

Abstract
Cortisol, the central stress hormone in humans, activates the glucocorticoid receptor (GR). Anti-inflammatory effects are the most important pharmaceutical effects mediated by the GR. Inasmuch as electrophilic cyclopentenone prostaglandin 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) has potent anti-inflammatory properties and activates the SUMOylation pathway, we have investigated the effect of 15d-PGJ2 on glucocorticoid signaling and receptor SUMOylation. To this end, we studied isogenic HEK293 cells expressing either wild-type GR or SUMOylation-defective GR. Interestingly, 15d-PGJ2 triggered SUMO-2 and -3 (SUMO-2/3) modification in the primary SUMOylation sites of the GR. Gene expression profiling and pathway analyses indicate that 15d-PGJ2 inhibits GR signaling in a genome-wide fashion that is significantly dependent on the GR SUMOylation sites. Chromatin immunoprecipitation assays showed that the repressive effect of 15d-PGJ2 on GR target gene expression occurs in parallel with the inhibition of receptor binding to the target gene chromatin. Furthermore, depletion of UBC9, the sole SUMO E2 conjugase, from HEK293 cells confirmed the involvement of active SUMOylation in the regulatory process. Taken together, our data indicate that GR SUMOylation modulates the glucocorticoid signaling during acute cell stress. Our data also suggest that GR SUMOylation modulates cross talk of the glucocorticoid signaling with other transcription factors that are responsive to cell stress.
INTRODUCTION
Mammals respond to stress by activating the hypothalamic-pituitary-adrenal axis, which causes secretion of primary stress hormones, namely, glucocorticoids (cortisol in humans and corticosterone in rodents) (1). The action of glucocorticoids is mediated by the glucocorticoid receptor (GR) (2, 3) that, upon ligand binding, moves to the nucleus and binds to short DNA sequences, glucocorticoid response elements (GREs), in target loci. The GR recruits and interacts with various coregulators that include histone-modifying and chromatin-remodeling activities, which leads to either enhancement or inhibition of target gene transcription (4, 5). Anti-inflammatory effects are among the most important effects mediated by the GR (6, 7) during short-term stress (8, 9). However, in prolonged stress, the effects of glucocorticoids can become proinflammatory (8, 9).
In addition to activating the GR, glucocorticoids induce posttranslational modifications (PTMs) of the receptor, including phosphorylation (10) and SUMOylation (11, 12). Small ubiquitin-related modifier proteins (SUMOs) can be covalently conjugated (SUMOylation) to specific lysine residues of several nuclear receptors (12–15). Humans express three SUMO paralogs, SUMO-1, -2, and -3, that can form isopeptide linkages with target proteins. SUMO-2 and -3 are essentially identical (and are called SUMO-2/3 here), but SUMO-1 is only ∼50% identical to SUMO-2/3 (16, 17). Prior to conjugation by UBC9 (E2 activity), the SUMOs require activation by SAE1 and -2 dimers (E1 activity) (18). Conjugation can be enhanced by SUMO ligases (E3 activities), such as protein inhibitor of activated STAT (PIAS) proteins (19). SUMO modifications are highly dynamic and are reversed by the presence of members of a family of SUMO-specific proteases (20). Our recent genome-wide analyses indicate that basal SUMOylation cycles of agonist-bound GR regulate the receptor's chromatin occupancy, playing an important role in controlling the antiproliferative effect of glucocorticoids (12).
Interestingly, various cell stress conditions, including electrophilic and oxidative stress, induce hyper-SUMOylation, i.e., accumulation of SUMO-2/3 to a number of proteins (21, 22, 23). Notably, a recent proteomic screening of SUMOylated proteins from pre- and postischemic brains of mice revealed hyper-SUMOylation of GR after ischemia (24). Cyclopentenone prostaglandin 15d-PGJ2, a product derived for the cyclo-oxygenase pathway involved in the resolution of inflammation (25), is a known activator of the anti-inflammatory and cytoprotective Kelch-like ECH-associated protein 1 (KEAP1)–nuclear factor erythroid 2-related factor 2 (NRF2) system (26). It is also an endogenous ligand for peroxisome proliferator-activated receptor γ (PPARγ) (27). The anti-inflammatory actions of 15d-PGJ2 are thought to mainly rely on its ability to activate the PPARγ and NRF2 and to inhibit proinflammatory transcription factors, such as nuclear factor κB(NF-κB) and activator protein 1 (AP-1) (28–30). In addition to inhibiting proinflammatory proteins, 15d-PGJ2 has been shown to inhibit estrogen receptor alpha (ERα) and androgen receptor (AR) activity (31, 32) as well as GR activity (33). Furthermore, 15d-PGJ2 also induces SUMOylation of the AR (32). Given that 15d-PGJ2 is anti-inflammatory and affects the activity of several nuclear receptors, we sought to determine its effects on glucocorticoid signaling and the role of GR SUMOylation. To this end, we used human A549 cells expressing endogenous GR as well as isogenic HEK293 cell lines stably expressing either wild-type GR or SUMOylation-defective GR. The results of the SUMOylation, transcriptome, and chromatin immunoprecipitation analyses indicate that the repressive effect of 15d-PGJ2-triggered cell stress on glucocorticoid signaling is modulated by GR SUMOylation.
MATERIALS AND METHODS
Cell culture.
Isogenic HEK293 cell lines stably expressing wild-type GR (wtGR) or GR3KR (VK277TE → VR277TE, IK293QE → IR293QE, VK703RE → VR703RE) were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco/Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 25 U/ml penicillin and 25 μg/ml streptomycin, and 100 μg/ml hygromycin B (12). A549 cells (from ATCC) were maintained in F-12K medium supplemented with 10% FBS, 25 U/ml penicillin, and 25 μg/ml streptomycin.
Antibodies and chemicals.
Primary antibodies from Santa Cruz Biotechnology (Santa Cruz, CA) were as follows: GR (sc-1003), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (sc-25778), UBC9 (sc-10759), and normal rabbit IgG (sc-2027). SUMO-1 (33-2400) was from Invitrogen Life Technologies (Carlsbad, CA), and SUMO-2/3 (M114-3) was from MBL International Corporation (Woburn, MA). 15-Deoxy-Δ12,14-prostaglandin J2 (18570) and its biotinylated form (10141) were from Cayman Chemicals (Ann Arbor, MI). Human tumor necrosis factor alpha (TNF-α) was a kind gift from Claude Libert (Ghent University).
Immunoprecipitation, immunoblotting, and biotin pulldown assay.
For SUMOylation assays, cells were harvested, lysed, immunoprecipitated, and analyzed by immunoblotting as described previously (32, 34). Stably GR-expressing HEK293 cells were treated with or without 100 nM dexamethasone (dex) in the presence or absence of biotinylated 15d-PGJ2 essentially as described previously (32). Biotin-bound proteins were affinity isolated with Streptavidin Plus UltraLink resin (Pierce, Rockford, IL), and bound complexes were analyzed by immunoblotting with anti-GR and anti-GAPDH antibodies.
Isolation of RNA and RT-qPCR analyses.
Stably GR-expressing HEK293 and A549 cells were seeded onto 6-well plates (300,000 cells per well) and grown for 36 h in steroid-depleted transfection medium. Subsequently, cells were treated with or without 100 nM dex for 6 h in the presence or absence of increasing concentrations (2.5, 5, and 10 μM) of 15d-PGJ2. For TNF-α experiments, the cells were treated with 1,000 IU/ml of TNF-α in the presence or absence of dex and/or 15d-PGJ2. Extraction of RNA and cDNA synthesis were performed as previously described (12). cDNA was used as a template in reverse transcription-quantitative PCR (RT-qPCR), which was carried out using LightCycler 480 SYBR green I Master (Roche Diagnostics GmbH) and a LightCycler 480 system (Roche) and with specific primers listed in Table SA1 in the supplemental material. Analyzed RPL13A mRNA levels were used to normalize the amounts of total RNA between the samples. Fold changes were calculated using the formula 2−(ΔΔCT), where ΔΔCT is ΔCT(treatment) − ΔCT(EtOH), ΔCT is CT(gene X) − CT(RPL13A), and CT is the cycle at which the threshold is crossed.
Microarray analysis.
Total RNA of dex-treated samples with or without treatment with 5 μM 15d-PGJ2 was analyzed on Sentrix HumanHT-12 v4 Expression BeadChips (Illumina, San Diego, CA) using the manufacturer's protocol at the Finnish Microarray and Sequencing Center (Turku, Finland). Microarray data were analyzed as previously described (12). Identification of dex target genes was based on our previous transcriptome analysis of dex-up- or -downregulated genes from the same cells (P < 0.01) (12) (GSE48328). Ingenuity pathway analysis (IPA) was used to identify pathways differently enriched and differences in upstream regulators between wtGR- and GR3KR-expressing cells. Heat maps were generated by using heatmap.2 in the R package gplots.
ChIP.
Stably GR-expressing HEK293 cells and A549 cells were seeded at ∼70% confluence into 175-cm2 bottles and allowed to grow in steroid-depleted transfection medium for 72 h prior to chromatin immunoprecipitation (ChIP). Cells were treated with either vehicle or 100 nM dex for 1 h in the presence or absence of 2.5, 5, or 10 μM 15d-PGJ2. ChIPs were performed as described previously (12). Quantitative PCR analyses were carried out with LightCycler 480 SYBR green I Master (Roche). Specific primers are listed in Table SA2 in the supplemental material. Results were calculated using the formula E−(ΔCT) × 19, where E (efficiency of target amplification) is a coefficient of DNA amplification by one PCR cycle for a particular primer pair and ΔCT is CT(ChIP-template) − CT(Input). Results are presented as fold increases over the value determined for IgG-precipitated samples.
RNA interference.
For silencing of UBC9, Dharmacon (Lafayette, CO) On-TARGETplus SMARTpool for human UBE2I (siUBC9) and a nontargeting pool (siNON-Target) were used. For small interfering RNA (siRNA) transfections, siUBC9 or siNON-Target (40 nM final concentration) was added with Lipofectamine RNAiMAX (Invitrogen) to 6-well plates. After 20 min of incubation at 22°C, wtGR or GR3KR HEK293 cells were seeded onto the plates (300,000 cells per well) in steroid-depleted transfection medium. The cells were grown with siRNAs for 96 h and treated with vehicle, with dex, or with dex in the presence of 5 μM 15d-PGJ2 for 6 h prior to extraction of RNA or proteins. For immunoprecipitations, cells were treated with dex for 2 h and 5 μM 15d-PGJ2 was added for 1 h before harvesting.
Microarray accession number.
Bead array data were submitted to the NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE54087.
RESULTS
GR activity is inhibited by 15d-PGJ2.
To study how 15d-PGJ2 influences GR activity and the role of GR SUMOylation, we treated isogenic wtGR- or SUMOylation-defective GR3KR-expressing HEK293 cells with increasing concentrations (0, 2.5, 5, and 10 μM) of 15d-PGJ2 and measured dexamethasone (dex)-induced expression of GR target genes CDKN1C, ELK1, and ARMC12 and GR chromatin binding to their regulatory regions by RT-qPCR and quantitative ChIP, respectively. Interestingly, both dex-induced expression of the genes and their receptor chromatin occupancy were dose-dependently and significantly inhibited by the prostaglandin in the wtGR but not in the GR3KR cells (Fig. 1A and B). It is of note that, although the expression of CDKN1C in GR3KR cells was lower than in wtGR cells (12), it was still induced in GR3KR-expressing cells by dex treatment (∼4-fold induction), but, in contrast to the wtGR cells, it was not downregulated in response to 15d-PGJ2 exposure in the GR3KR cells. The same holds true for ARMC12, which was also efficiently expressed in GR3KR-expressing cells by dex treatment (∼14-fold induction) and not downregulated in response to 15d-PGJ2 exposure. In the case of ELK1, the activity of GR3KR was notably equal to that of wtGR and yet GR3KR was not inhibited by 15d-PGJ2 exposure, whereas wtGR was significantly inhibited. In contrast, the expression of NRF2 target gene HMOX1 was augmented by 15d-PGJ2 in both cell lines (Fig. 1C), indicating that the prostaglandin exposure did not lead to a general repression of gene expression. However, since the fold induction of the HMOX1 gene expression by 15d-PGJ2 in the absence of dex clearly exceeded that seen in the presence of dex, it appears that the GR can antagonize the effect of the prostaglandin on the HMOX1 expression. This notion is in line with our ChIP sequencing data showing that GR binds to HMOX1 at ∼10 and ∼20 kb upstream of its transcription start site (12) (see Fig. SA1 in the supplemental material). To rule out the possibility that other lysine PTMs are targeted to the SUMOylation sites of GR, we mutated the +2-position glutamic acid residues of the N-terminal SUMOylation consensus sequences to alanine residues (E279, 295A; GR2EA). Indeed, 15d-PGJ2 had an effect on the expression of CDKN1C, ELK1, and ARMC12 in GR2EA-expressing cells comparable to that seen in GR3KR cells (see Fig. SA2). The target gene expression was significantly inhibited only in the HEK293 cells expressing wtGR, indicating that the repression is dependent on the SUMOylation sites of GR. The repressive effect of 15d-PGJ2 was not restricted to the wtGR HEK293 model cells, as the compound significantly repressed the expression and the GR chromatin occupancy of, e.g., CDKN1C in A549 cells containing endogenous GR (Fig. 1A and B). The expression of HMOX1 was augmented in response to the presence of 15d-PGJ2 also in the A549 cells (Fig. 1C).
FIG 1.

GR target gene expression and chromatin binding are dose-dependently inhibited by 15d-PGJ2. (A) wtGR-expressing HEK293 cells, GR3KR-expressing HEK293 cells, or A549 cells were exposed to vehicle or dex (100 nM) together with the indicated concentrations of 15d-PGJ2 for 6 h, and RNAs were isolated. RT-qPCR analyses were performed with specific primers for CDKN1C, ELK1, and ARMC12. (B) Cells were treated with vehicle or dex with increasing concentrations of 15d-PGJ2 for 1 h. Binding of GR to CDKN1C, ELK1, and ARMC12 regulatory elements was monitored by quantitative ChIP (qChIP) analyses with anti-GR antibody as described in Materials and Methods. Results are shown as values of fold increase compared to levels seen with the normal IgG-precipitated samples. (C) Cell lines were treated and analyzed as described for panel A but with specific primers for NRF2 target gene HMOX1. The effects of increasing 15d-PGJ2 concentrations on the expression of HMOX1 in the absence of dex are indicated. Results represent the means ± standard deviations (SD) of the results of three experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (for the differences in the results of dex treatment in the presence or absence of 5 μM 15d-PGJ2 [Student's t test]). ns, no significance.
15d-PGJ2 triggers GR SUMOylation.
Due to the fact that 15d-PGJ2 can be covalently conjugated to signaling proteins (29, 30), including the AR (32), we assessed whether the greater sensitivity of the wtGR compared to that of GR3KR to the prostaglandin exposure could be explained by its more robust adduct formation with 15d-PGJ2. However, this was not the case, as cell-permeative biotinylated 15d-PGJ2 was capable of forming complexes with both receptor forms in comparable manners (Fig. 2A). Biotinylated 15d-PGJ2 bound to both the DNA-binding domain (DBD) and the ligand-binding domain (LBD) of wtGR. However, the binding relative to the number of cysteine residues within the domain was more efficient with LBD than with DBD (see Fig. SA3 in the supplemental material). We next examined if 15d-PGJ2 induces GR SUMOylation. Immunoblotting of anti-GR antibody immunoprecipitates with anti-SUMO-1 and anti-SUMO-2/3 antibodies showed that 15d-PGJ2 triggered accumulation of SUMO-2/3 but not of SUMO-1 in dex-bound wtGR but not in dex-bound GR3KR (Fig. 2B), indicating that 15d-PGJ2 causes SUMO-2/3 conjugation to the major SUMOylation sites in the GR. The effect of dex on the SUMOylation most likely reflects the role of hormones in triggering dissociation of chaperone proteins from the GR and inducing nuclear translocation of the receptor (2). Therefore, the SUMOylation most likely takes place in the nucleus.
FIG 2.
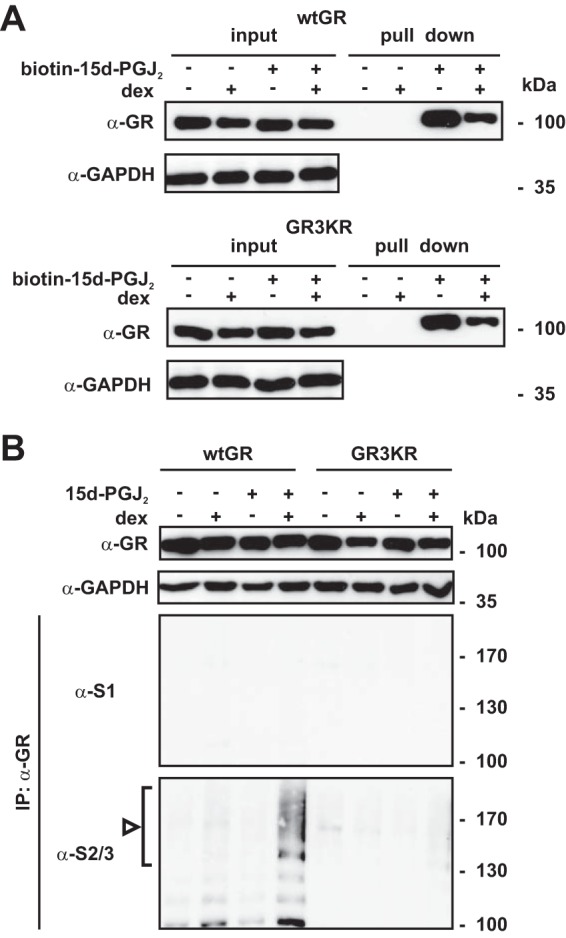
Agonist-bound GR is modified by SUMO-2/3 in response to 15d-PGJ2 exposure. (A) HEK293 cells stably expressing wtGR or GR3KR were grown in the presence or absence of 100 nM dex for 2 h and were subsequently exposed to biotinylated 15d-PGJ2 (10 μM) for 2 h as indicated. Cells were lysed, and the biotin–15d-PGJ2-bound proteins were subjected to affinity isolation with streptavidin beads. Formation of 15d-PGJ2 adducts was analyzed by immunoblotting with anti-GR antibody. (B) The cells were grown as described for panel A but were exposed to 15d-PGJ2. After 1 h, cells were lysed in a denaturing buffer and portions of lysates (20% of immunoprecipitation [IP] inputs) were analyzed by immunoblotting with anti-GR and anti-GAPDH antibodies or were subjected to immunoprecipitation with anti-GR antibody. The precipitates were analyzed by immunoblotting with anti-SUMO-1 (S1) and anti-SUMO-2/3 (S2/3) antibodies as indicated. The arrowhead and bracket indicate the position of SUMO-2/3-modified forms of GR.
Active SUMOylation modulates the inhibition of GR by 15d-PGJ2.
The results presented above suggested that SUMOylation is involved in the modulation of GR sensitivity by 15d-PGJ2. To confirm this notion, we silenced UBC9, the SUMO E2 conjugase, by the use of siRNAs in the HEK293 cells (Fig. 3A and B). As expected, depletion of the sole SUMO E2 activity abolished the 15d-PGJ2-triggered SUMO-2/3 modifications in the GR (Fig. 3A). Comparison of the effects of 15d-PGJ2 on the expression of CDKN1C, ELK1, and ARMC12 in the UBC9-depleted versus control siRNA-treated cells indicated that while the dex-induced expression was significantly inhibited by 15d-PGJ2 in the control siRNA-treated wtGR cells, there was no significant inhibition in the UBC9-depleted wtGR cells (Fig. 3C). Furthermore, the abolishment of dex-triggered repression of JUN by 15d-PGJ2 in the control siRNA-treated wtGR cells was not detectable in the UBC9-depleted cells. In the GR3KR cells, these genes remained inert with respect to the prostaglandin also after the UBC9 depletion. Together, these results strongly support the idea of a role of active SUMOylation of GR in the modulation of glucocorticoid signaling by 15d-PGJ2.
FIG 3.

SUMOylation modulates the inhibition of GR by 15d-PGJ2. HEK293 cells stably expressing wtGR- and GR3KR were transfected with 40 nM Dharmacon On-TARGETplus SMARTpool for human UBE2I (siUBC9) or Non-Targeting pool (siNON) by Lipofectamine RNAiMAX. After 96 h, the cells in the experiment represented in panel A were treated with vehicle, with dex, or with dex in the presence of 15d-PGJ2 as described in the Fig. 2B legend and the cells in the experiments represented in panels B and C were treated as described in the Fig. 1A legend with 5 μM 15d-PGJ2, and RNA and proteins were isolated. (A) Cells were lysed in a denaturing buffer, and portions of lysates (20% of the immunoprecipitation inputs) were analyzed by immunoblotting with anti-UBC9, anti-GR, and anti-GAPDH antibodies or were subjected to immunoprecipitation with anti-GR antibody and subsequent immunoblotting with anti-SUMO-2/3 (S2/3) antibody. The arrowhead and bracket indicate the position of SUMO-2/3-modified forms of GR. (B) Proteins were analyzed by immunoblotting with anti-UBC9 and anti-GAPDH antibodies. (C) RT-qPCR analyses were performed with specific primers for CDKN1C, ELK1, ARMC12, and JUN. Results represent the means ± SD of the results of three experiments. ***, P < 0.001; **, P < 0.01 (for the differences in the results of dex treatment in the presence or absence of 15d-PGJ2 [Student's t test]). ns, no significance.
Genome-wide effects of 15d-PGJ2 on glucocorticoid signaling are influenced by GR SUMOylation.
We next compared the genome-wide expression profiles of wtGR and GR3KR HEK293 cells that were exposed to dex with or without 15d-PGJ2 for 6 h by using Illumina HT-12 v4 Expression BeadChips. As shown in Fig. 4A, 15d-PGJ2 by and large affected (i.e., either inhibited or induced) different genes in the wtGR and the GR3KR cells. Only 194 genes were similarly affected in the two cell lines, whereas 908 and 1,708 genes were preferentially affected in the wtGR and the GR3KR cells by 15d-PGJ2, respectively. In addition, of the 194 genes affected by 15d-PGJ2 in both cell lines, over half (101 genes) had opposite effects on the two cell lines. Of the dex target genes in these cells (12)(GSE48328), 34 shared similar responses to 15d-PGJ2, while 98 and 141 genes were preferentially affected in wtGR and GR3KR cells, respectively (Fig. 3B). Subsequently, we grouped the dex-regulated genes influenced by 15d-PGJ2 by unsupervised hierarchical clustering (Fig. 4C to E). Interestingly, the dex-regulated genes affected by 15d-PGJ2 preferentially in the wtGR cells formed two distinct clusters in which the prostaglandin converted the effect of dex on gene expression; i.e., dex-upregulated genes became downregulated (cluster 3; e.g., CDKN1C and ELK1) or dex-downregulated genes lost their repression or became upregulated (cluster 1; e.g., FOS, JUN, and IL8) in the presence of 15d-PGJ2 (Fig. 4C). In the case of the genes preferentially affected in the GR3KR cells, the converting effect of 15d-PGJ2 was clear-cut only for the dex-downregulated genes (cluster 1) (Fig. 4E).
FIG 4.

Gene expression levels are differently affected by 15d-PGJ2 in wtGR- and GR3KR-expressing HEK293 cells. Both cell lines were treated with dex (100 nM) in the presence or absence of 15d-PGJ2 (5 μM) for 6 h, and RNAs were isolated for Illumina Bead array analysis as described in Materials and Methods. Fold inductions of >1.24 or <0.8 were considered to represent up- or downregulation, respectively, by 15d-PGJ2. (A) Venn diagram showing comparison of all genes influenced by 15d-PGJ2 exposure in the wtGR cells and the GR3KR cells. (B) Venn diagram showing comparison of the dex-regulated genes affected by 15d-PGJ2 in the wtGR and the GR3KR cells. Identification of dex target genes was based on our previous transcriptome analysis of dex-up- or -downregulated genes from the same cells (P < 0.01) (12) (GSE48328). Values inside the diagram indicate the number of dex-regulated genes influenced by 15d-PGJ2. (C to E) GR-type preferred or shared dex target genes affected by 15d-PGJ2 were grouped by unsupervised hierarchical clustering, resulting in 4 clusters for the wtGR-preferred genes (C), 3 clusters for the GR3KR-preferred genes (E), and 3 clusters for the wtGR- and GR3KR-shared genes (D). The genes were visualized as heat maps showing the differences between dex exposure and dex-plus-15d-PGJ2 coexposure. Red indicates upregulation, and green indicates downregulation.
Based on Ingenuity pathway analysis (IPA) of the gene expression data, 15d-PGJ2 most significantly influenced the following molecular and cellular functions: gene expression, cell growth and proliferation, cell death and survival, and cellular development (Fig. 5A and 6A). There was no marked difference between the wtGR and the GR3KR cells in the enrichment of these functions when either all the genes (Fig. 5) or only the dex-regulated genes (Fig. 6) affected by 15d-PGJ2 were analyzed. However, the enrichment of cell death and survival function was notably greater in wtGR cells versus GR3KR cells (P = 0.000002 versus P = 0.02, respectively). Interestingly, measurement of apoptosis indicated that the wtGR cells were significantly more prone to apoptosis caused by the cotreatment exposure to dex and 15d-PGJ2 than the GR3KR cells (see Fig. SA4 in the supplemental material). Furthermore, one of the major differences in the enrichment of the molecular and cellular functions between the wtGR and the GR3KR cells was the free radical scavenging; genes were significantly enriched only among those preferentially affected in the wtGR cells (P = 0.0006 versus P = 1 and P = 0.004 versus P = 1, respectively) (Fig. 5A and 6A). Of the canonical pathways, the glucocorticoid receptor signaling pathway was significantly enriched in the prostaglandin-affected dex target gene data of the wtGR cells versus the data of the GR3KR cells (P = 0.004 versus P = 0.61, respectively) (Fig. 6B). Moreover, the upstream regulator in IPA (URA/IPA) indicated that the action of GR (NR3C1) was inhibited in the presence of 15d-PGJ2 in the wtGR cells (Fig. 6C) but not in the GR3KR cells (Fig. 6E). In contrast, 15d-PGJ2 activated NRF2-mediated oxidative stress responses similarly in the wtGR and the GR3KR cells in the absence and presence of dex (Fig. 5B and D and 6D). Hypoxia-inducible factor 1α (HIF1A), activating transcription factor 4 (ATF4), and heat shock factor 1 (HSF1) in turn were responding differently to the prostaglandin in the wtGR and the GR3KR cells; the URA/IPA identified them as activated transcriptional regulators only in the group of the genes affected in the wtGR cells (Fig. 5C to E and 6C to E). In line with these data and supporting the notion that GR represses multiple stress-activated transcription factors, 15d-PGJ2 converted dex downregulation of HSF1 target genes HSPA1A and HSPA1B to upregulation in wtGR but not in GR3KR cells (see Fig. SA5 in the supplemental material). Similarly, AP1 and NF-κB were activated transcription factors among the prostaglandin-affected dex target genes of the wtGR cells (Fig. 6C to E).
FIG 5.

Ingenuity pathway analysis of genes affected by 15d-PGJ2 preferentially in the wtGR- or the GR3KR-expressing HEK293 cells. (A) Heat map showing unsupervised hierarchical clustering of the most significantly enriched molecular and cellular functions affected by 15d-PGJ2 exposure. The color key indicates the P value of the enrichment, where black depicts no enrichment and bright red depicts high enrichment. (B) Heat map showing unsupervised hierarchical clustering of the most significantly enriched canonical pathways affected by 15d-PGJ2 exposure. RXR, retinoid X receptor; TWEAK, TNF-related weak inducer of apoptosis; VDR, vitamin D receptor. (C to E) Heat maps (based on upstream regulator IPA) showing transcription factors that were influenced by 15d-PGJ2 (P < 0.01). The color key indicates the activation z-score, where values > 0 indicate activation and values < 0 indicate inhibition of a particular transcription factor by 15d-PGJ2 treatment.
FIG 6.

Ingenuity pathway analysis of glucocorticoid target genes affected by 15d-PGJ2 preferentially in the wtGR- or the GR3KR-expressing HEK293 cells. The gene expression data in Fig. 4B were subjected to IPA. (A) Heat map showing unsupervised hierarchical clustering of the most significantly enriched molecular and cellular functions of the dex-regulated genes affected by 15d-PGJ2 exposure. The color key indicates the P value of the enrichment, where black depicts no enrichment and bright red depicts high enrichment. (B) Heat map showing unsupervised hierarchical clustering of the most significantly enriched canonical pathways of the dex-regulated genes affected by 15d-PGJ2. ERK, extracellular signal-regulated kinase; HGF, hepatocyte growth factor; HMGB1, high-mobility group box 1; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; nNOS, neuronal nitric oxide synthase; FGF, fibroblast growth factor. (C to E) Heat maps (based on upstream regulator IPA) showing transcription factors that were influenced by 15d-PGJ2 (P < 0.01). The color key indicates the activation z-score, where values >0 indicate activation and values <0 indicate inhibition of a particular transcription factor by 15d-PGJ2 exposure.
Because the cross talk between NF-κB and GR under inflammatory conditions is well established (6, 7), we analyzed how cotreatment with 15d-PGJ2 and TNF-α influences GR-mediated transrepression of a proinflammatory target, IL8 (Fig. 7). The expression of IL8 evoked by treatment with TNF-α (in the absence of dex) was clearly higher in GR3KR cells than in wtGR cells. Addition of 15d-PGJ2 together with TNF-α resulted in synergistic activation of IL8 in GR3KR cells but not in wtGR cells. Although dex was able to transrepress TNF-α-induced IL8 to similar degrees in the two cell lines, interestingly, the repressive effect of dex in the presence of 15d-PGJ2 was clearly weaker in GR3KR cells than in wtGR cells. These results suggest that the SUMOylation modulates the anti-inflammatory action of GR. However, approaches at the genome-wide level are needed to confirm these notions.
FIG 7.
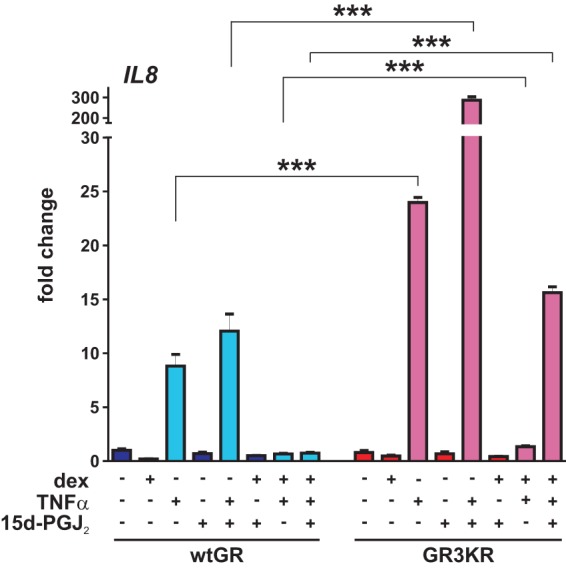
GR SUMOylation and 15d-PGJ2 influence TNF-α-induced expression of IL8. wtGR- and GR3KR-expressing HEK293 cells were treated with vehicle, dex (100 nM), 15d-PGJ2 (5 μM), or TNF-α (1,000 IU/ml) for 6 h as indicated by minus (−) and plus (+) signs, and RNAs were isolated. RT-qPCR analyses were performed with specific primers for IL8. Results represent the means ± SD of the results of three experiments. ***, P < 0.001 (for the differences between the wtGR and GR3KR cells in the results determined for TNF-α-treated samples [Student's t test]).
Taken together, these genome-wide data support the notion that, in addition to the glucocorticoid signaling, GR SUMOylation is capable of modulating cross talk of the GR with other transcription factors responsive to cell stress induced by 15d-PGJ2 (Fig. 8).
FIG 8.
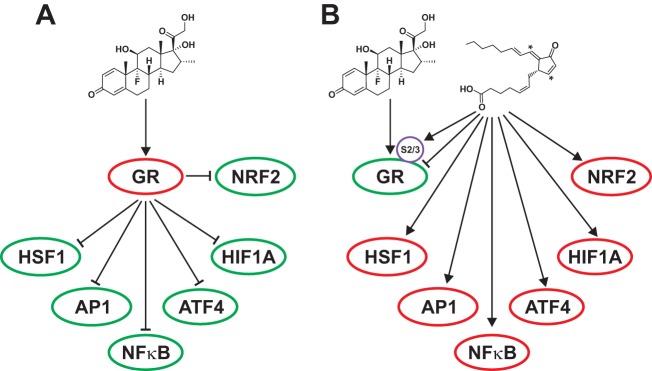
A model for the role of GR SUMOylation in the modulation of cross talk between glucocorticoid signaling and other transcription factors involved in stress and inflammation. (A) Activated GR results in the repression seen with AP1, NF-κB, HSF1, HIF1A, ATF4, and NRF2. (B) 15d-PGJ2 activates NRF2. It also triggers the hyper-SUMOylation of GR, resulting in alleviation of the GR-mediated repression of the cell stress-associated transcription factors. Green oval, inhibition; red oval, activation; arrow, activation; arrow with a blunt end, inhibition.
DISCUSSION
A large number of transcription factors, including GR, have been shown to be covalently modified by SUMOs, but the regulation of these modifications by cellular signals and their functional consequences for gene expression have been investigated only rarely in a systematic genome-wide fashion (13, 35). In this study, we examined the effect of 15d-PGJ2 on GR SUMOylation and glucocorticoid-regulated gene programs. 15d-PGJ2 has been shown to trigger cell signaling cascades via directly interacting with proteins containing reactive cysteine residues (29, 30, 36). 15d-PGJ2 principally activates the NRF2 pathway by modifying KEAP1, the inhibitory partner of NRF2, which results in stabilization and increased chromatin binding of the NRF2 and activation of genes such as HMOX1 that are involved in the cytoprotection against oxidative stress (25, 26, 28). 15d-PGJ2 has also been shown to be able to attenuate the action of transcription factors such as NF-κB, AP-1, ERα, and AR (29–32). Covalent adducts at cysteine residues, which can inhibit DNA binding, are involved in their inactivation. 15d-PGJ2 has also been previously shown to transiently attenuate GR signaling in monocytes/macrophages by a mechanism depending on an interaction of the 15d-PGJ2 cyclopentenone ring with cysteine residues in constituents of the GR activation pathway (33). For the repression of ERα by 15d-PGJ2, it was shown that 15d-PGJ2 is covalently attached to two cysteine residues in the DBD, thus affecting DNA binding (31). Our biotin–15d-PGJ2 pulldown with DBD and LBD fragments of GR suggested that 15d-PGJ2 binds to both domains of the GR, but the binding relative to the cysteine content was more pronounced in the LBD. This suggests that 15d-PGJ2 may also influence coregulator recruitment to the LBD. However, more-detailed proteomic screens are needed to confirm this notion. We show in this work that, in addition to the covalent adduct formation with the GR, the inhibitory effect of 15d-PGJ2 on the GR activity, with respect to both its target gene expression and chromatin binding, was linked to rapid hyper-SUMOylation of the GR. Notably, the SUMOylation-defective GR form was significantly less prone to the inhibition by the prostaglandin, even though it formed covalent adducts with 15d-PGJ2 as efficiently as the wtGR. The importance of an intact SUMOylation pathway for the sensitivity of GR signaling by 15d-PGJ2 was further proven by depletion of the sole SUMO E2 UBC9 ligase. Moreover, the GR EA mutant with the SUMO acceptor lysines intact showed no SUMOylation, which further demonstrated that the repression of GR signaling by 15d-PGJ2 is dependent on the SUMOylation consensus sequences. It is thus unlikely that another lysine modification is mediating the repressive effect of 15d-PGJ2. However, we cannot formally exclude the possibility that another lysine modification targeting the N-terminal region of GR in addition to SUMOylation is involved in the modulatory effect of 15d-PGJ2.
Transcriptome comparisons of our isogenic HEK293 cell models expressing the wtGR or the SUMOylation-defective GR indicated that the GR SUMOylation sites extensively modulate the effects of 15d-PGJ2 on gene expression. Pathway and upstream regulator analysis of the genome-wide expression data confirmed that the sensitivity of the GR to inhibition by 15d-PGJ2 is significantly dependent on the SUMOylation sites of the receptor. Pathway analysis further revealed that the free radical scavenging is one of the major significant differences among the molecular and cellular functions affected, suggesting that the GR SUMOylation participates in cross talk with the free radical scavenging system. However, activation of the NRF2-mediated oxidative stress response signaling by 15d-PGJ2 was not markedly influenced by the GR SUMOylation, even though the GR is capable of suppressing the HMOX1 NRF2-dependent antioxidant response (37).
Based on the upstream regulator analysis, the patterns of activation of ATF4, HSF1, and HIF1A upon 15d-PGJ2 exposure in turn differ between the wtGR and the SUMOylation-defective GR cells, being favored in the GR SUMOylation-competent cells. Also, other members of the ATF family, ATF2 and -3, were predicted to be activated in response to 15d-PGJ2 preferentially in the GR SUMOylation-competent cells. ATF4 and ATF3 are transcription factors integral to the unfolded protein response caused by endoplasmic reticulum stress (38, 39). ATF4 is also involved in cellular antioxidant protection (40), and its expression has been shown to be repressed by glucocorticoids (41). HSF1 is essential for organisms to survive during acute stress (42), and, interestingly, activation of GR signaling in stressed cells inhibits binding of HSF1 to the heat shock protein 70 promoter (43). In good agreement with the transcriptome data, the alleviation of GR-mediated repression of HSF1 by 15d-PGJ2 in GR SUMOylation-competent cells resulted in robust expression of HSF1 target genes HSPA1A and HSPA1B. HIF1A belongs to the hypoxia-inducible factor family that plays a critical role in hypoxic stress (44, 45). SUMOylation can also modulate the HIF1A action during hypoxia (46, 47), and glucocorticoids are capable of suppressing HIF1A expression through the induction of glucocorticoid-induced leucine zipper (48).
The upstream regulator analysis also predicted that the activation of NF-κB and AP1 in response to 15d-PGJ2 is favored by SUMOylation-competent GR. Interestingly, the NF-κB and the AP1 are also subject to regulation by SUMOylation (49–52). Both of them have been shown to mediate transcriptional responses to oxidative stress and inhibit the GR function (7, 53–56). Interestingly, the expression of IL8 in response to proinflammatory TNF-α and 15d-PGJ2 was altered by the GR SUMOylation, suggesting that the SUMOylation modulates the anti-inflammatory action of GR. However, more-detailed analyses are needed to further study the effects of GR SUMOylation on inflammatory responses.
The recent discovery of hyper-SUMOylated GR from postischemic mouse brains suggested that cell stress-induced SUMOylation attenuates the transcriptional activity of the GR (24). Those results, along with ours, strongly support the concept that the attenuation of GR activity is a protective stress response. Taken together, our genome-wide data support the notion that, in addition to the glucocorticoid signaling, the GR SUMOylation modulates the cross talk of GR with other transcription factors responsive to cell stress. In our model (Fig. 8), we suggest that under normal conditions, glucocorticoid-bound GR attenuates the action of cell stress-associated transcription factors, whereas during SUMOylation-triggering cell stress, the GR hyper-SUMOylation restricts the glucocorticoid signaling and the ability of the receptor to repress the action of the cell stress-associated transcription factors. The cross talk is likely to involve SUMOylation of other transcription factors also.
Supplementary Material
ACKNOWLEDGMENTS
We thank Merja Räsänen and Eija Korhonen for assistance with cell cultures and Claude Libert for the TNF-α. We also thank the Finnish Microarray and Sequencing Centre in Turku, Finland, for microarray services.
This work was supported by the Academy of Finland, the Emil Aaltonen Foundation, the North Savo Cancer Foundation, the Kuopio University Foundation, the Finnish Cultural Foundation's North Savo Regional Fund, the UEF Doctoral Programme in Molecular Medicine and the Sigrid Jusélius Foundation, and strategic funding of UEF.
Footnotes
Published ahead of print 30 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00748-14.
REFERENCES
- 1.Silverman MN, Sternberg EM. 2012. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann. N. Y. Acad. Sci. 1261:55–63. 10.1111/j.1749-6632.2012.06633.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB. 2007. Glucocorticoid receptor physiology. Rev. Endocr. Metab. Disord. 8:321–330. 10.1007/s11154-007-9059-8 [DOI] [PubMed] [Google Scholar]
- 3.Quax RA, Manenschijn L, Koper JW, Hazes JM, Lamberts SW, van Rossum EF, Feelders RA. 2013. Glucocorticoid sensitivity in health and disease. Nat. Rev. Endocrinol. 9:670–686. 10.1038/nrendo.2013.183 [DOI] [PubMed] [Google Scholar]
- 4.Biddie SC, John S, Hager GL. 2010. Genome-wide mechanisms of nuclear receptor action. Trends. Endocrinol. Metab. 21:3–9. 10.1016/j.tem.2009.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenfeld MG, Lunyak VV, Glass CK. 2006. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 20:1405–1428. 10.1101/gad.1424806 [DOI] [PubMed] [Google Scholar]
- 6.Smoak KA, Cidlowski JA. 2004. Mechanisms of glucocorticoid receptor signalling during inflammation. Mech. Ageing Dev. 125:697–706. 10.1016/j.mad.2004.06.010 [DOI] [PubMed] [Google Scholar]
- 7.Busillo JM, Cidlowski JA. 2013. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends. Endocrinol. Metab. 24:109–119. 10.1016/j.tem.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM. 2009. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron 64:33–39. 10.1016/j.neuron.2009.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorrells SF, Sapolsky RM. 2007. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav. Immun. 21:259–272. 10.1016/j.bbi.2006.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galliher-Beckley AJ, Williams JG, Cidlowski JA. 2011. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular signaling stress pathways with nuclear receptor signaling. Mol. Cell. Biol. 31:4663–4675. 10.1128/MCB.05866-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian S, Poukka H, Palvimo JJ, Jänne OA. 2002. Small ubiquitin-related modifier-1 (SUMO-1) modification of the glucocorticoid receptor. Biochem. J. 367:907–911. 10.1042/BJ20021085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paakinaho V, Kaikkonen S, Makkonen H, Benes V, Palvimo JJ. 2014. SUMOylation regulates the chromatin occupancy and anti-proliferative gene programs of glucocorticoid receptor. Nucleic Acids Res. 42:1575–1592. 10.1093/nar/gkt1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Treuter E, Venteclef N. 2011. Transcriptional control of metabolic and inflammatory pathways by nuclear receptor SUMOylation. Biochim. Biophys. Acta 1812:909–918. 10.1016/j.bbadis.2010.12.008 [DOI] [PubMed] [Google Scholar]
- 14.Rytinki M, Kaikkonen S, Sutinen P, Paakinaho V, Rahkama V, Palvimo JJ. 2012. Dynamic SUMOylation is linked to the activity cycles of androgen receptor in the cell nucleus. Mol. Cell. Biol. 32:4195–4205. 10.1128/MCB.00753-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knutson TP, Daniel AR, Fan D, Silverstein KAT, Covington KR, Fuqua SAW, Lange CA. 2012. Phosphorylated and sumoylation-deficient progesterone receptors drive proliferative gene signatures during breast cancer progression. Breast Cancer Res. 14:R95. 10.1186/bcr3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gill G. 2004. Sumo and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18:2046–2059. 10.1101/gad.1214604 [DOI] [PubMed] [Google Scholar]
- 17.Tatham MH, Jaffray E, Vaughan OA, Desterro JMP, Botting CH, Naismith JH, Hay RT. 2001. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276:35368–35374. 10.1074/jbc.M104214200 [DOI] [PubMed] [Google Scholar]
- 18.van Wijk SJL, Timmers HTM. 2010. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J. 24:981–993. 10.1096/fj.09-136259 [DOI] [PubMed] [Google Scholar]
- 19.Rytinki MM, Kaikkonen S, Pehkonen P, Jääskeläinen T, Palvimo JJ. 2009. PIAS proteins: pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 66:3029–3041. 10.1007/s00018-009-0061-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukhopadhyay D, Dasso M. 2007. Modification in reverse: the SUMO proteases. Trends Biochem. Sci. 32:286–295. 10.1016/j.tibs.2007.05.002 [DOI] [PubMed] [Google Scholar]
- 21.Tempé D, Piechaczyk M, Bossis G. 2008. SUMO under stress. Biochem. Soc. Trans. 36:874–878. 10.1042/BST0360874 [DOI] [PubMed] [Google Scholar]
- 22.Manza LL, Codreanu SG, Stamer SL, Smith DL, Wells KS, Roberts RL, Liebler DC. 2004. Global shifts in protein sumoylation in response to electrophile and oxidative stress. Chem. Res. Toxicol. 17:1706–1715. 10.1021/tx049767l [DOI] [PubMed] [Google Scholar]
- 23.Golebiowski F, Matic I, Tatham MH, Cole C, Yin Y, Nakamura A, Cox J, Barton GJ, Mann M, Hay RT. 2009. System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2:ra24. 10.1126/scisignal.2000282 [DOI] [PubMed] [Google Scholar]
- 24.Yang W, Sheng H, Thompson JW, Zhao S, Wang L, Miao P, Liu X, Moseley MA, Paschen W. 2014. Small ubiquitin-like modifier 3-modified proteome regulated by brain ischemia in novel small ubiquitin-like modifier transgenic mice: putative protective proteins/pathways. Stroke 45:1115–1122. 10.1161/STROKEAHA.113.004315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surh YJ, Na HK, Park JM, Lee HN, Kim W, Yoon IS, Kim DD. 2011. 15-Deoxy-delta12,14-prostaglandin J2, and electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem. Pharmacol. 82:1335–1351. 10.1016/j.bcp.2011.07.100 [DOI] [PubMed] [Google Scholar]
- 26.Kansanen E, Kivelä AM, Levonen AL. 2009. Regulation of Nrf2-dependent gene expression by 15-deoxy-delta12,14-prostaglandin J2. Free Radic. Biol. Med. 47:1310–1307. 10.1016/j.freeradbiomed.2009.06.030 [DOI] [PubMed] [Google Scholar]
- 27.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. 1995. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83:813–819. 10.1016/0092-8674(95)90194-9 [DOI] [PubMed] [Google Scholar]
- 28.Oeste CL, Pérez-Sala D. 2014. Modification of cysteine residues by cyclopentenone prostaglandins: interplay with redox regulation of protein function. Mass. Spectrom. Rev. 33:110–125. 10.1002/mas.21383 [DOI] [PubMed] [Google Scholar]
- 29.Cernuda-Morollón E, Pineda-Molina E, Cañada FJ, Pérez-Sala D. 2001. 15-Deoxy-delta12,14-prostaglandin J2 inhibition of NF-kappaB-DNA binding through covalent modification of the p50 subunit. J. Biol. Chem. 276:35530–35536. 10.1074/jbc.M104518200 [DOI] [PubMed] [Google Scholar]
- 30.Pérez-Sala D, Cernude-Morollón E, Cañada FJ. 2003. Molecular basis for the direct inhibition of AP-1 DNA binding by 15-deoxy-delta12,14-prostaglandin J2. J. Biol. Chem. 278:51251–51260. 10.1074/jbc.M309409200 [DOI] [PubMed] [Google Scholar]
- 31.Kim HJ, Kim JY, Menq Z, Wang LH, Liu F, Conrads TP, Burke TR, Veenstra TD, Farrar WL. 2007. 15-Deoxy-delta12,14-prostaglandin J2 inhibits transcriptional activity of estrogen receptor-alpha via covalent modification of DNA-binding domain. Cancer Res. 67:2595–2602. 10.1158/0008-5472.CAN-06-3043 [DOI] [PubMed] [Google Scholar]
- 32.Kaikkonen S, Paakinaho V, Sutinen P, Levonen AL, Palvimo JJ. 2013. Prostaglandin 15d-PGJ(2) inhibits androgen receptor signaling in prostate cancer cells. Mol. Endocrinol. 27:212–223. 10.1210/me.2012-1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheron A, Peltier J, Perez J, Bellocq A, Fourqueray B, Baud L. 2004. 15-Deoxy-delta12,14-prostaglandin J2 inhibits glucocorticoid binding and signaling in macrophages through a peroxisome proliferator-activated receptor gamma-independent process. J. Immunol. 172:7677–7683. 10.4049/jimmunol.172.12.7677 [DOI] [PubMed] [Google Scholar]
- 34.Rytinki MM, Kaikkonen S, Sutinen P, Palvimo JJ. 2011. Analysis of androgen receptor SUMOylation. Methods Mol. Biol. 776:183–197. 10.1007/978-1-61779-243-4_12 [DOI] [PubMed] [Google Scholar]
- 35.Cubeñas-Potts C, Matunis MJ. 2013. SUMO: a multifaceted modifier of chromatin structure and function. Dev. Cell 24:1–12. 10.1016/j.devcel.2012.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim DH, Kim EH, Na HK, Sun Y, Surh YJ. 2010. 15-Deoxy-delta(12,14)-prostaglandin J(2) stabilizes, but functionally inactivates p53 by binding to the cysteine 277 residue. Oncogene 29:2560–2576. 10.1038/onc.2010.8 [DOI] [PubMed] [Google Scholar]
- 37.Kratschmar DV, Calabrese D, Walsh J, Lister A, Birk J, Appenzeller-Herzog C, Moulin P, Goldring CE, Odermatt A. 2012. Suppression of the Nrf2-dependent antioxidant response by glucocorticoids and 11β-HSD1-mediated glucocorticoid activation in hepatic cells. PLoS One 7:e36774. 10.1371/journal.pone.0036774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. 2004. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 24:1365–1377. 10.1128/MCB.24.3.1365-1377.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kansanen E, Jyrkkänen HK, Levonen AL. 2012. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 52:973–982. 10.1016/j.freeradbiomed.2011.11.038 [DOI] [PubMed] [Google Scholar]
- 40.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11:619–633. 10.1016/S1097-2765(03)00105-9 [DOI] [PubMed] [Google Scholar]
- 41.Adams CM. 2007. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J. Biol. Chem. 282:16744–16753. 10.1074/jbc.M610510200 [DOI] [PubMed] [Google Scholar]
- 42.Akerfelt M, Morimoto RI, Sistonen L. 2010. Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11:545–555. 10.1038/nrm2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wadekar SA, Li D, Sánchez ER. 2004. Agonist-activated glucocorticoid receptor inhibits binding of heat shock factor 1 to the heat shock protein 70 promoter in vivo. Mol. Endocrinol. 18:500–508. 10.1210/me.2003-0215 [DOI] [PubMed] [Google Scholar]
- 44.Shay JE, Celeste Simon M. 2012. Hypoxia-inducible factors: crosstalk between inflammation and metabolism. Semin. Cell Dev. Biol. 23:389–394. 10.1016/j.semcdb.2012.04.004 [DOI] [PubMed] [Google Scholar]
- 45.Majmundar AJ, Wong WJ, Simon MC. 2010. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40:294–309. 10.1016/j.molcel.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng J, Kang X, Zhang S, Yeh ET. 2007. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 131:584–595. 10.1016/j.cell.2007.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK, Holsboer F, Arzt E. 2007. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell 131:309–323. 10.1016/j.cell.2007.07.044 [DOI] [PubMed] [Google Scholar]
- 48.Lim W, Park C, Shim MK, Lee YH, Lee YM, Lee Y. 2014. Glucocorticoids suppress hypoxia-induced COX-2 and hypoxia inducible factor-1α expression through the induction of glucocorticoid-induced leucine zipper. Br. J. Pharmacol. 171:735–745. 10.1111/bph.12491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mabb AM, Miyamoto S. 2007. SUMO and NF-kappaB ties. Cell. Mol. Life Sci. 64:1979–1996. 10.1007/s00018-007-7005-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Bridges R, Wortham A, Kulesz-Martin M. 2012. NF-κB repression by PIAS3 mediated RelA SUMOylation. PLoS One 7:e37636. 10.1371/journal.pone.0037636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bossis G, Malnou CE, Farras R, Andermarcher E, Hipskind R, Rodriguez M, Schmidt D, Muller S, Jariel-Encontre I, Piechaczyk M. 2005. Down-regulation of c-Fos/c-Jun AP-1 dimer activity by sumoylation. Mol. Cell. Biol. 25:6964–6979. 10.1128/MCB.25.16.6964-6979.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tempé D, Vives E, Brockly F, Brooks H, De Rossi S, Piechaczyk M, Bossis G. 2014. SUMOylation of the inducible (c-Fos:c-Jun)/AP-1 transcription complex occurs on target promoters to limit transcriptional activation. Oncogene 33:921–927. 10.1038/onc.2013.4 [DOI] [PubMed] [Google Scholar]
- 53.Zhou LZ, Johnson AP, Rando TA. 2001. NF kappa B and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic. Biol. Med. 31:1405–1416. 10.1016/S0891-5849(01)00719-5 [DOI] [PubMed] [Google Scholar]
- 54.Adcock IM, Cosio B, Tsaprouni L, Barnes PJ, Ito K. 2005. Redox regulation of histone deacetylases and glucocorticoid-mediated inhibition of the inflammatory response. Antioxid. Redox Signal. 7:144–152. 10.1089/ars.2005.7.144 [DOI] [PubMed] [Google Scholar]
- 55.Van Bogaert T, De Bosscher K, Libert C. 2010. Crosstalk between TNF and glucocorticoid receptor signaling pathways. Cytokine Growth Factor Rev. 21:275–286. 10.1016/j.cytogfr.2010.04.003 [DOI] [PubMed] [Google Scholar]
- 56.Asaba K, Iwasaki Y, Yoshida M, Asai M, Oiso Y, Murohara T, Hashimoto K. 2004. Attenuation by reactive oxygen species of glucocorticoid suppression on proopiomelanocortin gene expression in pituitary corticotroph cells. Endocrinology 145:39–42. 10.1210/en.2003-0375 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.