

Abstract
Both cyclin D1 and the transcription factor C/EBPβ are required for mammary epithelial cell differentiation; however, the pathway in which they operate is uncertain. Previous analyses of the patterns of gene expression in human tumors suggested a connection between cyclin D1 overexpression and C/EBPβ, but whether this represents a cancer-specific gain of function for cyclin D1 is unknown. C/EBPβ is an intronless gene encoding three protein isoforms—LAP1, LAP2, and LIP. Here, we provide evidence that cyclin D1 engages C/EBPβ in an isoform-specific manner. Cyclin D1 binds to LAP1, an event that activates the transcriptional function of LAP1 by relieving its autoinhibited state effected by intramolecular interactions. Reexpression of LAP1 but not LAP2 or LIP restores the ability of C/EBPβ-deficient mammary epithelial cells to differentiate and does so in a manner dependent on cyclin D1. And cyclin D1-mediated activation of LAP1 participates in mammary epithelial cell differentiation. Our findings indicate that cyclin D1 and C/EBPβ LAP1 operate in a common pathway to promote mammary epithelial cell differentiation.
INTRODUCTION
The transcription factor C/EBPβ (CEBPB, CCAAT/enhancer binding protein β) participates in the differentiation and specification of cell fate of diverse epithelial, mesenchymal, and hematopoietic cell lineages (1–3). In the mammary gland, loss of C/EBPβ results in a cell autonomous failure in lobuloalveolar development and milk protein synthesis during pregnancy (4, 5). Mammary gland development is governed by the action of steroid and peptide hormones that ultimately impinge on and orchestrate the activity of a number of transcription factors (6–8). Despite this insight, the pathway in which C/EBPβ operates during mammary epithelial cell (MEC) differentiation is poorly understood.
Difficulty in assigning C/EBPβ to a pathway derives in part from our lack of knowledge about the participation of the individual isoforms of C/EBPβ in mammary gland development. C/EBPβ is an intronless gene that encodes three isoforms—LAP1 (also called LAP*), LAP2 (also called LAP), and LIP—through the use of alternative translational start sites (9, 10); they will henceforth be referred to as LAP1, LAP2, and LIP. Although not absolute, LAP1 and LAP2 are generally thought to activate transcription, whereas LIP is believed to inhibit transcription by antagonizing the LAPs. In the cell types studied, it is thought that a specific ratio of the LAPs to LIP effects defined biological outcomes, e.g., differentiation—but only in a few instances has this been experimentally tested (11). In the analysis of MEC differentiation, focus has been placed on the involvement of LAP2 and LIP and the ratio between these two isoforms (5, 12). The contribution of LAP1, if any, to MEC differentiation has not been assessed.
How C/EBPβ is regulated during MEC differentiation has not been determined. It is known that the transcriptional activity of C/EBPβ is distinct in different cell types (13). Further, the activity of LAP1 and LAP2 are intrinsically autoinhibited (14, 15). Moreover, studies performed mostly in mesenchymal cells suggest that oncogenic Ras can promote mitogen-activated protein kinase-mediated phosphorylation of LAP1 and LAP2, an event that overcomes their autoinhibited state and allows for transcriptional activation (14, 16, 17). Nevertheless, the physiological signaling event that brings about activation of the transcriptional function of C/EBPβ during MEC differentiation is unknown.
The cyclin D1 gene (CCND1 or BCL1/PRAD1) is frequently amplified in human cancer, including breast cancer (18). Cyclin D1 also has a critical role in pregnancy-induced mammary gland development (19, 20), suggesting a link between development and tumorigenesis (19). Genetic analyses have pointed to a need to identify the effectors of cyclin D1 during mammary gland development (21, 22). With respect to tumorigenesis, we identified C/EBPβ as an effector in the transcriptional function of cyclin D1 and suggested that this activity represented a potentially important facet of cyclin D1 biology (23). We therefore undertook to determine the nature of the functional interaction between cyclin D1 and C/EBPβ. We used this information to assess whether the ability of overexpressed cyclin D1 to affect the transcriptional activity of C/EBPβ represents a tumor-specific gain of function or, in the absence of cyclin D1 overexpression, the cyclin D1-C/EBPβ pathway contributes to the normal physiological process of MEC differentiation.
MATERIALS AND METHODS
Cell culture.
MCF-7, C33A, and 293T cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 mg of streptomycin/ml, and 100 U of penicillin G/ml. SCp2 cells (24), a gift from Mina Bissell, were cultured in DMEM–F-12 supplemented with 5% FBS, 5 μg of insulin/ml, 100 mg of streptomycin/ml, and 100 U of penicillin G/ml. The C/EBPβ−/− mammary epithelial cell line (23) was cultured in DMEM–F-12 supplemented with 2% FBS, 10 mM HEPES, 5 μg of insulin/ml, 5 ng of epidermal growth factor/ml, 100 mg of streptomycin/ml, and 100 U of penicillin G/ml. All cell lines were cultured at 37°C and 5% CO2.
Plasmids.
pcDNA3.1-Cyclin D1-HA was generated from pRc/CMV-cyclin D1-HA (25) by subcloning the insert. pcDNA3.1-C/EBPβ (full-length human C/EBPβ LAP1 cDNA) and pcDNA3.1-C/EBPβΔSpl were gifts from Dan Tenen and Phil Auron. Plasmids directing the expression of human LAP2 and LIP (pcDNA3.1-LAP2 and pcDNA3.1-LIP) were generated by PCR. The inserts from these plasmids were subcloned into pBabe-puro and pWZL-blast (LAP1 and C/EBPβΔSpl) for use as retroviruses. The derivatives of LAP1 [LAP1(32A,34A), LAP1ΔCR5, LAP1ΔCR6, and LAP1ΔCR7] were generated by site-directed mutagenesis (QuikChange site-directed mutagenesis kit; Stratagene). The derivatives of C/EBPβΔSpl [C/EBPβΔSpl(Δ1-23) and C/EBPβΔSpl(Δ24-40) and the constructs resulting from alanine scanning mutagenesis of amino acids (aa) 24 to 40 in the context of C/EBPβΔSpl] were generated by site-directed mutagenesis. The Gal4 fusions using sequences derived from C/EBPβ [(1-40)-Gal4, (1-23)-Gal4, (24-40)-Gal4, CR5-Gal4, CR6-Gal4, CR7-Gal4, and CR7(Δ200-205)-Gal4] were generated by PCR, followed by subcloning into pCMX-Gal4(N), a gift from Ron Evans. The enhanced green fluorescent protein (EGFP) fusion using sequences derived from C/EBPβ [EGFP-(24-40)] and its derivatives [EGFP-(24-40;32A), EGFP-(24-40;34A), and EGFP-(24-40;32A,34A)] were generated by PCR, followed by subcloning into pCSH-EGFP-Myc, a gift from Andrew Kung. shRNAs in the lentiviral vector pLKO.1 targeting human and mouse cyclin D1 were obtained from The RNAi Consortium at the Broad Institute. The sequences of the shRNAs to human cyclin D1 are as follows: shD1-1, 5′-GCCAGGATGATAAGTTCCTTT-3′; and shD1-2, 5′-GATTGGAATAGCTTCTGGAAT-3′. The sequences of the shRNAs to murine cyclin D1 are as follows: shD1-3, 5′-CCACAGATGTGAAGTTCATTT-3′; and shD1-4, 5′-CTTTCTTTCCAGAGTCATCAA-3′. The sequence of the control shRNA, shCtrl, is 5′-CAACAAGATGAAGAGCACCAAT-3′. Lentiviral constructs directing the expression of murine LAP1, LAP2, and LIP were generated by PCR, followed by subcloning into HIV-Zsgreen (26), a gift from Zena Werb. The murine LAP1 derivative, LAP1s, which can only make the longest C/EBPβ isoform, LAP1, was generated by mutating the initiating methionines for LAP2 and LIP by site-directed mutagenesis. All plasmids were verified by DNA sequencing.
IP and immunoblotting.
Transfected cells were lysed in EBC200 buffer (50 mM Tris-HCl [pH 8.0], 200 mM NaCl, 0.5% NP-40, 0.5 mM phenylmethylsulfonyl fluoride, 1 mM NaF, 0.1 mM sodium orthovanadate) with protease inhibitor cocktail (Roche) and clarified by centrifugation. Lysates were subjected to immunoprecipitation (IP) with antibodies to Gal4 (sc-510) or C/EBPβ (sc-150) from Santa Cruz Biotechnology. Immune complexes were collected on protein A-Sepharose beads (GE Healthcare), washed four times with NETN buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% NP-40), and then boiled in Laemmli buffer. For the endogenous cyclin D1-C/EBPβ coimmunoprecipitation (co-IP) analysis, the cells were first cross-linked for 30 min at room temperature with 2 mM dithiobis(succinimidyl propionate) (DSP; Pierce/Thermo Scientific) and quenched for 10 min with 20 mM Tris (pH 7.5) before being processed as described above. The precipitates or cell lysates were resolved by SDS–12% PAGE and then transferred to polyvinylidene difluoride membranes (EMD Millipore). Immunoblotting (from whole-cell lysates or immunoprecipitates) was performed with antibodies to C/EBPβ, Gal4, cyclin D1 (Ab-3; Lab Vision/Fisher Scientific), Myc (9E10), hemagglutinin (HA) epitope (12CA5), only the LAP1 isoform (18F8, ab15049; Abcam), or α-tubulin (sc-8035; Santa Cruz Biotechnology), followed by incubation with appropriate peroxidase-conjugated secondary antibodies (GE Healthcare) and enhanced chemiluminescence detection (GE Healthcare). Densitometric analyses were performed with ImageJ software.
Reporter assay.
A wild-type promoter construct (pGL3-HSP70-2) was generated by PCR amplification of human genomic DNA spanning the cyclin D1 response element (CCCTGGAAT) in HSP70-2 (23) (HSP70-2 [HSPA1B], chromosome 6: 31795315 to 31795728 forward strand; Ensembl release GRCh37) and subcloned into pGL3-Basic. A mutant promoter construct was generated by introducing point mutations in the cyclin D1 response element (CCCTGGAAT→CCgTGcAAT [23]). Cells were transfected with the indicated plasmid constructs, together with pGL3-HSP70-2 (wild-type or mutant) and pRL-CMV (Promega), using PolyFect transfection reagent (Qiagen), and then the luciferase activity was measured with a dual fluorescence reporter assay kit (Promega). The relative reporter activity was calculated by normalizing the firefly luciferase activity with the Renilla luciferase activity.
ChIP.
Chromatin immunoprecipitation (ChIP) was carried out essentially as described previously (27). Cells grown in 10-cm dishes were cross-linked with 10 ml of a 1% formaldehyde–phosphate-buffered saline (PBS) solution at room temperature for 10 min and then quenched with 1 ml of 1.25 M glycine for 1 min, followed by two quick washes with cold PBS on ice. The cells were scraped into cold PBS and either stored at −80°C or used immediately for ChIP. Briefly, cells were lysed with ChIP lysis buffer (50 mM Tris-HCl [pH 8.0], 1% SDS, 10 mM EDTA, plus protease inhibitors [Roche]) on ice and sonicated 10 times at 30% amplitude for 15 s (Misonix sonicator 3000). The majority of DNA fragments were smaller than 500 bp. Samples were subjected to centrifugation at 16,000 × g for 10 min, and 200 μl of supernatant was mixed with 800 μl of ChIP dilution buffer (20 mM Tris-HCl [pH 8.0], 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, plus protease inhibitors). Lysates were precleared with protein A-Sepharose beads (GE Healthcare), 0.3 mg of salmon sperm DNA/ml, and 1 mg of bovine serum albumin/ml for 2 h at 4°C. Lysates were then incubated overnight at 4°C with specific antibodies to C/EPBβ (sc-150; Santa Cruz Biotechnology), cyclin D1 (Ab-3; Lab Vision/Fisher Scientific), and an appropriate rabbit IgG as a control. Immunocomplexes were incubated with protein A-Sepharose beads for 1 h and then washed with ChIP wash buffer (50 mM Tris [pH 7.5], 1 mM EDTA, 0.7% sodium deoxycholate, 1% NP-40, 0.5 M LiCl) six times, followed by two washes with Tris-EDTA (pH 8.0) for 10 min, each time at 4°C. The samples were eluted from the beads with 250 μl of 1% SDS–0.1 M NaHCO3 for 30 min, followed by incubation overnight at 65°C to reverse the cross-links. DNA was purified using Qiaex II kit and eluted in 40 μl of elution buffer (Qiagen). PCR was carried out using a Taq polymerase kit (Qiagen). The primer sequences are as follows: Hsc70+, 5′-TTCATTAGTGCAGTCGGCAAAGGC-3′; Hsc70−, 5′-GCTTCCCTCTCTCTTATACCTGT-3′; Hsc70 4kb downstream control+, 5′-TGGCACCATACAGTTGTCCTGACT-3′; and Hsc70 4kb downstream control−, 5′-TTCATAGCTCAGAACCAGCACCCT-3′.
Retrovirus and lentivirus.
Vesicular stomatitis virus protein G (VSV-G)-pseudotyped retroviruses were packaged by cotransfecting a given retroviral construct (pBabe-puro or pWZL-blast) with pMD.G and pMD.MLV, gifts from Jeng-Shin Lee and Richard Mulligan, into 293T cells. Viral supernatants were used for infections. VSV-G-pseudotyped lentiviruses were constructed in HIV-Zsgreen (26), packaged by cotransfection with pMD.G and pCMVΔR8.91 (28) into 293T cells, and concentrated by ultracentrifugation.
Primary mammary epithelial cells.
Primary mammary epithelial cells were isolated from 8-week-old C57BL/6 × 129/Sv mice as described previously (26) with the following modification. The digestion was performed using 10 ml/g of tissue collagenase/hyaluronidase buffer (DMEM–F-12, 5% FBS, 100 mg of streptomycin/ml, 100 U of penicillin G/ml, 300 U of collagenase IV [Sigma]/ml, and 100 U of hyaluronidase [Sigma]/ml) for 1 h at 37°C with shaking at 200 rpm. Research involving animals complied with protocols approved by the Tufts Medical Center and Tufts University Institutional Animal Care and Use Committee.
In vitro differentiation.
Stably infected SCp2 or C/EBPβ−/− mammary epithelial cells were plated on growth factor-reduced Matrigel (BD Biosciences) at a density of 7 × 105 cells per 35-mm-diameter dish, and differentiation was induced by the addition of insulin (Sigma; 5 μg/ml), hydrocortisone (Sigma; 1 μg/ml), and prolactin (Sigma; 3 μg/ml) as previously described (25).
RNA isolation and quantitative reverse transcription-PCR (RT-PCR).
Differentiated mammary epithelial cells were homogenized in TRIzol, and RNA was extracted with RNeasy minikit (Qiagen). cDNAs were synthesized using Superscript III first-strand synthesis system (Invitrogen), and quantitative real-time PCR was performed with SYBR green PCR master mix (Applied Biosystems). The primer sequences were as follows: β-casein+, 5′-GCTCAGGCTCAAACCATCTC-3′; β-casein−, 5′-TGTGGAAGGAAGGGTGCTAC-3′; WAP+, 5′-TGGCTTCTGCCCTTGGAATCTACT-3′; WAP−, 5′-GACACAGTCGACGTTGCAGCATTT-3′; GAPDH+, 5′-TCAACAGCAACTCCCACTCTTCCA-3′; and GAPDH−, 5′-ACCCTGTTGCTGTAGCCGTATTCA-3′.
RESULTS
Cyclin D1 activates the transcriptional function of C/EBPβ in an isoform-specific manner.
In order to assess whether the functional interaction between cyclin D1 and C/EBPβ brings about biological outcomes, we needed first to determine how cyclin D1 engages the three isoforms of C/EBPβ. Cyclin D1 was found to coimmunoprecipitate with LAP1 but not LAP2 or LIP from lysates of a MEC line derived from C/EBPβ-deficient mice (23) cotransfected with plasmids directing the expression of these proteins (Fig. 1A). Further, using a MEC line that expresses the C/EBPβ isoforms and cyclin D1, endogenous cyclin D1 coimmunoprecipitated with endogenous C/EBPβ (Fig. 1B). These observations suggest that cyclin D1 influences C/EBPβ in an isoform-specific manner.
FIG 1.

Cyclin D1 engages C/EBPβ in an isoform-specific manner. (A) Cyclin D1 interacts with LAP1 but not LAP2 or LIP. C/EBPβ−/− MECs were cotransfected with plasmids encoding the three C/EBPβ isoforms, HA-tagged cyclin D1 (D1-HA), or empty vector as indicated. After 24 h, whole-cell lysates were prepared, and complex formation was assessed by immunoprecipitation (IP) with antibody directed against C/EBPβ, followed by immunoblotting (IB) with anti-HA antibody. The relative amounts of cyclin D1-HA, LAP1, LAP2, and LIP in whole-cell lysates were assessed by IB with the same antibodies (left). That the C/EBPβ antibody can immunoprecipitate the three C/EBPβ isoforms with equal efficiency is also shown (right). The results are representative of at least five independent experiments. (B) Endogenous cyclin D1 and C/EBPβ interact. Whole-cell lysates were prepared from MCF-7 cells as described in Materials and Methods, and complex formation was assessed by IP with antibody directed against C/EBPβ, followed by IB with anti-cyclin D1 antibody. The levels of cyclin D1, LAP1, LAP2, and LIP in whole-cell lysates as input are also shown. The results are representative of five independent experiments. (C) Cyclin D1 is recruited to a cyclin D1-responsive promoter in a LAP1-dependent manner. C/EBPβ−/− MECs were infected with retroviruses directing the expression of LAP1, LAP2, LIP, or empty vector. Subsequently, cells were subjected to ChIP with antibody to C/EBPβ, cyclin D1, or IgG as a control. For the precipitated DNA, sequences spanning the cyclin D1 response element (C/EBPβ site) in the promoter of Hsc70 and 4 kb downstream of this element were analyzed by PCR; one-tenth of the lysate used for ChIP was also subjected to PCR (Input). The results are representative of at least four independent experiments. (D) Cyclin D1 effects transcriptional induction in a LAP1-dependent manner. MCF-7 cells were cotransfected with a wild-type (top left) or mutant (top right) HSP70-2 promoter-reporter construct and plasmids directing the expression of LAP1, LAP2, LIP, or empty vector (lanes 1 to 4), increasing amounts of cyclin D1-HA (indicated; lanes 5 to 8), or a saturating amount of cyclin D1-HA, together with LAP1, LAP2, or LIP (lanes 9 to 11). Normalized promoter activity was determined 24 h later, and the fold increases in promoter activity relative to empty vector were calculated. The results are means ± the standard deviations (SD) from three independent experiments. Also shown are the relative levels of endogenous and exogenous cyclin D1 and C/EBPβ under the conditions described above (bottom).
ChIP was used to assess where cyclin D1 might act to influence transcription. C/EBPβ−/− MECs were infected with virus directing the expression of LAP1, LAP2, LIP, or empty vector. Each of the three C/EBPβ isoforms was found to localize to a cyclin D1-responsive promoter (Fig. 1C). Endogenous cyclin D1 localization, however, was only observed in the presence of LAP1 (Fig. 1C), a finding consistent with the binding studies noted above. These observations suggest that cyclin D1 and LAP1 work together at cyclin D1 target gene promoters.
To begin to assess the functional relevance of these findings, an epithelial cell line was transfected with plasmids expressing LAP1, LAP2, or LIP, together with a cyclin D1-responsive promoter reporter in the presence or absence of a plasmid encoding cyclin D1. As expected, on their own, the C/EBPβ isoforms did not induce transcription (Fig. 1D). Introduction of cyclin D1 activated transcription through endogenous C/EBPβ. Moreover, only in the presence of coexpressed LAP1 was cyclin D1 able to further activate transcription (Fig. 1D). These observations, together with the findings noted above, suggest that cyclin D1 effects transcriptional induction only through the LAP1 isoform.
Cyclin D1 relieves the autoinhibited state of C/EBPβ LAP1.
C/EBPβ possesses a number of evolutionarily conserved regions (CR), with CR1 to -4 spanning the transactivation domain and CR5 to -7 encompassing the negative regulatory domain (Fig. 2A) (14). Initial insights into how cyclin D1 activates the transcriptional function of LAP1 were provided by the analyses of the LAP1 mutant, NF-IL-6ΔSpl (29), here referred to as C/EBPβΔSpl. This mutant lacks part of the transactivation domain and much of the negative regulatory domain (Fig. 2A). Our initial interest in C/EBPβΔSpl stemmed from our finding that this mutant, unlike wild-type LAP1, was transcriptionally active (Fig. 2B) (23). Deletion of sequences that include CR1 (aa 1 to 23) did not impact the ability of C/EBPβΔSpl to activate transcription, whereas a large loss of CR2 (aa 24 to 40) completely abolished transcriptional activity (Fig. 2B). These observations suggest that sequences shared by LAP1 and LAP2 (aa 24 to 40) are required for activation by C/EBPβΔSpl, whereas sequences unique to LAP1 (aa 1 to 23) are dispensable.
FIG 2.

Cyclin D1 relieves an autoinhibited state in C/EBPβ LAP1. (A) Schematic representation of the C/EBPβ isoforms, LAP1, LAP2, and LIP, and the LAP1 mutant, C/EBPβΔSpl. The evolutionarily conserved regions (CR) are indicated with amino acid coordinates corresponding to those reported (14). (B) Sequences in CR2 are required for transcriptional activation. MCF-7 cells were cotransfected with a reporter construct (HSP70-2) and plasmids encoding LAP1, LAP2, LIP, C/EBPβΔSpl (βSpl), derivatives of C/EBPβΔSpl lacking residues 24 to 40 [βSpl(Δ24-40)] or residues 1 to 23 [βSpl(Δ1-23)], or empty vector. Normalized promoter activity was determined 24 h later, and the fold increases in promoter activity relative to empty vector were calculated. The results are means ± the SD from three independent experiments. (C) Sequences in CR2 are involved in binding to cyclin D1. MCF-7 cells were cotransfected with plasmids encoding LAP1, LAP2, LIP, C/EBPβΔSpl (βSpl), derivatives of C/EBPβΔSpl [βSpl(Δ24-40) and βSpl(Δ1-23)], or empty vector, together with a plasmid encoding HA-tagged cyclin D1 (D1-HA) or empty vector. After 24 h, whole-cell lysates were prepared, and complex formation was assessed by IP with antibody to C/EBPβ, followed by IB with anti-HA antibody. The relative amounts of the C/EBPβ isoforms, C/EBPβΔSpl and its derivatives, and cyclin D1-HA in whole-cell lysates were assessed with the same antibodies. Densitometric analysis of immunoblots normalized to the anti-HA blot revealed that βSpl recovered 1.84 times more cyclin D1 than βSpl(Δ1-23). The results are representative of at least five independent experiments. (D) CR2 is sufficient for binding cyclin D1. MCF-7 cells were cotransfected with plasmids directing the expression of Gal4 fused to aa 1 to 40 [(1-40)-Gal4], 1 to 23 [(1-23)-Gal4], or 24 to 40 [(24-40)-Gal4] of LAP1, Gal4 alone, or empty vector together with a plasmid encoding HA-tagged cyclin D1 (D1-HA). After 24 h, whole-cell lysates were prepared, and complex formation was assessed by IP with antibody to Gal4 or HA, followed by IB with antibody to HA or Gal4, respectively. The relative amounts of the Gal4 fusions and cyclin D1-HA in whole-cell lysates were assessed by IP, followed by IB with antibody to Gal4 or the HA epitope. Densitometric analysis of immunoblots normalized to the anti-HA blot revealed that (1-40)-Gal4 recovered 2.45 times more cyclin D1 than (24-40)-Gal4. The results are representative of at least three independent experiments. (E) CR2 engages in intramolecular interactions within LAP1. MCF-7 cells were cotransfected with plasmids encoding LAP1, deletion mutants of LAP1 lacking aa 132 to 146, 154 to 192, or 199 to 242 designated LAP1ΔCR5, LAP1ΔCR6, and LAP1ΔCR7, respectively, together with Myc-tagged EGFP fused to aa 24 to 40 of LAP1 [EGFP-(24-40)] or EGFP alone. After 24 h, whole-cell lysates were prepared, and complex formation was assessed by IP with antibody to C/EBPβ, followed by IB with antibody to the Myc tag. The relative amounts of LAP1 and its deletion mutants or the EGFP fusion in whole-cell lysates were assessed with the same antibodies. The results are representative of at least three independent experiments. (F) CR2 binds CR7 and cyclin D1 disrupts this interaction. MCF-7 cells were cotransfected with plasmids directing the expression of Myc-tagged EGFP fused to aa 24 to 40 [EGFP-(24-40)], together with plasmids directing the expression of Gal4 fused with aa 132 to 153, 154 to 198, or 199 to 242 designated CR5-Gal4, CR6-Gal4, and CR7-Gal4, respectively, or Gal4 or CR7-Gal4 plus cyclin D1-HA (D1-HA). After 24 h, whole-cell lysates were prepared, and complex formation was assessed by IP with antibody to Gal4, followed by IB with antibody to the Myc tag. The relative amounts of the EGFP and Gal4 fusions in whole-cell lysates were assessed with the same antibodies, and the expression of cyclin D1-HA was assessed with antibody to the HA epitope. The results are representative of at least three independent experiments. (G) Potential intramolecular interactions within C/EBPβΔSpl cannot take place. Same analyses as in panel F, except that CR7-Gal4 and a derivative missing the first 6 aa of CR7 [CR7(Δ200-205)-Gal4] were analyzed.
We explored the relationship between the transcriptional activity of C/EBPβΔSpl and its derivatives and their interaction with cyclin D1. C/EBPβΔSpl and C/EBPβΔSpl(Δ1-23) were found to interact with cyclin D1, although the latter consistently displayed attenuated binding (Fig. 2C). In contrast, the transcriptionally inactive derivative, C/EBPβΔSpl(Δ24-40), failed to bind cyclin D1 (Fig. 2C).
The results presented above suggest that CR2 and possibly CR1 in C/EBPβ are important for the interaction of LAP1 with cyclin D1. To assess whether these conserved regions are sufficient for binding cyclin D1, a number of fusion proteins were constructed. When fused to Gal4, sequences in CR2 (aa 24 to 40), but not those encompassing CR1 (aa 1 to 23), were sufficient for interaction with cyclin D1 (Fig. 2D). A fusion protein spanning both CR1 and CR2 (aa 1 to 40) also bound cyclin D1. Similar to what was found with C/EBPβΔSpl and its derivatives (Fig. 2C), the inclusion of sequences from CR1 facilitated the interaction of CR2 with cyclin D1 (Fig. 2D). This “helper function” displayed by CR1 may underlie why cyclin D1 can interact with LAP1 but not LAP2.
Previous work has suggested that the activities of LAP1 and LAP2 are autoinhibited—a state effected by intramolecular interactions between their transactivation domain and negative regulatory domain (14, 15). Based on these reports, we entertained the possibility that cyclin D1 binding to LAP1 activates its transcriptional function, disrupting intramolecular interactions within LAP1. We sought to determine whether CR2 can interact with LAP1, and the answer was affirmative (Fig. 2E). Mapping the region in LAP1 that CR2 interacts with, we found that deletion of sequences in CR7 (aa 199 to 242) but not CR5 (aa 132 to 146) or CR6 (aa 154 to 192) abolished its interaction with CR2 (Fig. 2E). These findings suggest that CR2, which in the context of C/EBPβΔSpl is required for transcriptional activity, is able to engage in intramolecular interactions within LAP1.
Extending the findings noted above, we found that sequences spanning CR7 (aa 199 to 242) (as a fusion protein) were sufficient for interacting with CR2, whereas CR5 (aa 132 to 153) and CR6 (aa 154 to 198) scored negative in this assay (Fig. 2F). Further, cyclin D1 was able to prevent the association between CR2 and CR7 (Fig. 2F), suggesting that in the context of LAP1, its binding to cyclin D1 disrupts the interaction between CR2 and CR7. The internal deletion in LAP1 that generates C/EBPβΔSpl was such that the first six amino acids of CR7 are missing. In addition, a CR7 fusion protein lacking these residues failed to interact with CR2 (Fig. 2G), suggesting that the intramolecular interactions that normally occur in LAP1 do not take place in C/EBPβΔSpl.
Constitutively active and dominant-negative mutants of C/EBPβ LAP1.
To explore the functions of CR2 further, alanine scanning mutagenesis was performed in the context of C/EBPβΔSpl. These analyses revealed that the only two acidic amino acids in CR2, Glu32 and Asp34, were required for transcriptional activation (Fig. 3A). Further, mutation of both of these residues did not impact the ability of C/EBPβΔSpl to associate with cyclin D1 (see Fig. 4B), suggesting that this event is not required for the transcriptional activity of C/EBPβΔSpl.
FIG 3.

Constitutively active mutant of LAP1. (A) Two acidic amino acids in CR2 are required for transcriptional induction by C/EBPβΔSpl. Alanine scanning mutagenesis was performed on aa 24 to 40 in the context of C/EBPβΔSpl. MCF-7 cells were cotransfected with a reporter construct (HSP70-2) and plasmids directing the expression of C/EBPβΔSpl (βSpl), its mutant derivatives (alanine substitution is indicated), or empty vector. Normalized promoter activity was determined 24 h later, and the fold increases in promoter activity relative to empty vector were calculated. The results are means ± the SD from three independent experiments. (B) Two amino acids in CR2 are necessary for its interaction with CR7. MCF-7 cells were cotransfected with plasmids directing the expression of Gal4 fused to amino acids 199 to 242 of LAP1 (CR7-Gal4), together with plasmids directing the expression of Myc-tagged EGFP alone or fused to aa 24 to 40 of LAP1 [EGFP-(24-40)] or derivatives of EGFP-(24-40) bearing alanine substitutions at residues E32, D34, or both designated EGFP-(24-40;32A), EGFP-(24-40;34A), and EGFP-(24-40;32A,34A). After 24 h, whole-cell lysates were prepared, and complex formation was assessed by IP with antibody to Gal4, followed by IB with antibody to the Myc epitope tag. The relative amounts of the EGFP and Gal4 fusions in whole-cell lysates were assessed with the same antibodies. The results are representative of three independent experiments. (C) Model depicting the functional interaction between cyclin D1 and LAP1, and the action of a constitutively active mutant of LAP1. In the inactive state for LAP1, CR7 binding to CR2 masks E32 and D34 in CR2; the presence of either of these acidic amino acids is required for the interaction between CR2 and CR7. Cyclin D1 binding to CR2 in LAP1 disrupts the intramolecular interaction between CR2 and CR7. This event exposes E32 and D34, permitting them to participate in the interaction with a putative transcriptional coactivator (purple, left). In the constitutively active mutant of LAP1 (C/EBPβΔSpl), the intramolecular interaction between CR2 and CR7 do not take place. Consequently, E32 and D34 are constitutively exposed, and the presence of both E32 and D34 allows CR2 to interact with a putative transcriptional coactivator (right). (D) Alanine substitution at amino acids E32 and D34 in LAP1 relieves the autoinhibited state and enhances its interaction with cyclin D1. MCF-7 cells were cotransfected with plasmids encoding LAP1, a derivative of LAP1 bearing alanine substitutions as amino acids E32 and D34 [LAP1(32A,34A)], or empty vector together with a plasmid encoding HA-tagged cyclin D1 (D1-HA). After 24 h, whole-cell lysates were prepared, and complex formation was assessed by IP with antibody to C/EBPβ, followed by IB with anti-HA antibody. The relative amounts of LAP1, its mutant derivative, and cyclin D1 in whole-cell lysates were assessed with the same antibodies. The results are representative of three independent experiments. (E) C/EBPβΔSpl represents a constitutively active mutant of LAP1 that does not require cyclin D1 to effect transcription. C33A cells were infected with viruses directing the expression of one of two shRNA to cyclin D1 (shD1-1 and shD1-2) or an irrelevant shRNA (shCtrl). Subsequently, the cells were cotransfected with a reporter construct (HSP70-2), together with plasmids encoding cyclin D1-HA, C/EBPβΔSpl (βSpl), or empty vector. Normalized promoter activity was determined 24 h later, and the fold increases in promoter activity relative to empty vector were calculated. The results are means ± the SD from three independent experiments. The relative amounts of C/EBPβΔSpl and cyclin D1 under conditions where cyclin D1 was knocked down, as well as α-tubulin as a loading control, were assessed by IB (inset).
FIG 4.
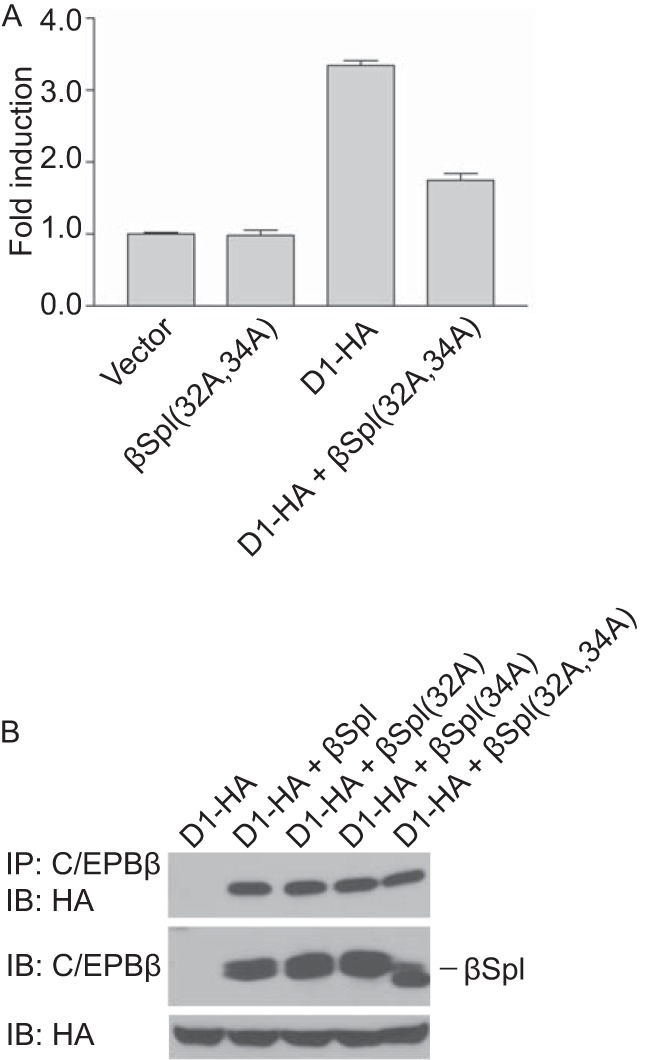
Dominant-negative mutant of LAP1. (A) C/EBPβΔSpl(32A,34A) blocks cyclin D1-mediated activation of LAP1. MCF-7 cells were cotransfected with a reporter construct (HSP70-2), together with empty vector, C/EBPβΔSpl(32A,34A), HA-tagged cyclin D1 (D1-HA), or both cyclin D1-HA and C/EBPβΔSpl(32A,34A). Normalized promoter activity was determined 24 h later, and the fold increases in promoter activity relative to empty vector were calculated. The results are means ± the SD from three independent experiments. (B) The mutation of amino acids E32 and D34 in C/EBPβΔSpl does not influence its ability to interact with cyclin D1. MCF-7 cells were cotransfected with plasmids encoding cyclin D1-HA, together with empty vector or plasmids directing the expression of C/EBPβΔSpl (βSpl) or derivative bearing alanine substitutions at residues E32, D34, or both designated βSpl(32A), βSpl(34A), and βSpl(32A,34A). After 24 h, whole-cell lysates were prepared, and complex formation was assessed by IP with antibody to C/EBPβ, followed by IB with anti-HA antibody. The relative amounts of C/EBPβΔSpl, its derivatives, and cyclin D1-HA in whole-cell lysates were assessed with the same antibodies. The results are representative of at least three independent experiments.
We also explored the possible involvement of Glu32 and Asp34 in the intramolecular interactions that take place in LAP1 between CR2 and CR7. We found that while alanine substitutions at either of these residues did not affect the ability of CR2 to interact with CR7, mutating both residues abolished binding of CR2 to CR7 (Fig. 3B). Together with the data presented above, these findings suggest a model whereby the binding of CR7 to CR2 masks key acidic amino acids (i.e., Glu32 and Asp34) that participate in transcriptional activation and that interaction of LAP1 with cyclin D1 exposes these residues (Fig. 3C). Consistent with this model, mutating Glu32 and Asp34 in LAP1 enhances its ability to associate with cyclin D1 (Fig. 3D), suggesting that CR2 is more accessible when its interaction with CR7 is disrupted.
The model put forth above and the data that support it suggest that the prominent function of cyclin D1 in activating the transcriptional function of LAP1 is to relieve its autoinhibited state by disrupting intramolecular interactions. Moreover, because the intramolecular interactions between CR2 and CR7 in C/EBPβΔSpl do not take place (Fig. 2G), this mutant of LAP1 remains transcriptionally active. In other words, cyclin D1 plus LAP1 is functionally equivalent to C/EBPβΔSpl. This suggests that while C/EBPβΔSpl can bind cyclin D1, it does not need to in order to bring about transcriptional induction.
To test this possibility, we assessed the effect of knocking down cyclin D1 on the transcriptional activity of C/EBPβΔSpl. To exclude possible secondary effects due to affects on the cell cycle, we used an epithelial cell line lacking functional retinoblastoma protein. Silencing cyclin D1 expression using short hairpin RNAs (shRNAs) had no impact on the ability of C/EBPβΔSpl to activate transcription (Fig. 3E). This suggests that C/EBPβΔSpl represents a constitutively active mutant of LAP1 that does not require cyclin D1 to induce the expression of cyclin D1 target genes (Fig. 3C).
We also sought to block the transcriptional function of cyclin D1. We reasoned that a transcriptionally inactive mutant of LAP1 might sequester cyclin D1, thereby blocking its ability to activate transcription. To this end, we used C/EBPβΔSpl(32A, 34A) (Fig. 3A). We found that expression of this LAP1 mutant was able to bind cyclin D1 and attenuate transcriptional induction by cyclin D1 (Fig. 4), suggesting that C/EBPβΔSpl(32A, 34A) represents a dominant-negative mutant of LAP1.
C/EBPβ isoform-specific contribution to mammary epithelial cell differentiation.
We attempted to determine whether cyclin D1′s C/EBPβ LAP1-dependent transcriptional function influenced normal biological outcomes—using mammary epithelial cell (MEC) differentiation as a defined endpoint (see Introduction). Given that studies on C/EBPβ and the biology of MECs have focused almost exclusively on the LAP2 and LIP isoforms, it was essential to determine whether the LAP1 isoform was expressed in MECs. We found that primary MECs do express LAP1, as evidenced by comigration with LAP1 expressed in a C/EBPβ-deficient cell line and reactivity with an antibody that detects only the LAP1 isoform (Fig. 5).
FIG 5.
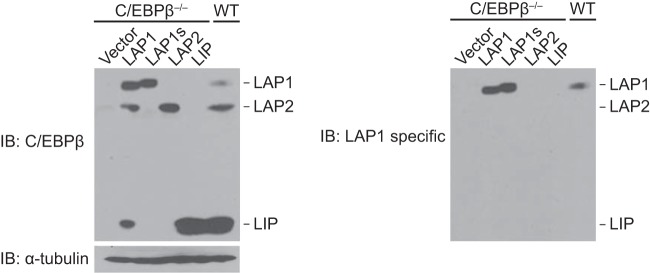
LAP1 is expressed in mammary epithelial cells. C/EBPβ−/− MECs were infected with lentivirus directing the expression of murine LAP1, LAP1s, LAP2, LIP, or empty vector. LAP1s is a derivative of LAP1 that harbors mutations in the full-length C/EBPβ cDNA such that the initiating methionines for LAP2 and LIP are mutated (only LAP1 is synthesized from LAP1s). Whole-cell lysates from these lines, together with that from primary MECs (WT), were resolved in a denaturing gel and subjected to IB with antibodies to C/EBPβ and α-tubulin (left panel). This same blot was stripped and reprobed with a LAP1-specific antibody (right panel).
Next, we assessed the ability of the three C/EBPβ isoforms to restore differentiation of C/EBPβ−/− MECs. Virus harboring a wild-type cDNA for C/EBPβ that expresses LAP1, LAP2, and LIP (denoted LAP1) was used to infect a C/EBPβ-deficient cell line. A derivative of C/EBPβ that directs the expression of only LAP1, denoted LAP1s, by mutating the initiating methionines for LAP2 and LIP was used to assess the contribution of LAP1 alone. Viruses with cDNAs directing the expression of LAP2 (that will also express LIP) and LIP were also used. Cells were plated on an extracellular matrix in the absence or presence of lactogenic hormones to induce their differentiation. Functional differentiation was assessed by measuring the transcript levels of two abundant milk proteins: β-casein and whey acidic protein (WAP).
In MECs expressing all three C/EBPβ isoforms, we observed robust induction of β-casein and WAP following culture under conditions that bring about differentiation (Fig. 6). In contrast, expression of LAP2 led to an induction of β-casein and WAP that was <15% of that observed with LAP1. LIP was unable to restore differentiation (Fig. 6). Expression of LAP1 alone (LAP1s) was capable of inducing differentiation and, remarkably, did so to a degree that was comparable to wild-type C/EBPβ (Fig. 6). Thus, to the extent that in vitro differentiation of MECs affords, these observations suggest that LAP1 is the predominant isoform that is responsible for MEC differentiation, and the ratio of LAP1 and LAP2 to LIP does not appear to play an appreciable role in this process.
FIG 6.

C/EBPβ isoform-specific contribution to mammary epithelial cell differentiation. (A) LAP1, but not LAP2 or LIP, rescues differentiation of C/EBPβ−/− MECs in a cyclin D1-dependent manner. C/EBPβ−/− MECs were infected with retroviruses directing the expression of LAP1, LAP2, LIP, or empty vector. LAP1s, a derivative of LAP1, from which only LAP1 can be synthesized was also included. Also included was LAP1 plus one of two shRNAs to cyclin D1 (shD1-3 and shD1-4) or an irrelevant shRNA (shCtrl). Infected cells were plated on an extracellular matrix (Matrigel) in the absence or presence of lactogenic hormones (insulin, prolactin, and hydrocortisone) and allowed to differentiate for 6 days. mRNA levels for β-casein normalized to GAPDH mRNA were determined by quantitative RT-PCR. The results are means ± the SD from three independent experiments. (B) Same analyses as in panel A, except whey acid protein (WAP) was analyzed. (C) For the experiments shown in panels A and B, the expression of the C/EBPβ isoforms, LAP1s, and degree of knockdown of cyclin D1 were assessed by IB using antibodies to C/EBPβ and cyclin D1. α-Tubulin was used as a loading control.
Our structure-function analysis of cyclin D1 and C/EBPβ suggests that cyclin D1 can activate the transcriptional function of LAP1. To begin to assess whether this pathway is relevant to MEC differentiation, we suppressed cyclin D1 expression using two different shRNAs in C/EBPβ-deficient MECs in which LAP1 had been re-expressed. We found that the ability of LAP1 to restore differentiation was inhibited upon knockdown of cyclin D1 (Fig. 6), suggesting that the function of LAP1 during differentiation is cyclin D1 dependent.
Cyclin D1-mediated activation of C/EBPβ LAP1 promotes mammary epithelial cell differentiation.
To more directly assess whether the function of cyclin D1 during MEC differentiation is, at least in part, to activate the transcriptional function of LAP1, we used the normal murine MEC line SCp2 (24). Endogenous cyclin D1 and C/EBPβ localized to a cyclin D1 responsive promoter by ChIP as a function of differentiation (Fig. 7A), suggesting that the output of transcriptional program effected by cyclin D1 and LAP1 increases with differentiation. Increased localization of these proteins to a cyclin D1 target gene in differentiated cells is consistent with the observations that cyclin D1 and to some extent C/EBPβ are induced during differentiation in vitro and in vivo (5, 25, 30).
FIG 7.

Cyclin D1-mediated activation of LAP1 participates in mammary epithelial cell differentiation. (A) Cyclin D1 and C/EBPβ localize to a cyclin D1-responsive promoter in differentiated MECs. The MEC line SCp2 were plated on an extracellular matrix (Matrigel) in the absence or presence of lactogenic hormones (insulin, prolactin, and hydrocortisone) and allowed to differentiate for 6 days. Subsequently, ChIP was performed with antibody to C/EBPβ, cyclin D1, or IgG as a control. Using the precipitated DNA, sequences spanning the cyclin D1 response element (C/EBPβ site) in the promoter of Hsc70 and 4 kb downstream of this element were analyzed by PCR; one-tenth of the lysate used for ChIP was also subjected to PCR (Input). The results are representative of at least two independent experiments. (B) The ability of cyclin D1 to activate the transcriptional function of LAP1 effects mammary epithelial cell differentiation. SCp2 cells were infected with retroviruses directing the expression of constitutively active LAP1, C/EBPβΔSpl (βSpl), dominant-negative LAP1, C/EBPβΔSpl(32A,34A) [βSpl(32A,34A)], one of two shRNAs to cyclin D1 (shD1-3 and shD1-4), or empty vector. Also, in the setting of cyclin D1 knockdown, βSpl was expressed. Infected cells were then differentiated, and the expression of β-casein was analyzed as described in Fig. 6A. The results are means ± the SD from three independent experiments. (C) Same analyses as in panel A, except whey acid protein (WAP) was analyzed. (D) For the experiments shown in panels A and B, the expression of the βSpl, βSpl(32A,34A), and the degree of knockdown of cyclin D1 was assessed by IB using antibodies to C/EBPβ and cyclin D1. α-Tubulin was used as a loading control.
Next, we introduced into SCp2 cells the dominant-negative C/EBPβ LAP1 mutant or silenced the expression of cyclin D1 with shRNAs and assessed the effect on differentiation. As expected, in cells where cyclin D1 was knocked down there was a marked reduction in the induction of β-casein and WAP relative to vector-infected cells (Fig. 7B, C, and D). Expression of the dominant-negative mutant of LAP1 also resulted in an inhibition of differentiation (Fig. 7B, C, and D), suggesting that the effect of loss of cyclin D1 can be phenocopied by expression of the dominant-negative LAP1 that can block the transcriptional function of cyclin D1.
As a complementary strategy, we sought to determine whether the constitutively activate mutant of LAP1 can functionally replace cyclin D1 during differentiation. Into MECs expressing shRNAs to cyclin D1 to silence its expression, we also expressed C/EBPβΔSpl. Upon differentiation, we observed that the constitutively active mutant of LAP1, which does not require cyclin D1 for activation, was able to reverse the inhibition of β-casein and WAP induction elicited by silencing cyclin D1 (Fig. 7B, C, and D). Together with the results noted above, these observations suggest that cyclin D1 and C/EBPβ LAP1 operate in the same pathway to promote MEC differentiation and that the ability of cyclin D1 to activate LAP1 figures prominently in this pathway.
DISCUSSION
While our previous work suggested the existence of a functional interaction between cyclin D1 and the transcription factor C/EBPβ (23), the nature of this connection and its ability to bring about a biological outcome were unknown. Here, we have provided evidence that cyclin D1 and C/EBPβ operate in a common pathway to promote MEC differentiation. Our studies are consistent with a model in which cyclin D1 relieves an autoinhibited state in LAP1 effected by intramolecular interactions, thereby allowing it to activate transcription. We have shown that LAP1, but not LAP2 or LIP, is the C/EBPβ isoform that promotes MEC differentiation. Moreover, the ability of cyclin D1 to activate the transcriptional function of LAP1 participates in MEC differentiation. Our findings suggest that the mammary gland defects observed in both cyclin D1- and C/EBPβ-deficient mice can be attributed, at least in part, to a block in the cyclin D1-LAP1 pathway.
Most studies on the regulation of the transcriptional output of C/EBPβ focus on two areas: the ratios between the isoforms and posttranslational modifications. Our findings suggest a third mode of activation—protein-protein interaction. Binding of cyclin D1 with one of the conserved regions in the N-terminal transactivation domain (CR2) disrupts the intramolecular interaction between CR2 and a conserved internal negative regulatory region (CR7) in LAP1. This event exposes residues in CR2 that are essential to the transactivation potential of LAP1. This mode of activation of LAP1 by cyclin D1 is not all that different from that proposed for one of the posttranslational modifications in C/EBPβ. Specifically, one of the most studied phosphorylation events in C/EBPβ occurs within CR7 (14, 16, 17, 31) and, like cyclin D1, this event relieves the autoinhibited state in C/EBPβ (32, 33). However, the effect of this phosphorylation event and cyclin D1 binding are distinct. Phosphorylation of CR2 can activate both LAP1 and LAP2 (14, 16, 17), while cyclin D1 selectively affects LAP1. Since LAP1 and LAP2 can have different target genes and they possess distinct requirements for transcription (34), there are likely situations where it is necessary to preferentially activate LAP1; cyclin D1 possesses this ability.
Although our analyses have focused on the interaction between CR2 and CR7 in LAP1 and the ability of cyclin D1 to disrupt this interaction, the model is likely incomplete. For example, it remains to formally demonstrate that cyclin D1 can induce a conformational change in LAP1. Further, there are other intermolecular interactions that take place in LAP1 beyond those described here. For example, it has been shown that the transactivation domain (CR1 to -4) can interact with both CR5 and CR7 (14), and LAP2 has been found to possess internal negative regulatory elements that control DNA binding and cell type specificity with respect to transcriptional activation (15). Whether these regulatory processes participate in activation of LAP1 by cyclin D1 remains to be determined.
Another aspect of the regulation of LAP1 activity is how physiological extracellular signals impinge on the cyclin D1-LAP1 pathway to bring about its activation. In the mammary gland, it is known that development is influenced by steroid and peptide hormones. And it has been shown that cyclin D1 is induced during pregnancy (30), and estrogen, progesterone, and prolactin can upregulate the expression of cyclin D1 (35–37). The importance of these signals to the activation of the pathway is demonstrated by our observation that LAP1 rescues the defects associated with loss of C/EBPβ only in the presence of lactogenic hormones. Regardless, it remains to be determined whether extracellular signals also impinge on LAP1 for its efficient activation by cyclin D1. Further, the possibility of steroid and peptide hormones working in parallel with the cyclin D1-LAP1 pathway cannot be ruled out. Efforts to address these issues will have implications on our understanding of the cancer-associated function of cyclin D1 effected by its amplification and overexpression (see below).
To the extent offered by the analyses of in vitro assays of MEC differentiation, LAP1 appears to be the prominent isoform of C/EBPβ that effects differentiation. Further, in our studies, the ratio of LAP1 to the other forms does not influence the ability of LAP1 to rescue defects associated with C/EBPβ loss. This does not rule out important roles for LAP2 and LIP, whose functions will likely be revealed by studies performed in vivo. In this regard, in limited analysis, no defects in mammary gland development were observed in mice lacking LAP2 or LIP (38, 39).
A cdk-independent function for cyclin D1 during mammary gland development was suggested by the observation that mice harboring a mutant of cyclin D1 deficient in activating cdk4 and cdk6, cyclin D1(K112E), display no overt defects in development (21), unlike those lacking cyclin D1 (19, 20). This same mutant of cyclin D1 retains the ability to induce transcription in a C/EBPβ-dependent manner (23). The findings presented here suggest that C/EBPβ LAP1 is an effector of cyclin D1 during mammary gland development.
Previously, through analyses involving the deconvolution of the patterns of gene expression in human cancer, we suggested that overexpressed cyclin D1 induces endogenous gene expression that is mediated by and dependent upon C/EBPβ (23). The analyses presented here validate our bioinformatic analysis in that they suggest that the functional interaction between cyclin D1 and C/EBPβ can effect a defined biological outcome. Further, they suggest that the oncogenic action of cyclin D1 does not represent a gain of function but rather is manifest as a deregulation of its normal function, at least in the breast. This suggests that we can build on the developmental studies reported here to provide insights into the oncogenic action of the cyclin D1-LAP1 pathway involved in breast cancer.
ACKNOWLEDGMENTS
We thank members of the Ewen lab and Phil Hinds for comments and critical review of the manuscript, and we thank Mina Bissell, Phil Auron, Dan Tenen, Zena Werb, Jeng-Shin Lee, Richard Mulligan, Dan Silver, Peter Sicinski, Nelson Brown, Ron Evans, and Andrew Kung for reagents and advice.
This study was supported by National Institutes of Health/National Cancer Institute grant R01 CA138944.
Footnotes
Published ahead of print 9 June 2014
REFERENCES
- 1.Nerlov C. 2010. Transcriptional and translational control of C/EBPs: the case for “deep” genetics to understand physiological function. Bioessays 32:680–686. 10.1002/bies.201000004 [DOI] [PubMed] [Google Scholar]
- 2.Nerlov C. 2007. The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell. Biol. 17:318–324. 10.1016/j.tcb.2007.07.004 [DOI] [PubMed] [Google Scholar]
- 3.Grimm SL, Rosen JM. 2003. The role of C/EBPβ in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia 8:191–204. 10.1023/A:1025900908026 [DOI] [PubMed] [Google Scholar]
- 4.Robinson GW, Johnson PF, Hennighausen L, Sterneck E. 1998. The C/EBPβ transcription factor regulates epithelial cell proliferation and differentiation in the mammary gland. Genes Dev. 12:1907–1916. 10.1101/gad.12.12.1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seagroves TN, Krnacik S, Raught B, Gay J, Burgess-Beusse B, Darlington GJ, Rosen JM. 1998. C/EBPβ, but not C/EBPα, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev. 12:1917–1928. 10.1101/gad.12.12.1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hennighausen L, Robinson GW. 2005. Information networks in the mammary gland. Nat. Rev. Mol. Cell. Biol. 6:715–725. 10.1038/nrm1714 [DOI] [PubMed] [Google Scholar]
- 7.Hynes NE, Watson CJ. 2010. Mammary gland growth factors: roles in normal development and in cancer. Cold Spring Harb. Perspect. Biol. 2:a003186. 10.1101/cshperspect.a003186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brisken C, O'Malley B. 2010. Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2:a003178. 10.1101/cshperspect.a003178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Descombes P, Schibler U. 1991. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67:569–579. 10.1016/0092-8674(91)90531-3 [DOI] [PubMed] [Google Scholar]
- 10.Calkhoven CF, Muller C, Leutz A. 2000. Translational control of C/EBPα and C/EBPβ isoform expression. Genes Dev. 14:1920–1932 [PMC free article] [PubMed] [Google Scholar]
- 11.Smink JJ, Begay V, Schoenmaker T, Sterneck E, de Vries TJ, Leutz A. 2009. Transcription factor C/EBPβ isoform ratio regulates osteoclastogenesis through MafB. EMBO J. 28:1769–1781. 10.1038/emboj.2009.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirai Y, Radisky D, Boudreau R, Simain M, Stevens ME, Oka Y, Takebe K, Niwa S, Bissell MJ. 2001. Epimorphin mediates mammary luminal morphogenesis through control of C/EBPβ. J. Cell Biol. 153:785–794. 10.1083/jcb.153.4.785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams SC, Cantwell CA, Johnson PF. 1991. A family of C/EBP-related proteins capable of forming covalently linked leucine dimers in vitro. Genes Dev. 5:1553–1567. 10.1101/gad.5.9.1553 [DOI] [PubMed] [Google Scholar]
- 14.Kowenz-Leutz E, Twamley G, Ansieau S, Leutz A. 1994. Novel mechanism of C/EBPβ (NF-M) transcriptional control: activation through derepression. Genes Dev. 8:2781–2791. 10.1101/gad.8.22.2781 [DOI] [PubMed] [Google Scholar]
- 15.Williams SC, Baer M, Dillner AJ, Johnson PF. 1995. CRP2 (C/EBPβ) contains a bipartite regulatory domain that controls transcriptional activation, DNA binding and cell specificity. EMBO J. 14:3170–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakajima T, Kinoshita S, Sasagawa T, Sasaki K, Naruto M, Kishimoto T. 1993. Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc. Natl. Acad. Sci. U. S. A. 90:2207–2211. 10.1073/pnas.90.6.2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu S, Yoon K, Sterneck E, Johnson PF, Smart RC. 2002. CCAAT/enhancer binding protein-β is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc. Natl. Acad. Sci. U. S. A. 99:207–212. 10.1073/pnas.012437299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463:899–905. 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge S, Weinberg RA. 1995. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82:621–630. 10.1016/0092-8674(95)90034-9 [DOI] [PubMed] [Google Scholar]
- 20.Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. 1995. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 9:2364–2372. 10.1101/gad.9.19.2364 [DOI] [PubMed] [Google Scholar]
- 21.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. 2006. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell 9:13–22. 10.1016/j.ccr.2005.12.019 [DOI] [PubMed] [Google Scholar]
- 22.Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, Sicinski P. 2006. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 9:23–32. 10.1016/j.ccr.2005.12.012 [DOI] [PubMed] [Google Scholar]
- 23.Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, Zahnow CA, Patterson N, Golub TR, Ewen ME. 2003. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell 114:323–334. 10.1016/S0092-8674(03)00570-1 [DOI] [PubMed] [Google Scholar]
- 24.Desprez P-Y, Roskelley C, Campisi J, Bissell MJ. 1993. Isolation of functional cell lines from a mouse mammary epithelial cell strain: the importance of basement membrane and cell-cell interaction. Mol. Cell Diff. 1:99–110 [Google Scholar]
- 25.Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, DiRenzo J, Pestell RG, Hinds PW, Dowdy SF, Brown M, Ewen ME. 1997. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol. Cell. Biol. 17:5338–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welm BE, Dijkgraaf GJ, Bledau AS, Welm AL, Werb Z. 2008. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell 2:90–102. 10.1016/j.stem.2007.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox E, Silver PA, Brown M. 2005. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122:33–43. 10.1016/j.cell.2005.05.008 [DOI] [PubMed] [Google Scholar]
- 28.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15:871–875. 10.1038/nbt0997-871 [DOI] [PubMed] [Google Scholar]
- 29.Tsukada J, Saito K, Waterman WR, Webb AC, Auron PE. 1994. Transcription factors NF-IL6 and CREB recognize a common essential site in the human prointerleukin 1β gene. Mol. Cell. Biol. 14:7285–7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA. 2008. Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene 27:1231–1242. 10.1038/sj.onc.1210738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanlon M, Sturgill TW, Sealy L. 2001. ERK2- and p90Rsk2-dependent pathways regulate the CCAAT/enhancer-binding protein-β interaction with serum response factor. J. Biol. Chem. 276:38449–38456. 10.1074/jbc.M102165200 [DOI] [PubMed] [Google Scholar]
- 32.Mo X, Kowenz-Leutz E, Xu H, Leutz A. 2004. Ras induces mediator complex exchange on C/EBPβ. Mol. Cell 13:241–250. 10.1016/S1097-2765(03)00521-5 [DOI] [PubMed] [Google Scholar]
- 33.Kowenz-Leutz E, Pless O, Dittmar G, Knoblich M, Leutz A. 2010. Crosstalk between C/EBPβ phosphorylation, arginine methylation, and SWI/SNF/Mediator implies an indexing transcription factor code. EMBO J. 29:1105–1115. 10.1038/emboj.2010.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kowenz-Leutz E, Leutz A. 1999. A C/EBPβ isoform recruits the SWI/SNF complex to activate myeloid genes. Mol. Cell 4:735–743. 10.1016/S1097-2765(00)80384-6 [DOI] [PubMed] [Google Scholar]
- 35.Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, Yasuda H, Smyth GK, Martin TJ, Lindeman GJ, Visvader JE. 2010. Control of mammary stem cell function by steroid hormone signalling. Nature 465:798–802. 10.1038/nature09027 [DOI] [PubMed] [Google Scholar]
- 36.Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, Stingl J, Waterhouse PD, Khokha R. 2010. Progesterone induces adult mammary stem cell expansion. Nature 465:803–807. 10.1038/nature09091 [DOI] [PubMed] [Google Scholar]
- 37.Brisken C, Ayyannan A, Nguyen C, Heineman A, Reinhardt F, Jan T, Dey SK, Dotto GP, Weinberg RA. 2002. IGF-2 is a mediator of prolactin-induced morphogenesis in the breast. Dev. Cell 3:877–887. 10.1016/S1534-5807(02)00365-9 [DOI] [PubMed] [Google Scholar]
- 38.Uematsu S, Kaisho T, Tanaka T, Matsumoto M, Yamakami M, Omori H, Yamamoto M, Yoshimori T, Akira S. 2007. The C/EBPβ isoform 34-kDa LAP is responsible for NF-IL-6-mediated gene induction in activated macrophages, but is not essential for intracellular bacteria killing. J. Immunol. 179:5378–5386. 10.4049/jimmunol.179.8.5378 [DOI] [PubMed] [Google Scholar]
- 39.Wethmar K, Begay V, Smink JJ, Zaragoza K, Wiesenthal V, Dorken B, Calkhoven CF, Leutz A. 2010. C/EBPβΔuORF mice—a genetic model for uORF-mediated translational control in mammals. Genes Dev. 24:15–20. 10.1101/gad.557910 [DOI] [PMC free article] [PubMed] [Google Scholar]