_ml], carnitine palmitoyl-transferase [Cpt1a; Mm00550439_m1], cytochrome P450 4a10 [Cyp4a10; Mm01188913_g1], Cyp4a14 [Mm00484132_m1], and Cyp2e1 [Mm00491127_m1]), ER stress proteins (Atf4 [Mm00515325_g1], Atf6 [Mm00520279_m1], Chop [Mm01135937_g1], growth arrest and DNA-damage-inducible protein 34 [Gadd34; Mm01205601_g1], Grp78 [Mm00517690_g1], and Xbp1s and Xbp1unspliced [56]), inflammation proteins (cyclooxygenase 2 [Cox2; Mm00478377_g1], interleukin-1β [IL-1β; Mm99999061_mH], IL-6 [Mmoo446190_m1], myeloperoxidase [Mpo; Mm01298424_m1], nitric oxide synthase 2 [Nos2; Mm00440485_m1], NF-κB p65 [RelA; Mm00501346_m1], and tumor necrosis factor alpha [TNF-α; Mm00443259_g1]), antioxidant proteins (cystine/glutamate antiporter light chain xCT [Slc7a11; Mm00442530_m1], Gclc [50], Gclm [50], sulfiredoxin [Srxn1; Mm00769566_m1], and thioredoxin reductase [Txnrd1; Mm00443675_m1]), drug-metabolizing enzymes (Gsta1 [50], Gsta2 [50], Gsta4 [50], Gstm1 [50], and Gstm2 [50] subunits and Nqo1 [57]), and the general stress enzyme heme oxygenase 1 (Hmox1; Mm00516006_m1).
Western blotting.
Tissue proteins were resolved by SDS-PAGE in 10% polyacrylamide gels before they were transferred onto Immobilon-P membranes. The immobilized polypeptides were probed using a variety of commercial antibodies or in-house rabbit antiserum, and following extensive washing of the blots, the cross-reacting bands were identified using horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG and visualized by enhanced chemiluminescence (50, 55). Equal protein loading of samples was assessed using glyceraldehyde-3-phosphate dehydrogenase (GAPDH), actin, or PCNA.
Cells.
Primary hepatocytes were prepared from the livers of RC diet-fed 10-week-old mice by the method of Foretz et al. (58). The livers were first washed with perfusion buffer (137 mM NaCl, 7 mM KCl, 0.7 mM Na2HPO4, and 10 mM HEPES, adjusted to pH 7.65) containing 0.5 mM EDTA to remove blood before they were treated with 400 μg/ml collagenase in the perfusion buffer. Following isolation, primary hepatocytes were plated in M199 medium plus GlutaMax (Invitrogen) supplemented with 10% fetal calf serum (FCS), 1% penicillin-streptomycin, 0.1% bovine serum albumin (BSA), 10 nM insulin, 200 nM triiodothyronine, and 500 nM dexamethasone.
Metabolic studies.
Experiments were performed to determine whether the rates of mitochondrial respiration or glycolysis differed in Nrf2−/− and Nrf2+/+ hepatocytes upon treatment with fatty acids or glucose using a Seahorse Bioscience XF 24 analyzer. Freshly prepared primary hepatocytes were plated in XF 24-well plates at a density of 1 × 104 cells per well in M199 medium containing 2 mM GlutaMax, which was supplemented with 10% FCS, 1% penicillin-streptomycin, 0.1% BSA, 10 nM insulin, 200 nM triiodothyronine, and 500 nM dexamethasone. The cells were cultured overnight at 37°C before being washed with XF 24 Dulbecco's modified Eagle's medium (DMEM) assay medium supplemented with 5.5 mM glucose and 2 mM GlutaMax prior to incubation for 1 h in the same medium (without CO2 preincubation). Thereafter, the plates were loaded onto an XF 24 analyzer to measure the oxygen consumption rate (OCR) at regular intervals as cells alone (baseline) and after injecting BSA conjugated with 375 μM palmitic acid or 375 μM oleic acid in the presence of 1 mM carnitine. The values were normalized against the total protein content of the cells in each well.
Statistics.
Comparisons between the biochemical and molecular biology results from the four experimental groups were made using a paired or unpaired two-tailed Student's t test, a one-sample Student's t test, or analysis of variance (ANOVA) with a Bonferroni post hoc analysis as appropriate. Analysis of covariance (ANCOVA) was used to examine the relationship between body weight and oxygen consumption. The results are means and standard errors of the mean (SEM), and P values of ≤0.05 were considered statistically significant. Comparisons between the histology NAS results were made using the Kruskal-Wallis H test.
RESULTS
Whole-body metabolic response to a high-fat diet is modified by Nrf2.
Nrf2−/− mice fed an RC diet starting from 8 to 10 weeks of age did not differ from wild-type mice in average length (data not shown) or body mass (Fig. 1A); Nrf2−/− mice appeared consistently lighter, although this difference did not reach significance. When fed an HF diet, mass accumulation was less apparent in Nrf2−/− mice (Fig. 1B and C), with a mean increase in body mass of 14.5 ± 1.3 g (n = 10) and 10.7 ± 1.1 g (n = 11; P < 0.05) for Nrf2+/+ and Nrf2−/− mice, respectively. At 28 weeks of age, qMR scanning revealed that RC-fed Nrf2−/− and Nrf2+/+ mice had identical body fat levels and that HF-fed Nrf2−/− and Nrf2+/+ mice increased their relative fat mass, but significantly less for the Nrf2−/− mice (Fig. 1D).
FIG 1.
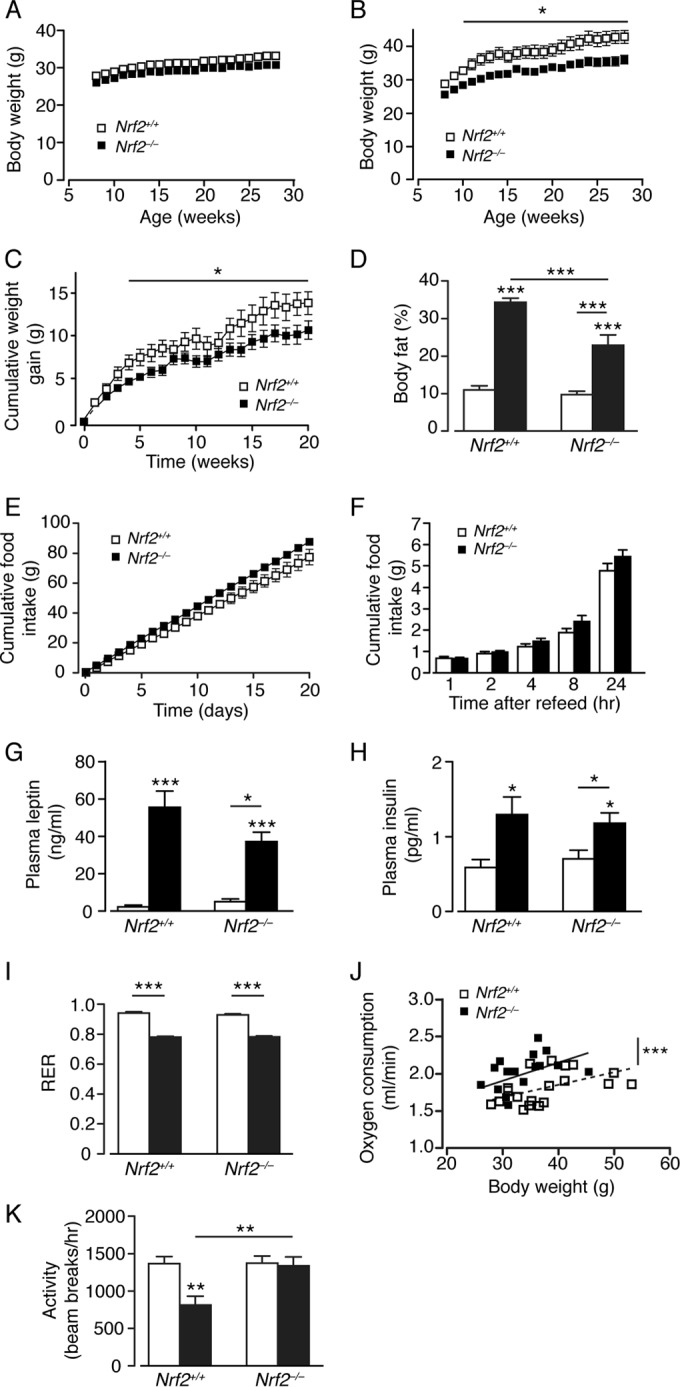
Nrf2−/− mice on an HF diet exhibit reduced adiposity and higher energy expenditure than Nrf2+/+ mice. (A and B) Body mass curves of male age-matched Nrf2+/+ and Nrf2−/− mice fed on an RC (A) or HF (B) diet for 20 weeks from 8 weeks of age (10 or 11 mice per group). (C) Body mass gains of Nrf2+/+ and Nrf2−/− mice fed on an HF diet. (D) Percentages of body fat in Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (12 or 13 mice per group). (E) Cumulative food intake of age-matched male Nrf2+/+ and Nrf2−/− mice fed on RC over a period of 20 weeks (6 mice per group). (F) Food intake after overnight fast in age-matched Nrf2+/+ and Nrf2−/− mice fed on an HF diet (5 mice per group). (G and H) Fed plasma leptin (G) and insulin (H) levels from Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (9 to 11 mice per group). (I and J) RER (I) and oxygen consumption (J) (the lines show fitted regressions) for Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (7 to 13 mice per group). (K) Effects of genotype and diet on locomotor activity for Nrf2+/+ and Nrf2−/− mice (7 to 13 mice per group). (D, G to I, and K) White bars, RC fed; black bars, HF fed. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Daily food intake was slightly greater for RC-fed Nrf2−/− than for Nrf2+/+ mice (Fig. 1E), but no difference in food consumption was observed in HF-fed Nrf2−/− and Nrf2+/+ mice by determining compensatory feeding following an overnight fast (Fig. 1F). Plasma leptin and insulin levels were similar in RC-fed Nrf2−/− and Nrf2+/+ mice, but consistent with their increased body fat content, HF-fed Nrf2−/− and Nrf2+/+ mice displayed increased plasma leptin and insulin levels, with a nonsignificant trend for reduced leptin levels in Nrf2−/− mice (Fig. 1G and H). No difference was observed in the respiratory exchange ratio between Nrf2−/− and Nrf2+/+ mice, though it was lowered by the HF diet in both (Fig. 1I). Indirect calorimetry showed that Nrf2−/− mice exhibit higher O2 consumption, indicating that loss of Nrf2 increases energy expenditure (Fig. 1J). HF feeding decreased the ambulatory activity of Nrf2+/+ mice in comparison to RC-fed Nrf2+/+ mice, whereas ambulatory activity was unchanged by diet in Nrf2−/− mice (Fig. 1K). Thus, although Nrf2−/− mice display sensitivity to HF feeding by increasing body fat content, the magnitude of this effect was less than was observed in Nrf2+/+ mice, likely due to the increased energy expenditure of Nrf2−/− mice.
Insulin sensitivity is maintained in Nrf2−/− mice fed the high-fat diet.
As NASH is associated with insulin resistance, we examined glucose homeostasis. On an RC diet, fasting blood glucose levels were similar in Nrf2+/+ and Nrf2−/− mice, and HF feeding for 16 weeks raised fasted glucose levels in both groups, indicative of decreased liver insulin sensitivity (Fig. 2A). Although HF feeding produced a significant increase in fed glucose levels in both Nrf2−/− and Nrf2+/+ mice, fed glucose levels were lower in the mutant than in wild-type mice for both RC and HF diets (Fig. 2B). Intraperitoneal glucose tolerance tests (GTT) showed that RC-fed Nrf2−/− and Nrf2+/+ mice had comparable glucose disposal (Fig. 2C and E), whereas HF-fed Nrf2−/− mice displayed significantly better glucose disposal from the peripheral circulation than diet- and age-matched Nrf2+/+ mice (Fig. 2D and E). Also, HF-fed Nrf2−/− mice exhibited a greater decrease in blood glucose during insulin tolerance tests (ITT) than Nrf2+/+ mice, an outcome not observed for RC-fed mice (Fig. 2F and G); importantly, the ITT for HF-fed Nrf2−/− mice had to be curtailed after 60 min, as ∼50% of them did not mount a counterregulatory response to insulin-induced hypoglycemia and had to be given glucose to recover.
FIG 2.

Nrf2−/− mice display improved glucose homeostasis and insulin sensitivity with enhanced skeletal muscle and liver insulin signaling. (A and B) Fasted (A) and fed (B) blood glucose levels in Nrf2+/+ and Nrf2−/− mice after 16 weeks on an RC or HF diet (5 or 6 mice per group). (C) GTT on Nrf2+/+ (n = 11) and Nrf2−/− (n = 9) mice, 28 weeks old, fed RC. (D) GTT on Nrf2+/+ and Nrf2−/− mice fed an HF diet for 20 weeks from 8 weeks of age (11 mice per group). (E) Quantification of the area under the curve (AUC) for the total glycemic excursions shown in panels C and D by genotype and diet. (F) ITT on Nrf2+/+ (n = 11) and Nrf2−/− (n = 9) mice, 28 weeks old, fed RC. (G) ITT on Nrf2+/+ (n = 11) and Nrf2−/− (n = 10) mice fed an HF diet for 20 weeks from 8 weeks of age. Note the curtailed time course of tolerance tests on Nrf2−/− mice owing to continual decline in blood glucose and the requirement to intervene with glucose administration. (H and I) Representative immunoblots of total PKB and insulin-stimulated phosphorylation of PKB (also called Akt) at Ser473 (p-PKB) from the liver (H) and skeletal muscle (I) of Nrf2+/+ and Nrf2−/− mice (7 months old) fed RC (10 to 19 mice per group). Quantification of the immunoblot data is shown, and in each case, p-PKB/PKB was normalized with respect to RC-fed Nrf2+/+ levels. White bars, RC fed; black bars, HF fed. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further assess peripheral insulin signaling strength in Nrf2−/− and Nrf2+/+ mice, we measured the levels of protein kinase B phosphorylated at Ser473 (p-PKB; also called Akt) in liver and skeletal muscle following intraperitoneal injection of insulin. In nonstimulated tissues, p-PKB levels did not differ in the two groups of mice. However, in response to insulin, Nrf2−/− mice displayed significantly higher p-PKB levels in liver and skeletal muscle than Nrf2+/+ mice (Fig. 2H and I). Therefore, Nrf2−/− mice exhibit greater peripheral insulin sensitivity than Nrf2+/+ mice, and consequently, when challenged chronically with the HF diet, Nrf2−/− mice retain better insulin-mediated glucose disposal than HF-fed Nrf2+/+ mice.
Nrf2−/− mice develop a NASH phenotype when fed a high-fat diet.
Hematoxylin and eosin (H&E) staining of liver sections was assessed by an expert histopathologist, using the NAS scoring system developed for human disease. Individual scores for fat, inflammation, and fibrosis were also analyzed. Livers from RC-fed Nrf2+/+ mice showed normal hepatocyte architecture and no evidence of steatosis. However, livers of HF-fed Nrf2+/+ mice developed hepatic steatosis without significant histological evidence of either inflammation or fibrosis that is consistent with simple steatosis of the human NAFLD spectrum but not with NASH (Fig. 3A and Table 1). Livers from RC-fed Nrf2−/− mice displayed signs of microvesicular steatosis but no inflammation. However, livers from HF-fed Nrf2−/− mice showed significant microvesicular and macrovesicular steatosis, neutrophil infiltration, apoptotic bodies, and disruption of hepatic architecture that are consistent with NASH. Cirrhosis was observed only in livers of HF-fed Nrf2−/− mice. In conclusion, HF-fed Nrf2+/+ and HF-fed Nrf2−/− livers contained similar levels of fat that were significantly higher than in RC-fed Nrf2−/− livers, and all contained more fat than RC-fed Nrf2+/+ livers. Moreover, the HF-fed Nrf2−/− livers had significantly more inflammation than any other group. All of these observations are based upon scores that are relative to each other and to the normal, rather than absolute values.
FIG 3.
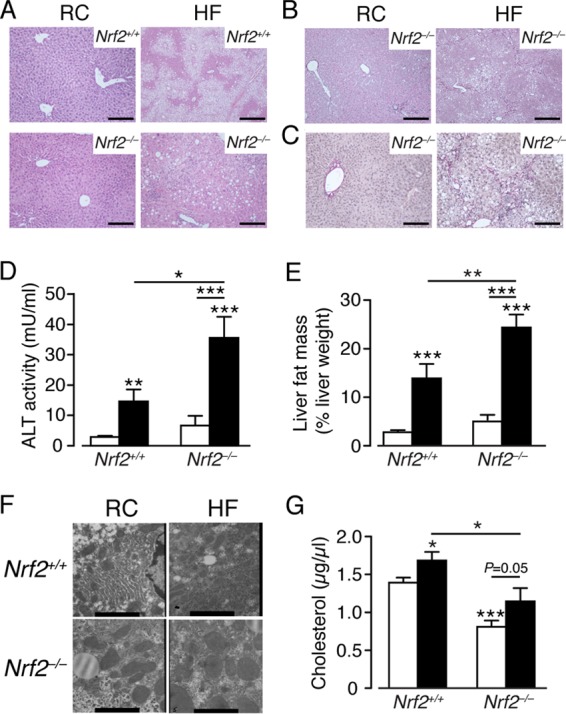
Histological and biochemical evidence of steatohepatitis and fibrosis in the livers of Nrf2−/− mice fed a high-fat diet. (A) Representative H&E-stained images of livers from Nrf2+/+ and Nrf2−/− mice after 24 weeks on an RC or HF diet from 8 to 10 weeks of age. (B and C) Representative images of reticulin staining (B) and Van Gieson's staining (C) in livers from Nrf2−/− mice after 24 weeks on an RC or HF diet. Bars, 100 μm. (D) ALT activity in plasma of Nrf2+/+ and Nrf2−/− mice after 24 weeks on an RC or HF diet (8 to 11 mice per group). (E) Magnetic resonance analysis of fat content in livers from Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (12 or 13 mice per group). (F) Representative electron microscope images of liver sections from Nrf2+/+ and Nrf2−/− mice on an RC or HF diet; bars, 1,731 nm. (G) Cholesterol levels in plasma from Nrf2+/+ and Nrf2−/− mice on RC and HF diets (9 to 11 mice per group). (D, E, and G) White bars, RC fed; black bars, HF fed. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
TABLE 1.
Histology of livers from Nrf2+/+ and Nrf2−/− mice fed a high-fat diet for 24 weeks
| Parameter | Valuef (n) |
|
|---|---|---|
| RC diet | HF diet | |
| NASa (maximum 8) | ||
| Nrf2+/+ mice | 0.5 (10) | 3.45 (11) |
| Nrf2−/− mice | 2.0 (9) | 4.9 (10) |
| Steatosis component of NASb (0–3) | ||
| Nrf2+/+ mice | 0.9 (10) | 2.55 (11) |
| Nrf2−/− mice | 1.33 (9) | 2.4 (10) |
| Inflammatory component of NASc (0–3) | ||
| Nrf2+/+ mice | 0 (10) | 0.82 (11) |
| Nrf2−/− mice | 0.66 (9) | 2.3 (10) |
| Ballooning component of NASd (0–2) | ||
| Nrf2+/+ mice | 0 (10) | 0 (11) |
| Nrf2−/− mice | 0 (9) | 0.3 (10) |
| Fibrosis stagee (0–3) | ||
| Nrf2+/+ mice | 0 (10) | 0 (11) |
| Nrf2−/− mice | 0 (9) | 1 (10) |
In Nrf2+/+ mice, the NAS is significantly higher in livers of HF-fed mice than in RC-fed animals (Kruskal-Wallis H test; P = 0.002), and the same is seen in livers of HF-fed Nrf2−/− mice compared to RC-fed Nrf2−/− livers. The HF-fed Nrf2−/− mice had a significantly higher NAS than HF-fed Nrf2+/+ mice (Kruskal-Wallis H test; P = 0.03).
Both HF-fed Nrf2+/+ and Nrf2−/− mice had more steatosis than their RC-fed counterparts (Kruskal-Wallis H test; P = 0.03). No significant difference was observed between Nrf2+/+ and Nrf2−/− mice fed the same diet.
The inflammatory component of the NAS was significantly higher in livers of HF-fed Nrf2−/− mice than in either HF-fed Nrf2+/+ or RC-fed Nrf2−/− mice (Kruskal-Wallis H test; P = 0.002).
No statistical differences were observed for ballooning between the experimental groups.
The median values are shown, and fibrosis was observed only in livers of HF-fed Nrf2−/− mice.
The significance of differences in the NAS scores of livers from RC-fed and HF-fed Nrf2+/+ and Nrf2−/− mice was assessed using the Kruskal-Wallis H test. The differences between the NAS results of the 4 experimental groups were highly significant (Kruskal-Wallis H test; P = 0.002), with both HF diet mice having higher NAS values than their chow-fed counterparts. Moreover, a significant difference was observed between the two HF-fed groups, with the Nrf2−/− animals having the higher score (Kruskal-Wallis H test; P = 0.03), despite the fact that the steatosis components of NAS were similar in the two HF-fed groups, which blurs the difference in their NAS results. The difference in the NAS between HF-fed Nrf2+/+ and Nrf2−/− animals is smaller than the difference in inflammation and the degree of NASH between them; this represents a limitation of the NAS ranking system, which simply adds the 3 components together. Therefore, the apparent relative increase of steatosis in HF-fed Nrf2+/+ livers compared to HF-fed Nrf2−/− livers masks the increased inflammation score in the latter, because hepatocytes lose fat as they become inflamed. Fibrosis, which is not part of the NAS, was not observed in the livers of RC-fed animals or in HF-fed Nrf2+/+ livers, but it was seen in HF-fed Nrf2−/− livers (amounting to cirrhosis in 2 of 10 animals).
Reticulin staining revealed that HF-fed Nrf2+/+ livers contained significantly increased amounts of type III collagen compared with their RC-fed counterparts and that RC-fed Nrf2−/− mouse livers had levels of type III collagen similar to those of Nrf2+/+ mice on the same diet (data not shown). However, livers from HF-fed Nrf2−/− mice contained larger amounts of collagen than those of HF-fed Nrf2+/+ mice (Fig. 3B). Van Gieson's staining of hepatic sections revealed normal levels of collagen in RC-fed Nrf2−/− livers but large increases in collagen deposition in HF-fed Nrf2−/− livers from the central vein through hepatocytes to the portal triad (Fig. 3C). Increased liver pathology in HF-fed Nrf2−/− mice was accompanied by elevated plasma ALT activity (Fig. 3D).
qMR analysis revealed that RC-fed Nrf2−/− and RC-fed Nrf2+/+ livers had comparable levels of fat and that HF feeding increased fat deposition in both genotypes, with Nrf2−/− livers containing the most fat among the four groups (Fig. 3E). Ultrastructural analysis showed normal architecture for RC-fed Nrf2+/+ livers, with fat globules present in hepatocytes from HF-fed Nrf2+/+ and RC-fed Nrf2−/− mice, whereas hepatocytes of HF-fed Nrf2−/− mice were characterized by swollen mitochondria with reduced crista and disrupted membranes (Fig. 3F). HF feeding did not increase plasma triglycerides or FA levels in either genotype (data not shown) but increased plasma cholesterol in both genotypes; Nrf2−/− mice exhibited lower cholesterol on either diet than Nrf2+/+ mice (Fig. 3G), and this is most likely due to inactivation of the redox-sensitive HMG CoA reductase in the mutant mouse by higher ROS levels (59).
Overall, a clear progression was seen in the animal models across the NAFLD disease spectrum, with RC-fed Nrf2+/+ mice representing normality; RC-fed Nrf2−/− mice and HF-fed Nrf2+/+ mice developing steatosis, worse in the latter; and HF-fed Nrf2−/− mice developing the complete NASH phenotype with fibrosis. We therefore sought, in the experiments described below, to determine molecular changes that account for the greater degree of NAFLD, inflammation, hepatocellular death, and oxidative stress in mice lacking Nrf2.
Induction of lipogenesis genes by a high-fat diet is accentuated in Nrf2−/− livers.
First, we examined the mRNA levels of the following transcription factors that contribute to liver steatosis: Srebf1 and Srebf2, which regulate cholesterol biosynthesis; Mlxipl, which regulates triglyceride synthesis genes in a glucose-dependent manner; PPARγ, which promotes storage of FA; LXRα and LXRβ, which control lipid homeostasis and inflammation; Frx, which regulates cholesterol and bile acid production; and Shp, which represses LXR (for reviews, see references 60 to 64). In Nrf2+/+ livers, the HF diet increased mRNA levels for Srebf1, Srebf2, Mlxip, PPARγ, and LXRα but had no significant effect on mRNA for Shp and reduced mRNAs for LXRβ and FXR (Fig. 4A). Loss of Nrf2 had a moderate impact on liver expression of these transcription factors, with a modest increase in Mlxipl and more significant decreases in mRNAs for Srebf2 and Shp. However, HF-fed Nrf2−/− livers exhibited significant increases in mRNAs for Srebf1, Srebf2, Mlxipl, and PPARγ, with no change in mRNA for LXRα and reduced levels of mRNAs for Shp, LXRβ, and FXR (Fig. 4A). Western blotting revealed that the active form of Srebp-1 (encoded by Srebf1) was most abundant in the nuclei of HF-fed Nrf2−/− livers but was also elevated in the nuclei of RC-fed Nrf2−/− and HF-fed Nrf2+/+ livers compared with RC-fed Nrf2+/+ livers (Fig. 4B). Thus, the HF diet increased expression in the liver of transcription factors that are associated with FA uptake, FA oxidation, FA storage, lipogenesis, and inflammation in wild-type and Nrf2−/− mice, but in all cases, the effect was greater in Nrf2−/− than in Nrf2+/+ livers. Also, the HF diet decreased expression of FXR, which controls the synthesis of bile acids, as well as the expression of the nuclear receptor repressor Shp, which is regulated by Fxr (60), and again, this was most apparent in Nrf2−/− livers.
FIG 4.

Expression of many lipid-associated transcription factors and lipogenic enzymes is upregulated in HF-fed Nrf2−/− livers to a greater extent than in HF-fed Nrf2+/+ livers. (A) Quantitative-PCR analysis of mRNAs for transcription factors that regulate lipid and glucose metabolism and storage in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (6 to 12 mice per group). (B) Immunoblots of Srebp-1c in nuclear subcellular fractions (nSREBP-1c) and cytosolic subcellular fractions (cSREBP-1c) from livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. (C and D) Quantitative-PCR analysis of mRNA for enzymes involved in lipogenesis (C) and FA catabolism (D) in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (10 to 12 mice per group). (E) Quantitative-PCR analysis of mRNA for cytochrome P450 enzymes that oxidize fatty acids in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (10 to 12 mice per group). In each case, the data were normalized with respect to RC-fed Nrf2+/+ levels for each gene. White bars, RC fed; black bars, HF fed. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Second, mRNAs for proteins involved in lipogenesis were examined. They included Acly, which catalyzes production of acetyl-CoA that is required for lipid biosynthesis; the FA synthesis Acaca gene, which encodes AccI that produces malonyl-CoA; the FA synthase Fasn; the FA elongases Elovl5 and Elovl6; the desaturase Scd1; and the FA uptake and transport proteins Fabp1 and Fabp5 (61, 64). In Nrf2+/+ mice, HF feeding increased mRNAs for Acly, Acaca, Elovl5, Elovl6, Fasn, and Fabp5 but had no effect on mRNA for Fabp1 and reduced mRNA for Scd1 (Fig. 4C). Livers from RC-fed Nrf2−/− mice showed a modest decrease in mRNA for Acaca and an increase in Elovl6 and Fabp5, with no change in the other transcripts. However, HF-fed Nrf2−/− livers demonstrated increases in mRNAs for Acly, Acaca, Fasn, Elovl5, Elovl6, and Fabp5 that were significantly greater than those observed in HF-fed Nrf2+/+ livers but had no effect on Fabp1. The HF diet appeared to downregulate mRNA for Scd1 expression independently of genotype (Fig. 4C). Thus, while the HF diet increased the expression of several key enzymes/proteins associated with lipogenesis, this induction was accentuated in Nrf2−/− livers.
Lipid catabolism genes are downregulated in Nrf2−/− livers.
Expression of Pparα, which supports uptake, utilization, and catabolism of FA, was increased in HF-fed Nrf2+/+ livers (Fig. 4D). The expression of Pparα in RC-fed Nrf2−/− livers was lower than in wild-type controls. However, induction of Pparα in HF-fed Nrf2−/− livers exceeded that seen in wild-type mice. Similarly, expression of the Acacb gene, which encodes Acc2 and results in raised malonyl-CoA, causing inhibition of Cpt1a, the rate-limiting step in FA uptake into mitochondria, was increased by the HF diet in Nrf2+/+ livers and to a much greater extent in HF-fed Nrf2−/− livers. In addition, Acacb gene expression was lower in RC-fed Nrf2−/− livers than in RC-fed Nrf2+/+ livers.
RC- and HF-fed Nrf2−/− livers had much reduced mRNA levels for Cpt1a, which is required for β-oxidation of long-chain FA, compared to RC-fed Nrf2+/+ livers, with the HF diet reducing Cpt1a mRNA in Nrf2+/+ livers to a much lesser extent than in Nrf2−/− livers (Fig. 4D). The expression of Acaa1, which contributes to the β-oxidation of FA in peroxisomes, was diminished in HF-fed Nrf2+/+ livers, but it was further lowered in HF-fed Nrf2−/− livers. Thus, the increased expression of Acacb in HF-fed Nrf2−/− livers, coupled with the exaggerated loss of Cpt1a and Acaa1 expression, may contribute to the increased hepatic steatosis in these animals.
We monitored Cyp isoenzymes that oxidize medium-chain FA. Loss of Nrf2 had no effect on Cyp2e1 or Cyp4a10 but significantly reduced Cyp4a14 expression in livers of RC-fed mice (Fig. 4E). However, the HF diet did not decrease the expression of these Cyp isoenzymes in Nrf2−/− livers to levels lower than those observed in Nrf2+/+ livers, so it seems improbable that they account for the higher level of steatosis in mutant livers than in wild-type livers.
Loss of Nrf2 decreases inhibitory phosphorylation of acetyl-CoA carboxylase.
The above-mentioned data indicate the HF diet combined with loss of Nrf2 results in decreased hepatic mitochondrial and peroxisomal FA oxidation. One consequence of lowered FA β-oxidation is reduced ketogenesis (82). Consistent with this prediction, RC- and HF-fed Nrf2−/− mice exhibited lower plasma β-hydroxybutyrate levels than similarly fed Nrf2+/+ mice (Fig. 5A). FA β-oxidation is regulated by AMPK. Activation of mouse Ampk arises from phosphorylation of Thr172 in its α-subunit, which can lead subsequently to inactivation of AccI and Acc2 by phosphorylation at Ser79 and Ser218, respectively. In turn, inhibition of AccI and Acc2 increases levels of malonyl-CoA, which is a substrate for Fasn and a potent allosteric inhibitor of Cpt1a. Immunoblotting revealed no difference in AccI and/or Acc2 protein (referred to here as Acc) levels by diet or genotype but reduced levels of Ampk protein in Nrf2−/− livers (Fig. 5B and C). Moreover, the amounts of phosphorylated Acc (p-Acc) and Ampk (p-Ampk) were reduced in Nrf2-null livers, regardless of diet, with the p-Acc/Acc ratio severely diminished in Nrf2−/− compared to Nrf2+/+ livers for both diets. In contrast, the p-Ampk/Ampk ratio was unaltered by genotype and diminished by HF feeding in both Nrf2+/+ and Nrf2−/− mouse livers. Thus, loss of Nrf2 diminishes the phosphorylation status of Acc, strongly suggesting elevation in its activity and greater production of malonyl-CoA, an important FA building block and an inhibitor of FA oxidation.
FIG 5.
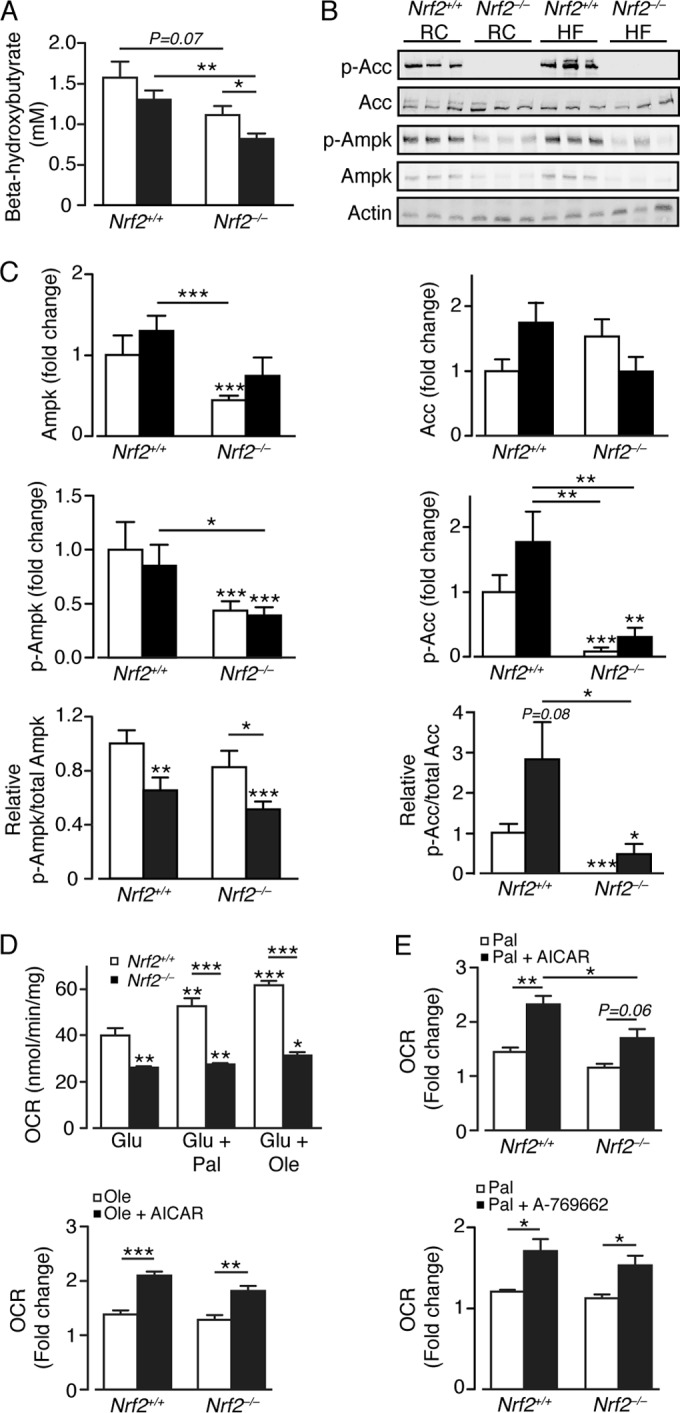
Loss of Nrf2 diminishes phosphorylation of acetyl-CoA carboxylase and decreases the oxidation of free fatty acids. (A) Plasma β-hydroxybutyrate levels in Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (9 to 11 mice per group). White bars, RC fed; black bars, HF fed. (B) Representative immunoblots for Acc, p-Acc, Ampk, and p-Ampk, with actin as a loading control, from livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. (C) Quantification of the immunoblot data for panel B, with p-Ampk/Ampk and p-Acc/Acc ratios (9 or 10 mice per group). The data were normalized with respect to RC-fed Nrf2+/+ levels for each protein or ratio. White bars, RC fed; black bars, HF fed. (D) OCRs for primary hepatocytes obtained from livers of Nrf2+/+ (n = 3) and Nrf2−/− (n = 4) mice on RC diets, presented with the following substrates: glucose alone (Glu), followed by glucose plus palmitate (Glu + Pal) or glucose + oleate (Glu + Ole). (E) Fold change in OCR on addition of Pal or Ole for primary Nrf2+/+ and Nrf2−/− hepatocytes treated or not with AICAR or A-769662. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Nrf2 influences the utilization of glucose and fatty acids in hepatocytes.
We next examined the ability of primary hepatocytes from Nrf2−/− and Nrf2+/+ mice to utilize glucose and FA as substrates. Nrf2-null hepatocytes possessed a significantly lower basal glucose oxidation rate and were less able to oxidize palmitate or oleate than Nrf2+/+ hepatocytes (Fig. 5D). Treatment of Nrf2−/− hepatocytes with the AMPK activator AICAR or Abbott (A-769662) increased their ability to oxidize palmitate and oleate, proportionally equivalent to that observed for Nrf2+/+ hepatocytes (Fig. 5E). Together, these results indicate that loss of Nrf2 severely diminishes oxidation by the liver of glucose and FA and that this is partly due to a reduction in hepatic Ampk activity in the mouse liver.
The UPR is perturbed in Nrf2-null mouse liver under basal conditions.
In Nrf2+/+ livers, the HF diet stimulated a substantial increase in the ER-resident stress sensor proteins Perk and Ire1 (Fig. 6A). Also, an increase in Atf6 p90 but not Atf6 p50 was observed, suggesting the HF diet did not activate the ATF6 pathway in wild-type livers. Stimulation of Perk and Ire1 in Nrf2+/+ livers was associated with induction of Chop and the spliced mRNA form Xbp1s but not Gadd34 (Fig. 6A and B). Remarkably, in RC-fed Nrf2−/− livers, substantial increases in Xbp1s and Atf6 p50 were observed, suggesting that the Ire1 and Atf6 ER stress pathways are activated in the mutant mice even under basal conditions. In addition, a modest increase in p-eIF2α, and a more obvious increase in Chop, was observed in RC-fed Nrf2−/− livers suggesting that the PERK pathway was also activated, though a decrease in p-Perk and Ire1 was observed relative to the wild-type control. Surprisingly, livers from HF-fed Nrf2−/− mice did not exhibit an overt UPR, as evidenced by failure to increase mRNA and/or protein levels of Atf6 p90, Ire1, Atf4, Atf6 p50, Grp78, and Xbp1s compared with HF-fed Nrf2+/+ and/or RC-fed Nrf2−/− livers, and we suppose this failure occurs either because the UPR cannot be maintained as a consequence of extensive liver damage or because triggering of the UPR is fundamentally altered in Nrf2−/− livers. The only exception to this trend was the observation that Chop mRNA levels were slightly increased in the livers of HF-fed Nrf2−/− mice (Fig. 6B).
FIG 6.

ER stress pathways are perturbed in the livers of Nrf2−/− mice. (A) Representative immunoblots from analysis of ER stress and UPR-associated proteins in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. Quantification of the immunoblot data for each protein is shown by genotype and diet (3 mice per group). (B) Quantitative-PCR analysis of mRNAs of ER stress markers from livers of Nrf2+/+ and Nrf2−/− mice on RC and HF diets (5 to 8 mice per group). In each case, the data were normalized with respect to RC-fed Nrf2+/+ levels for each protein and gene. White bars, RC fed; black bars, HF fed. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Loss of Nrf2 exacerbates inflammation in the liver caused by a high-fat diet.
As activation of PERK and IRE1 during ER stress can stimulate inflammatory responses mediated by NF-κB and JNK (15, 16), we evaluated whether the HF diet differentially stimulates inflammatory responses in wild-type and Nrf2−/− livers. First, we measured Mpo, because it is indicative of activated Kupffer cells (65). The HF diet increased Mpo mRNA in both genotypes, with loss of Nrf2 significantly enhancing Mpo expression under both dietary regimes (Fig. 7A). In Nrf2+/+ livers, the HF diet increased mRNAs for the inflammatory markers IL-1β and TNF-α, though it modestly decreased that for IL-6 (Fig. 7B). Compared to wild-type control livers, RC-fed Nrf2−/− livers exhibited no increase in TNF-α, IL-1β, and IL-6 expression. However, the HF diet induced their expression to levels significantly higher than it did in Nrf2+/+ mice, suggesting an exacerbated inflammatory response in the knockout mice (Fig. 7B).
FIG 7.

A high-fat diet elicits an exaggerated inflammatory response in the livers of Nrf2−/− mice. (A) Quantitative-PCR analysis of mRNA for Mpo in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (10 to 12 mice per group). (B) Quantitative-PCR analysis of levels of mRNAs for the inflammatory markers TNF-α, IL-1β, and IL-6 in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (10 to 12 mice per group). (C) Representative immunoblots for nuclear levels of the p65 subunit of NF-κB in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. PCNA was used as a loading control. Quantification of the immunoblot data for NF-κB p65 is shown (6 mice per group), along with quantitative-PCR analysis of mRNA for RelA (10 to 12 mice per group). (D) Quantitative-PCR analysis of mRNAs for the NF-κB Cox2 and Nos2 target genes in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (10 to 12 mice per group). (E) Representative immunoblot for phosphorylated JNK (p-JNK) and total JNK in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. Quantification of the immunoblot data for p-JNK is shown (3 mice per group). (F) Staining for nitrotyrosine in liver sections from Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (the arrows indicate cells giving a positive nitrotyrosine stain; bars, 100 μm). In each case, the data were normalized with respect to RC-fed Nrf2+/+ levels for each protein or gene. White bars, RC fed; black bars, HF fed. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The nuclear abundance of the NF-κB p65 RelA subunit was increased in livers of HF-fed Nrf2+/+ and RC-fed Nrf2−/− mice, with the HF diet raising NF-κB p65 levels in Nrf2−/− livers even further (Fig. 7C). The HF diet also produced pronounced increases in RelA mRNA in Nrf2−/− mice (Fig. 7C). The expression of the NF-κB target genes Cox2 and Nos2 was increased by 12- and 6-fold, respectively, in HF-fed Nrf2+/+ livers and by 23- and 60-fold, respectively, in livers of HF-fed Nrf2−/− mice (Fig. 7D). We also measured JNK activation. As expected, p-JNK levels were increased by the HF diet in Nrf2+/+ livers and were elevated in RC-fed Nrf2−/− livers. Significantly, p-JNK levels were highest in Nrf2−/− livers (Fig. 7E).
As Nos2 was highly expressed in HF Nrf2−/− livers, it is possible that excessive nitric oxide levels contribute to inflammation. We therefore examined the possibility that nitrotyrosine levels were high in livers from Nrf2−/− mice. As anticipated, staining for nitrotyrosine was greater in HF Nrf2−/− livers than in the other livers examined (Fig. 7F).
Collectively, these results indicate that the HF diet stimulates a mild inflammatory response in livers of Nrf2+/+ mice. However, Nrf2−/− mice exhibited mild liver inflammation even under basal conditions, and this was markedly aggravated by the HF diet.
Livers from high-fat-fed Nrf2−/− mice are subject to oxidative stress and hepatocellular injury.
We examined whether the HF diet provokes oxidative stress in the liver by measuring GSH, GSSG, MDA, and the levels of oxidized protein. In Nrf2+/+ mice, the HF diet produced an increase in GSSG (Fig. 8A) and MDA (Fig. 8B), but no change was observed in protein carbonyl levels, suggesting that the extent of oxidative stress in wild-type mice was not sufficient to cause irreversible damage (Fig. 8C). In comparison, the livers of RC-fed Nrf2−/− mice contained significantly increased GSSG (Fig. 8A), but the levels of MDA and protein carbonyls did not differ from those in RC-fed Nrf2+/+ livers. Most strikingly, the HF-fed Nrf2−/− livers contained elevated GSSG and large increases in MDA and protein carbonyls compared with either RC- or HF-fed Nrf2+/+ mice (Fig. 8A to C), indicating that the higher oxidative-stress load produced by the HF diet in the mutant mouse exceeded its intrinsic antioxidant capacity.
FIG 8.

The livers of Nrf2−/− mice fed a high-fat diet are subject to oxidative stress and exhibit an increased level of apoptosis and DNA damage. (A) Ratio of GSSG to GSH in the livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (6 mice per group). (B) Lipid peroxidation product MDA levels in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (8 mice per group). (C) Levels of oxidized protein shown as a representative OxyBlot using 20 μg of protein from each experimental group. Lanes: 1, molecular weight standard; 2, 4, 6, and 8, negative controls (no 2,4-dinitrophenylhydrazine); 3, RC-fed Nrf2+/+; 5, RC-fed Nrf2−/−; 7, HF-fed Nrf2+/+; 9, HF-fed Nrf2−/−. The graph shows densitometric measurements of lanes 3, 5, 7, and 9 (normalized to the RC-fed Nrf2+/+ amount) in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (8 to 11 mice per group). (D) Representative immunoblots of full-length (FL) and cleaved caspase 9 and caspase 3 and phosphorylated H2AX in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. (E) DNA fragmentation in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet was examined by TUNEL assay (the arrows indicate apoptotic/necrotic cells; bars, 100 μm), with the mean percentages of TUNEL-positive cells shown (5 mice per group). (F) Immunoblot for HMGB1 in nuclear and cytosolic fractions of Nrf2+/+ and Nrf2−/− livers from RC- or HF-fed mice. Quantification of the immunoblot data for each protein is shown by genotype and diet (3 mice per group). In each case, the data were normalized with respect to RC-fed Nrf2+/+ levels for each protein. White bars, RC fed; black bars, HF fed. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Molecular mass markers (C, D, and F) are in kDa.
The Nrf2−/− livers exhibited evidence of apoptosis and necrosis. Thus, in HF-fed Nrf2−/− livers but not HF-fed Nrf2+/+ livers, increases in cleaved caspases 3 and 9 were observed, as was an increase in a marker for DNA double-strand breaks, p-H2AX (Fig. 8D). RC-fed Nrf2−/− mouse livers also demonstrated evidence of apoptosis, with increased levels of cleaved caspases 3 and 9 and p-H2AX. TUNEL analysis revealed a marked increase in DNA fragmentation in HF-fed Nrf2+/+ and RC-fed Nrf2−/− livers and that cleaved DNA was present in the cytoplasm of a substantial number of hepatocytes (Fig. 8E). This pattern of staining is consistent with necrotic cell death and was observed to a much greater degree in HF-fed Nrf2−/− livers. Immunoblotting for the nuclear nonhistone protein HMGB1 in cytosolic fractions also suggested release in RC- and HF-fed Nrf2−/− livers of nuclear protein into the cytoplasm, which suggests loss of nuclear membrane integrity in a significant portion of hepatocytes that lack Nrf2 (Fig. 8F).
Nrf2−/− livers fail to adapt to high-fat diet-stimulated oxidative stress.
Consistent with the notion that the HF diet elicits an antioxidant response (37, 39), mRNA levels for the cystine/glutamate antiporter Slc7a11 and the Gclc and Gclm subunits, which together catalyze the rate-limiting step in glutathione synthesis, were increased in HF-fed Nrf2+/+ livers (Fig. 9A). An increase in mRNA for Gsr1, which catalyzes reduction of GSSG to GSH, was not observed. The HF diet also increased the amount of Gclc protein but not Gclm protein in Nrf2+/+ livers (Fig. 9B). In contrast, RC-fed Nrf2−/− livers contained smaller amounts of mRNA for Gclc and Gclm than their Nrf2+/+ counterparts, and also reduced levels of Gclc protein, with diminished or no increase following administration of the HF diet, suggesting that the basal expression of these genes is compromised in the mutant mice and that they mount a blunted antioxidant response. An exception to this outcome was the level of Gclm protein, which was relatively unchanged by genotype and exhibited a small increase in response to the HF diet. The failure of Gclm protein to reflect changes in Gclm mRNA is probably due to posttranscriptional regulation of the subunit, but at present, no definitive explanation exists for this anomaly. The hypothesis that Nrf2−/− livers have less intrinsic antioxidant capacity than Nrf2+/+ livers is supported by the fact that levels of mRNAs for Txnrd1, which reduces oxidized thioredoxin, and Srxn1, which reactivates overoxidized peroxiredoxin, are significantly lower in RC- and HF-fed Nrf2−/− livers (Fig. 9C).
FIG 9.

A high-fat diet stimulates an antioxidant response in Nrf2+/+ livers that is attenuated in Nrf2−/− livers. (A) Quantitative-PCR analysis of mRNAs for glutathione homeostasis genes in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (7 to 12 mice per group). (B) Representative immunoblots of Gclc and Gclm proteins in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. Quantification of the immunoblot data for each protein is shown by genotype and diet (3 mice per group). (C) Quantitative PCR of mRNAs for Txnrd1 and Srxn1 in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. (D) Quantitative PCR of mRNA for “prototypic” ARE-driven genes from livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet (10 to 12 mice per group). (E) Representative immunoblots of Gsta4, Gstm1-2, and Gsta1-2 in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. Quantification of the immunoblot data for each protein is shown by genotype and diet (3 mice per group). (F) Representative immunoblots of Nqo1 and Hmox1 in livers of Nrf2+/+ and Nrf2−/− mice on an RC or HF diet. Quantification of the immunoblot data for each protein is shown by genotype and diet (6 mice per group). In each case, the data were normalized with respect to RC-fed Nrf2+/+ levels for each protein or gene. White bars, RC fed; black bars, HF fed. The results are means and SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Molecular mass markers (B, E, and F) are in kDa.
To examine whether the HF diet induces prototypic ARE-driven genes, we measured Gst subunit mRNA levels and Gst, Nqo1, and Hmox1 proteins. In Nrf2+/+ livers, the HF diet produced ∼2- to 3-fold increases in mRNA levels for Gsta1, Gsta2, Gsta4, Gstm1, and Gstm2 and similar increases in the amounts of Gsta1/2, Gsta4, Gstm1/2, Hmox1, and Nqo1 proteins (Fig. 9C to E), suggesting that livers of wild-type mice can adapt to the oxidative stress presented by consumption of an HF diet. As expected, in RC-fed Nrf2−/− mouse livers, levels of these mRNAs and proteins were lower than in RC-fed Nrf2+/+ livers, indicating that Nrf2 controls their basal expression. However, the HF diet stimulated a modest induction of the majority of these genes in the mutant mice, albeit from a lower basal level (Fig. 9D to F), suggesting that Nrf2−/− livers are intrinsically more sensitive to oxidative stress and are less able to adapt to such insult.
DISCUSSION
NASH may arise through a “two-step” process in which insulin resistance first produces steatosis and subsequently oxidative stress drives inflammation (66, 67). As loss of Nrf2 increases both expression of lipogenesis genes and sensitivity to oxidative stress, it seems likely to contribute to the etiology of NASH. We therefore examined whether Nrf2−/− mice become insulin resistant when fed an HF diet and whether this results in their being more sensitive than wild-type mice to developing NASH.
The development of NASH in Nrf2−/− mice does not require insulin resistance.
Upon HF feeding, we found Nrf2−/− mice exhibit partial protection against obesity, increased energy expenditure, and better glucose disposal and insulin sensitivity than HF-fed Nrf2+/+ mice. Such outcomes, although conflicting with the majority of Nrf2 gain-of-function studies (37, 68, 69), are in agreement with other recent studies of long-term HF-fed Nrf2−/− mice (39, 43). The increased strength of insulin signaling in both Nrf2−/− livers and skeletal muscle possibly reflects impaired antioxidant capacity in the mutant animal that results in an increase in H2O2 levels, which in turn increases inactivation of protein tyrosine phosphatase 1B that antagonizes insulin signaling (70). The situation in Nrf2−/− mice may resemble that in glutathione peroxidase 1 knockout mice, where a relative decrease in the ability to eliminate H2O2 enhances insulin sensitivity (71). Although further work is required to demonstrate why HF-fed Nrf2−/− mice are more sensitive to insulin than their wild-type counterparts, our study demonstrates clearly that insulin resistance is not a prerequisite for NASH in Nrf2−/− mice. This is an unexpected result, because it is commonly believed that ER stress and inflammation, both of which occur in Nrf2−/− livers, are inexorably linked to insulin resistance (16).
Steatosis in Nrf2−/− mice is associated with multiple changes in lipid metabolism genes.
The greater degree of steatosis in livers of Nrf2−/− mice fed the HF diet for 24 weeks than in livers of similarly treated Nrf2+/+ mice is multifactorial. In part, it probably arises because the mutant animals upregulate the FA uptake protein Fabp5 and the FA synthesis enzymes Acly, Acaca, Fasn, and Elovl6 to a greater extent than Nrf2+/+ mice. It is also likely that the greater decrease in Cpt1a, which is rate limiting for FA β-oxidation, in Nrf2−/− livers than in Nrf2+/+ livers contributes to increased steatosis in the mutant animals.
The heightened upregulation of Fabp5, Acly, Acaca, Fasn, and Elovl6 in Nrf2−/− livers in response to the HF diet is likely to be due to the increased expression of the transcription factors Srebf1, Srebf2, Mlxipl, and PPARγ compared with that in HF Nrf2+/+ livers. Furthermore, decreased expression of Shp, which inhibits the cholesterol and FA homeostasis transcription factor LXRα, may also contribute to steatosis. Consistent with the latter idea, Kay et al. (45) reported that loss of Nrf2 greatly exacerbates stimulation of NASH by the LXRα agonist T0901317, whereas activation of Nrf2 by administration of sulforaphane inhibited NASH caused by the LXRα agonist. Indeed, they proposed that activation of Nrf2 resulted in FXR-mediated induction of Shp, which subsequently inhibited LXRα by forming an inactive LXRα-Shp heterodimer, and prevented T0901317 from stimulating NASH.
AMPK signaling is impaired in Nrf2−/− mice.
An unexpected finding was that Acc is no longer subject to inhibitory phosphorylation in the livers of RC- and HF-fed Nrf2−/− mice. The resulting increase in Acc is likely to augment malonyl-CoA levels, thus driving lipogenesis and decreasing FA oxidation. This was a surprising result, because AMPK, the kinase predominantly responsible for inhibitory Acc phosphorylation, is activated by H2O2 through phosphorylation by LKB1 (72, 73), and it is probable that H2O2 is more abundant in Nrf2−/− than in Nrf2+/+ livers. Immunoblotting suggested that the lack of Acc phosphorylation in Nrf2−/− livers results from reduction in AMPK activity. However, treatment with the AMPK activators AICAR and A-769662 enhanced the ability of Nrf2−/− hepatocytes to oxidize FA, suggesting the kinase can be activated in livers from knockout mice. Further work is required to establish how Nrf2 regulates this kinase. Intriguingly, the findings that Ampkβ1−/− mice are protected from HF diet-induced steatosis (74) and that Ampkβ1 (i.e., Prkab1) is upregulated in Nrf2−/− mice (31) suggest that it may contribute to exacerbation of steatosis in Nrf2−/− liver. Furthermore, reduced Ampk activity in Nrf2−/− livers also results in increased activity of Srebp-1c and Srebp-2, which drive lipogenesis.
Nrf2 suppresses ER stress and inflammation.
We found that Xbp1s, Chop, p-elf2α, and Atf6 protein levels and Chop, Xbp1, and Grp78 transcripts are upregulated in the livers of RC-fed Nrf2−/− mice, suggesting they may experience ER stress under basal conditions. It has not been reported previously that Nrf2−/− livers are subject to ER stress, but as we examined mice that are 32 to 34 weeks of age, it is possible this phenotype is apparent only in older mice, not the younger animals that are more commonly studied. Notably, Nrf2−/− fibroblasts have previously been reported to upregulate Chop under basal conditions (75), which agrees with our findings.
As ROS facilitates triggering of the UPR, and the antioxidant butylated hydroxyanisole can reduce ER stress (76, 77), it might be anticipated that Nrf2−/− mice are susceptible to ER stress. Evidence suggests that the UPR, along with ROS and release of Ca2+ from the ER, is linked to inflammation through activation of NF-κB and JNK (15, 16). Therefore, the increase in NF-κB and p-JNK proteins and expression of Cox2 and Nos2 in RC-fed Nrf2−/− livers is likely partly due to constitutive activation of the UPR in livers of mutant mice. Furthermore, the overexpression of Chop in Nrf2−/− livers may contribute to metabolic dysregulation, inflammation, and fibrosis, as these conditions have been linked to increased Chop levels in the mouse (78).
Our finding that the nuclear protein HMGB1 is present in the cytosolic fraction of HF Nrf2−/− livers is interesting, because upon release from cells, it can function as a damage-associated molecular-pattern polypeptide that contributes to inflammatory processes (79, 80). Importantly, signaling by HMGB1 is influenced by its redox status, and this might be expected to differ markedly in Nrf2−/− and wild-type mice.
Surprisingly, in HF-fed Nrf2−/− livers there was an overall reduction in the levels of ER stress markers. This apparent failure to maintain the UPR in livers of HF-fed Nrf2−/− mice might be associated with chronic damage owing to the duration of the experiment. Further experiments are required to allow the time course of HF diet-stimulated ER stress in wild-type and Nrf2−/− livers to be documented.
Concluding comments.
The results presented here show that knockout of Nrf2 renders mice more sensitive to NASH when placed on an HF diet. Our data indicate that the increase in steatosis in Nrf2−/− mice placed on the HF diet is due to a heightened level of induction of lipogenesis genes and suppression of β-oxidation genes compared with wild-type animals. Administration of the HF diet stimulated hepatic oxidative stress, and as livers of Nrf2−/− mice are unable to adapt to such insults, this leads to failure to maintain normal homeostatic levels of the glutathione- and thioredoxin-based antioxidant systems, which may stimulate inflammation. Unexpectedly, we found that the UPR is perturbed in the livers of Nrf2−/− mice on the RC control diet, suggesting that ER stress is triggered in these animals even under basal conditions. It is therefore possible that disturbance of the UPR in the livers of Nrf2−/− mice contributes to the rapid development of NASH when they are placed on an HF diet. In humans, promoter polymorphisms exist in the NRF2 gene (NFE2L2) that alter its activity (81), and it will be interesting in the future to determine whether they influence lipid metabolism, oxidative stress, inflammation, or ER stress in humans.
ACKNOWLEDGMENTS
This work was supported by grants from the Medical Research Council (MR/J001465/1), the Wellcome Trust (086989), and Diabetes UK (12/0004458), and we thank them for their support.
We thank Michael T. Lotze (University of Pittsburgh) for advice on HMGB1, Frank A. Carey (University of Dundee) for advice on hepatocyte ultrastructure, and Philip J. Coates (University of Dundee) for advice on histology.
Footnotes
Published ahead of print 23 June 2014
REFERENCES
- 1.Browning JD, Horton JD. 2004. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114:147–152. 10.1172/JCI200422422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrell GC, Larter CZ. 2006. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43:S99–S112. 10.1002/hep.20973 [DOI] [PubMed] [Google Scholar]
- 3.Cohen JC, Horton JD, Hobbs HH. 2011. Human fatty liver disease: old questions and new insights. Science 332:1519–1523. 10.1126/science.1204265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. 2004. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461. 10.1126/science.1103160 [DOI] [PubMed] [Google Scholar]
- 5.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. 2011. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473:528–531. 10.1038/nature09968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchs M, Sanyal AJ. 2012. Lipotoxicity in NASH. J. Hepatol. 56:291–293. 10.1016/j.jhep.2011.05.019 [DOI] [PubMed] [Google Scholar]
- 7.Schroeder-Gloeckler JM, Rahman SM, Janssen RC, Qiao L, Shao J, Roper M, Fischer SJ, Lowe E, Orlicky DJ, McManaman JL, Palmer C, Gitomer WL, Huang W, O'Doherty RM, Becker TC, Klemm DJ, Jensen DR, Pulawa LK, Eckel RH, Friedman JE. 2007. CCAAT/enhancer-binding protein β deletion reduces adiposity, hepatic steatosis, and diabetes in Lepr(db/db) mice. J. Biol. Chem. 282:15717–15729. 10.1074/jbc.M701329200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee AH, Scapa EF, Cohen DE, Glimcher LH. 2008. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320:1492–1496. 10.1126/science.1158042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. 2008. Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 7:520–532. 10.1016/j.cmet.2008.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferré P, Foufelle F. 2009. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 119:1201–1215. 10.1172/JCI37007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, Katze MG, Hussain MM, Song B, Swathirajan J, Wang J, Yau GD, Kaufman RJ. 2008. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 15:829–840. 10.1016/j.devcel.2008.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chikka MR, McCabe DD, Tyra HM, Rutkowski DT. 2013. C/EBP homologous protein (CHOP) contributes to suppression of metabolic genes during endoplasmic reticulum stress in the liver. J. Biol. Chem. 288:4405–4415. 10.1074/jbc.M112.432344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rolo AP, Teodoro JS, Palmeira CM. 2012. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 52:59–69. 10.1016/j.freeradbiomed.2011.10.003 [DOI] [PubMed] [Google Scholar]
- 14.Farrell GC, van Rooyen D, Gan L, Chitturi S. 2012. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver 6:149–171. 10.5009/gnl.2012.6.2.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang K, Kaufman RJ. 2008. From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462. 10.1038/nature07203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flamment M, Hajduch E, Ferré P, Foufelle F. 2012. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 23:381–390. 10.1016/j.tem.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 17.Cao SS, Kaufman RJ. 2014. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 21:396–413 . 10.1089/ars.2014.5851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iredale JP, Thompson A, Henderson NC. 2013. Extracellular matrix degradation in liver fibrosis: biochemistry and regulation. Biochim. Biophys. Acta 1832:876–883. 10.1016/j.bbadis.2012.11.002 [DOI] [PubMed] [Google Scholar]
- 19.Kensler TW, Wakabayashi N, Biswal S. 2007. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47:89–116. 10.1146/annurev.pharmtox.46.120604.141046 [DOI] [PubMed] [Google Scholar]
- 20.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. 2010. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 13:1713–1748. 10.1089/ars.2010.3221 [DOI] [PubMed] [Google Scholar]
- 21.Suzuki T, Motohashi H, Yamamoto M. 2013. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 34:340–346. 10.1016/j.tips.2013.04.005 [DOI] [PubMed] [Google Scholar]
- 22.McMahon M, Itoh K, Yamamoto M, Hayes JD. 2003. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 278:21592–21600. 10.1074/jbc.M300931200 [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. 2004. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24:7130–7139. 10.1128/MCB.24.16.7130-7139.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. 2004. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24:10941–10953. 10.1128/MCB.24.24.10941-10953.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumagai Y, Yamamoto M. 2009. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 29:493–502. 10.1128/MCB.01080-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMahon M, Lamont DJ, Beattie KA, Hayes JD. 2010. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. U. S. A. 107:18838–18843. 10.1073/pnas.1007387107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hourihan JM, Kenna JG, Hayes JD. 2013. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 19:465–481. 10.1089/ars.2012.4944 [DOI] [PubMed] [Google Scholar]
- 28.Higgins LG, Kelleher MO, Eggleston IM, Itoh K, Yamamoto M, Hayes JD. 2009. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 237:267–280. 10.1016/j.taap.2009.03.005 [DOI] [PubMed] [Google Scholar]
- 29.Yates MS, Tran QT, Dolan PM, Osburn WO, Shin S, McCulloch CC, Silkworth JB, Taguchi K, Yamamoto M, Williams CR, Liby KT, Sporn MB, Sutter TR, Kensler TW. 2009. Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 30:1024–1031. 10.1093/carcin/bgp100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitteringham NR, Abdullah A, Walsh J, Randle L, Jenkins RE, Sison R, Goldring CE, Powell H, Sanderson C, Williams S, Higgins L, Yamamoto M, Hayes J, Park BK. 2010. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J. Proteomics 73:1612–1631. 10.1016/j.jprot.2010.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu KC, Cui JY, Klaassen CD. 2011. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 123:590–600. 10.1093/toxsci/kfr183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy NM, Potteti HR, Mariani TJ, Biswal S, Reddy SP. 2011. Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am. J. Respir. Cell Mol. Biol. 45:1161–1168. 10.1165/rcmb.2011-0144OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chowdhry S, Nazmy MH, Meakin PJ, Dinkova-Kostova AT, Walsh SV, Tsujita T, Dillon JF, Ashford ML, Hayes JD. 2010. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 48:357–371. 10.1016/j.freeradbiomed.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 34.Sugimoto H, Okada K, Shoda J, Warabi E, Ishige K, Ueda T, Taguchi K, Yanagawa T, Nakahara A, Hyodo I, Ishii T, Yamamoto M. 2010. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 298:G283–G294. 10.1152/ajpgi.00296.2009 [DOI] [PubMed] [Google Scholar]
- 35.Zhang YK, Yeager RL, Tanaka Y, Klaassen CD. 2010. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 245:326–334. 10.1016/j.taap.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka Y, Aleksunes LM, Yeager RL, Gyamfi MA, Esterly N, Guo GL, Klaassen CD. 2008. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J. Pharmacol. Exp. Ther. 325:655–664. 10.1124/jpet.107.135822 [DOI] [PubMed] [Google Scholar]
- 37.Shin S, Wakabayashi J, Yates MS, Wakabayashi N, Dolan PM, Aja S, Liby KT, Sporn MB, Yamamoto M, Kensler TW. 2009. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 620:138–144. 10.1016/j.ejphar.2009.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang J, Tabbi-Anneni I, Gunda V, Wang L. 2010. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 299:G1211–G1221. 10.1152/ajpgi.00322.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chartoumpekis DV, Ziros PG, Psyrogiannis AI, Papavassiliou AG, Kyriazopoulou VE, Spandidos DA, Habeos IG. 2011. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes 60:2465–2473. 10.2337/db11-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka Y, Ikeda T, Yamamoto K, Ogawa H, Kamisako T. 2012. Dysregulated expression of fatty acid oxidation enzymes and iron-regulatory genes in livers of Nrf2-null mice. J. Gastroenterol. Hepatol. 27:1711–1717. 10.1111/j.1440-1746.2012.07180.x [DOI] [PubMed] [Google Scholar]
- 41.Zhang YK, Wu KC, Liu J, Klaassen CD. 2012. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol. Appl. Pharmacol. 264:305–314. 10.1016/j.taap.2012.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C, Cui Y, Li C, Zhang Y, Xu S, Li X, Li H, Zhang X. 2013. Nrf2 deletion causes “benign” simple steatosis to develop into nonalcoholic steatohepatitis in mice fed a high-fat diet. Lipids Health Dis. 12:165. 10.1186/1476-511X-12-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okada K, Warabi E, Sugimoto H, Horie M, Gotoh N, Tokushige K, Hashimoto E, Utsunomiya H, Takahashi H, Ishii T, Yamamoto M, Shoda J. 2013. Deletion of Nrf2 leads to rapid progression of steatohepatitis in mice fed atherogenic plus high-fat diet. J. Gastroenterol. 48:620–632. 10.1007/s00535-012-0659-z [DOI] [PubMed] [Google Scholar]
- 44.Chartoumpekis DV, Ziros PG, Zaravinos A, Iskrenova RP, Psyrogiannis AI, Kyriazopoulou VE, Sykiotis GP, Habeos IG. 2013. Hepatic gene expression profiling in Nrf2 knockout mice after long-term high-fat diet-induced obesity. Oxid. Med. Cell Longev. 2013:340731. 10.1155/2013/340731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kay HY, Kim WD, Hwang SJ, Choi HS, Gilroy RK, Wan YJ, Kim SG. 2011. Nrf2 inhibits LXRα-dependent hepatic lipogenesis by competing with FXR for acetylase binding. Antioxid. Redox Signal. 15:2135–2146. 10.1089/ars.2010.3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu J, Kulkarni SR, Donepudi AC, More VR, Slitt AL. 2012. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 61:3208–3218. 10.2337/db11-1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.More VR, Xu J, Shimpi PC, Belgrave C, Luyendyk JP, Yamamoto M, Slitt AL. 2013. Keap1 knockdown increases markers of metabolic syndrome after long-term high fat diet feeding. Free Radic. Biol. Med. 61C:85–94. 10.1016/j.freeradbiomed.2013.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cazanave SC, Wang X, Zhou H, Rahmani M, Grant S, Durrant DE, Klaassen CD, Yamamoto M, Sanyal AJ. 2014. Degradation of Keap1 activates BH3-only proteins Bim and PUMA during hepatocyte lipoapoptosis. Cell Death Differ. 21:1303–1312. 10.1038/cdd.2014.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236:313–322. 10.1006/bbrc.1997.6943 [DOI] [PubMed] [Google Scholar]
- 50.Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GJ, Itoh K, Yamamoto M, Hayes JD. 2002. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 365:405–416. 10.1042/BJ20020320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meakin PJ, Harper AL, Hamilton DL, Gallagher J, McNeilly AD, Burgess LA, Vaanholt LM, Bannon KA, Latcham J, Hussain I, Speakman JR, Howlett DR, Ashford ML. 2012. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem. J. 441:285–296. 10.1042/BJ20110512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander-Tetri BA, NASH Clinical Research Network 2011. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology 53:810–820. 10.1002/hep.24127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ritchie KJ, Walsh S, Sansom OJ, Henderson CJ, Wolf CR. 2009. Markedly enhanced colon tumorigenesis in Apc(Min) mice lacking glutathione S-transferase Pi. Proc. Natl. Acad. Sci. U. S. A. 106:20859–20864. 10.1073/pnas.0911351106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Save V, Hall PA, Coates PJ. 2004. Detecting and quantifying apoptosis in tissue sections. Methods Mol. Biol. 282:67–84. 10.1385/1-59259-812-9:067 [DOI] [PubMed] [Google Scholar]
- 55.Kelly VP, Ellis EM, Manson MM, Chanas SA, Moffat GJ, McLeod R, Judah DJ, Neal GE, Hayes JD. 2000. Chemoprevention of aflatoxin B1 hepatocarcinogenesis by coumarin, a natural benzopyrone that is a potent inducer of aflatoxin B1-aldehyde reductase, the glutathione S-transferase A5 and P1 subunits, and NAD(P)H:quinone oxidoreductase in rat liver. Cancer Res. 60:957–969 http://cancerres.aacrjournals.org/content/60/4/957 [PubMed] [Google Scholar]
- 56.Zheng Z, Zhang C, Zhang K. 2011. Measurement of ER stress response and inflammation in the mouse model of nonalcoholic fatty liver disease. Methods Enzymol. 489:329–348. 10.1016/B978-0-12-385116-1.00019-4 [DOI] [PubMed] [Google Scholar]
- 57.Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. 2003. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem. J. 374:337–348. 10.1042/BJ20030754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. 2010. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120:2355–2369. 10.1172/JCI40671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cappel RE, Gilbert HF. 1993. Oxidative inactivation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase and subunit cross-linking involve different dithiol/disulfide centers. J. Biol. Chem. 268:342–348 [PubMed] [Google Scholar]
- 60.Claudel T, Staels B, Kuipers F. 2005. The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler. Thromb. Vasc. Biol. 25:2020–2030. 10.1161/01.ATV.0000178994.21828.a7 [DOI] [PubMed] [Google Scholar]
- 61.Nguyen P, Leray V, Diez M, Serisier S, Le Bloc'h J, Siliart B, Dumon H. 2008. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 92:272–283. 10.1111/j.1439-0396.2007.00752.x [DOI] [PubMed] [Google Scholar]
- 62.Strable MS, Ntambi JM. 2010. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 45:199–214. 10.3109/10409231003667500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. 2012. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 56:952–964. 10.1016/j.jhep.2011.08.025 [DOI] [PubMed] [Google Scholar]
- 64.Serviddio G, Bellanti F, Vendemiale G. 2013. Free radical biology for medicine: learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 65:952–968. 10.1016/j.freeradbiomed.2013.08.174 [DOI] [PubMed] [Google Scholar]
- 65.Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, Malle E, Greve JW, Buurman WA. 2009. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am. J. Pathol. 175:1473–1482. 10.2353/ajpath.2009.080999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Day CP, James OF. 1998. Steatohepatitis: a tale of two “hits”? Gastroenterology 114:842–845. 10.1016/S0016-5085(98)70599-2 [DOI] [PubMed] [Google Scholar]
- 67.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. 2008. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic. Biol. Med. 44:1259–1272. 10.1016/j.freeradbiomed.2007.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu Z, Shao W, Chiang Y, Foltz W, Zhang Z, Ling W, Fantus IG, Jin T. 2011. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 (NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia 54:922–934. 10.1007/s00125-010-2001-8 [DOI] [PubMed] [Google Scholar]
- 69.Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, Sugawara A, Kensler TW, Yamamoto M. 2013. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 33:2996–3010. 10.1128/MCB.00225-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tiganis T. 2011. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol. Sci. 32:82–89. 10.1016/j.tips.2010.11.006 [DOI] [PubMed] [Google Scholar]
- 71.Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, Stepto N, Wu B, Mitchell CA, Tonks NK, Watt MJ, Febbraio MA, Crack PJ, Andrikopoulos S, Tiganis T. 2009. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 10:260–272. 10.1016/j.cmet.2009.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blättler SM, Rencurel F, Kaufmann MR, Meyer UA. 2007. In the regulation of cytochrome P450 genes, phenobarbital targets LKB1 for necessary activation of AMP-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 104:1045–1050. 10.1073/pnas.0610216104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. 2010. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11:554–565. 10.1016/j.cmet.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dzamko N, van Denderen BJ, Hevener AL, Jørgensen SB, Honeyman J, Galic S, Chen ZP, Watt MJ, Campbell DJ, Steinberg GR, Kemp BE. 2010. AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J. Biol. Chem. 285:115–122. 10.1074/jbc.M109.056762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cullinan SB, Diehl JA. 2004. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 279:20108–20117. 10.1074/jbc.M314219200 [DOI] [PubMed] [Google Scholar]
- 76.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. 2008. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U. S. A. 105:18525–18530. 10.1073/pnas.0809677105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. 2009. Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 10:13–26. 10.1016/j.cmet.2009.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.DeZwaan-McCabe D, Riordan JD, Arensdorf AM, Icardi MS, Dupuy AJ, Rutkowski DT. 2013. The stress-regulated transcription factor CHOP promotes hepatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS Genet. 9:e1003937. 10.1371/journal.pgen.1003937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kang R, Zhang Q, Zeh HJ, III, Lotze MT, Tang D. 2013. HMGB1 in cancer: good, bad, or both? Clin. Cancer Res. 19:4046–4057. 10.1158/1078-0432.CCR-13-0495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen R, Hou W, Zhang Q, Kang R, Fan XG, Tang D. 2013. Emerging role of high-mobility group box 1 (HMGB1) in liver diseases. Mol. Med. 19:357–366. 10.2119/molmed.2013.00099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cho HY. 2013. Genomic structure and variation of nuclear factor (erythroid-derived 2)-like 2. Oxid. Med. Cell Longev. 2013:286524. 10.1155/2013/286524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Winder WW, Hardie DG. 1999. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am. J. Physiol. 277:E1–E10 [DOI] [PubMed] [Google Scholar]