

Abstract
Caveolae are specialized microdomains on membranes that are critical for signal transduction, cholesterol transport, and endocytosis. Caveolin-1 (CAV1) is a multifunctional protein and a major component of caveolae. Cav1 is directly activated by hypoxia-inducible factor (HIF). HIFs are heterodimers of an oxygen-sensitive α subunit, HIF1α or HIF2α, and a constitutively expressed β subunit, aryl hydrocarbon receptor nuclear translocator (ARNT). Whole-genome expression analysis demonstrated that Cav1 is highly induced in mouse models of constitutively activated HIF signaling in the intestine. Interestingly, Cav1 was increased only in the colon and not in the small intestine. Currently, the mechanism and role of HIF induction of CAV1 in the colon are unclear. In mouse models, mice that overexpressed HIF1α or HIF2α specifically in intestinal epithelial cells demonstrated an increase in Cav1 gene expression in the colon but not in the duodenum, jejunum, or ileum. HIF2α activated the Cav1 promoter in a HIF response element-independent manner. myc-associated zinc finger (MAZ) protein was essential for HIF2α activation of the Cav1 promoter. Hypoxic induction of CAV1 in the colon was essential for intestinal barrier integrity by regulating occludin expression. This may provide an additional mechanism by which chronic hypoxia can activate intestinal inflammation.
INTRODUCTION
Tissue hypoxia is caused by inadequate oxygen delivery, leading to activation of hypoxia-inducible factor (HIF). A large set of HIF target genes has been identified as being critical in the control of metabolism, cell proliferation/survival, angiogenesis, and iron uptake (1). HIF is a heterodimer of an oxygen-sensitive α subunit, HIF1α, HIF2α, or HIF3α, and a constitutively expressed β subunit, aryl hydrocarbon receptor nuclear translocator (ARNT) (2–4). Hypoxic signaling plays important physiological and pathological roles in the intestine (5). HIF signaling is essential for maintaining iron homeostasis during iron deprivation, regulating the intestinal inflammatory response in colitis, and increasing colon cancer progression. Studies assessing the overlapping and distinct roles of HIF1α and HIF2α demonstrate that HIF1α protects the epithelial barrier from acute and chronic intestinal inflammation by activating expression of a large set of barrier protective genes, such as CD55, intestinal trefoil factors, MUC3, and CD73 (6–8). Disruption of intestinal epithelial HIF1α leads to increased injury and inflammation, whereas activation of HIF signaling decreases injury and inflammation in acute models of colitis (9–12). In contrast, disruption of HIF2α decreases the intestinal inflammatory response in acute models of colitis, while chronic activation of HIF2α in the intestine leads to spontaneous colitis (13, 14). Moreover, activation of HIF2α markedly potentiates experimental colon carcinogenesis (15, 16). Contrary to HIF1α, the mechanisms by which HIF2α increases intestinal inflammation remain unclear.
Whole-genome mRNA expression analysis identified caveolin-1 (CAV1) to be highly induced in intestinal epithelial cells of mice that overexpress HIF (16). CAV1 is the major structural protein in the formation of caveolae, which are small (50- to 100-nm) flask-shaped membrane structures on the plasma membrane (17). Caveolae are present in most mammalian cell types and play important physiological roles in regulating diverse biological processes, including signaling transduction and endocytosis (18, 19). CAV1 is a critical homeostatic protein in inflammation and cancer. Several studies have demonstrated that disruption of CAV1 can increase the inflammatory response and cancer progression (20–22). On the contrary, other studies have suggested that disruption of CAV1 reduces inflammation and cancer progression (22, 23). A recent study demonstrated CAV1 to be a direct HIF2α target gene and serve a critical role in hypoxia-induced cell growth and epidermal growth factor receptor (EGFR) signaling in renal carcinoma-derived cell lines (24). However, the functional role and precise mechanism of regulation of CAV1 following HIF activation in the intestine have not been determined. CAV1 is specifically upregulated in the colon in a HIF2α-dependent manner. However, activation of HIF2α in the small intestine did not increase CAV1 expression. The data from the present study demonstrated that HIF2α activates Cav1 expression through a noncanonical mechanism dependent on myc-associated zinc finger (MAZ) protein. Interestingly, unlike what was observed in renal carcinoma cell lines, hypoxia-induced CAV1 did not regulate EGFR or cell proliferation. Increased expression of CAV1 led to a decrease in occludin expression, thus revealing a possible role for HIF2α in regulating barrier function and intestinal inflammation.
MATERIALS AND METHODS
Animal experiments.
Vhlflox/flox, VhlΔIE (an intestine-specific disruption of Vhl), VhlΔIE/Hif1αΔIE, VhlΔIE/Hif2αΔIE, Hif1αLSL/LSL, Hif2αLSL/LSL, and Hif2αLSL/+ mice were previously described (13, 25, 26). For methyl-β-cyclodextrin (MβCD; Sigma, St. Louis, MO) treatment, the mice were gavaged or intrarectally treated with 200 μl of 10 mM MβCD or saline vehicle for 6 h. Colon permeability was assessed with fluorescein isothiocyanate (FITC)-labeled dextran. Mice were either untreated, treated with dextran sulfate sodium (DSS) for 3 days, or treated with DSS for 3 days and placed back on regular drinking water for 6 days. For the infliximab experiment, mice were injected twice with 10 mg/kg of body weight infliximab (Remicade; Janssen Biotech, Inc., Horsham, PA) at 1 day and 4 h prior to the FITC-dextran assay. Mice were gavaged with 0.5 mg/g body weight of FITC-dextran for 6 h, and serum fluorescence for FITC was assessed. Ex vivo colon culture was done as described previously (16). The colons were incubated in dimethyl sulfoxide (DMSO) or MG132 (10 μM) for 1 h, and Western blot analysis was performed as described below. All mice were maintained in standard cages in a light- and temperature-controlled room and given standard chow and water ad libitum. All animal studies were carried out in accordance with the Institute of Laboratory Animal Resources guidelines under protocols approved by the Committee on the Use and Care of Animals at the University of Michigan.
Cell culture.
Human colorectal carcinoma (HCT116) cells obtained from ATCC were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Life Technologies) and 1% antibiotic-antimyotic (Life Technologies). For hypoxia experiments, HCT116 cells at 60% confluence were cultured in a hypoxia chamber flushed with premixed gas (1% O2, 5% CO2, and 94% N2).
Plasmid construction.
The 1.3-kb mouse Cav1 promoter construct was previously described (27). 5′, 3′, and mutated constructs were generated, and the sequences were verified through the University of Michigan DNA Sequencing Core. The primer sequences are listed in Table S1 in the supplemental material.
Transient-transfection and luciferase assay.
HCT116 cells were seeded into a 24-well plate at a cell density of 5 × 104 cells per well. Cav1 promoter constructs were cotransfected with the HIF1α, HIF2α, and/or MAZ expression vector by polyethylenimine (PEI; Polysciences Inc., Warrington, PA) (28). For hypoxic experiments, Cav1 promoter constructs were transfected into HCT116 cells that were cultured in a hypoxia chamber for 24 h. Cells were lysed in reporter lysis buffer (Promega, Madison, WI), and firefly luciferase activity was measured and normalized to β-galactosidase (β-Gal) activity.
Protein isolation and Western blotting.
HCT116 cells or scraped mucosal cells of the colon were lysed to obtain whole-cell extracts or membrane extracts as described previously (25, 26). Protein was resolved on 10% SDS-polyacrylamide gels and transferred to a polyvinylidene difluoride membrane. Following blocking with 3% nonfat milk, the blots were incubated with primary antibodies against GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and CAV1 (Santa Cruz Biotechnology, Santa Cruz, CA), HIF2α (Novus Biological, Littleton, CO), occludin (Life Technologies), EGFR, phospho-EGFR (pEGFR), phospho-ERK (pERK), and extracellular signal-regulated kinase 1/2 (ERK1/2) (Cell Signaling Technologies, Boston, MA).
RNA isolation and quantitative reverse transcription-PCR.
Total RNA was extracted and reverse transcribed as previously described (29). The primers used for quantitative PCR are listed in Table S1 in the supplemental material.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed with HIF2α and ARNT antibody in VhlΔIE mice and HCT116 cells treated with hypoxia for 24 h, as described before (26). The primers used for the ChIP assay are listed in Table S1 in the supplemental material.
Immunohistochemistry.
Frozen sections of the colon (Vhlflox/flox and VhlΔIE mice) were stained with antibodies for CAV1 (Santa Cruz Biotechnology), E-cadherin (E-Cad), and occludin (Life Technologies), as previously described (16).
Statistical analysis.
Statistical analysis was performed using GraphPad Prism software (La Jolla, CA). Two-way analysis of variance was used to compare the means between the control and experimental groups. The Student t test was performed to compare statistical differences between the control and experimental groups. Data are presented as means ± standard deviations (SDs).
RESULTS
Activation of HIF signaling in intestinal epithelial cells increases Cav1 expression in the colon but not the small intestine.
HIFs are regulated through an oxygen-dependent degradation domain (ODD). Under normoxia, ODD is hydroxylated by prolyl hydroxylases, followed by recognition and proteasomal degradation via the von Hippel-Lindau tumor suppressor (VHL) and E3 ubiquitination (30–32). Therefore, disruption of Vhl enables constitutive stabilization and activation of HIF under normoxia. To assess the role of HIF in regulating CAV1 expression, mice with an intestine-specific disruption of Vhl (VhlΔIE) were used (14). Cav1 mRNA expression (Fig. 1A) and CAV1 protein (Fig. 1B) were highly induced in the colons of VhlΔIE mice compared to the levels of induction in their littermate controls (Vhlflox/flox mice). Immunofluorescence staining demonstrated that the increase of CAV1 in the colons of VhlΔIE mice was from epithelial cells (Fig. 1C). The Cav1 mRNA level was not increased in the small intestine of VhlΔIE mice compared to that in their littermate controls (Fig. 1D and E).
FIG 1.

The hypoxic signaling pathway increases Cav1 expression in the colon. (A) Gene expression normalized to β-actin expression. (B) Western analysis of CAV1 in the colons of VhlΔIE and littermate control (Vhlflox/flox [VhlF/F]) mice. (C) CAV1 staining in the colons of VhlΔIE and littermate control (Vhlflox/flox) mice. The nucleus was stained with DAPI (4′,6-diamidino-2-phenylindole), and the epithelial cells were stained with E-cadherin (E-Cad) (bottom). Magnifications are indicated on the left. (D and E) Cav1 mRNA expression level in the jejunum (D) and ileum (E) of VhlΔIE mice normalized to the level of β-actin expression. Eight or nine mice were assessed per each group. Each bar represents the mean value ± SD. The results for bars with different letters are statistically significantly different (P < 0.005); N.S., not significant.
HIF2α specifically increases CAV1 expression in colon epithelial cells.
To characterize the role of HIF-dependent pathways on CAV1 expression in the colon, mice with a compound disruption of Vhl and Hif1α, Vhl and Hif2α, and Vhl and Arnt were examined. Disruption of Vhl and Hif2α (VhlΔIE/Hif2αΔIE) or Vhl and Arnt (VhlΔIE/ArntΔIE) abrogated the induction of CAV1 mRNA, whereas disruption of Vhl and Hif1α (VhlΔIE/Hif1αΔIE) had no effect on the Cav1 mRNA level compared to that in VhlΔIE mice (Fig. 2A). Disruption of Vhl also increased the CAV1 protein level, while disruption of both Vhl and Hif2α abrogated the induction (Fig. 2B). To directly assess the role of HIF1α and HIF2α in the increase of Cav1 in the colon, mice with an intestine-specific overexpression of HIF1α (Hif1αLSL/LSL mice) or HIF2α (Hif2αLSL/LSL mice) were used (13). Cav1 expression was strongly induced in the colon but not in the small intestine of Hif2αLSL/LSL mice. Intestinal overexpression of HIF1α did not activate the expression of Cav1 (Fig. 2C and D). These data clearly demonstrate that Cav1 expression is increased in a HIF2α- and cell type-dependent manner.
FIG 2.

HIF2α increases Cav1 expression in the colon. (A) Gene expression normalized to β-actin expression. (B) Western analysis of Cav1 in the colons of VhlΔIE/Hif1αΔIE, VhlΔIE/Hif2αΔIE, or VhlΔIE/ArntΔIE mice and littermate control mice (Vhlflox/flox/Hif1αflox/flox, Vhlflox/flox/Hif2αflox/flox, or Vhlflox/flox/Arntflox/flox). (C and D) Cav1 gene expression normalized to β-actin expression in the colon (C) or small intestine (D) of Hif1αLSL/LSL and Hif2αLSL/LSL mice and littermate controls (wild type [WT]). Eight or nine mice were assessed per each group. Each bar represents the mean value ± SD. The results for bars with different letters are statistically significantly different (P < 0.005); N.S., not significant.
HIF2α regulates Cav1 expression through a noncanonical pathway.
HIF1α and HIF2α directly activate genes through binding to promoters that contain a HIF response element (HRE), such as those encoding erythropoietin (33) and transferrin (34). To further understand the mechanism by which HIF2α activates Cav1 expression in the colon, a 1.3-kb Cav1 promoter luciferase reporter containing three canonical hypoxia-response elements was characterized. Colon-derived HCT116 cells were transfected with the Cav1 promoter luciferase construct and incubated under normoxia or in a hypoxia chamber containing 1% O2 for 24 h. A significant increase in luciferase activity compared to that in the transfected cells incubated under normoxia was observed (Fig. 3A). Consistent with the in vivo data, cotransfection of HIF2α under normoxic conditions robustly increased Cav1 promoter activity, whereas HIF1α did not affect Cav1 promoter activity (Fig. 3A). To delineate the role of putative HREs in HIF2α-mediated induction of Cav1 promoter activity, 5′ deletion constructs of the Cav1 promoter were generated and characterized. The HIF2α response was localized to a 114-bp region of the proximal promoter (Fig. 3B and C). This region contained a single putative HRE. Mutation of the HRE did not abolish HIF2α-mediated induction of the Cav1 promoter (Fig. 3D). However, in the absence of a functional HRE, HIF2α and ARNT were still recruited to the Cav1 promoter in the VhlΔIE mice and HCT116 cells treated with hypoxia, as assessed by chromatin immunoprecipitation (ChIP) assays (Fig. 3E to H).
FIG 3.

HIF2α activates the Cav1 promoter through an HRE-independent mechanism. (A to D) A luciferase assay was performed in HCT116 cells cotransfected with HIF1α and/or HIF2α or cultured in a hypoxia chamber with a 1.3-kb (A), 650-bp (B), 114-bp (C), or 114-bp mutated (mut) HRE (D) Cav1 reporter construct. (E and F) ChIP assays for HIF2α in VhlΔIE mice and littermate controls (E) or in HCT116 cells (F) following 24 h of hypoxia treatment. (G and H) ChIP assays for ARNT in VhlΔIE mice and littermate controls (G) or in HCT116 cells (H) following 24 h of hypoxia treatment. Promoter luciferase values were normalized to those for β-Gal, and each bar represents the mean value ± SD. The results of the ChIP analysis were normalized to the input, and each bar represents the mean value ± SD. Four mice were assessed for each group. The results for bars with different letters are statistically significantly different (P < 0.005).
myc-associated zinc finger (MAZ) transcription factor is critical for HIF2α regulation of the Cav1 promoter.
The Cav1 promoter is a TATA-less promoter containing a GC-rich sequence. Previous work demonstrated that HIF2α can activate GC-rich elements through a MAZ protein, also known as serum amyloid A-activating factor 1 (SAF1) (13). To assess if the GC-rich sequences were critical for HIF2α induction of Cav1, constructs with a deletion of the GC-rich sequence located at the 5′ end of the promoter (99-bp GC-del) and serial 3′ deletions (96 bp, 64 bp, and 24 bp) were generated (Fig. 4A). Compared to the luciferase activity of the 114-bp promoter, luciferase activity decreased with the 3′ truncations, and the basal promoter activity was completely abolished with the deletion of the GC-rich sequence (Fig. 4A), indicating that the GC-rich sequence is critical for promoter activity. 3′ deletions (96 bp, 64 bp, and 24 bp) were also generated, and the promoter constructs were cotransfected with HIF2α. The 96-bp and 24-bp promoter constructs were significantly induced to a level comparable to that of the 114-bp construct (Fig. 4B). The 64-bp promoter construct was also induced by HIF2α, but to a lesser extent. This suggested that the GC-rich sequence may be critical for HIF2α induction. To understand if the GC-rich binding transcription factor MAZ can activate the Cav1 promoter, MAZ was overexpressed and luciferase activity was measured. MAZ increased the promoter activity in the 3′-truncated constructs (Fig. 4C). To further confirm the role of MAZ in HIF2α-induced Cav1 promoter activation, HCT116 cells with MAZ knockdown by short hairpin RNA (shRNA) were examined (13). Basal and HIF2α-induced Cav1 promoter activity was significantly attenuated in the HCT116 cells with MAZ knockdown compared to that in cells with scrambled shRNA (Fig. 4D). Cotransfection of MAZ and HIF2α further induced Cav1 promoter activity compared to that in cells transfected with HIF2α or MAZ only (Fig. 4E). A novel splice variant of MAZ, SAF3, could also potentiate HIF2α-induced Cav1 promoter activity (Fig. 4E) (35). To assess if MAZ was essential for HIF2α recruitment to the Cav1 promoter, ChIP assays were assessed in MAZ knockdown or scrambled cells. Hypoxia led to the robust recruitment of HIF2α in HCT116 cells with scrambled shRNA, which was completely abrogated in cells with shRNA knockdown of MAZ (Fig. 4F). Overexpression of MAZ and HIF2α demonstrated that these transcription factors are predominantly colocalized in the nucleus (Fig. 4G). To understand if HIF2α and MAZ interact, HIF2α tagged with streptavidin binding peptide (NTAP) was cotransfected with empty vector or MAZ in HEK293T cells. Cells were precipitated with streptavidin, and Western analysis demonstrated a protein interaction between HIF2α, MAZ, and ARNT (Fig. 4H).
FIG 4.

HIF2α activates the Cav1 promoter through MAZ. (A) Luciferase (Luc) analysis of the Cav1 promoter deletion constructs. (B) Deletion analysis of the 114-bp proximal Cav1 promoter following cotransfection with HIF2α. (C) Cav1 promoter activity following cotransfection with MAZ. (D) Luciferase activity of the proximal 114-bp Cav1 promoter in MAZ knockdown and control (scrambled) HCT116 cells. (E) Luciferase activity of the 114-bp Cav1 promoter cotransfected with HIF2α in MAZ knockdown and control (scrambled) HCT116 cells. (F) ChIP assay for HIF2α in HCT116 cells with MAZ knockdown following 24 h of hypoxia treatment. (G) HIF2α and MAZ staining following cotransfection of the HIF2α and MAZ expression vector in HCT116 cells. (H) Coimmunoprecipitation (Co-IP) of HIF2α and MAZ in HEK293T cells. IP-Strp, immunoprecipitation with streptavidin. Promoter luciferase values were normalized to those for β-Gal, and each bar represents the mean value ± SD. The results of the ChIP analysis were normalized to the input, and each bar represents the mean value ± SD. The results for bars with different letters are statistically significantly different (P < 0.05); N.S., not significant.
Overexpression of HIF2α in the colon does not alter EGFR signaling or proliferation.
EGFR was shown to interact with CAV1 (36). Hypoxia upregulated CAV1 expression, which promoted ligand-independent EGFR signaling in renal cell carcinoma-derived cell lines (24). Overexpression of HIF2α in the colon did not alter the levels of phosphorylated EGFR and its downstream ERK signaling pathway (Fig. 5A). In addition, adenovirus-mediated overexpression of CAV1 did not alter cell growth in HCT116 cells (Fig. 5B and C).
FIG 5.
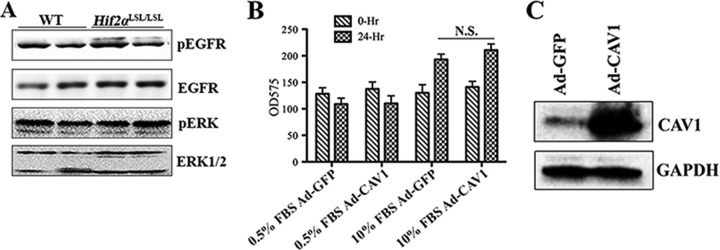
Induction of CAV1 does not increase EGFR signaling or cell growth. (A) Western analysis of phospho-EGFR (pEGFR), total EGFR, phospho-ERK (pERK), and total ERK signaling in the colons of Hif2αLSL/LSL and littermate control (wild-type) mice. (B) 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay in HCT116 cells with CAV1 overexpression in medium containing 0.5% or 10% FBS. N.S., not significant. (C) Western blot of CAV1 expression in HCT116 cells infected with adenovirus expressing CAV1. Ad-CAV1, adenovirus expressing CAV1; Ad-GFP, adenovirus expressing green fluorescent protein.
HIF2α increases colon barrier permeability.
CAV1 is critical in the regulation of occludin, a major tight-junction protein important in intestinal epithelial barrier function (37). Occludin expression was significantly decreased in the colons of VhlΔIE mice compared to that in their littermate control mice (Fig. 6A). The decrease in expression was not observed in the VhlΔIE/Hif2αΔIE mice (Fig. 6A). Decreased expression of occludin was observed on the apical surface of colonic epithelial cells from VhlΔIE mice compared to the level of expression in their littermate controls (Fig. 6B). To assess if the decrease in occludin expression correlated with a change in the barrier function, Vhlflox/flox and VhlΔIE mice were treated with regular drinking water, DSS in the drinking water for 3 days, or DSS in the drinking water for 3 days and regular drinking water for 6 days following DSS treatment. DSS treatment led to an increase in intestinal barrier permeability. The VhlΔIE mice had a decrease in basal barrier function compared to their littermate controls, which was further potentiated following DSS treatment (Fig. 6C). The increase in barrier permeability was abolished in mice with a compound disruption (Vhl and Hif2α), demonstrating a role for HIF2α in barrier permeability (Fig. 6D). Furthermore, occludin expression was decreased in Hif2αLSL/+ mice (Fig. 6E). These mice overexpressed HIF2α from a single allele and did not have overt intestinal inflammation (13). The tumor necrosis factor alpha (TNF-α) gene (Tnfa) is a direct target gene of HIF2α and can regulate occludin expression and barrier permeability (13, 37). To determine whether TNF-α is involved in the HIF2α-induced barrier defect, Hif2αLSL/+ mice were examined. Although no overt inflammation was observed, these mice still had increased Tnfa mRNA expression (13). Consistent with the decrease in occludin expression, Hif2αLSL/+ mice also demonstrated increased barrier permeability. However, the increase in intestinal permeability was not decreased following treatment with a clinically used TNF-α antagonist, infliximab (Fig. 6F).
FIG 6.

HIF2α in the colon decreased occludin expression and increased colon barrier permeability. (A) Occludin expression level in the colons of VhlΔIE and VhlΔIE/Hif2αΔIE mice. (B) Occludin staining in colon sections from VhlΔIE and Vhlflox/flox mice. (C) FITC-dextran colon barrier permeability assay in VhlΔIE and Vhlflox/flox mice which were untreated, treated with DSS for 3 days, or treated with DSS for 3 days and placed back on regular drinking water for 6 days. (D) FITC-dextran colon barrier permeability assay in untreated VhlΔIE/Hif2αΔIE and Vhlflox/flox/Hif2αflox/flox mice. (E) CAV1 and occludin expression level in the colons of Hif2αLSL/+ mice or their littermate controls (wild type). (F) FITC-dextran colon barrier permeability assay in Hif2αLSL/+ mice pretreated with infliximab. Four to six mice were assessed per each group. Each bar represents the mean value ± SD. The results for bars with different letters are statistically significantly different (P < 0.05); N.S., not significant.
CAV1 expression regulates occludin levels in the intestine of VhlΔIE mice.
TNF-α triggered inflammation-induced CAV1-mediated endocytosis of occludin and disruption of the epithelial paracellular tight junction (16, 37). Our data suggested that a constitutive increase in Cav1 expression may lead to a decrease in occludin expression. To further assess this possibility, two colon-derived cell lines, Caco-2 and HCT116, which differentially express CAV1, were assessed. Caco-2 cells have low levels of expression of CAV1 and a higher level of occludin expression than HCT116 cells (Fig. 7A). Forced expression of CAV1 in Caco-2 cells decreased occludin expression (Fig. 7B), whereas knockdown of CAV1 in HCT116 cells increased occludin expression (Fig. 7C). These data suggested that overexpression of CAV1 independently of TNF-α signaling can also regulate occludin. MβCD is a water-soluble cyclic heptasaccharide compound that decreases CAV1 expression and inhibits its endocytic shuttling of occludin (37). To understand if the decrease in occludin following HIF overexpression was due to CAV1, mice were treated with MβCD by gavage (Fig. 7D) or intrarectally (Fig. 7E) and CAV1 and occludin expression was measured. Administration of MβCD led to a significant decrease in CAV1 expression and a concomitant increase in occludin expression in colonic membrane extracts. To assess the mechanism of CAV1 inhibition of occludin expression, occludin mRNA was assessed in the colons of VhlΔIE mice. No change in occludin mRNA expression was observed in the VhlΔIE mice compared to that in their littermate controls (Fig. 7F). Occludin has been demonstrated to be ubiquitinated and degraded to regulate junction permeability (38). To assess if a chronic elevation in CAV1 could increase proteasome-mediated degradation, the colons of VhlΔIE and Vhlflox/flox mice were excised and incubated in medium containing the proteasome inhibitor MG132 or DMSO. Inhibition of the proteasome abolished the reduction of membrane-bound occludin in the colons of VhlΔIE mice (Fig. 7G). Together, these data demonstrate that CAV1 is critical for hypoxic dysregulation of occludin.
FIG 7.

CAV1 expression regulates occludin expression in the colon. (A) Expression of CAV1 and occludin in Caco-2 and HCT116 cells. (B) Western analysis in Caco-2 cells 24 h following adenovirus-mediated overexpression of CAV1 compared to the results for the control (adenovirus expressing green fluorescent protein). (C) Western analysis in HCT116 cells 48 h following an shRNA-mediated decrease in CAV1 compared to the results for the control (Scramble). Expression of CAV1 and occludin in the colons of VhlΔIE mice treated with MβCD by gavage (D) or intrarectally (E). Veh, vehicle. (F) Occludin gene expression normalized to the level of β-actin gene expression. (G) Membrane occludin and CAV1 expression in the colons of VhlΔIE and littermate control mice (Vhlflox/flox) treated ex vivo with MG132 (10 μM) or vehicle for 1 h. Four to six mice were assessed per each group. Each bar represents the mean value ± SD. N.S., not significant.
DISCUSSION
The intestinal mucosa and barrier tight junction play an important role in the host immune and inflammatory response. Moreover, recent work has demonstrated that colon tumor tight junctions are inherently dysregulated, leading to an increase in permeability and a major mechanism leading to activation of the tumor-elicited inflammatory response (39). Previous work demonstrated that inflammatory foci in patients with inflammatory bowel disease and in colon tumors have increased HIF2α expression in the epithelium compared to that in the epithelium of normal intestines (13). Mice with HIF2α overexpression in intestinal epithelial cells had increased inflammation and colon tumors (13, 16). In the current investigation, CAV1, a structural protein of caveolae, was robustly induced by HIF2α specifically in the colon. CAV1 is integral in inflammation-induced dysregulation of the intestinal barrier function. TNF-α or a core family member, LIGHT, induced intestinal barrier defects by increasing the internalization of occludin (37, 40). The present study demonstrates that HIF2α activation in intestinal epithelial cells leads to a decrease in occludin expression and that inhibition of CAV1 by MβCD leads to an increase in occludin expression. HIF2α-induced CAV1 regulated occludin degradation, as the decrease in occludin expression was reversed in VhlΔIE mice following incubation of the colon with a proteasome inhibitor. Interestingly, a recent study also demonstrated that disruption of HIF2α signaling could lead to barrier defects through the regulation of creatine metabolism (41). Together with the findings of the present study, this suggests that HIF2α is a homeostatic factor in the regulation of tight junctions and either disruption or chronic activation can lead to barrier dysregulation.
CAV1 has a dual role in the progression of colon cancer. Consistent with the growth-promoting and growth-inhibitory role of CAV1 in colon cancer, studies have shown that the CAV1 level can be decreased or CAV1 can be overexpressed in colon adenocarcinomas (20, 42, 43). There are data that clearly demonstrate the inhibitory role of CAV1 in cell transformation and tumor formation. Studies in carcinoma cells, including colon cancer (HT29 and DLD-1), breast cancer (ZR75), and embryonic kidney (HEK293T) cells, demonstrate that CAV1 plays an essential role in oncogenic transformation and tumor growth suppression (23, 44, 45). Recently, it was shown that disruption of Cav1 promotes colorectal carcinogenesis in mouse models (46). However, studies in cancer patients and animal models have shown that in prostate, breast, lung, and pancreatic cancer, an increase in CAV1 expression is correlated with a poor prognosis and reduced survival (47–51). Several studies have shown a role for CAV1 in tumor invasion and metastasis (52, 53). Recent studies in renal cell carcinoma cells showed that HIF-mediated induction of CAV1 expression is correlated with activation of EGFR signaling in a ligand-independent manner (24). Hypoxic activation of CAV1 increased the levels of phosphorylation of EGFR and activation of downstream signaling pathways, such as phosphorylation of ERK and MEK (24). EGFR and its downstream signaling pathways are the critical regulators of key cellular events in tumorigenesis (54–56). In the current work, Cav1 mRNA and CAV1 protein are highly induced in mouse colon following an increase in HIF2α signaling. However, EGFR and its downstream signaling pathways were not altered in vivo. This may be due to the tissue-specific functions of Cav1 or differences in signaling between cultured cells and in tissues in vivo.
Previous work demonstrated that HIF2α increased CAV1 expression directly by regulating CAV1 promoter activity through a canonical HRE (24). Consistent with the published data, the present study also demonstrated that activation of HIF2α (but not HIF1α) in intestinal epithelial cells leads to a robust activation of CAV1. Interestingly, the HIF2α-dependent increase in CAV1 was localized to the colon but not the small intestine. This suggested other levels of regulation, in addition to the HRE. Similar to the regulation of other genes by HIF2α, the HRE was dispensable and MAZ was required for HIF2α-mediated induction (13). The ChIP analysis demonstrated that HIF2α is recruited to the Cav1 promoter in a MAZ-dependent manner. MAZ was first characterized as the myc-associated zinc finger protein transcription factor induced during inflammation (57, 58). MAZ can bind to GC-rich DNA sequences, and the GC-rich sequence on the promoter of Cav1 was required for HIF2α-mediated induction. Moreover, overexpression of MAZ potentiated the HIF2α-mediated induction of Cav1 and knockdown of MAZ abolished HIF2α activation of Cav1. Coimmunoprecipitation studies demonstrate that HIF2α and MAZ can interact, suggesting a model in the colon where MAZ, HIF2α, and ARNT form a complex at the GC-rich sequence of the Cav1 promoter to activate transcription (Fig. 8). However, MAZ ChIP assays to definitively verify this model were inconclusive due to the specificity and sensitivity of the MAZ antibodies in HCT116 cells or mouse colon. In addition, MAZ is expressed in the colon and small intestine and therefore does not completely address the tissue specificity of Cav1 induction by HIF2α. Colon and colon-derived cell lines may contain cell type-specific transcriptional regulators that are critical for hypoxic induction of Cav1.
FIG 8.

Proposed model of hypoxia-induced dysregulation of the intestinal barrier function. Intestinal HIF2α and MAZ activate Cav1 expression, which mediates internalization of occludin, leading to decreased expression of occludin on the cellular membrane. Ub, ubiquitin.
In summary, MAZ-HIF2α-mediated signaling increased Cav1 expression, promoted disruption of intestinal tight junctions, and increased barrier permeability in vivo. MAZ binding was required for hypoxia-mediated induction of CAV1 in HCT116 cells. The proposed novel regulatory mechanism may be critical in driving inflammation in hypoxic foci in inflammatory bowel disease and colon cancer.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants (CA148828 and DK095201 to Y.M.S.), the University of Michigan Gastrointestinal Peptide Center (Y.M.S.), a pilot grant from the University of Michigan GI Spore (CA130810 to Y.M.S.), the Crohn's Colitis Foundation of America (grant number 276556 to X.X.), and the National Cancer Institute Intramural Research Program (F.J.G.).
Footnotes
Published ahead of print 2 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00324-14.
REFERENCES
- 1.Semenza GL. 2012. Hypoxia-inducible factors in physiology and medicine. Cell 148:399–408. 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith TG, Robbins PA, Ratcliffe PJ. 2008. The human side of hypoxia-inducible factor. Br. J. Haematol. 141:325–334. 10.1111/j.1365-2141.2008.07029.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kallio PJ, Okamoto K, O'Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. 1998. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 17:6573–6586. 10.1093/emboj/17.22.6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pugh CW, Chang GW, Cockman M, Epstein AC, Gleadle JM, Maxwell PH, Nicholls LG, O'Rourke JF, Ratcliffe PJ, Raybould EC, Tian YM, Wiesener MS, Wood M, Wykoff CC, Yeates KM. 1999. Regulation of gene expression by oxygen levels in mammalian cells. Adv. Nephrol. Necker Hosp. 29:191–206 [PubMed] [Google Scholar]
- 5.Pandey BD, Nabeshima T, Pandey K, Rajendra SP, Shah Y, Adhikari BR, Gupta G, Gautam I, Tun MM, Uchida R, Shrestha M, Kurane I, Morita K. 2013. First isolation of dengue virus from the 2010 epidemic in Nepal. Trop. Med. Health 41:103–111. 10.2149/tmh.2012-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. 2001. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 193:1027–1034. 10.1084/jem.193.9.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louis NA, Hamilton KE, Kong T, Colgan SP. 2005. HIF-dependent induction of apical CD55 coordinates epithelial clearance of neutrophils. FASEB J. 19:950–959. 10.1096/fj.04-3251com [DOI] [PubMed] [Google Scholar]
- 8.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. 2002. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Invest. 110:993–1002. 10.1172/JCI0215337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. 2008. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 134:156–165. 10.1053/j.gastro.2007.10.012 [DOI] [PubMed] [Google Scholar]
- 10.Tambuwala MM, Cummins EP, Lenihan CR, Kiss J, Stauch M, Scholz CC, Fraisl P, Lasitschka F, Mollenhauer M, Saunders SP, Maxwell PH, Carmeliet P, Fallon PG, Schneider M, Taylor CT. 2010. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 139:2093–2101. 10.1053/j.gastro.2010.06.068 [DOI] [PubMed] [Google Scholar]
- 11.Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. 2008. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 134:145–155. 10.1053/j.gastro.2007.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. 2004. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Invest. 114:1098–1106. 10.1172/JCI200421086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xue X, Ramakrishnan S, Anderson E, Taylor M, Zimmermann EM, Spence JR, Huang S, Greenson JK, Shah YM. 2013. Endothelial PAS domain protein 1 activates the inflammatory response in the intestinal epithelium to promote colitis in mice. Gastroenterology 145:831–841. 10.1053/j.gastro.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, Gonzalez FJ. 2008. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology 134:2036–2048. 10.1053/j.gastro.2008.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue X, Shah YM. 2013. Hypoxia-inducible factor-2alpha is essential in activating the COX2/mPGES-1/PGE2 signaling axis in colon cancer. Carcinogenesis 34:163–169. 10.1093/carcin/bgs313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue X, Taylor M, Anderson E, Hao C, Qu A, Greenson JK, Zimmermann EM, Gonzalez FJ, Shah YM. 2012. Hypoxia-inducible factor-2alpha activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. 72:2285–2293. 10.1158/0008-5472.CAN-11-3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palade GE. 1953. An electron microscope study of the mitochondrial structure. J. Histochem. Cytochem. 1:188–211. 10.1177/1.4.188 [DOI] [PubMed] [Google Scholar]
- 18.Williams TM, Lisanti MP. 2005. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am. J. Physiol. Cell Physiol. 288:C494–C506. 10.1152/ajpcell.00458.2004 [DOI] [PubMed] [Google Scholar]
- 19.Parton RG, Simons K. 2007. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8:185–194. 10.1038/nrm2122 [DOI] [PubMed] [Google Scholar]
- 20.Bender FC, Reymond MA, Bron C, Quest AF. 2000. Caveolin-1 levels are down-regulated in human colon tumors, and ectopic expression of caveolin-1 in colon carcinoma cell lines reduces cell tumorigenicity. Cancer Res. 60:5870–5878 [PubMed] [Google Scholar]
- 21.Cantiani L, Manara MC, Zucchini C, De Sanctis P, Zuntini M, Valvassori L, Serra M, Olivero M, Di Renzo MF, Colombo MP, Picci P, Scotlandi K. 2007. Caveolin-1 reduces osteosarcoma metastases by inhibiting c-Src activity and Met signaling. Cancer Res. 67:7675–7685. 10.1158/0008-5472.CAN-06-4697 [DOI] [PubMed] [Google Scholar]
- 22.Chidlow JH, Jr, Greer JJ, Anthoni C, Bernatchez P, Fernandez-Hernando C, Bruce M, Abdelbaqi M, Shukla D, Granger DN, Sessa WC, Kevil CG. 2009. Endothelial caveolin-1 regulates pathologic angiogenesis in a mouse model of colitis. Gastroenterology 136:575–584. 10.1053/j.gastro.2008.10.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams TM, Medina F, Badano I, Hazan RB, Hutchinson J, Muller WJ, Chopra NG, Scherer PE, Pestell RG, Lisanti MP. 2004. Caveolin-1 gene disruption promotes mammary tumorigenesis and dramatically enhances lung metastasis in vivo. Role of Cav-1 in cell invasiveness and matrix metalloproteinase (MMP-2/9) secretion. J. Biol. Chem. 279:51630–51646. 10.1074/jbc.M409214200 [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J, Roos FC, Chen Y, Finak G, Milosevic M, Wilson BC, Teh BT, Park M, Irwin MS, Ohh M. 2012. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc. Natl. Acad. Sci. U. S. A. 109:4892–4897. 10.1073/pnas.1112129109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor M, Qu A, Anderson ER, Matsubara T, Martin A, Gonzalez FJ, Shah YM. 2011. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 140:2044–2055. 10.1053/j.gastro.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. 2009. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 9:152–164. 10.1016/j.cmet.2008.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kathuria H, Cao YX, Ramirez MI, Williams MC. 2004. Transcription of the caveolin-1 gene is differentially regulated in lung type I epithelial and endothelial cell lines. A role for ETS proteins in epithelial cell expression. J. Biol. Chem. 279:30028–30036. 10.1074/jbc.M402236200 [DOI] [PubMed] [Google Scholar]
- 28.Yan X, Zhang Y, Zhang H, Wang PG, Chu X, Wang X. 2014. Amphiphilic polyethylenimine (PEI) as highly efficient non-viral gene carrier. Org. Biomol. Chem. 12:1975–1982. 10.1039/c3ob42279h [DOI] [PubMed] [Google Scholar]
- 29.Ramakrishnan SK, Taylor M, Qu A, Ahn SH, Suresh MV, Raghavendran K, Gonzalez FJ, Shah YM. 2014. Loss of Von Hippel-Lindau (VHL) increases systemic cholesterol level through targeting HIF-2alpha and regulation of bile acid homeostasis. Mol. Cell. Biol. 34:1208–1220. 10.1128/MCB.01441-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. 2001. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472. 10.1126/science.1059796 [DOI] [PubMed] [Google Scholar]
- 31.Salceda S, Caro J. 1997. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 272:22642–22647 [DOI] [PubMed] [Google Scholar]
- 32.Huang LE, Gu J, Schau M, Bunn HF. 1998. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U. S. A. 95:7987–7992. 10.1073/pnas.95.14.7987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. 1996. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 271:17771–17778. 10.1074/jbc.271.30.17771 [DOI] [PubMed] [Google Scholar]
- 34.Rolfs A, Kvietikova I, Gassmann M, Wenger RH. 1997. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J. Biol. Chem. 272:20055–20062. 10.1074/jbc.272.32.20055 [DOI] [PubMed] [Google Scholar]
- 35.Ray A, Dhar S, Shakya A, Ray P, Okada Y, Ray BK. 2009. SAF-3, a novel splice variant of the SAF-1/MAZ/Pur-1 family, is expressed during inflammation. FEBS J. 276:4276–4286. 10.1111/j.1742-4658.2009.07136.x [DOI] [PubMed] [Google Scholar]
- 36.Couet J, Sargiacomo M, Lisanti MP. 1997. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J. Biol. Chem. 272:30429–30438 [DOI] [PubMed] [Google Scholar]
- 37.Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, II, Raleigh DR, Guan Y, Watson AJ, Montrose MH, Turner JR. 2010. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 189:111–126. 10.1083/jcb.200902153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murakami T, Felinski EA, Antonetti DA. 2009. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 284:21036–21046. 10.1074/jbc.M109.016766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G, Karin M. 2012. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491:254–258. 10.1038/nature11465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarz BT, Wang F, Shen L, Clayburgh DR, Su L, Wang Y, Fu YX, Turner JR. 2007. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology 132:2383–2394. 10.1053/j.gastro.2007.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glover LE, Bowers BE, Saeedi B, Ehrentraut SF, Campbell EL, Bayless AJ, Dobrinskikh E, Kendrick AA, Kelly CJ, Burgess A, Miller L, Kominsky DJ, Jedlicka P, Colgan SP. 2013. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc. Natl. Acad. Sci. U. S. A. 110:19820–19825. 10.1073/pnas.1302840110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fine SW, Lisanti MP, Galbiati F, Li M. 2001. Elevated expression of caveolin-1 in adenocarcinoma of the colon. Am. J. Clin. Pathol. 115:719–724. 10.1309/YL54-CCU7-4V0P-FDUT [DOI] [PubMed] [Google Scholar]
- 43.Mori Y, Cai K, Cheng Y, Wang S, Paun B, Hamilton JP, Jin Z, Sato F, Berki AT, Kan T, Ito T, Mantzur C, Abraham JM, Meltzer SJ. 2006. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterology 131:797–808. 10.1053/j.gastro.2006.06.006 [DOI] [PubMed] [Google Scholar]
- 44.Williams TM, Lisanti MP. 2004. The caveolin genes: from cell biology to medicine. Ann. Med. 36:584–595. 10.1080/07853890410018899 [DOI] [PubMed] [Google Scholar]
- 45.Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP. 2004. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes 53:1261–1270. 10.2337/diabetes.53.5.1261 [DOI] [PubMed] [Google Scholar]
- 46.Friedrich T, Richter B, Gaiser T, Weiss C, Janssen KP, Einwachter H, Schmid RM, Ebert MP, Burgermeister E. 2013. Deficiency of caveolin-1 in Apc(min/+) mice promotes colorectal tumorigenesis. Carcinogenesis 34:2109–2118. 10.1093/carcin/bgt142 [DOI] [PubMed] [Google Scholar]
- 47.Williams TM, Lee H, Cheung MW, Cohen AW, Razani B, Iyengar P, Scherer PE, Pestell RG, Lisanti MP. 2004. Combined loss of INK4a and caveolin-1 synergistically enhances cell proliferation and oncogene-induced tumorigenesis: role of INK4a/CAV-1 in mammary epithelial cell hyperplasia. J. Biol. Chem. 279:24745–24756. 10.1074/jbc.M402064200 [DOI] [PubMed] [Google Scholar]
- 48.Williams TM, Lisanti MP. 2004. The caveolin proteins. Genome Biol. 5:214. 10.1186/gb-2004-5-3-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Satoh T, Yang G, Egawa S, Addai J, Frolov A, Kuwao S, Timme TL, Baba S, Thompson TC. 2003. Caveolin-1 expression is a predictor of recurrence-free survival in pT2N0 prostate carcinoma diagnosed in Japanese patients. Cancer 97:1225–1233. 10.1002/cncr.11198 [DOI] [PubMed] [Google Scholar]
- 50.Yang G, Truong LD, Wheeler TM, Thompson TC. 1999. Caveolin-1 expression in clinically confined human prostate cancer: a novel prognostic marker. Cancer Res. 59:5719–5723 [PubMed] [Google Scholar]
- 51.Ho CC, Huang PH, Huang HY, Chen YH, Yang PC, Hsu SM. 2002. Up-regulated caveolin-1 accentuates the metastasis capability of lung adenocarcinoma by inducing filopodia formation. Am. J. Pathol. 161:1647–1656. 10.1016/S0002-9440(10)64442-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bailey KM, Liu J. 2008. Caveolin-1 up-regulation during epithelial to mesenchymal transition is mediated by focal adhesion kinase. J. Biol. Chem. 283:13714–13724. 10.1074/jbc.M709329200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cokakli M, Erdal E, Nart D, Yilmaz F, Sagol O, Kilic M, Karademir S, Atabey N. 2009. Differential expression of caveolin-1 in hepatocellular carcinoma: correlation with differentiation state, motility and invasion. BMC Cancer 9:65. 10.1186/1471-2407-9-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chou YT, Lin HH, Lien YC, Wang YH, Hong CF, Kao YR, Lin SC, Chang YC, Lin SY, Chen SJ, Chen HC, Yeh SD, Wu CW. 2010. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res. 70:8822–8831. 10.1158/0008-5472.CAN-10-0638 [DOI] [PubMed] [Google Scholar]
- 55.Fraguas S, Barberan S, Cebria F. 2011. EGFR signaling regulates cell proliferation, differentiation and morphogenesis during planarian regeneration and homeostasis. Dev. Biol. 354:87–101. 10.1016/j.ydbio.2011.03.023 [DOI] [PubMed] [Google Scholar]
- 56.Gazdar AF, Minna JD. 2008. Deregulated EGFR signaling during lung cancer progression: mutations, amplicons, and autocrine loops. Cancer Prev. Res. (Phila.) 1:156–160. 10.1158/1940-6207.CAPR-08-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ray A, Ray BK. 1996. A novel cis-acting element is essential for cytokine-mediated transcriptional induction of the serum amyloid A gene in nonhepatic cells. Mol. Cell. Biol. 16:1584–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ray A, Ray BK. 1998. Isolation and functional characterization of cDNA of serum amyloid A-activating factor that binds to the serum amyloid A promoter. Mol. Cell. Biol. 18:7327–7335 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.