

Abstract
The functional interplay between cancer cells and marrow stromal cells (MSCs) has attracted a great deal of interest due to the MSC tropism for tumors but remains to be fully elucidated. In this study, we investigated human MSC-secreted paracrine factors that appear to have critical functions in cancer stem cell subpopulations. We show that MSC-conditioned medium reduced the cancer stem cell-enriched subpopulation, which was detected as a side population and quiescent (G0) cell cycle fraction in human lung cancer cells by virtue of fibroblast growth factor 10 (FGF10). This reduction of the stem cell-enriched fraction was also observed in lung cancer cells supplemented with recombinant human FGF10 protein. Moreover, supplementary FGF10 attenuated the expression of stemness genes encoding transcription factors, such as OCT3/4 and SOX2, and crippled the self-renewal capacity of lung cancer cells, as evidenced by the impaired formation of floating spheres in the suspension culture. We finally confirmed the therapeutic potential of the FGF10 treatment, which rendered lung cancer cells prone to a chemotherapeutic agent, probably due to the reduced cancer stem cell subpopulation. Collectively, these results add further clarification to the molecular mechanisms underlying MSC-mediated cancer cell kinetics, facilitating the development of future therapies.
INTRODUCTION
Despite therapeutic advances, cancer-related death remains common, mainly because of the property of cancer cell populations to restore themselves after treatment (1). Accumulating evidence indicates that such cancer cell characteristics are derived from a small subpopulation with distinct stem-like properties capable of self-renewal, expelling cellular toxins, and maintaining a quiescent state (2–4). This subpopulation is defined as cancer stem cells, and it has been proposed that quiescent cancer stem cells can resist cytotoxic drugs that target cycling cancer cells, with the help of high drug efflux capacities and sustain the long-term self-renewal that potentially leads to eventual relapse after the completion of therapy (5–8).
The functional traits of cancer stem cells are sustained in the tumor microenvironment, where the importance of marrow stromal cells (MSCs) (also referred to as mesenchymal stem cells) has been highlighted by their tumor-homing potential (7, 9, 10). In spite of extensive studies, the impact of MSCs on tumor progression remains unclear; some investigations have reported the MSC-mediated promotion of tumor growth, while others have shown that MSCs rather alleviate tumor progression (9, 11, 12). MSCs are functionally characterized by their ability not only to differentiate into several mesenchymal cell lineages but also to secrete a vast array of paracrine factors, including growth factors, cytokines, proangiogenic factors, exosomes, and even extracellular matrix components (10, 11). Some factors are perceived to influence tumor growth in general (11). Thus, the inconsistent findings on MSCs in cancer progression are thought to result from the complexity of tumor cell heterogeneity and the diverse paracrine effectors secreted from MSCs (9, 11).
In the present study, we hypothesized that MSCs can release a paracrine factor that affects the cellular kinetics of cancer stem cells and thereby likely exert paradoxical effects on the growth of tumors, which are variably composed of cancer stem and non-stem cells. To evaluate this concept, we examined cancer cells exposed to conditioned medium (CM) from human bone marrow-derived MSCs by using assays for the side population and the G0 cell cycle state, which take advantage of the active efflux capacity and the quiescent property in cancer stem cells. Our data show that the MSC CM reduces the stem cell fraction of lung cancer cells but not that of non-lung cancer cells, via fibroblast growth factor 10 (FGF10) released from MSCs.
MATERIALS AND METHODS
Cancer cell lines and culture conditions.
The human lung cancer cell lines A549, NCI-H1299, and NCI-H1975 were obtained from the American Type Culture Collection (Manassas, VA). The human breast cancer cell line MCF-7 and human cervical cancer cell line HeLa were obtained from the Riken Bioresource Center (Tsukuba, Japan). All cancer cells were maintained at 37°C in 5% CO2 with full cancer medium—i.e., Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (Nichirei, Tokyo, Japan), 100 U/ml penicillin (Life Technologies, Carlsbad, CA), and 100 μg/ml streptomycin (Life Technologies).
CM from MSCs.
Primary human MSCs were maintained at 37°C in 5% CO2 with minimum essential medium alpha (Life Technologies) supplemented with 17% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (Life Technologies) unless otherwise noted (13). One million MSCs at passage 1 were obtained from the Texas A&M Health Science Center for the Preparation and Distribution of Adult Stem Cells (Temple, TX) and were incubated at passage 2 in a 150-mm-diameter dish for 24 h. Only adherent (i.e., viable) cells were recovered and then replaced in a 150-mm-diameter dish at a density of 60 cells/cm2. The MSCs at passage 3 were then cultured for 9 days with the medium changed every 3 days. After 9 days, confluent MSCs were incubated with 30 ml of the full cancer medium. In parallel, the full cancer medium was incubated in empty culture dishes without MSCs for preparation of mock-conditioned medium (mock CM). After 48 h, the culture supernatant was recovered, passed through a 0.45-μm-pore filter to eliminate the cell debris, and stored at 4°C until use. Human MSCs were obtained from three donors: a 21-year-old female (donor 1), a 22-year-old male (donor 2), and a 24-year-old male (donor 3). MSCs from donor 1 were used in this study unless otherwise noted.
Cancer cell treatment.
One million cancer cells were seeded in a 100-mm-diameter dish and cultured for 24 h. The cancer cells were washed with phosphate-buffered saline (PBS [pH 7.4]) two times and were treated with 10 ml of conditioned medium for 48 h. In some experiments, instead of the conditioned medium, the full cancer medium containing 50 ng/ml human FGF1 (R&D Systems, Minneapolis, MN), 50 ng/ml human FGF3 (R&D Systems), 50 ng/ml human FGF7 (R&D Systems), 50 ng/ml human FGF10 (R&D Systems), 50 ng/ml human FGF22 (R&D Systems), or an equal volume of vehicle (PBS) was used for the cancer cell treatment. Where indicated, 5 μg/ml rabbit polyclonal anti-FGF10 antibody (Abcam, Cambridge, United Kingdom) or 5 μg/ml control rabbit IgG (Wako Pure Chemical, Osaka, Japan) was added to the conditioned medium before the cancer cell treatment was started.
Side population assay.
Cancer cells were collected and suspended in high-glucose DMEM (Sigma-Aldrich) containing 2% bovine serum albumin (Wako Pure Chemical) and 10 mM HEPES at a density of 106 cells/ml and then were stained with 5 μg/ml of Hoechst 33342 (Sigma-Aldrich) at 37°C for 90 min. After discrimination of the dead cells stained with 2 μg/ml of propidium iodide (Sigma-Aldrich), the Hoechst 33342-stained live cells were analyzed using a FACSAria II cell sorter and FACSDiva software (BD Biosciences, San Jose, CA).
Cell cycle assay.
Cancer cells were collected and stained in a nucleic acid staining solution (0.1 M phosphate-citrate buffer [pH 4.8] with 0.9% NaCl, 0.5% bovine serum albumin, 5 mM EDTA, and 0.02% saponin) containing 10 μg/ml 7-aminoactinomycin D (7-AAD) (Imgenex, San Diego, CA) for 20 min. The cells were washed twice with PBS and were further stained in the nucleic acid staining solution containing 1 μg/ml pyronin Y (Sigma-Aldrich) for 5 min at 4°C. The cell cycle fractions were identified and quantified using an Epics XL cytometer and EXPO32 ADC software (Beckman Coulter, Miami, FL).
Animal studies.
BALB/c nu/nu mice were purchased from Charles River Japan (Yokohama, Japan), and were housed under specific-pathogen-free conditions. For limiting dilution analyses, conditioned medium-treated H1299 cells in the side population fraction were subcutaneously injected to 4-week-old female mice (day 0). The macroscopic tumor formation was examined at day 50. To evaluate the growth kinetics of tumors, 5 × 106 unfractionated H1299 cells were injected. The size of each tumor was assessed every other day and recorded as the tumor volume in mm3 (length by width2 × 0.52) (14). Where indicated, the tumor-bearing mice were treated with repeated intraperitoneal administration of cisplatin (4 mg/kg body weight; Wako Pure Chemical). All procedures were performed according to protocols approved by Tohoku University's Institutional Committee for the Use and Care of Laboratory Animals.
Immunohistochemistry.
Paraffin-embedded lung sections from 3 lung cancer patients (Tohoku University Hospital, Sendai, Japan) were deparaffinized and rehydrated. After antigen unmasking for 10 min at 95 to 100°C in 10 mM sodium citrate buffer (pH 6.0), the specimens were stained with fluorescein isothiocyanate (FITC)-conjugated anti-human CD90 antibody (clone 5E10; BD Biosciences) and phycoerythrin (PE)-conjugated anti-FGF10 antibody (Bioss, Woburn, MA). CD90 is commonly regarded as an essential marker of human MSCs (15, 16). Nuclei were counterstained by using Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories, Burlingame, CA). The Institutional Review Board at Tohoku University Graduate School of Medicine (Sendai, Japan) approved the study.
RT-PCR.
For reverse transcription-PCR (RT-PCR), total cellular RNA was extracted from cancer cells or MSCs at passage 3 with an RNeasy Plus kit (Qiagen, Valencia, CA) and reverse transcribed to cDNA with the SuperScript III first-strand synthesis system (Life Technologies). The generated cDNA was amplified by semiquantitative and quantitative PCR (qPCR). For semiquantitative PCR, products were amplified using Platinum Taq DNA polymerase (Life Technologies), resolved on a 2% agarose gel, and detected by ethidium bromide staining; the semiquantitative amplification conditions were 94°C for 2 min, followed by 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s. For quantitative PCR, a DNA Engine Opticon 2 system (Bio-Rad Laboratories, Hercules, CA) and a SYBR GreenER qPCR SuperMix Universal kit (Life Technologies) were used according to the suggestions of the manufacturer; the quantitative data were normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression, and the relative gene expression was determined as a factor by which the normalized expression of the sample was changed from that of the reference. The following primer pairs were used in this study: FGFR1B (FGF receptor 1B gene), 5′-AATGTGACAGAGGCCCAGAG-3′ and 5′-GGAGTCAGCAGACACTGT-3′; FGFR2B, 5′-ACTCGGGGATAAATAGTTCCAA-3′ and 5′-CCTTACATATATATTCCCCAGCAT-3′; FGF1, 5′-CACATTCAGCTGCAGCTCAG-3′ and 5′-TGCTTTCTGGCCATAGTGAGTC-3′; FGF2, 5′-CTTCTTCCTGCGCATCCACC-3′ and 5′-CACATACCAACTGGTGTATTTC-3′; FGF3, 5′-TGAACAAGAGGGGACGACTCTATG-3′ and 5′-AGTCTCGAAGCCTGAACGTGAG-3′; FGF7, 5′-GAAGACTCTTCTGTCGAACAC-3′ and 5′-TATTGCCATAGGAAGAAAGTGG-3′; FGF10, 5′-GCGGAGCTACAATCACCTTC-3′ and 5′-GGAAGAAAGTGAGCAGAGGTG-3′; FGF22, 5′-AGCATCCTGGAGATCCGCTC-3′ and 5′-GCTGTGAGGCGTAGGTGTTG-3′; ACTB (β-actin), 5′-GTGGGGCGCCCCAGGCACCA-3′ and 5′-CTCCTTAATGTCACGCACGATTTC-3′; OCT3/4, 5′-ACATGTGTAAGCTGCGGCC-3′ and 5′-GTTGTGCATAGTCGCTGCTTG-3′; SOX2, 5′-CGATGCCGACAAGAAAACTT-3′ and 5′-CAAACTTCCTGCAAAGCTCC-3′; LGR4, 5′-GGCTGAGTGCTTTGCAGTCT-3′ and 5′-CCTCCGTCAAGCTGTTGTC-3′; and GAPDH, 5′-GAGTCAACGGATTTGGTCGT-3′ and 5′-TTGATTTTGGAGGGATCTCG-3′.
Western blotting.
MSCs at passage 3 and those cocultured for 48 h with A549 lung cancer cells separated with a 0.4-μm-pore membrane were lysed in radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology, Danvers, MA) containing protease inhibitor cocktail (Sigma-Aldrich). The whole-cell extract was separated in a 15% Tris-glycine gel (Novex, San Diego, CA) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Life Technologies). After treatment with PVDF blocking reagent (Toyobo, Osaka, Japan), the membrane was probed with a primary antibody against FGF10 or β-actin (clone AC-15; Sigma-Aldrich) and a secondary horseradish peroxidase (HRP)-conjugated antibody against the primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The signals were visualized using the ECL enhanced chemiluminescence detection system (GE Healthcare, Piscataway, NJ).
Sphere-forming assay.
Using an ultra-low-attachment 96-well plate, 100 A549 cells were seeded and cultured in 100 μl of serum-free DMEM–F-12 containing 10 ng/ml human recombinant epidermal growth factor (EGF) (R&D Systems) and 10 ng/ml human recombinant basic fibroblast growth factor (bFGF) (R&D Systems) with 50 ng/ml recombinant human FGF10 (rFGF10) or an equal volume of vehicle (PBS) on day 1. On days 4 and 7, 50 μl of the medium with FGF10 or vehicle was added for replenishment. On day 10, the numbers of spheres over 100 μm in diameter were counted in six wells.
Cell reproliferation monitoring after cytotoxic treatment.
In a 96-well plate, 5 × 103 A549 lung cancer cells were seeded and cultured 1 day before the start of the experiment. On the next day (day 0), the medium was replaced by the full cancer medium containing 80 μM cisplatin (Wako Pure Chemical) with 20 ng/ml of recombinant human FGF10 or an equal volume of vehicle (PBS), and the cells were cultured for 2 days. The A549 cells were washed with PBS two times and were further cultured in the full cancer medium for another 10 days. The number of viable cells was determined by a colorimetric method using MTS (3-4,5-dimethylthiazol-2-yl-5-3-carboxymethoxyphenyl-2-4-sulfophenyl-2H-tetrazolium, inner salt) (CellTiter 96, Promega, Madison, WI) per the manufacturer's instructions.
Statistical analysis.
Data sets were compared by Student's unpaired two-tailed t test. P values of <0.05 were considered statistically significant. Error bars in the graphical data represent means ± standard errors.
RESULTS
An MSC-derived soluble factor decreases lung cancer cells with stem cell phenotypes.
At first, we explored the biological effect of MSCs on the cancer stem cell kinetics, analyzing the side population of various cancer cells treated with conditioned medium from human MSCs (MSC CM) (Fig. 1A and B). The side population possessed the characteristic low-fluorescence profile imparted by the dye exclusion property of the ATP-binding cassette (ABC) transporters, having become a crucial hallmark for the stem cell trait (5, 7, 8). In our flow cytometry, 11% of the H1299 lung cancer cells treated with MSC CM were detected in the side population, whereas 19% of the control H1299 cells were detected in the side population (Fig. 1A). The decrease in the side population fraction of H1299 cells was statistically significant between the MSC CM and control CM treatments, and a similarly significant but modest decrease was also observed in cells of both lung cancer lines A549 and H1975 that had been treated with MSC CM (H1299, P < 0.01; A549, P < 0.05; H1975, P < 0.01) (Fig. 1B). However, the MSC CM treatment did not significantly decrease the side population fractions of MCF-7 breast cancer cells and HeLa cervical cancer cells compared with the control CM treatment (MCF-7, P > 0.3; HeLa, P > 0.3) (Fig. 1B).
FIG 1.
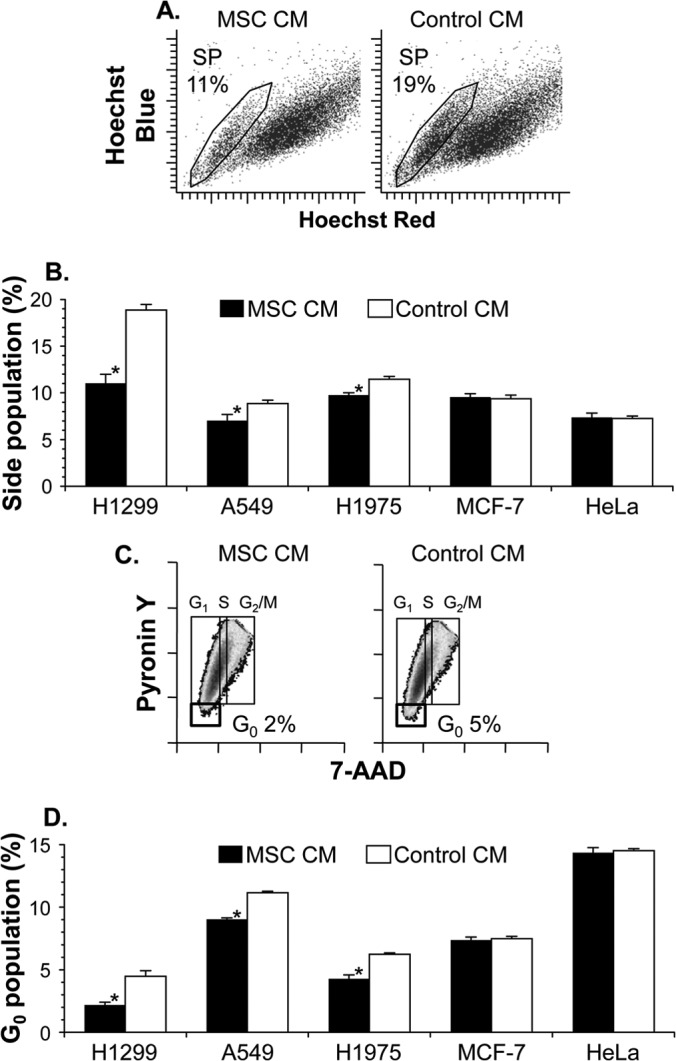
Conditioned medium of human MSCs reduces the proportion of lung cancer stem cells. Lung cancer cells (H1299, A549, and H1975), breast cancer cells (MCF-7), and cervical cancer cells (HeLa) were cultured for 48 h with conditioned medium of MSCs (MSC CM) or mock-conditioned medium (control CM). After the culture, cells were stained with Hoechst 33342 or 7-AAD–pyronin Y to analyze the side population (A and B) or cell cycle (C and D), respectively. (A) Representative side population subset (SP) with the percentage in H1299 cells. (B) Proportion of cancer cells in the side population. (C) Representative phase distribution of the cell cycle with the percentage of H1299 cells in the G0 phase. (D) Proportion of cancer cells in the G0 phase. For panels B and D, the cell proportion is reported as the mean percentage ± standard error per group (n = 3). Asterisks indicate significant differences compared with the control CM.
Consistent results were achieved in a cell cycle analysis to evaluate the number of quiescent cells (Fig. 1C and D). Cell cycle quiescence is another hallmark of stem cells, as the G0 phase was characterized by lower levels of both DNA (7-AAD) staining and RNA (pyronin Y) staining in the two-parameter flow cytometric analysis. When treated with MSC CM, 2% of H1299 lung cancer cells were in the G0 phase, whereas 5% of control H1299 cells were in the G0 phase (Fig. 1C). A lower G0 cell fraction mediated by the MSC CM treatment was also observed in A549 and H1975 lung cancer cells but not in MCF-7 and HeLa non-lung cancer cells (H1299, P < 0.005; A549, P < 0.005; H1975, P < 0.005; MCF-7, P > 0.05; HeLa, P > 0.3) (Fig. 1D). Notably, in H1299 cells with the side population phenotype, the MSC CM treatment increased the gene expression of cyclin-dependent kinases 4 (CDK4) and 6 (CDK6), which help trigger progression of cell cycle entry and thereby decreased the G0 proportion in the side population cells (MSC CM versus control CM, 7.7% versus 37%; CDK4, P < 0.0005; CDK6, P = 0.08) (data not shown).
This is also confirmed in vivo by the three analyses comprising limiting dilution, the growth model, and the treatment model for tumors that arose from H1299 lung cancer cells treated with MSC CM (Fig. 2 and Fig. 3). In a limiting dilution analysis to determine the fraction of tumor-initiating cells, as few as 5 × 103 side population cells of H1299 lung cancer cells treated with control CM were sufficient to form a tumor in 2 of 4 injections into BALB/c nu/nu mice, but side population cells treated with MSC CM failed to form a tumor in all 4 injections (Fig. 2). With 5 × 104 H1299 cells in the side population fraction, all 4 injections of cells treated with control CM formed tumors, whereas only 1 injection of side population cells treated with MSC CM formed a more slowly growing tumor (Fig. 2). With 5 × 106 unfractionated H1299 cells injected, all injections formed tumors, and tumors from cells treated with MSC CM grew much more slowly than those from cells treated with control CM (Fig. 3A). Strikingly, even after the intraperitoneal repeated administration of the chemotherapeutic agent cisplatin, the tumor growth kinetics of H1299 cells treated with MSC CM were also lower than those of cells treated with control CM (Fig. 3B). Taken together, these results show that MSCs produce a soluble factor that reduces the stem cell subpopulation of lung cancers.
FIG 2.
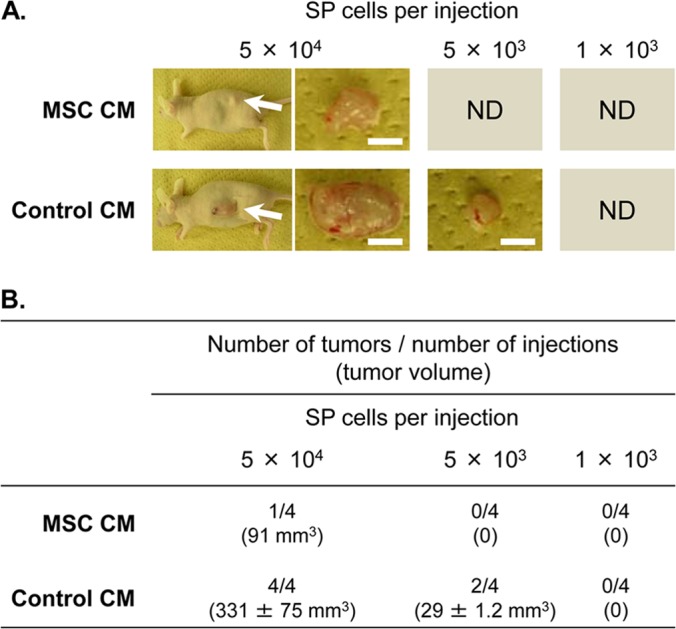
Treatment with conditioned medium of MSCs decreases cancer stem cell frequency. The side population (SP) fraction was isolated from H1299 lung cancer cells that had been treated with conditioned medium of MSCs (MSC CM) or mock-conditioned medium (control CM). For limiting dilution analyses, the indicated numbers of side population cells were subcutaneously injected into female BALB/c nu/nu mice (n = 4 per group). (A) Representative macroscopic images of resulting tumors (indicated by arrows) 50 days after the injection. Scale bar, 5 mm. ND, not detected. (B) Tumor development from side population cells 50 days after the injection. The tumor volume is reported as the mean ± standard error number of detected tumors.
FIG 3.
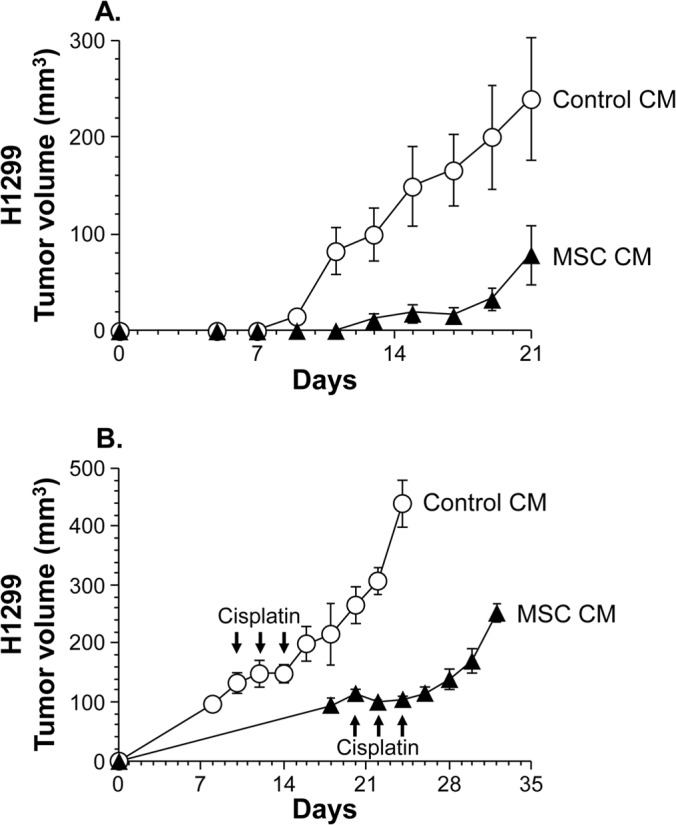
Treatment with conditioned medium of MSCs delays the tumor growth of lung cancer cells and renders them chemosensitive in vivo. Female BALB/c nu/nu mice were injected subcutaneously with H1299 lung cancer cells (5 × 106) that had been treated with conditioned medium of MSCs (MSC CM) or mock-conditioned medium (control CM). (A) Tumor growth model. On and after day 5, the size of each tumor was assessed every other day and is reported as the average tumor volume ± standard error per group (n = 3). (B) Treatment model. When the average tumor volume reached 100 mm3, the treatment was started with intraperitoneal repeated administration of cisplatin (4 mg/kg body weight; MSC CM on days 20, 22, and 24 and control CM on days 10, 12, and 14). The size of each tumor was assessed every other day and is reported as the average tumor volume ± standard error per group (n = 4).
FGF10 is defined as a candidate factor to decrease lung cancer stem cells.
To further clarify the molecular mechanisms regulating the stem cell subpopulation of lung cancers, we examined the FGF signaling components (Fig. 4), as FGFs from lung mesenchyme have been implicated in the lung morphogenesis of early embryo development as locally acting paracrine cues (17, 18). In the semiquantitative RT-PCR analysis for the mRNA expression of epithelium-specific FGF receptors (FGFRs [i.e., FGFR1b and -2b splice isoforms]), the expression of the FGFR1b isoform was observed in all types of cancer cells to a greater or lesser extent, whereas that of the FGFR2b isoform was not observed in any types of cancer cells examined in this study (Fig. 4A). We next investigated the mRNA expression of FGFs, which can be specific ligands for triggering the FGFR1b isoform, using semiquantitative RT-PCR in human MSCs from three unrelated donors. Among FGFs with binding affinity for FGFR1b, expression of FGF1, FGF7, and FGF10 mRNA was detected in MSCs from every donor, but expression of FGF2, FGF3, and FGF22 mRNA was not detected in MSCs from any donor (Fig. 4B). In H1299 cells with side population phenotypes, only FGF1 mRNA expression was detected, whereas FGF3, FGF7, FGF10, and FGF22 mRNA expression was not (data not shown). We therefore examined whether the stem cell subpopulation could be reduced in lung cancers by the addition of these proteins, except for FGF2, which specifically binds to FGFR2c. Recombinant human FGF10 (rFGF10) but not other FGFs caused a 50% reduction of the side population fraction in H1299 lung cancer cells compared with the control vehicle (rFGF10, P < 0.05; rFGF1, P < 0.05, but increase; rFGF3, P < 0.01, but increase; rFGF7, P > 0.05; rFGF22, P < 0.005, but increase) (Fig. 4C and D) (data not shown). In A549 and H1975 lung cancer cells, rFGF10, but not rFGF1, also reduced the side population fraction significantly but to a lesser extent, whereas neither rFGF10 nor rFGF1 significantly affected the side population in MCF-7 and HeLa cells (rFGF10, A549 and H1975, P < 0.05; rFGF10, MCF-7 and HeLa, P > 0.05; rFGF1, all cell types, P > 0.05) (Fig. 4D). In clinical specimens from three lung cancer patients, immunohistochemical analyses using CD90 as an essential marker of MSCs demonstrated that FGF10-expressing MSCs accumulated in the lung cancer regions but not in the normal lung regions (Fig. 5). Thus, we focused on the biological role of MSC-derived FGF10 in decreasing lung cancer stem cells.
FIG 4.
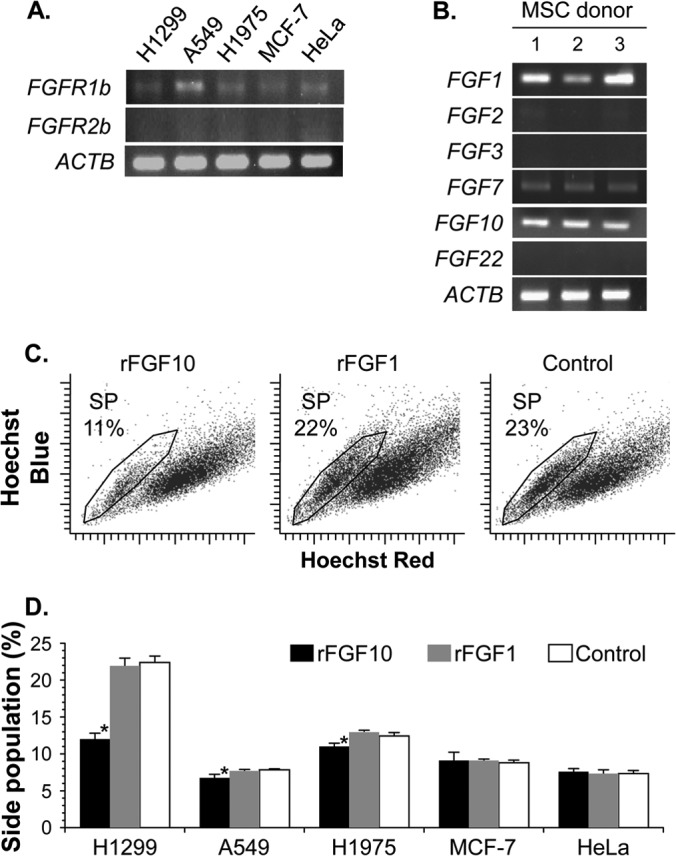
FGF signaling reduces the proportion of lung cancer stem cells. (A) Semiquantitative RT-PCR analysis of epithelial-specific FGF receptor isoforms FGFR1b and FGFR2b, in five cancer cells. (B) Semiquantitative RT-PCR analysis for FGFR1b-reacting FGFs (coded for by the FGF1, FGF2, FGF3, FGF7, FGF10, and FGF22 genes) in MSCs from three unrelated donors. For panels A and B, expression of β-actin mRNA (ACTB) was used as a control. (C and D) Functional analysis of recombinant FGF proteins. Cancer cells were cultured for 48 h with recombinant FGF10, FGF1, or vehicle (PBS [control]) and were stained with Hoechst 33342 to analyze the side population. A representative side population subset (SP) with its percentage in H1299 lung cancer cells (C) and proportions of five cancer cells in the side population (D) are shown. The cell proportion is reported as the mean percentage ± standard error per group (n = 3). Asterisks indicate significant differences compared with the control vehicle.
FIG 5.
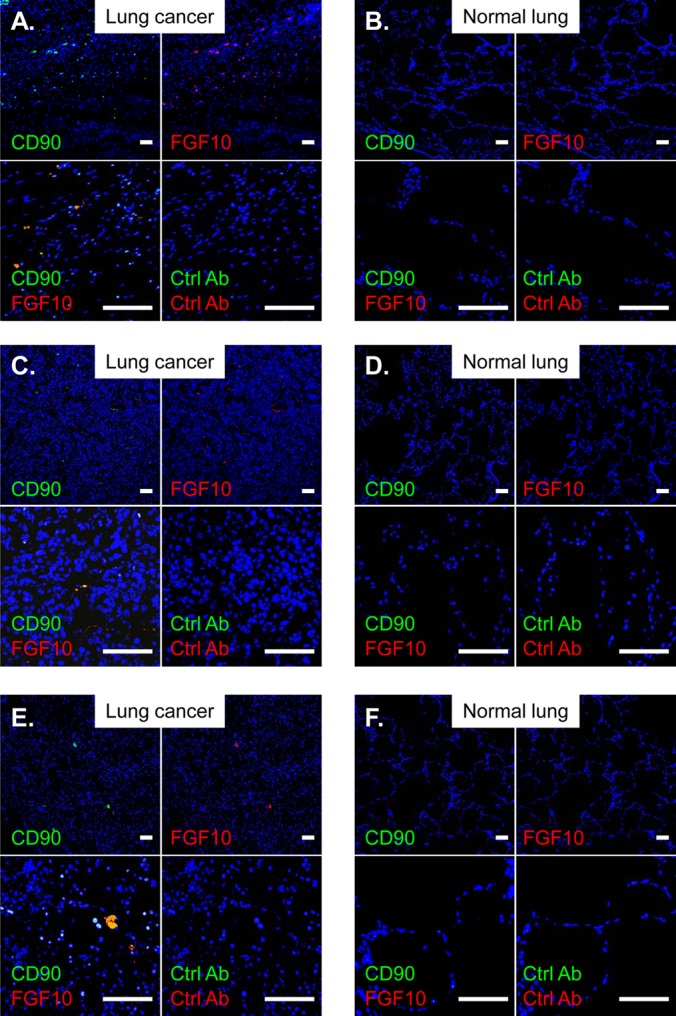
MSCs expressing FGF10 accumulate in lung tumors. (A and B) Specimen from a 59-year-old woman with squamous lung cancer. (C and D) Specimen from an 81-year-old man with lung adenocarcinoma. (E and F) Specimen from a 59-year-old woman with squamous lung cancer. Lung specimens from lung cancer patients were stained with FITC- and PE-conjugated antibodies specific for CD90 (an essential marker for MSCs) and FGF10, respectively. Nuclei were counterstained with DAPI. Scale bars, 100 μm. The lung cancer regions and the normal lung regions are shown in panels A, C, and E and panels B, D, and F, respectively. In all panels, controls included the isotype-matched control antibodies (Ctrl Ab).
FGF10 production in MSCs and its regulatory function for cancer stem cells.
To confirm FGF10 production in MSCs even in the microenvironment of a tumor, we analyzed by Western blotting the whole-cell extract from MSCs under coculture conditions with or without cancer cells. The amounts of FGF10 were comparable among MSCs from three unrelated donors, regardless of whether the MSCs had been cocultured with H1299/A549 lung cancer cells or not, suggesting that the FGF10 production of MSCs was not affected by the cocultured cancer cells (Fig. 6A). We then tested whether FGF10 produced in MSCs was functionally active or not by neutralizing FGF10 in conditioned medium from the MSC monoculture (MSC CM). The addition of a neutralizing antibody against FGF10 to MSC CM caused a significant 2-fold increase of the side population fraction in H1299 lung cancer cells compared with that of the control antibody (P < 0.005) (Fig. 6B and C). We observed essentially similar results in A549 and H1975 lung cancer cells but not in MCF-7 and HeLa non-lung cancer cells (A549, P < 0.05; H1975, P < 0.01; MCF-7, P > 0.1; HeLa, P > 0.1) (Fig. 6C). In this experimental setting, no significant differences in the side population fractions were detected in all cancer cells used between the presence and absence of control antibody (all cell lines, P > 0.05) (data not shown). The data suggest that, even in the tumor microenvironment, MSCs produce active FGF10, which appeared to have a regulatory function in lung cancer stem cells.
FIG 6.
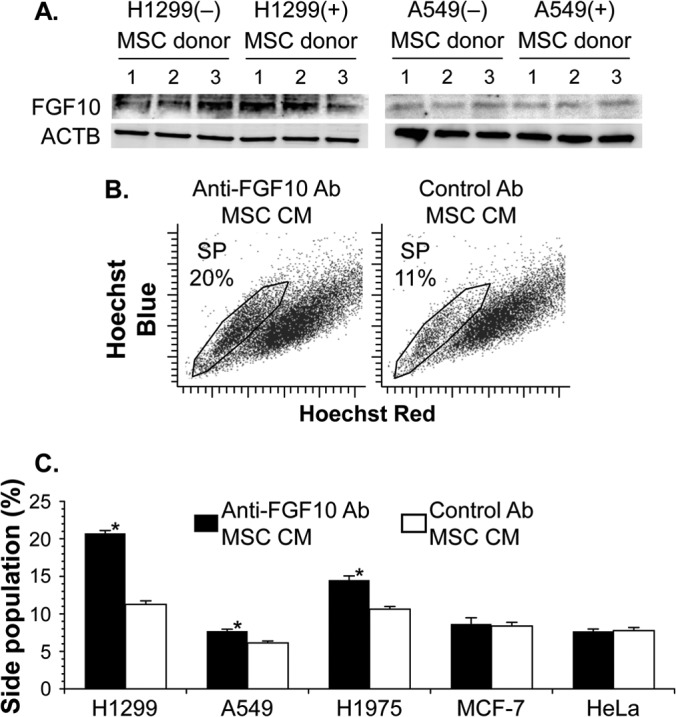
MSCs produce FGF10 affecting lung cancer stem cells. (A) Western blot analysis of FGF10 in transwell-cocultured MSCs with H1299 or A549 lung cancer cells. Controls included MSCs cultured without lung cancer cells. MSCs were from three unrelated donors, and β-actin production (ACTB) was used as a control. (B and C) Functional analysis of FGF10 production. The side population was analyzed in cancer cells stained with Hoechst 33342 after the 48-h culture with conditioned medium of MSCs in the presence of anti-FGF10 neutralizing antibody or control antibody. A representative side population subset (SP) with the percentage in H1299 lung cancer cells (B) and proportions of five cancer cells in the side population (C) are shown. The cell proportion is reported as the mean percentage ± standard error per group (n = 3). Asterisks indicate significant differences compared with the control antibody.
Recombinant FGF10 alone regulates lung cancer stem cells.
To understand the importance of FGF10 in influencing the stem cell kinetics of lung cancers, we further analyzed various aspects of the stem cell features, including self-renewing clonogenicity, the gene expression profile, and cell cycle quiescence (Fig. 7). When cultured with recombinant FGF10 or the vehicle under serum-free nonadherent culture conditions, FGF10-treated H1299 and A549 human lung cancer cells formed significantly fewer clonal spheres than control cells (H1299, 6.0 versus 13.0 spheres formed from 100 cells, P < 0.001; A549, 5.4 versus 12.6 spheres formed from 100 cells, P < 0.001) (Fig. 7A and B). Additionally, the FGF10 treatment of H1299 and A549 cells decreased the mRNA expression of the stemness genes OCT3/4 and SOX2, both of which encode key transcription factors in the maintenance of cancer stem cells as well as pluripotent embryonic stem cells, compared with the control treatment with vehicle (OCT3/4, H1299, P = 0.08; OCT3/4, A549, P < 0.005; SOX2, both cells, P < 0.05) (Fig. 7C). As determined by flow cytometry analysis of the cell cycle distribution, the fraction of 7-AADlow pyronin Ylow quiescent cells in the G0 phase was significantly attenuated by recombinant FGF10 in lung cancer cells, including H1299, A549, and H1975 cells, but was not affected in non-lung cancer cells, including MCF-7 and HeLa cells (FGF10 compared to vehicle: H1299, P < 0.005; A549, P < 0.005; H1975, P < 0.005; MCF-7, P > 0.05; HeLa, P > 0.2) (Fig. 7D and E). Conversely, these various aspects of the stem cell feature suppressed by conditioned medium from MSCs (MSC CM) were significantly recovered in lung cancer cells by the addition of a neutralizing antibody against FGF10 (compared with control antibody by number of spheres, P < 0.001; OCT3/4, P = 0.07; SOX2, P = 0.07; H1299, P < 0.005; A549, P < 0.005; H1975, P < 0.001; MCF-7, P > 0.05; HeLa, P > 0.05) (data not shown). Together, these findings support the notion that FGF10 essentially contributes to the regulatory function of MSC for the stem cell subpopulation in lung cancers.
FIG 7.

Additive FGF10 reduces the proportion of lung cancer stem cells. Cancer cells were cultured in the presence of recombinant FGF10 or vehicle (PBS [control]) for 10 days (A and B) or 48 h (C to E). (A and B) Sphere-forming assay of H1299 and A549 lung cancer cells. Representative spherical images of H1299 cells (A) and the number of formed spheres in a well (B) are shown. Scale bars, 200 μm. (C) OCT3/4 and SOX2 gene expression in H1299 and A549 cells. By quantitative RT-PCR, the levels of gene expression in FGF10-treated cells were analyzed relative to those in control cells. (D) Representative phase distribution of cell cycle with the percentage of H1299 lung cancer cells in the G0 phase. (E) Proportion of five cancer cells in the G0 phase. For panels D and E, after the FGF10 treatment, cancer cells were stained with 7-AAD–pyronin Y for the cell cycle analysis. Data are presented as the mean ± standard error per group (n = 5 for panel B and n = 3 for panels C and E). Asterisks indicate significant differences compared with the control vehicle.
Anticancer implications of FGF10 in combination with a cytotoxic agent.
Finally, we sought to clarify the pharmaceutical potential of FGF10 for cancer therapy (Fig. 5). Based on accumulating evidence that cancer stem cells are responsible for disease recurrence due to their chemoresistance (19), we hypothesized that FGF10 would enhance the overall chemosensitivity of cancer cells by reducing the cancer stem cell fraction. To evaluate this concept, when treating A549 lung cancer cells with the cytotoxic drug cisplatin (days 0 to 2), we used recombinant FGF10 or vehicle as an adjunctive treatment and thereafter monitored the cell viability status as a percentage of the initial value on day 0 (Fig. 8A). The adjunctive FGF10 caused a significant decrease in the viability of cisplatin-treated A549 cancer cells on days 2 to 16 compared with the control vehicle (P < 0.05) (Fig. 8A). Of note, control A549 cells that had been treated with cisplatin alone recovered substantial viability on day 4, whereas those that had been adjunctively treated with FGF10 displayed only 83% cell viability even on day 12. Additionally, control A549 cells treated with cisplatin alone showed an approximately 3-fold increase in the mRNA expression levels of the stemness genes OCT3/4 and SOX2 on day 4 compared to those on day 0, whereas A549 cells treated with FGF10 as well as cisplatin expressed both genes at comparable levels on days 0 and 4 (Fig. 8B). Thus, the levels of stemness gene expression, including LGR4, were significantly lower in A549 cells after the FGF10 adjunctive treatment than those in control A549 cells (OCT3/4, P < 0.05; SOX2, P < 0.05; LGR4, P < 0.005) (Fig. 8B). Similar in vitro results were observed with H1299 lung cancer cells (days 4 to 26, P < 0.05; OCT3/4, P < 0.05; SOX2, P < 0.05; LGR4, P = 0.07) (data not shown).
FIG 8.
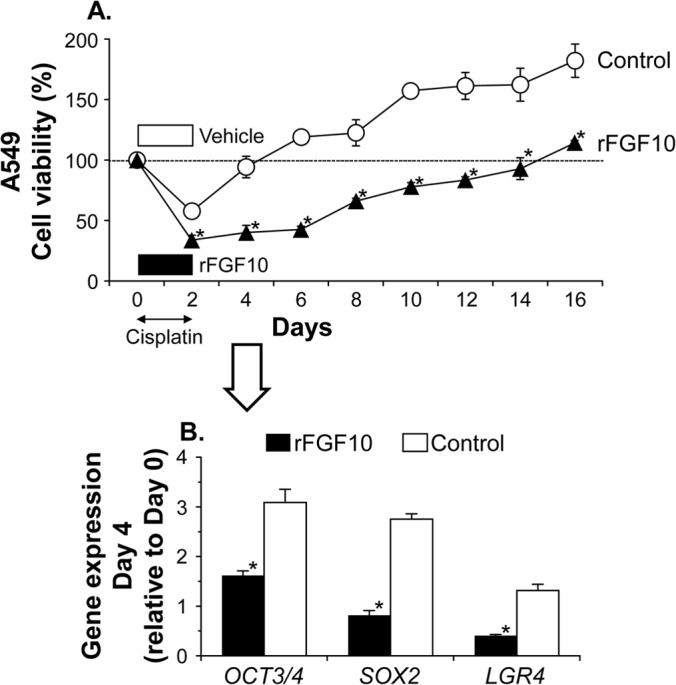
FGF10 suppresses reproliferation after an anticancer treatment. A549 lung cancer cells were treated with the anticancer drug cisplatin (CDDP) in the presence of recombinant FGF10 or vehicle (PBS [control]) for 2 days. (A) Time course of reproliferation after the anticancer treatment. The cell density relative to that at start of the experiment (day 0, indicated by the dotted horizontal line) was monitored by a colorimetric assay using MTS. (B) OCT3/4, SOX2, and LGR4 gene expression in A549 cells on day 4. The level of expression relative to that on day 0 was analyzed by quantitative RT-PCR. Data are presented as the mean ± standard error per group (n = 3). Asterisks indicate significant differences compared with the control vehicle.
DISCUSSION
In this study, we demonstrated that treatment with MSC-conditioned medium resulted in decreased proportions of both lung cancer cells with a side population phenotype and those in the quiescent G0 cell cycle state. The lung cancer-specific suppression of the cancer stem cell-enriched fraction was caused by FGF10 secreted from MSCs, as evidenced by the finding that the MSC-mediated decrease was impeded by the neutralization of FGF10 and restored by recombinant FGF10 protein. Of note, the addition of FGF10 to the lung cancer cell culture was also found to decrease the number of self-renewal cancer cells with the ability to grow as floating spheres in serum-free medium. Consistent with the reduced cancer stem cell fraction, the mRNA expression of stemness markers, such as OCT3/4 and SOX2, was significantly blunted in lung cancer cells treated with recombinant FGF10 protein. Indeed, the FGF10 treatment rendered the lung cancer cells more responsive to an anticancer agent, most likely by reducing the chemoresistant stem cell component.
Cancer stem cells are characterized as quiescent cells with self-renewal and high drug efflux capacities, which thereby retain resistance to anticancer agents (2, 3, 20). These conventional cytotoxic drugs exert killing effects on rapidly proliferating cells but not on quiescent cells, such as cancer stem cells. Their chemoresistance is also enhanced by increased levels of drug efflux pumps. Thus, even after the chemotherapy-induced eradication of the majority of tumor cells, the self-renewal capacity of cancer stem cells enables them to repopulate tumors.
This concept of cancer stem cells has fueled the notion that the pool of cancer stem cells within a tumor may have important implications for the development of better therapeutic approaches (5, 6, 8). Indeed, several studies have aimed to induce cancer stem cells out of the cell cycle quiescent state for their complete eradication following chemotherapy (7). For instance, in chronic myeloid leukemia, arsenic inhibition of the promyelocytic leukemia protein tumor suppressor enhanced the cell cycle entry of leukemia-initiating cells and increased the proapoptotic effect of chemotherapy on them (21). In acute myeloid leukemia (AML), treatment with granulocyte colony-stimulating factor (G-CSF) made AML stem cells susceptible to chemotherapy by inducing them to divide (22). Such pharmacological action controlling cancer stemness is now shared with retinoic acids that have been efficaciously used for patients of acute promyelocytic leukemia and high-risk neuroblastoma (23, 24). In this context, we have identified FGF10 as a stemness regulator specific for lung cancer cells in conditioned medium from MSC, with evidence supporting the view that FGF10 could be successfully applied for induction of lung cancer stem cells out of quiescence and making them more susceptible to antiproliferative chemotherapy.
The paracrine FGF-FGFR interactions are known for their main roles in epithelial-mesenchymal communication for tissue patterning and organogenesis (25–27). The mammalian FGF family comprises 18 ligands, which are generally produced in either epithelial or mesenchymal tissue and distinctly engaged by their cognate receptors on the opposite tissue. This unidirectional FGF signaling safeguards normal physiological conditions against morbid autocrine stimulation, and the importance of the FGF signaling axis has been highlighted by ligand-promiscuous stimulation and ectopic expression of FGFRs in pathological settings, including cancer stem cells (25, 26, 28). For instance, breast cancer cells were found to enhance the cancer stem cell property through pretreatment of FGF9, which generally signals from epithelium to mesenchyme (29). In pancreatic ductal adenocarcinoma, the ectopic expression of the EGFR2 mesenchymal isoform was observed in 83 of 117 cases and associated with stem cell-like features of the tumor cells (30).
Unlike the aberrant FGF-FGFR signaling that appears to enrich the cancer stem cell component, triggering FGFRs in line with the homeostasis between cancer-originating epithelium and mesenchyme may conversely reduce the fraction. FGF10 is a paracrine ligand produced from mesenchyme and is thought to act on the epithelium as an indispensable morphogenetic factor of the lungs, as evidenced by the finding that FGF10-deficient mouse embryos exhibit lung agenesis (31, 32). These lung-specific functions of FGF10 may provide further insight into molecular mechanisms by which FGF10, but not other FGFs with binding affinity for FGFR1b, can affect cancer stem cells derived only from lung. In this way, our data show that FGF10 from mesenchyme-derived MSCs preferentially activates the epithelial-type FGFR1b of lung cancer cells, resulting in the reduction of cancer stem cells. Previous studies have also shown that a recombinant form of FGF10 had no effect on the growth of a certain lung cancer cell line and that forced overexpression of FGF10 rather disrupted lung morphogenesis, causing pulmonary adenomas (33, 34). Furthermore, FGF10-expressing cells have been shown to represent a pool of resident MSCs in mouse lung, while our immunohistochemical data did not demonstrate FGF10-expressing MSCs in normal human lung (35). The universality of our concept that recruited, or resident, MSCs in lung cancer affect cancer stem cell kinetics awaits further studies using various tumor cells and experimental conditions.
ACKNOWLEDGMENTS
We thank Mitsu Takahashi for technical assistance and Brent Bell for reading the manuscript.
These studies were supported, in part, by Grants-in-Aid for Scientific Research and the Project for Development of Innovative Research on Cancer Therapeutics from the Ministry of Education, Culture, Sports, Science and Technology (Tokyo, Japan), the Core Research for Evolutional Science and Technology Program, the Adaptable and Seamless Technology Transfer Program from the Japan Science and Technology Agency (Tokyo, Japan), and the Urgent Research Grant Program from the Astellas Foundation for Research on Metabolic Disorders (Tokyo, Japan).
Footnotes
Published ahead of print 27 May 2014
REFERENCES
- 1.Bunn PA., Jr 2012. Worldwide overview of the current status of lung cancer diagnosis and treatment. Arch. Pathol. Lab. Med. 136:1478–1481. 10.5858/arpa.2012-0295-SA [DOI] [PubMed] [Google Scholar]
- 2.Gottschling S, Schnabel PA, Herth FJ, Herpel E. 2012. Are we missing the target? Cancer stem cells and drug resistance in non-small cell lung cancer. Cancer Genomics Proteomics 9:275–286 http://cgp.iiarjournals.org/content/9/5/275.long [PubMed] [Google Scholar]
- 3.Jiang W, Peng J, Zhang Y, Cho WC, Jin K. 2012. The implications of cancer stem cells for cancer therapy. Int. J. Mol. Sci. 13:16636–16657. 10.3390/ijms131216636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu X, Chen H, Wang X. 2012. Can lung cancer stem cells be targeted for therapies? Cancer Treat. Rev. 38:580–588. 10.1016/j.ctrv.2012.02.013 [DOI] [PubMed] [Google Scholar]
- 5.Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, Laino L, De Francesco F, Papaccio G. 2013. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB J. 27:13–24. 10.1096/fj.12-218222 [DOI] [PubMed] [Google Scholar]
- 6.Marjanovic ND, Weinberg RA, Chaffer CL. 2013. Cell plasticity and heterogeneity in cancer. Clin. Chem. 59:168–179. 10.1373/clinchem.2012.184655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baccelli I, Trumpp A. 2012. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 198:281–293. 10.1083/jcb.201202014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang DG. 2012. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 22:457–472. 10.1038/cr.2012.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergfeld SA, DeClerck YA. 2010. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 29:249–261. 10.1007/s10555-010-9222-7 [DOI] [PubMed] [Google Scholar]
- 10.Keating A. 2012. Mesenchymal stromal cells: new directions. Cell Stem Cell 10:709–716. 10.1016/j.stem.2012.05.015 [DOI] [PubMed] [Google Scholar]
- 11.Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F., 3rd 2011. Concise review. Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells 29:11–19. 10.1002/stem.559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torsvik A, Bjerkvig R. 2013. Mesenchymal stem cell signaling in cancer progression. Cancer Treat. Rev. 39:180–188. 10.1016/j.ctrv.2012.03.005 [DOI] [PubMed] [Google Scholar]
- 13.Kanehira M, Kikuchi T, Ohkouchi S, Shibahara T, Tode N, Santoso A, Daito H, Ohta H, Tamada T, Nukiwa T. 2012. Targeting lysophosphatidic acid signaling retards culture-associated senescence of human marrow stromal cells. PLoS One 7:e32185. 10.1371/journal.pone.0032185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaini J, Andarini S, Tahara M, Saijo Y, Ishii N, Kawakami K, Taniguchi M, Sugamura K, Nukiwa T, Kikuchi T. 2007. OX40 ligand expressed by DCs costimulates NKT and CD4+ Th cell antitumor immunity in mice. J. Clin. Invest. 117:3330–3338. 10.1172/JCI32693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mafi P, Hindocha S, Mafi R, Griffin M, Khan WS. 2011. Adult mesenchymal stem cells and cell surface characterization—a systematic review of the literature. Open Orthop. J. 5:253–260. 10.2174/1874325001105010253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nery AA, Nascimento IC, Glaser T, Bassaneze V, Krieger JE, Ulrich H. 2013. Human mesenchymal stem cells: from immunophenotyping by flow cytometry to clinical applications. Cytometry A 83:48–61. 10.1002/cyto.a.22205 [DOI] [PubMed] [Google Scholar]
- 17.Metzger RJ, Krasnow MA. 1999. Genetic control of branching morphogenesis. Science 284:1635–1639. 10.1126/science.284.5420.1635 [DOI] [PubMed] [Google Scholar]
- 18.Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV. 2000. The molecular basis of lung morphogenesis. Mech. Dev. 92:55–81. 10.1016/S0925-4773(99)00325-1 [DOI] [PubMed] [Google Scholar]
- 19.Maugeri-Sacca M, Vigneri P, De Maria R. 2011. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 17:4942–4947. 10.1158/1078-0432.CCR-10-2538 [DOI] [PubMed] [Google Scholar]
- 20.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. 2004. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. U. S. A. 101:14228–14233. 10.1073/pnas.0400067101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, Rosenblatt J, Avigan DE, Teruya-Feldstein J, Pandolfi PP. 2008. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 453:1072–1078. 10.1038/nature07016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A, Taniguchi S, Shultz LD, Ishikawa F. 2010. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol. 28:275–280. 10.1038/nbt.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de The H, Chen Z. 2010. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat. Rev. Cancer 10:775–783. 10.1038/nrc2943 [DOI] [PubMed] [Google Scholar]
- 24.Masetti R, Biagi C, Zama D, Vendemini F, Martoni A, Morello W, Gasperini P, Pession A. 2012. Retinoids in pediatric onco-hematology: the model of acute promyelocytic leukemia and neuroblastoma. Adv. Ther. 29:747–762. 10.1007/s12325-012-0047-3 [DOI] [PubMed] [Google Scholar]
- 25.Beenken A, Mohammadi M. 2009. The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 8:235–253. 10.1038/nrd2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goetz R, Mohammadi M. 2013. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 14:166–180. 10.1038/nrm3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miraoui H, Marie PJ. 2010. Fibroblast growth factor receptor signaling crosstalk in skeletogenesis. Sci. Signal. 3:re9. 10.1126/scisignal.3146re9 [DOI] [PubMed] [Google Scholar]
- 28.Acevedo VD, Ittmann M, Spencer DM. 2009. Paths of FGFR-driven tumorigenesis. Cell Cycle 8:580–588. 10.4161/cc.8.4.7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, Kuperwasser C. 2010. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc. Natl. Acad. Sci. U. S. A. 107:21737–21742. 10.1073/pnas.1007863107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishiwata T, Matsuda Y, Yamamoto T, Uchida E, Korc M, Naito Z. 2012. Enhanced expression of fibroblast growth factor receptor 2 IIIc promotes human pancreatic cancer cell proliferation. Am. J. Pathol. 180:1928–1941. 10.1016/j.ajpath.2012.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS. 1998. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 12:3156–3161. 10.1101/gad.12.20.3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. 1999. Fgf10 is essential for limb and lung formation. Nat. Genet. 21:138–141. 10.1038/5096 [DOI] [PubMed] [Google Scholar]
- 33.Alderson R, Gohari-Fritsch S, Olsen H, Roschke V, Vance C, Connolly K. 2002. In vitro and in vivo effects of repifermin (keratinocyte growth factor-2, KGF-2) on human carcinoma cells. Cancer Chemother. Pharmacol. 50:202–212. 10.1007/s00280-002-0493-8 [DOI] [PubMed] [Google Scholar]
- 34.Clark JC, Tichelaar JW, Wert SE, Itoh N, Perl AK, Stahlman MT, Whitsett JA. 2001. FGF-10 disrupts lung morphogenesis and causes pulmonary adenomas in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L705–L715 http://ajplung.physiology.org/content/280/4/L705 [DOI] [PubMed] [Google Scholar]
- 35.El Agha E, Herold S, Al Alam D, Quantius J, MacKenzie B, Carraro G, Moiseenko A, Chao CM, Minoo P, Seeger W, Bellusci S. 2014. Fgf10-positive cells represent a progenitor cell population during lung development and postnatally. Development 141:296–306. 10.1242/dev.099747 [DOI] [PMC free article] [PubMed] [Google Scholar]