

Abstract
Much of the work on nuclear lamins during the past 15 years has focused on mutations in LMNA (the gene for prelamin A and lamin C) that cause particular muscular dystrophy, cardiomyopathy, partial lipodystrophy, and progeroid syndromes. These disorders, often called “laminopathies,” mainly affect mesenchymal tissues (e.g., striated muscle, bone, and fibrous tissue). Recently, however, a series of papers have identified important roles for nuclear lamins in the central nervous system. Studies of knockout mice uncovered a key role for B-type lamins (lamins B1 and B2) in neuronal migration in the developing brain. Also, duplications of LMNB1 (the gene for lamin B1) have been shown to cause autosome-dominant leukodystrophy. Finally, recent studies have uncovered a peculiar pattern of nuclear lamin expression in the brain. Lamin C transcripts are present at high levels in the brain, but prelamin A expression levels are very low—due to regulation of prelamin A transcripts by microRNA 9. This form of prelamin A regulation likely explains why “prelamin A diseases” such as Hutchinson-Gilford progeria syndrome spare the central nervous system. In this review, we summarize recent progress in elucidating links between nuclear lamins and neurobiology.
INTRODUCTION
The nuclear lamina has attracted considerable scrutiny from biochemists, cell biologists, and geneticists. Much of this attention has been stimulated by the discovery that mutations in LMNA (the gene for the A-type lamins, prelamin A and lamin C) cause multiple human diseases, including muscular dystrophy, cardiomyopathy with conduction system disease, partial lipodystrophy, and progeroid syndromes (1–3). These diseases, often called “laminopathies,” are largely confined to mesenchymal tissues. For the past decade, many laboratories have worked to define disease mechanisms and to devise therapeutic strategies. These efforts have been summarized in many reviews (2–11).
While “LMNA diseases” have attracted most of the research efforts, a new topic has slowly emerged in the field—nuclear lamin biology in the brain. Three lines of investigation have contributed to the emergence of this topic. The first was work by developmental biologists that uncovered a role for the B-type lamins (lamins B1 and B2) in neuronal migration in the developing brain (12–14). The second was work showing that a demyelinating disorder, autosomal dominant leukodystrophy, is caused by LMNB1 gene duplications (15, 16). The third was work by clinical investigators to understand the spectrum of disease phenotypes in children with Hutchinson-Gilford progeria syndrome (17, 18), a precocious aging syndrome caused by a toxic form of prelamin A (19). For years, the field marveled that children with progeria have multisystem aging-like phenotypes but are spared common features of physiologic aging in the central nervous system, notably, senile dementia. Recent studies have identified a likely mechanism—regulation of prelamin A by a brain-specific microRNA (17, 18).
PROTEINS OF THE NUCLEAR LAMINA
The nuclear lamina in somatic cells is mainly composed of four nuclear lamins (lamins A, C, B1, and B2), which form a filamentous meshwork lining the inner nuclear membrane. Prelamin A (the precursor to mature lamin A) and lamin C are products of the LMNA gene and are produced by alternative splicing (20). Prelamin A and lamin C are identical through the first 566 amino acids but have distinct carboxyl-terminal sequences (20). Lamin C contains sequences from exons 1 to 10 and terminates with six unique amino acids not found in prelamin A. Prelamin A contains sequences from two additional exons (exons 11 and 12) and terminates with 98 unique residues (20). Lamins B1 and B2 are produced from separate genes, LMNB1 and LMNB2 (21, 22). The nuclear lamina interacts with chromatin and nuclear membrane proteins and is important for providing structural support for the cell nucleus and tethering the nucleus to the cytoskeleton (7, 23, 24).
Prelamin A terminates with a “CaaX” motif (CSIM), which triggers three posttranslational modifications: farnesylation of the carboxyl-terminal cysteine, endoproteolytic release of the last three amino acids of the protein, and carboxymethylation of the newly exposed farnesylcysteine. Subsequently, the last 15 residues of prelamin A, including the farnesylcysteine methyl ester, are removed by a zinc metalloprotease, ZMPSTE24, releasing mature lamin A (25–27). The generation of mature lamin A is utterly dependent on protein farnesylation; when protein farnesylation is inhibited, the subsequent processing steps do not occur, and nonfarnesylated prelamin A accumulates in cells (28).
Prelamin A's farnesylcysteine methyl ester is generally assumed to associate with the inner nuclear membrane and facilitate the delivery of lamin A to the nuclear lamina (29). However, studies of Lmna knock-in mice have suggested that farnesylation and methylation might have little impact on the targeting of lamin A to the nuclear rim (10). Coffinier et al. (30) created Lmna knock-in mice that produce mature lamin A directly, bypassing prelamin A synthesis and all of the subsequent posttranslational processing steps. These mice were quite healthy, even after 2 years of observation, and the targeting of mature lamin A to the nuclear rim in mouse tissues appeared normal. Also, in Lmna knock-in mice that produce a nonfarnesylated version of prelamin A, there was no obvious defect in the delivery of the nonfarnesylated prelamin A to the nuclear rim (31).
Lamin C does not have a CaaX motif and therefore is not farnesylated or methylated. Lamins B1 and B2 terminate with a CaaX motif and undergo farnesylation and methylation. Unlike the situation with prelamin A, however, the farnesylcysteine methyl ester in lamins B1 and B2 is not removed by a subsequent endoproteolytic cleavage step.
Lamins B1 and B2 are expressed at high levels in virtually all cells from the earliest stages of development (32). Lamins A and C are expressed at low levels early in development (33), but expression levels increase late in development (14, 34). The low levels of Lmna expression early in development (14, 34), along with the fact that Lmna knockout mice survive development (35), have led to the notion that lamins A and C are mainly important for differentiated cells (4).
Sullivan et al. (35) created mice lacking both lamin A and lamin C (Lmna−/−). Although the mice survived embryonic development, they succumbed to muscular dystrophy and cardiomyopathy by 4 to 5 weeks of age. Embryonic fibroblasts exhibited striking abnormalities in nuclear shape. Subsequent studies revealed that the knockout mice expressed trace amounts of an internally truncated prelamin A (36). However, that product appeared to be relatively unimportant because the phenotypes of a newer line of Lmna−/− mice were similar (37). To gain insights into unique roles of prelamin A and lamin C, Fong et al. (38) and Davies et al. (31) created knock-in mice that produced exclusively lamin C or exclusively prelamin A. However, both mouse models were healthy and free of pathology, limiting decisive insights into the unique functions for the two Lmna isoforms.
Until recently, dogma held that the B-type lamins were essential proteins with many crucial functions in the cell nucleus (4), ranging from roles in DNA replication and gene transcription (39–41) to the formation of the mitotic spindle (42) and the organization of heterochromatin (43). A paper holding that cultured cells undergo growth arrest and apoptosis after small interfering RNA (siRNA) inhibition of LMNB1 and LMNB2 expression lent credence to this view (44). However, recent studies with genetically modified mice have undercut this view—at least in the case of peripheral tissues. Mice lacking both lamin B1 and lamin B2 in keratinocytes had no skin pathology, had normal keratinocyte proliferation rates, and had no abnormalities in the nuclear envelope or in heterochromatin distribution (45). Also, an absence of both lamin B1 and lamin B2 in hepatocytes did not lead to liver pathology (46). One obvious explanation for these findings is that expression of lamins A and C is sufficient for the vitality of keratinocytes and hepatocytes. However, Kim et al. (47) recently reported that mouse embryonic stem cells lacking all nuclear lamins were able to proliferate and differentiate—and even form large teratomas in mice. Those studies were surprising and at face value further undermine the view that lamins play important roles in mitotic spindle assembly, DNA replication, and other vital functions in the cell nucleus. Additional efforts to define the consequence of a complete deficiency of nuclear lamins in specific cell types are needed.
A ROLE FOR B-TYPE LAMINS IN BRAIN DEVELOPMENT
The first clue about the in vivo importance of B-type lamins in mammals came from lamin B2 knockout mice (12). Lmnb2-deficient embryos (Lmnb2−/−) were virtually normal in size during development, but they died shortly after birth (12) with obvious neuropathology. The cerebellum was small and devoid of sulci; the cerebral cortex was also small, and the layering of cortical neurons was abnormal. Further studies revealed that the neuronal layering abnormality was present as early as embryonic day 16.5 (E16.5) (Fig. 1A). Because this pathology was similar to pathology in mice with well-characterized defects in neuronal migration (48–50), Coffinier et al. (12) performed immunochemistry studies with layer-specific neuronal markers and bromodeoxyuridine (BrdU) birth-dating studies. In E19.5 Lmnb2−/− embryos, many NeuN-positive neurons accumulated in lower levels of the cortical plate. In wild-type littermates, most NeuN-positive neurons migrated past Ctip2-positive neurons into the superficial layers of the cortex. Also, FoxP1-positive neurons in Lmnb2−/− embryos accumulated in lower levels of the cortical plate, and few neurons reached the more superficial layers. The neuronal migration defect was confirmed by BrdU birth-dating studies. In wild-type mice, most neurons that originated at E13.5 (i.e., when the BrdU was injected) were found deep in the cortical plate at E18.5. Neurons that originated at later time points (and which had little BrdU) migrated past the BrdU-positive neurons into more-superficial layers of the cortical plate. In contrast, most BrdU-positive neurons in Lmnb2−/− embryos were located in superficial layers of the cortical plate, suggesting that newer, BrdU-negative neurons lacked the capacity to migrate into more-superficial layers (12). Neuronal migration defects were also documented in an independently created line of Lmnb2−/− mice (51).
FIG 1.
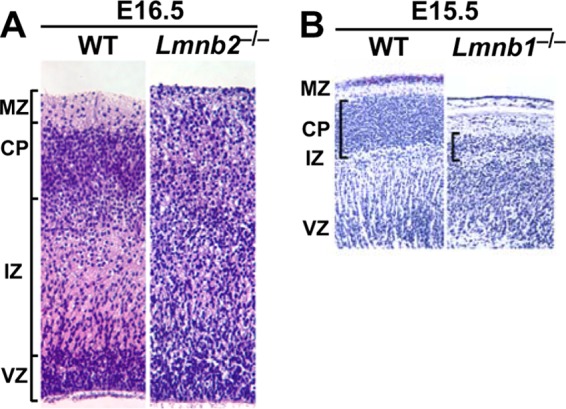
Defects in neuronal migration and neuronal survival in Lmnb1 and Lmnb2 knockout mice. (A and B) Hematoxylin and eosin staining of cerebral cortex sections from E16.5 wild-type (WT) and Lmnb2 knockout (Lmnb2−/−) embryos (A) and sections from E15.5 WT and Lmnb1 knockout (Lmnb1−/−) embryos (B). (Panel A was reprinted from reference 12 and panel B was reprinted from reference 14 with permission of the publishers.) MZ, marginal zone; CP, cortical plate; IZ, intermediate zone; VZ, ventricular zone.
In hindsight, a role for the nuclear lamina in neuronal migration was not surprising. Neuronal migration depends on cytoplasmic motors pulling the cell nucleus forward toward the centrosome in the leading edge of the cell (nucleokinesis) (52). The trailing portion of the migrating neuron is then remodeled, and the net effect is forward movement of the cell. Repeated cycles of this process make it possible for neurons to traverse from the ventricular zone to the cortical plate. When Lmnb2−/− mice were initially created (12), the cytoplasmic proteins required for “tugging” on the cell nucleus had already been investigated in considerable detail (49, 53, 54), but the identity of nuclear proteins required for this process was unclear. Finding defective neuronal migration in Lmnb2−/− embryos implied that neuronal migration depends on B-type lamins.
The cytoplasmic motors required for nuclear translocation cannot interact directly with the nuclear lamina, simply because the nuclear lamina is located within the nucleus. Instead, the forces on the cell nucleus are almost certainly exerted through the LINC (Linker of nucleoskeleton and cytoskeleton complex) (13, 55). The LINC complex involves KASH proteins (e.g., nesprins) in the outer nuclear membrane and SUN proteins in the inner nuclear membrane. On the cytoplasmic side, the nesprins interact with elements of the cytoskeleton, while on the nucleoplasmic side, the SUN proteins interact with the nuclear lamina (13). Nuclear translocation and neuronal migration depend on the LINC complex; the absence of SUN1 and SUN2, or the removal of the KASH domain from nesprin 1 and nesprin 2, results in neuronal migration defects similar to those in Lmnb2−/− mice (56).
Coffinier et al. (12) suggested that the neuronal migration defects in Lmnb2−/− mice were a consequence of reduced integrity of the nuclear lamina—leading to impaired nuclear translocation. Studies on the brain pathology in Lmnb2−/− mice supported this model (14). Many of the cell nuclei in the cortical plate neurons of Lmnb2−/− embryos were markedly “stretched out” (comet shaped), and they suggested that this abnormality was due to the deformational stresses during nuclear translocation. Presumably, the cytoplasmic motors in Lmnb2−/− neurons simply “stretched out” a weakened nuclear envelope rather than moving the nucleus into the leading edge of the cell.
In the report on Lmnb2−/− mice, Coffinier et al. (12) speculated that lamin B1 might also play a role in brain development. At that time, Lmnb1 knockout mice from a gene-trap ES cell clone had already been described (57). The Lmnb1−/− embryos, which expressed a lamin B1–β-galactosidase fusion protein, were small and died shortly after birth with an abnormally shaped cranium. More-detailed studies by Coffinier et al. (14) revealed that Lmnb1 knockout embryos had a small brain with abnormal layering of neurons in the cerebral cortex (Fig. 1B). Immunohistochemical studies with antibodies against Otx1 and TBR1 indicated a neuronal migration defect, and that was confirmed with BrdU birth-dating studies (14). Another group, using an independent line of Lmnb1−/− mice, reached similar conclusions (51).
The brain pathology in the Lmnb1−/− embryos was more severe than in the Lmnb2−/− embryos; there were fewer neurons in the cortical plate, and numbers of neuronal progenitors were reduced (14). Also, nuclear shape abnormalities were different. There were almost no comet-shaped nuclei in Lmnb1−/− embryos, but a large fraction of Lmnb1−/− neurons contained a single large nuclear bleb. Also, lamin B2 was asymmetrically distributed and much of it was located in the nuclear bleb. The authors suggested that reduced integrity of the nuclear envelope during nucleokinesis may have contributed to the formation of the nuclear bleb (14).
As discussed previously by Coffinier and coworkers and Young and coworkers (12, 13, 58), the discovery that lamins B1 and B2 are essential for neuronal migration in the mammalian brain was foreshadowed by a publication by Patterson et al. on eye development in Drosophila (59). They found that a Drosophila lamin and Klarsicht, a KASH domain-containing protein, were essential for photoreceptor nuclear migration in the Drosophila eye. They speculated that the lamin-Klarsicht interaction was relevant to human laminopathies, such as muscular dystrophy and cardiomyopathy.
A ROLE FOR THE B-TYPE LAMINS IN NEURONAL SURVIVAL
To explore the importance of lamin B1 and lamin B2 in neurons postnatally, Coffinier et al. (14) used recombination techniques involving Cre and loxP to generate forebrain-specific Lmnb1 knockout mice and forebrain-specific Lmnb2 knockout mice. Mice from both models survived, but they had a small forebrain with neuronal layering abnormalities. In both models, the number of forebrain neurons in adult mice was markedly reduced (more so than in the knockout embryos), suggesting that the loss of either B-type lamin impairs neuronal survival. Coffinier et al. (14) went on to breed forebrain-specific Lmnb1 and Lmnb2 double-knockout mice. Those mice had complete atrophy of the forebrain and no detectable neurons in the forebrain, indicating that the loss of both B-type lamins is not compatible with neuronal survival. This situation is obviously different than with keratinocytes and hepatocytes, where the loss of B-type lamins had no significant consequences (45, 46). Of note, Lmna is expressed in the adult mouse brain, but it is not expressed in neurons during development (14). We suspect that the complete absence of nuclear lamins in the forebrain neurons of forebrain-specific Lmnb1-Lmnb2 knockout embryos “seals their fate” and places them on a pathway to cell death. The general idea that nuclear lamins are relevant to cell survival in response to stress is supported by other studies. For example, lamin A expression promotes survival of cells forced to migrate through 3-μm micropores (60). Other studies have shown that nuclear lamin expression is a key determinant of cell stiffness and the ability of cells to migrate through micropores (61–65).
The mouse studies leave little doubt that the two B-type lamins play important roles in brain development and neuronal survival in mice, but what is the situation in humans? Surprisingly, no one has yet identified LMNB1 or LMNB2 loss-of-function mutations in humans with neurodevelopmental abnormalities. There have been suggestions that LMNB1 polymorphisms may be associated with neural tube defects (66, 67), but more data are needed to be confident in that association. In the NHLBI exome sequencing database (http://evs.gs.washington.edu/EVS/), there are many rare LMNB1 and LMNB2 missense mutations, and there were several LMNB1 frameshift or splicing mutations. However, homozygous LMNB1 or LMNB2 mutations have not yet been encountered in humans, likely because the relevant patient populations have not yet been screened. Sooner or later, we suspect that LMNB1 and LMNB2 loss-of-function mutations will be encountered in human fetuses with neurodevelopmental defects, and we would not be surprised to find missense mutations in outpatient populations in neurology clinics.
INVESTIGATING THE IMPORTANCE OF THE FARNESYL LIPID ANCHOR IN LAMIN B1 AND LAMIN B2
Lamins B1 and B2 are farnesylated proteins, and it is likely that the farnesyl lipid is embedded in the inner nuclear membrane, thereby attaching the nuclear lamina to the inner nuclear membrane. Jung et al. (68) speculated that a tight association between the nuclear lamina and nuclear membranes might be crucial for migrating neurons. To test that idea, they created knock-in mice expressing nonfarnesylated versions of lamin B1 and lamin B2 (by replacing the cysteine of the CaaX motif with a serine). The targeted missense mutations worked as planned, abolishing lamin B1 and lamin B2 farnesylation. Absent farnesylation led to lower steady-state levels of both B-type lamins (68).
The “nonfarnesylated lamin B2” knock-in mice survived and were free of neuropathology or behavioral abnormalities, but the situation was very different for mice expressing nonfarnesylated lamin B1. Like Lmnb1−/− mice, the “nonfarnesylated lamin B1” mice died soon after birth with neuronal layering abnormalities in the cerebral cortex. However, the nonfarnesylated lamin B1 appeared to retain partial function. The size of newborn nonfarnesylated lamin B1 mice was nearly normal, whereas Lmnb1−/− embryos were extremely small (14, 57). Also, the brain pathology in the nonfarnesylated lamin B1 mice was milder than in Lmnb1−/− mice.
The neurons expressing nonfarnesylated lamin B1 exhibited a remarkable nuclear shape abnormality—dumbbell-shaped nuclei in which the nuclear lamina was separated from the bulk of the chromosomal DNA (Fig. 2A) (68). The nuclear lamina was “bunched up” in one end of the dumbbell within the leading edge of the cell, while most of the chromatin was found at the other end of the dumbbell and was “naked”—in that it was entirely free of a nuclear lamina. Both ends of the dumbbell were surrounded by nuclear membranes, as judged by staining for the inner nuclear membrane protein LAP2β. Dumbbell-shaped nuclei were detected in the midbrain and cortex and were also found in cultured neurons as they migrated away from neurospheres. Jung et al. (68) proposed that the dumbbell-shaped nuclei and “naked chromatin” were the consequence of weakened interactions between the nuclear lamina and the inner nuclear membrane (Fig. 2B). They suggested that the absence of lamin B1 farnesylation abolishes the hydrophobic interactions that normally affix the nuclear lamina to the inner nuclear membrane. They further suggested that migrating neurons from the “nonfarnesylated lamin B1 mice” would retain the ability to pull the nuclear lamina forward into the leading edge of the cell but that the chromatin would not “come along” and would instead “leak” into the potential space between the inner nuclear membrane and the nuclear lamina. Neurons expressing nonfarnesylated lamin B1 also had a honeycombed nuclear lamina, which likely facilitated the escape of genomic DNA from the nuclear lamina. Of note, the nonfarnesylated version of lamin B1 appeared (by immunofluorescence microscopy) to be located mainly at the nuclear rim—even in cells with a honeycombed nuclear lamina. In wild-type neurons, DNA cannot escape the bounds of the nuclear lamina because the farnesyl lipids anchor the nuclear lamina to the inner nuclear membrane and because of the more tightly woven pattern of the nuclear lamina.
FIG 2.
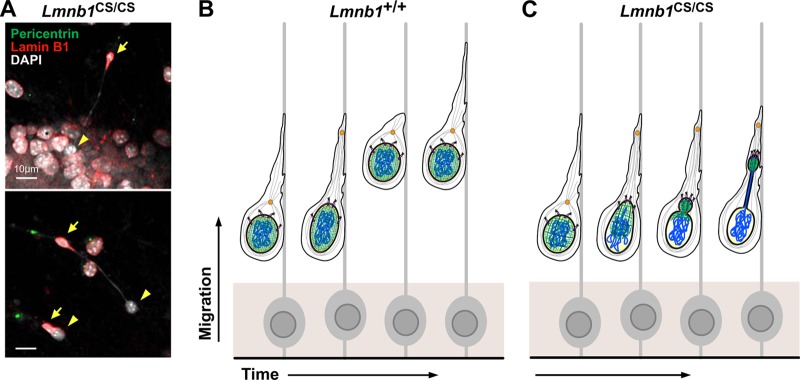
Nuclear abnormalities in Lmnb1CS/CS mouse embryonic fibroblasts (MEFs) and Lmnb1CS/CS neurons. (A) Immunofluorescence microscopy of migrating Lmnb1CS/CS neurons in cell culture, stained with antibodies against pericentrin (green) and lamin B1 (red). In the dumbbell-shaped Lmnb1CS/CS nuclei, lamin B1 was always in the leading edge of the cells (arrows), leaving “naked chromatin” behind in the trailing edge (arrowheads). (B and C) A model for the neuronal migration defect in Lmnb1CS/CS embryos. Unlike wild-type neurons (Lmnb1+/+) (B), the nuclear lamina (green) in Lmnb1CS/CS neurons (C) is not tightly affixed to the inner nuclear membrane because of the absence of the farnesyl lipid anchor on lamin B1 (red) (creating a potential space between the nuclear lamina and the inner nuclear membrane). Also, the nuclear lamina meshwork in Lmnb1CS/CS cells is not as tightly woven, exhibiting a honeycomb distribution. During neuronal migration, the nuclear lamina is pulled forward by the LINC complex (SUN1/2, purple; nesprin-1/2, pink) by microtubule (gray strands)-associated dynein motors (yellow) in the direction of the centrosome (orange) in the leading edge of the cell. However, the chromatin (blue) escapes from the bounds of the nuclear lamina into the space between the nuclear lamina and inner nuclear membrane. (Reprinted from reference 68 with permission of the publisher.)
INVESTIGATING THE FUNCTIONAL REDUNDANCY OF LAMINS B1 AND B2 IN BRAIN DEVELOPMENT
Lamins B1 and B2 are ∼60% identical at the amino acid level (11), and both are expressed ubiquitously beginning at the earliest stages of development (32). Also, both proteins play crucial roles in neuronal migration and neuronal survival (12). The similarities in lamin B1 and lamin B2 sequences and expression patterns, along with the similar neurodevelopmental abnormalities in the knockout mice, naturally raise the issue of whether the two proteins have redundant functions. To address that issue, Lee and coworkers (69) generated “reciprocal knock-in mice,” which made it possible to determine if increased expression of one of the B-type lamins would compensate for the loss of the other. In the first knock-in mouse (Lmnb1B2/B2 mice), they inserted a lamin B2 cDNA into exon 1 of Lmnb1, resulting in lamin B2 expression from the Lmnb1 locus while eliminating Lmnb1 transcripts. In the other mouse (Lmnb2B1/B1 mice), they inserted a lamin B1 cDNA into exon 1 of Lmnb2, resulting in lamin B1 expression from the Lmnb2 locus while eliminating Lmnb2 transcripts.
The Lmnb1B2/B2 mice worked as planned; Lmnb2 transcript levels in the cerebral cortex were >2.5-fold higher than in wild-type mice (reflecting the production of Lmnb2 transcripts from both Lmnb2 and Lmnb1) (69). Lamin B2 levels in the cortex of Lmnb1B2/B2 embryos were ∼3-fold higher than in wild-type embryos. Despite increased levels of lamin B2 expression, the Lmnb1B2/B2 mice were small and died shortly after birth with neuronal layering abnormalities in the cerebral cortex, indicating that increased lamin B2 production cannot prevent the developmental abnormalities associated with lamin B1 deficiency (69). However, the body and brain weights of Lmnb1B2/B2 embryos were higher than in Lmnb1−/− embryos, and the density of cortical neurons in Lmnb1B2/B2 embryos was greater—implying that increased lamin B2 production partially compensates for the loss of lamin B1 (69).
The story with Lmnb2B1/B1 mice was similar. Those mice, which expressed increased amounts of lamin B1 and no lamin B2, manifested neurodevelopmental abnormalities (69) that were virtually identical to those in Lmnb2−/− mice (12). Thus, surplus lamin B1 could not fully compensate for the loss of lamin B2. Again, however, there was evidence for partial compensation. The pathology in the cerebral cortex of Lmnb1B2/B2 Lmnb2B1/B1 embryos was milder than in Lmnb1B2/B2 embryos, implying that lamin B1 synthesis from the Lmnb2B1 allele was capable of lessening the severity of the developmental defects found in Lmnb1B2/B2 embryos.
LAMIN B1 GENE DUPLICATIONS AND AUTOSOMAL DOMINANT LEUKODYSTROPHY
A major advance in the human genetics of B-type lamins was the discovery that an adult-onset demyelinating disorder, autosomal dominant leukodystrophy (ADLD), is caused by LMNB1 duplications (15, 16). ADLD was first identified in a large American-Irish kindred (70) but was soon identified in multiple families of various ethnicities (15). ADLD typically begins between the fourth and sixth decades with autonomic dysfunction (manifested by orthostatic hypotension, impotence, and bladder abnormalities), followed by progressive signs of cerebellar and pyramidal disease. Cerebellar disease is generally manifested by ataxia and tremors, while the pyramidal signs include weakness in the extremities and spasticity. ADLD is diagnosed by magnetic resonance imaging (MRI) scanning, which reveals widespread and symmetrical loss of white matter, most prominently in the frontal and parietal lobes and cerebellum (71). At autopsy, patients with ADLD have vacuolated white matter with a loss of myelin but minimal or no loss of neurons or oligodendrocytes (the cells that produce myelin) (15, 16, 71). As revealed by Western blotting, however, brain tissue from ADLD patients has reduced amounts of oligodendrocyte and myelin proteins (72). There is little astrogliosis in ADLD brains, and there are no inflammatory infiltrates (distinguishing ADLD from multiple sclerosis) (15, 71).
The ADLD gene was first localized to chromosome 5q31 (73) and later narrowed to a 1.5-Mb segment within that region (74). In 2006, Padiath et al. (16) showed that ADLD is caused by head-to-tail duplication events involving LMNB1. The LMNB1 duplication led to increased expression of lamin B1 at both the RNA and protein levels. Recently, Giorgio et al. (75) characterized 16 LMNB1 gene duplication events in 20 ADLD families (Fig. 3). The minimum duplication required for ADLD is ∼72 kb—spanning the entire LMNB1 gene and including 9.9 kb of sequences upstream from LMNB1 and 1.8 kb downstream. The expression of the duplicated copy of the gene is equivalent to that of the other copies of the gene. In one instance, the LMNB1 duplication event involved recombination between Alu sequences, but most of the duplication events appeared to involve short (<6-bp) regions of homology upstream and downstream of LMNB1 (75). Interestingly, one family with clinical features of ADLD manifested lamin B1 overexpression without a gene duplication event, presumably from a LMNB1 regulatory abnormality (76).
FIG 3.

Genomic rearrangements in autosomal dominant leukodystrophy (ADLD) families. The modified output from the University of California, Santa Cruz (UCSC), genome browser shows LMNB1 gene duplications in 20 ADLD families (16 unique LMNB1 duplications). The duplications are marked in blue, with the exception of the BR1 duplication/inversion, which is in yellow; a “triplicated” segment is marked in green. Duplications marked with asterisks have sequence insertions at their duplication junctions and show a clustering of their centromeric (cen.) breakpoints within a 25-kb segment. The minimal critical region duplicated in ADLD of ∼75 kb is also shown. (Reprinted from reference 75 with permission of the publisher.)
IMPACT OF LAMIN B1 OVEREXPRESSION IN CULTURED CELLS AND IN DROSOPHILA AND MOUSE MODELS
Cell culture studies have shown that overexpression of B-type lamins leads to increased nuclear membrane formation and nuclear shape abnormalities (blebbing) (16, 77, 78). Padiath et al. (16) showed that lamin B1 overexpression in neurons or glia of Drosophila results in lethality, while overexpression in the eye leads to a degenerative abnormality. Subsequent studies by Lin and Fu (72) showed that overexpression of lamin B1 in oligodendrocyte cell lines results in an arrest of oligodendrocyte differentiation along with reduced expression of oligodendrocyte markers.
Recently, Heng et al. (79) examined the effects of lamin B1 overexpression by creating transgenic mice with a 177-kb bacterial artificial chromosome (BAC) clone spanning the Lmnb1 gene. The transgenic mice manifested cognitive abnormalities, as judged by the Morris water maze test, and progressive motor impairment, as judged by rotarod and balance beam tests. Electroencephalography uncovered frequent spontaneous seizures (79). Myelination defects were not apparent in 12-month-old BAC transgenic mice but were evident by 24 months of age, as judged by electron microscopy. The ultrastructural defects consisted mainly of “outfoldings, extensions, and invaginations” in the myelin sheath; axon disintegration was also observed. Heng et al. (79) also created transgenic mice that overexpressed lamin B1 in oligodendrocytes. Those mice developed a rapidly progressive motor abnormality leading to death by 12 months of age. The mice also had seizure activity. These abnormalities were accompanied by evidence of demyelination and axonal degeneration (79). Lamin B1 overexpression in oligodendrocytes also led to reduced expression of the major myelin protein, proteolipid protein (PLP), at both the protein and RNA levels (79). Reduced expression of PLP was attributed to reduced binding of a transcriptional activator, Yin Yang 1, to the PLP promoter.
THE IMPORTANCE OF miR-23 FOR LAMIN B1 REGULATION AND MYELIN FORMATION
LMNB1 is predicted to contain multiple binding sites for microRNA 23 (miR-23), which is among the most highly expressed microRNAs in oligodendrocytes. miR-23 overexpression led to reduced lamin B1 expression in reporter assays as well as reduced amounts of lamin B1 protein in cells, as judged by Western blotting. Lentiviral transduction of miR-23 into primary glial cultures led to increased expression of oligodendrocyte markers, whereas lamin B1 expression had the opposite effects (72). Of note, miR-23 expression appeared to mitigate the oligodendrocyte maturation defects elicited by lamin B1 overexpression. The impact of miR-23 on oligodendrocyte differentiation and myelin formation was recently tested in transgenic mice that overexpress miR-23a driven by an oligodendrocyte-specific promoter (80, 81). Interestingly, the miR-23 transgenic mice manifested a variety of neurologic abnormalities accompanied by increased myelination in the corpus callosum and increased expression of myelin-specific proteins (80). Electron microscopy revealed hypermyelination of axons and other abnormalities in myelin formation (80).
HGPS AND miR-9 REGULATION OF PRELAMIN A EXPRESSION IN THE BRAIN
Hutchinson-Gilford progeria syndrome (HGPS) is a pediatric progeroid syndrome characterized by multiple disease phenotypes resembling premature aging (e.g., thin skin, osteoporosis, alopecia, and atherosclerotic coronary heart disease) (82). However, some common phenotypes with normal aging, for example, senile dementia, are absent. For this reason, HGPS and other progeroid syndromes are often referred to as “segmental aging syndromes” (83–85).
HGPS is caused by a de novo point mutation in exon 11 of LMNA that alters mRNA splicing and leads to the synthesis of a mutant prelamin A (progerin) containing an internal deletion of 50 amino acids (19, 86). Lamin C synthesis is not affected. The internal deletion does not affect prelamin A's CaaX motif; hence, progerin is farnesylated and methylated (25, 87–89). However, the site for the subsequent ZMPSTE24 cleavage step (the step that would ordinarily release mature lamin A) is eliminated by the internal deletion; hence, progerin retains its farnesyl lipid anchor. Progerin causes misshapen nuclei in cultured cells and is solely responsible for the disease phenotypes of HGPS. Lmna knock-in mice that synthesize progerin have many of the same phenotypes as those found in children with HGPS (8, 10, 90).
For many years, the absence of primary neurological disease in HGPS was a mystery. Jung et al. (18) hypothesized that the explanation might be straightforward—that the level of prelamin A/lamin A expression in the brain might simply be lower than in other tissues. Indeed, surveys of nuclear lamin expression in different tissues supported this explanation. In most tissues, the amounts of lamin A and lamin C are roughly equivalent, but the brain produces mainly lamin C and little lamin A (18). By immunohistochemistry, lamin C is expressed at high levels in neurons and glia of the mouse brain, while lamin A expression in the brain is virtually absent—except in capillary endothelial cells and the meningeal cells (18). The findings were similar at the RNA level—high levels of lamin C transcripts and low levels of prelamin A transcripts. Most scientists would have wagered that the distinct pattern of lamin A/lamin C expression in the brain was caused by alternative splicing, but studies with “lamin A-only” knock-in mice (30) showed that this was not the case. The targeted mutation in the lamin A-only mice eliminates lamin C splicing; all of the output from Lmna in these mice is channeled into prelamin A transcripts. The lamin A-only mice produced large amounts of lamin A in peripheral tissues, but lamin A expression was negligible in the brain (18). Similarly, progerin-only knock-in mice (where all of the output of Lmna is channeled into progerin transcripts) produced large amounts of progerin in peripheral tissues but only trace amounts in the brain (18). These findings eliminated alternative splicing as a potential explanation for low levels of prelamin A/lamin A expression in the brain. Subsequent studies showed that prelamin A expression in the brain is regulated by miR-9, which is expressed at high levels in the brain (18). miR-9 binds to a single site in prelamin A's 3′ untranslated region (UTR) and reduces prelamin A expression. When the miR-9 binding site is mutated, miR-9 has no effect on prelamin A expression. Overexpression of miR-9 in fibroblasts or HeLa cells reduces levels of prelamin A transcripts and lamin A protein but has no effect on lamin C (Fig. 4A) (18). Nissan et al. (91) reported similar findings and also showed that miR-9 regulation of prelamin A occurs in neurons generated from induced pluripotent stem cells.
FIG 4.
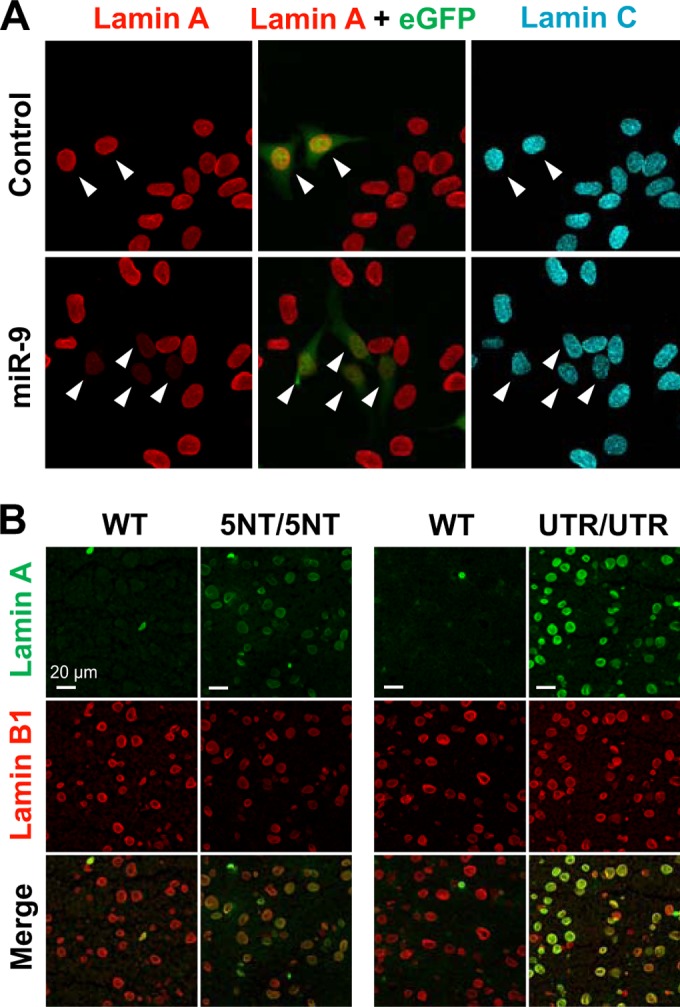
Downregulation of lamin A expression by miR-9. (A) Immunofluorescence microscopy of HeLa cells transfected with an empty vector (control) or a miR-9 expression vector. Transfected cells could be identified by enhanced green fluorescent protein (eGFP) expression (arrowheads). (Reprinted from reference 18 with permission of the publisher.) (B) Immunofluorescence microscopy of the cerebral cortex of LmnaPLAO-5NT/PLAO-5NT (5NT/5NT), LmnaPLAO-UTR/PLAO-UTR (UTR/UTR), and wild-type (WT) littermates, stained with antibodies against lamin A (green) and lamin B1 (red). Scale bar, 20 μm. (Reprinted from reference 17 with permission of the publisher.)
The regulation of prelamin A expression by a brain-specific microRNA suggested a possible explanation for the absence of neurodegenerative disease in HGPS. However, there were legitimate questions about the in vivo significance of the cell culture studies, mainly because microRNA regulation of target transcripts can be context dependent (92–95). Another issue was whether other prelamin A sequences, aside from the miR-9 binding site, might be relevant to the regulation of prelamin A. To assess the in vivo relevance of miR-9-mediated prelamin A regulation in the brain, Jung et al. (17) created two Lmna knock-in mouse lines with mutations in prelamin A's 3′ UTR. One knock-in line, Lmna5NT/5NT, contained a 5-nucleotide (5NT) mutation in the miR-9 binding site in prelamin A's 3′ UTR. In the other knock-in line (LmnaUTR/UTR), prelamin A's 3′ UTR was replaced with lamin C's 3′ UTR. The levels of prelamin A transcripts and lamin A protein in the brain of Lmna5NT/5NT and LmnaUTR/UTR mice were far higher than in control mice (17). By immunohistochemistry, cortical neurons in Lmna5NT/5NT and LmnaUTR/UTR mice contained large amounts of lamin A (Fig. 4B) (17). In contrast, lamin A expression in the brain of control mice was mainly confined to capillary endothelial cells. Interestingly, the expression of lamin A in the brain was higher in LmnaUTR/UTR mice than in Lmna5NT/5NT mice (17), implying that additional 3′ UTR sequences (aside from the miR-9 site) play a role in regulating prelamin A expression. The existence of additional regulatory sequences is not particularly surprising because large segments of prelamin A's 3′ UTR—and not just the miR-9 binding site—have been conserved in mammalian evolution.
The Lmna5NT/5NT and LmnaUTR/UTR models provided proof that miR-9 has a crucial role in regulating prelamin A in the mammalian brain. As yet, however, the “physiologic rationale” for the preferential synthesis of lamin C in the brain is not clear. The Lmna5NT/5NT and LmnaUTR/UTR mice produced large amounts of prelamin A/lamin A in the brain, and yet there was no obvious neuropathology or behavioral abnormality in those mice. Thus, the explanation for the preferential synthesis of lamin C in neurons remains elusive. One possibility is that lamin C is better suited for interacting with the spectrum of nuclear envelope proteins that are expressed in neurons and glia (17). Another possibility is that lamin A expression in the brain, while innocuous in laboratory mice, is poorly suited to extenuating situations, for example, in the setting of injury or metabolic stress (17). It is also possible that prelamin A has subtle adverse effects on the function of B-type lamins.
In our opinion, understanding the “rationale” for the preferential expression of lamin C in the brain will require a far better understanding of the biochemical properties of lamins A and C and their binding partners within the nucleus. Also, while miR-9 plays a key role in lamin A expression and helps to explain the absence of neurodegenerative disease in HGPS, this argument is not entirely complete, simply because we still do not know whether progerin—if it were to be expressed in neurons—would be toxic to neurons or glia. Other cell types, for example, hepatocytes, produce abundant amounts of progerin and yet manifest little or no toxicity. In the future, it would be worthwhile determining whether progerin expression would be toxic in the brain, and if so, which cell types would be most affected.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants (HL86683 [L.G.F.], HL089781 [L.G.F.], AG035626 [S.G.Y.], and 5T32HL007895-15 [J.M.L.]).
Footnotes
Published ahead of print 19 May 2014
REFERENCES
- 1.Muchir A, Worman HJ. 2004. The nuclear envelope and human disease. Physiology (Bethesda) 19:309–314. 10.1152/physiol.00022.2004 [DOI] [PubMed] [Google Scholar]
- 2.Worman HJ, Bonne G. 2007. “Laminopathies”: a wide spectrum of human diseases. Exp. Cell Res. 313:2121–2133. 10.1016/j.yexcr.2007.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Worman HJ, Fong LG, Muchir A, Young SG. 2009. Laminopathies and the long strange trip from basic cell biology to therapy. J. Clin. Invest. 119:1825–1836. 10.1172/JCI37679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. 2006. Nuclear lamins: laminopathies and their role in premature ageing. Physiol. Rev. 86:967–1008. 10.1152/physrev.00047.2005 [DOI] [PubMed] [Google Scholar]
- 5.Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. 2005. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 6:21–31. 10.1038/nrm1550 [DOI] [PubMed] [Google Scholar]
- 6.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. 2008. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22:832–853. 10.1101/gad.1652708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke B, Stewart CL. 2013. The nuclear lamins: flexibility in function. Nat. Rev. Mol. Cell Biol. 14:13–24. 10.1038/nrm3488 [DOI] [PubMed] [Google Scholar]
- 8.Young SG, Fong LG, Michaelis S. 2005. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res. 46:2531–2558. 10.1194/jlr.R500011-JLR200 [DOI] [PubMed] [Google Scholar]
- 9.Davies BS, Coffinier C, Yang SH, Barnes RH, II, Jung HJ, Young SG, Fong LG. 2011. Investigating the purpose of prelamin A processing. Nucleus 2:4–9. 10.4161/nucl.2.1.13723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young SG, Yang SH, Davies BS, Jung HJ, Fong LG. 2013. Targeting protein prenylation in progeria. Sci. Transl. Med. 5:171ps173. 10.1126/scitranslmed.3005229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies BS, Fong LG, Yang SH, Coffinier C, Young SG. 2009. The posttranslational processing of prelamin A and disease. Annu. Rev. Genomics Hum. Genet. 10:153–174. 10.1146/annurev-genom-082908-150150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coffinier C, Chang SY, Nobumori C, Tu Y, Farber EA, Toth JI, Fong LG, Young SG. 2010. Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc. Natl. Acad. Sci. U. S. A. 107:5076–5081. 10.1073/pnas.0908790107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coffinier C, Fong LG, Young SG. 2010. LINCing lamin B2 to neuronal migration: growing evidence for cell-specific roles of B-type lamins. Nucleus 1:407–411. 10.4161/nucl.1.5.12830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coffinier C, Jung HJ, Nobumori C, Chang S, Tu Y, Barnes RH, II, Yoshinaga Y, de Jong PJ, Vergnes L, Reue K, Fong LG, Young SG. 2011. Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol. Biol. Cell 22:4683–4693. 10.1091/mbc.E11-06-0504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Padiath QS, Fu YH. 2010. Autosomal dominant leukodystrophy caused by lamin B1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol. 98:337–357. 10.1016/S0091-679X(10)98014-X [DOI] [PubMed] [Google Scholar]
- 16.Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. 2006. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 38:1114–1123. 10.1038/ng1872 [DOI] [PubMed] [Google Scholar]
- 17.Jung HJ, Tu Y, Yang SH, Tatar A, Nobumori C, Wu D, Young SG, Fong LG. 2014. New Lmna knock-in mice provide a molecular mechanism for the ‘segmental aging' in Hutchinson-Gilford progeria syndrome. Hum. Mol. Genet. 23:1506–1515. 10.1093/hmg/ddt537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BS, Yang SH, Barnes RH, II, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG. 2012. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. U. S. A. 109:E423–E431. 10.1073/pnas.1111780109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. 2003. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423:293–298. 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin F, Worman HJ. 1993. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 268:16321–16326 [PubMed] [Google Scholar]
- 21.Lin F, Worman HJ. 1995. Structural organization of the human gene (LMNB1) encoding nuclear lamin B1. Genomics 27:230–236. 10.1006/geno.1995.1036 [DOI] [PubMed] [Google Scholar]
- 22.Biamonti G, Giacca M, Perini G, Contreas G, Zentilin L, Weighardt F, Guerra M, Della Valle G, Saccone S, Riva S, Falaschi A. 1992. The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S-phase. Mol. Cell. Biol. 12:3499–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. 2006. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 281:25768–25780. 10.1074/jbc.M513511200 [DOI] [PubMed] [Google Scholar]
- 24.Ji JY, Lee RT, Vergnes L, Fong LG, Stewart CL, Reue K, Young SG, Zhang Q, Shanahan CM, Lammerding J. 2007. Cell nuclei spin in the absence of lamin B1. J. Biol. Chem. 282:20015–20026. 10.1074/jbc.M611094200 [DOI] [PubMed] [Google Scholar]
- 25.Corrigan DP, Kuszczak D, Rusinol AE, Thewke DP, Hrycyna CA, Michaelis S, Sinensky MS. 2005. Prelamin A endoproteolytic processing in vitro by recombinant ZMPSTE24. Biochem. J. 387:129–138. 10.1042/BJ20041359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, van Bruggen N, Carano RAD, Michaelis S, Griffey SM, Young SG. 2002. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. U. S. A. 99:13049–13054. 10.1073/pnas.192460799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pendás AM, Zhou Z, Cadiñanos J, Freije JMP, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodríguez F, Tryggvason K, Lopéz-Otín C. 2002. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 31:94–99. 10.1038/ng871 [DOI] [PubMed] [Google Scholar]
- 28.Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. 1994. The processing pathway of prelamin A. J. Cell Sci. 107:61–67 [DOI] [PubMed] [Google Scholar]
- 29.Barrowman J, Hamblet C, George CM, Michaelis S. 2008. Analysis of prelamin A biogenesis reveals the nucleus to be a CaaX processing compartment. Mol. Biol. Cell 19:5398–5408. 10.1091/mbc.E08-07-0704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coffinier C, Jung HJ, Li Z, Nobumori C, Yun UJ, Farber EA, Davies BS, Weinstein MM, Yang SH, Lammerding J, Farahani JN, Bentolila LA, Fong LG, Young SG. 2010. Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J. Biol. Chem. 285:20818–20826. 10.1074/jbc.M110.128835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies BS, Barnes RH, II, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG. 2010. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum. Mol. Genet. 19:2682–2694. 10.1093/hmg/ddq158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart C, Burke B. 1987. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell 51:383–392. 10.1016/0092-8674(87)90634-9 [DOI] [PubMed] [Google Scholar]
- 33.Eckersley-Maslin MA, Bergmann JH, Lazar Z, Spector DL. 2013. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 4:53–60. 10.4161/nucl.23384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Röber RA, Weber K, Osborn M. 1989. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105:365–378 [DOI] [PubMed] [Google Scholar]
- 35.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. 1999. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147:913–919. 10.1083/jcb.147.5.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jahn D, Schramm S, Schnolzer M, Heilmann CJ, de Koster CG, Schutz W, Benavente R, Alsheimer M. 2012. A truncated lamin A in the Lmna -/- mouse line: implications for the understanding of laminopathies. Nucleus 3:463–474. 10.4161/nucl.21676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, Herrmann H, Blum H, Engelkamp D, Stewart CL, Leonhardt H, Joffe B. 2013. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152:584–598. 10.1016/j.cell.2013.01.009 [DOI] [PubMed] [Google Scholar]
- 38.Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, Cote N, Gavino B, Qiao X, Chang SY, Young SR, Yang SH, Stewart CL, Lee RT, Bennett CF, Bergo MO, Young SG. 2006. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Invest. 116:743–752. 10.1172/JCI27125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moir RD, Montag-Lowy M, Goldman RD. 1994. Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J. Cell Biol. 125:1201–1212. 10.1083/jcb.125.6.1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, Goldman RD. 2008. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 22:3409–3421. 10.1101/gad.1735208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang CW, Maya-Mendoza A, Martin C, Zeng K, Chen S, Feret D, Wilson SA, Jackson DA. 2008. The integrity of a lamin-B1-dependent nucleoskeleton is a fundamental determinant of RNA synthesis in human cells. J. Cell Sci. 121:1014–1024. 10.1242/jcs.020982 [DOI] [PubMed] [Google Scholar]
- 42.Tsai MY, Wang S, Heidinger JM, Shumaker DK, Adam SA, Goldman RD, Zheng Y. 2006. A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science 311:1887–1893. 10.1126/science.1122771 [DOI] [PubMed] [Google Scholar]
- 43.Belmont AS, Zhai Y, Thilenius A. 1993. Lamin B distribution and association with peripheral chromatin revealed by optical sectioning and electron microscopy tomography. J. Cell Biol. 123:1671–1685. 10.1083/jcb.123.6.1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. 2001. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 114(Pt 24):4557–4565 [DOI] [PubMed] [Google Scholar]
- 45.Yang SH, Chang SY, Yin L, Tu Y, Hu Y, Yoshinaga Y, de Jong PJ, Fong LG, Young SG. 2011. An absence of both lamin B1 and lamin B2 in keratinocytes has no effect on cell proliferation or the development of skin and hair. Hum. Mol. Genet. 20:3537–3544. 10.1093/hmg/ddr266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang SH, Jung HJ, Coffinier C, Fong LG, Young SG. 2011. Are B-type lamins essential in all mammalian cells? Nucleus 2:562–569. 10.4161/nucl.2.6.18085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim Y, Zheng X, Zheng Y. 2013. Proliferation and differentiation of mouse embryonic stem cells lacking all lamins. Cell Res. 23:1420–1423. 10.1038/cr.2013.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gupta A, Tsai LH, Wynshaw-Boris A. 2002. Life is a journey: a genetic look at neocortical development. Nat. Rev. Genet. 3:342–355. 10.1038/nrg799 [DOI] [PubMed] [Google Scholar]
- 49.Wynshaw-Boris A. 2007. Lissencephaly and LIS1: insights into the molecular mechanisms of neuronal migration and development. Clin. Genet. 72:296–304. 10.1111/j.1399-0004.2007.00888.x [DOI] [PubMed] [Google Scholar]
- 50.Gambello MJ, Hirotsune S, Wynshaw-Boris A. 1999. Murine modelling of classical lissencephaly. Neurogenetics 2:77–86. 10.1007/s100480050056 [DOI] [PubMed] [Google Scholar]
- 51.Kim Y, Sharov AA, McDole K, Cheng M, Hao H, Fan CM, Gaiano N, Ko MS, Zheng Y. 2011. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 334:1706–1710. 10.1126/science.1211222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solecki DJ, Govek EE, Tomoda T, Hatten ME. 2006. Neuronal polarity in CNS development. Genes Dev. 20:2639–2647. 10.1101/gad.1462506 [DOI] [PubMed] [Google Scholar]
- 53.Wynshaw-Boris A, Gambello MJ. 2001. LIS1 and dynein motor function in neuronal migration and development. Genes Dev. 15:639–651. 10.1101/gad.886801 [DOI] [PubMed] [Google Scholar]
- 54.Vallee RB, Tsai JW. 2006. The cellular roles of the lissencephaly gene LIS1, and what they tell us about brain development. Genes Dev. 20:1384–1393. 10.1101/gad.1417206 [DOI] [PubMed] [Google Scholar]
- 55.Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. 2006. Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 172:41–53. 10.1083/jcb.200509124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X, Lei K, Yuan X, Wu X, Zhuang Y, Xu T, Xu R, Han M. 2009. SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron 64:173–187. 10.1016/j.neuron.2009.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K. 2004. Lamin B1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. U. S. A. 101:10428–10433. 10.1073/pnas.0401424101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Young SG, Jung HJ, Coffinier C, Fong LG. 2012. Understanding the roles of nuclear A- and B-type lamins in brain development. J. Biol. Chem. 287:16103–16110. 10.1074/jbc.R112.354407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patterson K, Molofsky AB, Robinson C, Acosta S, Cater C, Fischer JA. 2004. The functions of Klarsicht and nuclear lamin in developmentally regulated nuclear migrations of photoreceptor cells in the Drosophila eye. Mol. Biol. Cell 15:600–610. 10.1091/mbc.E03-06-0374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harada T, Swift J, Irianto J, Shin JW, Spinler KR, Athirasala A, Diegmiller R, Dingal PC, Ivanovska IL, Discher DE. 2014. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 204:669–682. 10.1083/jcb.201308029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swift J, Harada T, Buxboim A, Shin JW, Tang HY, Speicher DW, Discher DE. 2013. Label-free mass spectrometry exploits dozens of detected peptides to quantify lamins in wildtype and knockdown cells. Nucleus 4:450–459. 10.4161/nucl.27413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE. 2013. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341:1240104. 10.1126/science.1240104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rowat AC, Jaalouk DE, Zwerger M, Ung WL, Eydelnant IA, Olins DE, Olins AL, Herrmann H, Weitz DA, Lammerding J. 2013. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J. Biol. Chem. 288:8610–8618. 10.1074/jbc.M112.441535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Friedl P, Wolf K, Lammerding J. 2011. Nuclear mechanics during cell migration. Curr. Opin. Cell Biol. 23:55–64. 10.1016/j.ceb.2010.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dahl KN, Engler AJ, Pajerowski JD, Discher DE. 2005. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophys. J. 89:2855–2864. 10.1529/biophysj.105.062554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson A, Partridge D, Malhas A, De Castro SC, Gustavsson P, Thompson DN, Vaux DJ, Copp AJ, Stanier P, Bassuk AG, Greene ND. 2013. Is LMNB1 a susceptibility gene for neural tube defects in humans? Birth Defects Res. A Clin. Mol. Teratol. 97:398–402. 10.1002/bdra.23141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Castro SC, Malhas A, Leung KY, Gustavsson P, Vaux DJ, Copp AJ, Greene ND. 2012. Lamin b1 polymorphism influences morphology of the nuclear envelope, cell cycle progression, and risk of neural tube defects in mice. PLoS Genet. 8:e1003059. 10.1371/journal.pgen.1003059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jung HJ, Nobumori C, Goulbourne CN, Tu Y, Lee JM, Tatar A, Wu D, Yoshinaga Y, de Jong PJ, Coffinier C, Fong LG, Young SG. 2013. Farnesylation of lamin B1 is important for retention of nuclear chromatin during neuronal migration. Proc. Natl. Acad. Sci. U. S. A. 110:E1923–E1932. 10.1073/pnas.1303916110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee JM, Tu Y, Tatar A, Wu D, Nobumori C, Jung HJ, Yoshinaga Y, Coffinier C, de Jong PJ, Fong LG, Young SG. 26 March 2014. Reciprocal knock-in mice to investigate the functional redundancy of lamin B1 and lamin B2. Mol. Biol. Cell 10.1091/mbc.E14-01-0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eldridge R, Anayiotos CP, Schlesinger S, Cowen D, Bever C, Patronas N, McFarland H. 1984. Hereditary adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. N. Engl. J. Med. 311:948–953. 10.1056/NEJM198410113111504 [DOI] [PubMed] [Google Scholar]
- 71.Melberg A, Hallberg L, Kalimo H, Raininko R. 2006. MR characteristics and neuropathology in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. Am. J. Neuroradiol. 27:904–911 http://www.ajnr.org/content/27/4/904.long [PMC free article] [PubMed] [Google Scholar]
- 72.Lin ST, Fu YH. 2009. miR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis. Model. Mech. 2:178–188. 10.1242/dmm.001065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coffeen CM, McKenna CE, Koeppen AH, Plaster NM, Maragakis N, Mihalopoulos J, Schwankhaus JD, Flanigan KM, Gregg RG, Ptacek LJ, Fu YH. 2000. Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum. Mol. Genet. 9:787–793. 10.1093/hmg/9.5.787 [DOI] [PubMed] [Google Scholar]
- 74.Marklund L, Melin M, Melberg A, Giedraitis V, Dahl N. 2006. Adult-onset autosomal dominant leukodystrophy with autonomic symptoms restricted to 1.5 Mbp on chromosome 5q23. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141:608–614. 10.1002/ajmg.b.30342 [DOI] [PubMed] [Google Scholar]
- 75.Giorgio E, Rolyan H, Kropp L, Chakka AB, Yatsenko S, Gregorio ED, Lacerenza D, Vaula G, Talarico F, Mandich P, Toro C, Pierre EE, Labauge P, Capellari S, Cortelli P, Vairo FP, Miguel D, Stubbolo D, Marques LC, Gahl W, Boespflug-Tanguy O, Melberg A, Hassin-Baer S, Cohen OS, Pjontek R, Grau A, Klopstock T, Fogel B, Meijer I, Rouleau G, Bouchard JP, Ganapathiraju M, Vanderver A, Dahl N, Hobson G, Brusco A, Brussino A, Padiath QS. 2013. Analysis of LMNB1 duplications in autosomal dominant leukodystrophy provides insights into duplication mechanisms and allele-specific expression. Hum. Mutat. 34:1160–1171. 10.1002/humu.22348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brussino A, Vaula G, Cagnoli C, Panza E, Seri M, Di Gregorio E, Scappaticci S, Camanini S, Daniele D, Bradac GB, Pinessi L, Cavalieri S, Grosso E, Migone N, Brusco A. 2010. A family with autosomal dominant leukodystrophy linked to 5q23.2–q23.3 without lamin B1 mutations. Eur. J. Neurol. 17:541–549. 10.1111/j.1468-1331.2009.02844.x [DOI] [PubMed] [Google Scholar]
- 77.Ralle T, Grund C, Franke WW, Stick R. 2004. Intranuclear membrane structure formations by CaaX-containing nuclear proteins. J. Cell Sci. 117:6095–6104. 10.1242/jcs.01528 [DOI] [PubMed] [Google Scholar]
- 78.Favreau C, Dubosclard E, Ostlund C, Vigoroux C, Capeau J, Wehnert M, Higuet D, Worman H, Courvalin J-C, Buendia B. 2003. Expression of lamin A mutated in the carboxyl-terminal tail generates an aberrant nuclear phenotype similar to that observed in cells from patients with Dunnigan-type partial lipodystrophy and Emery-Dreifuss muscular dystrophy. Exp. Cell Res. 282:14–23. 10.1006/excr.2002.5669 [DOI] [PubMed] [Google Scholar]
- 79.Heng MY, Lin ST, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptacek LJ, Fu YH. 2013. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J. Clin. Invest. 123:2719–2729. 10.1172/JCI66737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin ST, Huang Y, Zhang L, Heng MY, Ptacek LJ, Fu YH. 2013. MicroRNA-23a promotes myelination in the central nervous system. Proc. Natl. Acad. Sci. U. S. A. 110:17468–17473. 10.1073/pnas.1317182110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin ST, Heng MY, Ptacek LJ, Fu YH. 2014. Regulation of Myelination in the central nervous system by nuclear lamin B1 and non-coding RNAs. Transl. Neurodegener. 3:4. 10.1186/2047-9158-3-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DeBusk FL. 1972. The Hutchinson-Gilford progeria syndrome. J. Pediatr. 80:697–724. 10.1016/S0022-3476(72)80229-4 [DOI] [PubMed] [Google Scholar]
- 83.Navarro CL, Cau P, Levy N. 2006. Molecular bases of progeroid syndromes. Hum. Mol. Genet. 15(Spec No 2):R151–R161. 10.1093/hmg/ddl214 [DOI] [PubMed] [Google Scholar]
- 84.Martin GM. 1985. Genetics and aging; the Werner syndrome as a segmental progeroid syndrome. Adv. Exp. Med. Biol. 190:161–170. 10.1007/978-1-4684-7853-2_5 [DOI] [PubMed] [Google Scholar]
- 85.Martin GM. 1982. Syndromes of accelerated aging. Natl. Cancer Inst. Monogr. 60:241–247 [PubMed] [Google Scholar]
- 86.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Lévy N. 2003. Lamin A truncation in Hutchinson-Gilford progeria. Science 300:2055. 10.1126/science.1084125 [DOI] [PubMed] [Google Scholar]
- 87.Yang SH, Andres DA, Spielmann HP, Young SG, Fong LG. 2008. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J. Clin. Invest. 118:3291–3300. 10.1172/JCI35876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang SH, Chang SY, Andres DA, Spielmann HP, Young SG, Fong LG. 2010. Assessing the efficacy of protein farnesyltransferase inhibitors in mouse models of progeria. J. Lipid Res. 51:400–405. 10.1194/jlr.M002808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fong LG, Vickers TA, Farber EA, Choi C, Yun UJ, Hu Y, Yang SH, Coffinier C, Lee R, Yin L, Davies BS, Andres DA, Spielmann HP, Bennett CF, Young SG. 2009. Activating the synthesis of progerin, the mutant prelamin A in Hutchinson-Gilford progeria syndrome, with antisense oligonucleotides. Hum. Mol. Genet. 18:2462–2471. 10.1093/hmg/ddp184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. 2006. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Invest. 116:2115–2121. 10.1172/JCI28968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nissan X, Blondel S, Navarro C, Maury Y, Denis C, Girard M, Martinat C, De Sandre-Giovannoli A, Levy N, Peschanski M. 2012. Unique preservation of neural cells in Hutchinson-Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep. 2:1–9. 10.1016/j.celrep.2012.05.015 [DOI] [PubMed] [Google Scholar]
- 92.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bushati N, Cohen SM. 2007. microRNA functions. Annu. Rev. Cell Dev. Biol. 23:175–205. 10.1146/annurev.cellbio.23.090506.123406 [DOI] [PubMed] [Google Scholar]
- 94.Gao FB. 2010. Context-dependent functions of specific microRNAs in neuronal development. Neural Dev. 5:25. 10.1186/1749-8104-5-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Coolen M, Bally-Cuif L. 2009. MicroRNAs in brain development and physiology. Curr. Opin. Neurobiol. 19:461–470. 10.1016/j.conb.2009.09.006 [DOI] [PubMed] [Google Scholar]