

Abstract
Cross talk between the actin cytoskeleton and microtubules (MT) has been implicated in the amplification of agonist-induced Rho signaling, leading to increased vascular endothelial permeability. This study tested the involvement of actin-MT cross talk in the mechanisms of barrier enhancement induced by hepatocyte growth factor (HGF) and evaluated the role of the adaptor protein IQGAP1 in integrating the MT- and actin-dependent pathways of barrier enhancement. IQGAP1 knockdown by small interfering RNA attenuated the HGF-induced increase in endothelial barrier properties and abolished HGF-activated cortical actin dynamics. IQGAP1 reduction abolished HGF-induced peripheral accumulation of Rac cytoskeletal effector cortactin and cortical actin remodeling. In addition, HGF stimulated peripheral MT growth in an IQGAP1-dependent fashion. HGF also induced Rac1-dependent IQGAP1 association with the MT fraction and the formation of a protein complex containing end-binding protein 1 (EB1), IQGAP1, and cortactin. Decreasing endogenous IQGAP1 abolished HGF-induced EB1-cortactin colocalization at the cell periphery. In turn, expression of IQGAP1ΔC (IQGAP1 lacking the C-terminal domain) attenuated the cortactin association with EB1 and suppressed HGF-induced endothelial cell peripheral actin cytoskeleton enhancement. These results demonstrate for the first time the MT-actin cross talk mechanism of HGF-induced endothelial barrier enhancement and suggest that IQGAP1 functions as a hub linking HGF-induced signaling to MT and actin remodeling via EB1-IQGAP1-cortactin interactions.
INTRODUCTION
Vascular permeability under physiologic and pathological conditions is controlled by a spectrum of circulating vasoactive mediators. Hepatocyte growth factor (HGF) is a multifunctional, mesenchyme-derived, pleiotropic factor secreted by several cell types that regulates different biological processes, including the maintenance of vascular barrier integrity (1–4). Increased HGF levels have been detected in inflamed lungs and suggested to serve as a compensatory mechanism to help protect lung vascular integrity under acute lung injury conditions and attenuate the devastating consequences of lung inflammation and tissue injury (5). Barrier-protective effects of HGF have been observed in pulmonary and cerebral endothelial cell (EC) monolayers (6, 7).
Agonist-mediated control of endothelial cytoskeleton and monolayer permeability involves multiple cytoskeletal signaling proteins. Agonist-induced EC hyperpermeability often develops as a result of receptor-mediated activation of Rho GTPase and Rho-associated kinase, which leads to increased myosin light chain (MLC) phosphorylation, actin stress fiber formation, actomyosin contraction, and cell retraction (8). In turn, EC monolayer recovery and actin reorganization are characterized by cytoskeletal relaxation, dissolution of stress fibers, activation of cortical actin polymerization, and actin filament branching, leading to increased cytoskeletal motility and resealing of intercellular gaps. These events are regulated by Rac GTPase-dependent mechanisms (9).
HGF binding to its receptor, c-Met, stimulates receptor tyrosine kinase activity and activates multiple downstream signaling kinases and small GTPases. Activated Rac1 GTPase stimulates the cytoskeletal effectors cortactin, Arp2/3, and others, which leads to cortical actin thickening, assembly of adherens junctions, and enhancement of the endothelial barrier (2, 9). Emerging evidence suggests a role for cross talk between cytoskeletal elements such as microtubules (MT) and the actin network in the regulation of Rho GTPase signaling and the maintenance of cell monolayer integrity. For example, partial disassembly of the peripheral MT pool by barrier-disruptive agents leads to release of the MT-associated Rho activator GEF-H1, leading to a further increase in Rho signaling, actin remodeling, and EC barrier dysfunction (10). This mechanism of MT-actin cytoskeletal cross talk has been shown to be involved in endothelial hyperpermeability induced by transforming growth factor beta (TGF-β), tumor necrosis factor alpha (TNF-α), lipopolysaccharide (LPS), thrombin, and EC exposure to a high-magnitude cyclic stretch (11–14). However, the role of signaling cross talk between MT and the actin cytoskeleton in the enhancement or restoration of EC barrier properties remains virtually unexplored. Recent studies connected the increased extension of MT to the cell cortical region with activation of cortical actin polymerization and cortical cytoskeletal enhancement (15). The mechanistic links between peripheral MT extension and cortical actin cytoskeleton enhancement and the proteins integrating signals from MT to initiate Rac-dependent actin polymerization remain unknown.
IQGAP1 is a multifunctional regulatory protein involved in cytoskeletal remodeling and assembly of cell junctions. IQGAP1 controls MT and actin cytoskeletal dynamics via interactions with the small GTPases Rac1 and Cdc42. IQGAP1 contains a calponin homology domain (CHD), four calmodulin-binding IQ domains, a RasGAP-related domain (GRD), and a RasGAP C-terminal domain (16). IQGAP1 interacts with several proteins, including signaling molecules like Cdc42 and Rac1 GTPases, calmodulin, β-catenin, E-cadherin, actin filaments, MT-associated plus-end tracking proteins (CLIP170 and CLASP-2), adenomatous polyposis coli, and others (17, 18). The interactions with the actin cytoskeleton, MT, and adherens junction proteins (19–21) suggest a role for IQGAP1 in MT-actin functional cross talk and local regulation of Rac/Cdc42 signaling. This study examined the role of IQGAP1 in MT peripheral growth and MT-actin cytoskeleton interactions as a potential mechanism of HGF-induced barrier enhancement in the vascular endothelium.
MATERIALS AND METHODS
Cell culture and reagents.
Human HGF was obtained from R&D Systems (Minneapolis, MN). The cell-permeating c-Met kinase inhibitor N-[3-fluoro-4-(7-methoxy-4-quinolinyl)phenyl]-1-(2-hydroxy-2-methylpropyl)-5-methyl-3-oxo-phenyl-2,3-dihydro-1H-pyrazole carboxamide was from EMD Millipore (Billerica, MA). Texas Red-conjugated phalloidin and Alexa Fluor 488 were purchased from Molecular Probes (Eugene, OR). End-binding protein 1 (EB1) and Rac1 antibodies were purchased from BD Transduction Laboratories (San Diego, CA), IQGAP1, anti-Myc tag antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), cortactin and phospho-Y421-cortactin antibodies were from Millipore (Billerica, MA), and diphosphorylated MLC antibody was obtained from Cell Signaling (Beverly, MA). Unless otherwise specified, all biochemical reagents, including β-actin and β-tubulin antibodies, were obtained from Sigma (St. Louis, MO). Human pulmonary artery endothelial cells (HPAEC) were obtained from Lonza (East Rutherford, NJ) and used for experiments at passages 5 to 7.
siRNA and DNA transfections.
HPAEC were treated with predesigned IQGAP1-specific small interfering RNA (siRNA). A set of three Stealth Select siRNA duplexes was purchased from Invitrogen (Carlsbad, CA) in a ready-to-use, desalted, deprotected, annealed, double-stranded form. Predesigned Rac1-specific siRNA of standard purity was ordered from Ambion (Austin, TX). Transfection of EC with siRNA was performed as previously described (22). Nonspecific, nontargeting RNA (Dharmacon, Lafayette, CO) was used as a control treatment. Seventy-two hours after transfection, cells were harvested and used for experiments. Plasmids encoding Myc-tagged full-length IQGAP1 and an IQGAP1ΔC mutant isoform lacking amino acids 1502 to 1657 (23) were used for transient transfections according to the protocols described previously (12, 24). After 24 h of transfection, cells were treated with the agonist of interest and used for experiments.
Analysis of EC permeability.
EC permeability to macromolecules was monitored by the express permeability testing assay (XPerT) recently developed by our group (25) and now available from Millipore (Vascular Permeability Imaging Assay, catalog number 17-10398). This assay is based on the high-affinity binding of a non-cell-permeating, avidin-conjugated, fluorescein isothiocyanate (FITC)-labeled tracer to biotinylated extracellular matrix proteins immobilized on the bottoms of culture dishes covered with EC monolayers. FITC-avidin binding to the bottom of a matrix-coated culture dish increases if the EC barrier is compromised by treatment with a barrier-disruptive agonist. In permeability visualization experiments, 15 min after EC stimulation with HGF, an FITC-avidin solution was added directly to the culture medium for 3 min before termination of the experiment. Unbound FITC-avidin was washed out with phosphate-buffered saline (PBS; pH 7.4, 37°C), cells were fixed with 3.7% formaldehyde in PBS (10 min, room temperature), and visualization of FITC-avidin on the bottoms of coverslips was performed with the Nikon Eclipse TE 300 imaging system (Nikon, Tokyo, Japan) equipped with a digital camera (DKC 5000; Sony, Tokyo, Japan); 10× objective lenses were used. Images were processed with Photoshop 7.0 software (Adobe Systems, San Jose, CA). For permeability assays in 96-well plates, HPAEC were seeded onto biotinylated gelatin-coated 96-well plates, transfected with nonspecific or IQGAP1-specific siRNA, and grown for 72 h prior to testing. FITC-avidin solution was added directly to the culture medium 3 min before termination of the experiment. After washing away of unbound FITC-avidin, the fluorescence of matrix-bound FITC-avidin was measured on a Victor X5 multilabel plate reader (PerkinElmer, Waltham, MA). Measurements of transendothelial electrical resistance (TER) across confluent HPAEC monolayers were performed with an electrical cell-substrate impedance-sensing system (Applied Biophysics, Troy, NY) as previously described (24, 26).
Immunofluorescence and live-cell imaging.
Endothelial cells plated on glass coverslips were treated with the agonist of interest, fixed in 3.7% formaldehyde in PBS for 10 min at 4°C, washed three times with PBS, permeabilized with 0.1% Triton X-100 in PBS-Tween (PBST) for 30 min at room temperature, and blocked with 2% bovine serum albumin (BSA) in PBST for 30 min. Incubations with antibodies to the Myc tag or cortactin were performed in blocking solution (2% BSA in PBST) for 1 h at room temperature and followed by staining with Alexa 488-conjugated secondary antibodies. Actin filaments were stained with Texas Red-conjugated phalloidin. After immunostaining, slides were analyzed with a Nikon video imaging system (Nikon Instech Co.) as described elsewhere (26, 27). For live imaging of green fluorescent protein (GFP)-cortactin, cells were plated on MatTek dishes (MatTek, Ashland, MA) and transfected with a GFP-cortactin plasmid. Time-lapse images were acquired with a 100×, numerical aperture (NA) 1.45 oil objective in a 3I Marianas Yokogawa-type spinning-disk confocal system equipped with a CO2 chamber and a heated stage. For time-lapse MT plus-end tracking, cells were transfected with GFP-EB1 and images were acquired at 2-s intervals for 40 to 60 s with a 100×, NA 1.45 oil objective in a manner similar to that used in experiments with GFP-cortactin. Twenty consecutive images obtained under each condition were used for projection analysis with ImageJ software. For MT quantification, cells were fixed with −20°C methanol and immunostaining was carried out with β-tubulin as described previously (15, 28). For tracking analysis of peripheral MT tip growth, EB1 in the cell margin area (2 to 10 μm from the cell border) was tracked with the Manual Tracking plug-in of the ImageJ software. The median track length was calculated with the Excel software.
Isolation of MT-enriched fractions.
MT-enriched fractions were isolated as previously described (29). After agonist stimulation, cells were incubated with extraction buffer containing PEM [100 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES; pH 6.75), 1 mM EGTA, 1 mM MgSO4], 0.5% NP-40, and a protease and phosphatase inhibitor cocktail for 10 min at room temperature. The cytosolic fraction containing soluble tubulin was collected at room temperature by centrifugation at 12,000 rpm for 15 min. The attached cells containing polymerized MT were incubated on ice for 30 min to induce MT depolymerization and tubulin release into the soluble fraction. Cells were scraped into PEM containing a protease and phosphatase inhibitor cocktail and passed several times through a 26-gauge needle. The cell debris was removed by centrifugation at 2,000 × g for 2 min at 4°C. Collection of the tubulin fraction corresponding to polymerized tubulin was performed after centrifugation at 92,000 × g for 30 min at 4°C to remove other cytoskeletal components (F-actin and intermediate filaments). Depolymerized tubulin from the supernatant was repolymerized in the presence of 20 μM paclitaxel (originally named taxol) and 1 mM GTP at 37°C for 1 h. Sedimentation of polymerized MT with associated proteins was performed by centrifugation at 92,000 × g for 30 min at room temperature over a 20% sucrose gradient in PEM containing a protease and phosphatase inhibitor cocktail and 20 μM paclitaxel. This approach allowed the reconstitution of MT-bound proteins with the exact fraction of tubulin corresponding to the pool of polymerized MT from control and agonist-stimulated cells. MT pellets were separated by SDS-PAGE. Sample loading normalized to the tubulin content was used for Western blot analysis to evaluate the relative amounts of MT-associated GEF-H1 in control and stimulated cells.
Coimmunoprecipitation, differential protein fractionation, and immunoblotting.
After agonist stimulation, cells were washed in cold PBS and lysed on ice with cold TBS–NP-40 lysis buffer (20 mM Tris [pH 7.4], 150 mM NaCl, 1% NP-40) supplemented with a protease and phosphatase inhibitor cocktail (Roche, Indianapolis, IN). Clarified lysates were then incubated with antibodies to Myc, IQGAP1, EB1, or cortactin overnight at 4°C and washed three or four times with TBS–NP-40 lysis buffer, and the complexes were analyzed by Western blotting with appropriate antibodies. In fractionation studies, cytosolic and membrane fractions were isolated with a subcellular protein fractionation kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's protocol. For analysis of the protein phosphorylation profile, cells were stimulated and lysed and then protein extracts were separated by SDS-PAGE, transferred to polyvinylidene fluoride membrane, and probed with specific antibodies. Equal protein loading was verified by reprobing membranes with an antibody to β-actin or the specific protein of interest.
Statistical analysis.
Results are expressed as mean values ± standard deviations (SDs) of three to five independent experiments. Stimulated samples were compared to controls by unpaired Student t tests. For multiple-group comparisons, a one-way analysis of variance, followed by the Fisher's post hoc test, was used. A P value of <0.05 was considered statistically significant.
RESULTS
IQGAP1 knockdown attenuates HGF-induced barrier enhancement and peripheral actin remodeling.
HGF exhibits a potent barrier-enhancing effect on EC monolayers, as reflected by a pronounced increase in TER (Fig. 1A). Interestingly, removal of HGF did not significantly affect the HGF-induced TER increase in the first hour, but the effect of HGF washout became evident at later times. This result suggests the sustained nature of HGF-induced cytoskeletal remodeling and strengthening of intercellular junctions, leading to EC barrier enhancement. The involvement of IQGAP1 in HGF-induced barrier enhancement mechanisms was tested by an siRNA-induced IQGAP1 knockdown approach. Seventy-two hours after transfection with nonspecific or IQGAP1-specific siRNA, EC were stimulated with HGF, and EC monolayer barrier properties were evaluated by measurements of TER. HGF treatment of EC monolayers transfected with nonspecific RNA duplexes increased TER levels (Fig. 1B). In contrast, IQGAP1 knockdown strongly attenuated the HGF-induced barrier enhancement response in a dose-dependent fashion.
FIG 1.

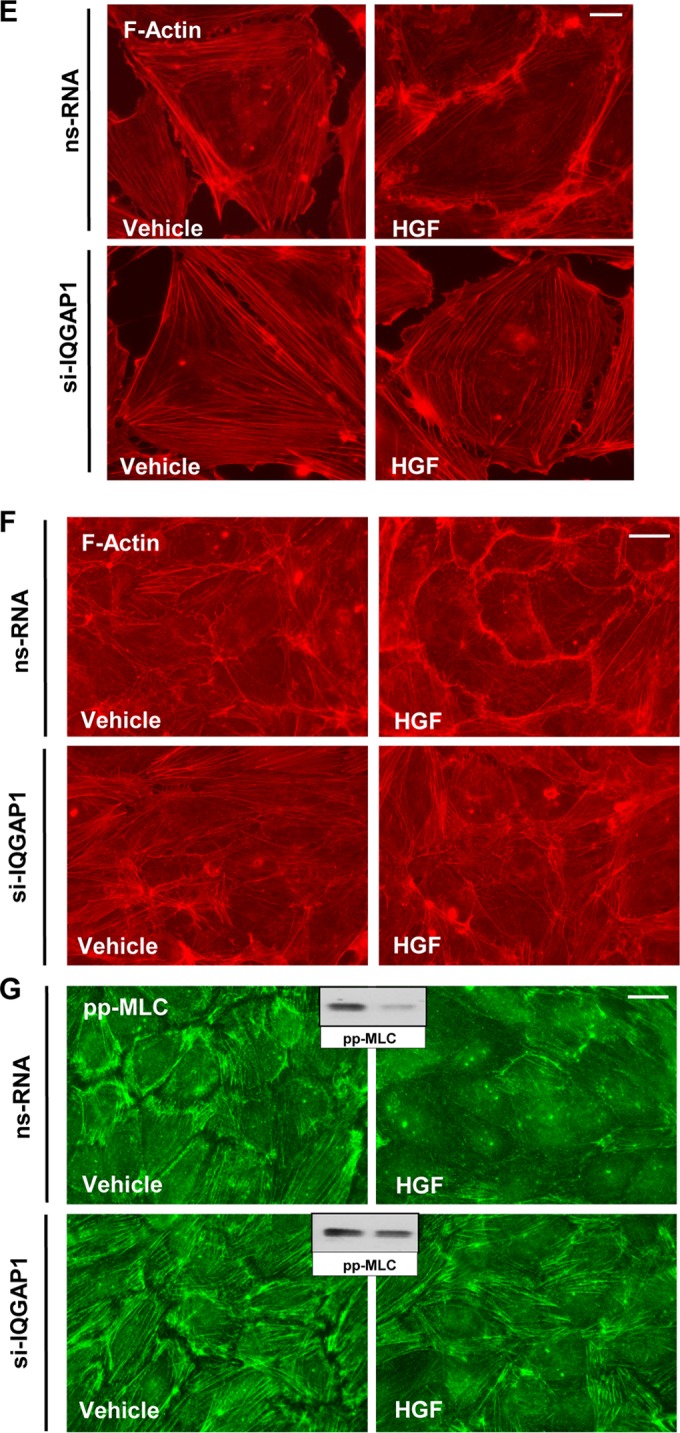
IQGAP1 knockdown attenuates HGF-induced EC barrier enhancement. (A to D) Permeability measurements. (A) TER measurements in EC stimulated with HGF (50 ng/ml) with or without HGF washout (marked by arrow). (B) EC transfected with 50 or 100 nM IQGAP1-specific siRNA (si-IQGAP1) or nonspecific RNA (nsRNA) were stimulated with HGF (50 ng/ml), and TER was monitored over time. The bar graph depicts EC permeability changes at the time point corresponding to the maximal TER increase. Results are represented as mean ± SD; *, P < 0.05; n = 3. The inset shows a quantitative analysis of IQGAP1 levels detected by immunoblotting in siRNA-treated EC. (C) HPAEC transfected with IQGAP1-specific siRNA or nonspecific RNA were stimulated with the vehicle or HGF (50 ng/ml); this was followed by the addition of FITC-avidin (25 μg/ml, 3 min). Unbound FITC-avidin was removed, and the FITC fluorescence signal on the coverslip bottom was visualized by fluorescence microscopy as described in Materials and Methods. Results are representative of four independent experiments. Bar = 5 μm. (D) Bar graph depicting a quantitative analysis of XPerT permeability assay data obtained in parallel experiments with 96-well plates. Results are presented as mean ± SD; *, P < 0.05; n = 6. (E and F) Sparsely seeded EC (E) or EC monolayers (F) transfected with IQGAP1-specific siRNA (si-IQGAP1) or nonspecific RNA were stimulated with HGF (50 ng/ml, 10 min). Immunofluorescence visualization of the actin cytoskeleton was performed by Texas Red-phalloidin staining. Results are representative of three independent experiments. Bars = 5 (E) and 10 (F) μm. (G) Immunofluorescence visualization of diphosphorylated MLC (pp-MLC) was performed by staining with the corresponding antibody. Results are representative of three independent experiments. Bar = 10 μm. Insets show Western blot analyses of pp-MLC in control and HGF-stimulated HPAEC.
The effects of IQGAP1 knockdown on HGF-induced EC barrier enhancement were further tested in a novel permeability assay (XPerT) that allows the visualization of local areas in EC monolayers with increased EC permeability to macromolecules. HGF significantly decreased basal EC monolayer permeability to FITC-labeled avidin (Fig. 1C, upper panels). The HGF barrier-enhancing effect in EC monolayers was abrogated by IQGAP1 knockdown (Fig. 1C, lower panels). The bar graph in Fig. 1D shows results of quantitative XPerT permeability assays performed in the 96-well plate format.
The effects of IQGAP1 knockdown on HGF-induced actin cytoskeletal remodeling were tested by immunofluorescence staining of EC with Texas Red-phalloidin. In control cells, HGF induced accumulation of F-actin at the cell periphery, reflecting activation of cortical actin polymerization (Fig. 1E). This process was inhibited in EC by IQGAP1 knockdown. Similar inhibitory effects of IQGAP1 knockdown on HGF-induced peripheral F-actin accumulation were observed in confluent EC monolayers (Fig. 1F). HGF stimulation also decreased the phosphorylation of MLC in nontransfected EC. siRNA-induced IQGAP1 knockdown did not significantly change the diphospho-MLC pattern under basal conditions but did attenuate the reduction of diphospho-MLC immunoreactivity caused by HGF (Fig. 1G).
IQGAP1 knockdown abolishes HGF-induced cortical actin dynamics.
Cortactin plays an essential role in cortical actin cytoskeletal dynamics. HGF-induced cortactin accumulation at the cell leading edge, signifying increased actin polymerization, was investigated by live microscopy. EC treated with nonspecific RNA and transfected with GFP-tagged cortactin were stimulated with HGF, and peripheral accumulation of GFP-cortactin was tracked over time (Fig. 2A, top panels). In similar experiments, EC were treated with IQGAP1-specific siRNA prior to transfection with GFP-cortactin and HGF stimulation (Fig. 2A, bottom panels). Treatment with HGF induced prominent cortactin accumulation at the edges of control cells (Fig. 2, arrows), and this effect was abolished in cells with IQGAP1 knockdown.
FIG 2.

IQGAP1 mediates HGF-induced cortactin activation. HPAEC transfected with IQGAP1-specific siRNA (si-IQGAP1) or nonspecific RNA (nsRNA) (100 nM, 72 h) were stimulated with HGF (50 ng/ml). (A) Live-cell imaging of control and IQGAP1-depleted cells expressing GFP-cortactin. Snapshots depict HGF-induced cortical dynamics at the periphery of control and IQGAP1-depleted cells. Arrows indicate cortactin accumulation at cell edges. Results are representative of three independent experiments. Bar = 5 μm. (B) HGF-induced cortactin phosphorylation in control and IQGAP1-depleted EC was evaluated by Western blotting with phospho-Y421-cortactin antibody. Equal protein loading was confirmed by probing the membrane with a cortactin antibody. siRNA-induced IQGAP1 knockdown was confirmed by probing whole-cell lysates with an IQGAP1 antibody. (C) Cells transfected with nonspecific RNA or IQGAP1-specific siRNA were treated with HGF for 5, 10, or 15 min, and membrane translocation of cortactin was analyzed by Western blot analysis of EC membrane fractions. siRNA-induced IQGAP1 knockdown was confirmed by probing whole-cell lysates with IQGAP1 antibody. Results are representative of four independent experiments. (D) Control and IQGAP1-depleted pulmonary EC cultures were stimulated with HGF (50 ng/ml, 10 min). HGF-induced cortactin translocation was evaluated by immunofluorescence staining of formaldehyde-fixed EC. Results are representative of three independent experiments. Bar = 5 μm.
Rac activation triggers signaling pathways leading to increased tyrosine phosphorylation of cortactin (30). We observed that knockdown of IQGAP1 almost completely abolished HGF-induced cortactin phosphorylation (Fig. 2B). Similarly, in cells with reduced IQGAP1, the ability of HGF translocation to the membrane/cytoskeletal compartment was impaired. (Fig. 2C). In agreement with these results, HGF induced cortactin localization in the submembrane regions at the periphery of control cells and IQGAP1 knockdown abrogated this effect (Fig. 2D).
IQGAP1 is required for stimulation of peripheral MT growth by HGF.
To assess a possible role for IQGAP1 in the HGF-induced changes in peripheral MT density, control and HGF-stimulated EC were fixed with methanol and subjected to immunofluorescence staining with an antibody to β-tubulin. Microscopic analysis of MT structures showed a more expanded peripheral MT network in HGF-treated EC than in cells treated with the vehicle (Fig. 3A, top panels). This effect was abolished when HGF-stimulated cells were transfected with IQGAP1-specific siRNA (Fig. 3A, bottom panels). The insets in Fig. 3A represent higher-magnification images of the MT network in the peripheral regions of HGF-stimulated control EC and cells with IQGAP1 knockdown. Quantitative analysis of peripheral MT density showed no differences between peripheral MT in control cells and those in IQGAP1 knockdown cells. However, IQGAP1 knockdown significantly attenuated the HGF-induced increase in peripheral MT in lung EC (Fig. 3A, inset).
FIG 3.

IQGAP1 promotes HGF-induced peripheral MT network formation. (A) Cells grown on coverslips were transfected with nonspecific RNA (ns-RNA) or IQGAP1-specific siRNA (si-IQGAP1) and stimulated with HGF (50 ng/ml, 10 min); this was followed by immunofluorescence staining of MT with an antibody against β-tubulin. The magnified images of the insets (right side) show details of MT structure. Bar = 5 μm. The bar graph presents a quantitative analysis of peripheral MT density in control and IQGAP1-depleted EC. Results are presented as mean ± SD; *, P < 0.05; n = 4 independent experiments, 10 cells per condition. Veh, vehicle. (B) Live-cell imaging of HPAEC transfected with IQGAP1-specific siRNA or nonspecific RNA and labeled with GFP-EB1. Projection analysis of 20 consecutive images before and after HGF (50 ng/ml) treatment shows changes in GFP-EB1 track length (left panels). Bar = 2 μm. The panels on the right show quantification of GFP-EB1 track length. Each pair of dots represents the median track length in a cell before and after HGF treatment. Results are representative of three independent experiments.
Effects of HGF on MT peripheral growth in control and IQGAP1 knockdown EC were further evaluated by live microscopy of GFP-labeled MT plus-end binding protein EB1 and tracking of EB1-positive MT tips. HGF stimulated MT peripheral growth, as illustrated by EB1 projections obtained from live images and quantitative analysis of microscopy data (Fig. 3B, top panels). Knockdown of IQGAP1 abrogated HGF-induced MT peripheral growth (Fig. 3B, bottom panels).
HGF induces Rac1-dependent IQGAP1 association with the MT fraction.
The role of IQGAP1 in HGF-induced control of MT dynamics and the nature of IQGAP1 interaction with MT remain incompletely understood. These effects were evaluated by biochemical analysis of MT fractions from control and HGF-stimulated pulmonary EC. HGF induced a rapid association of IQGAP1 with the MT fraction (Fig. 4A). This effect was abolished in EC pretreated with a pharmacologic inhibitor of the HGF receptor (c-Met) (Fig. 4B). HGF-induced association of IQGAP1 with the MT fraction was also abrogated in cells with knockdown of Rac1 (Fig. 4C). These data suggest that c-Met receptor activation by HGF induces Rac1-dependent association of IQGAP1 with MT.
FIG 4.
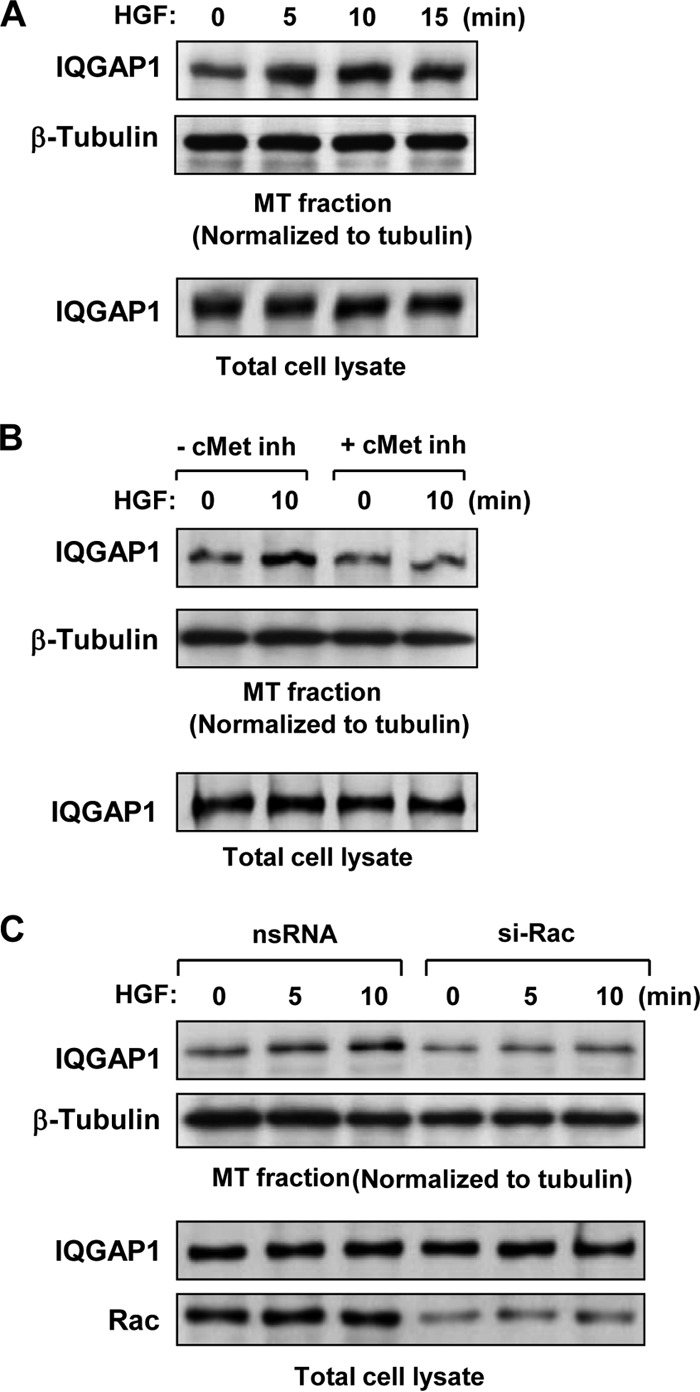
HGF stimulates Rac1-dependent association of IQGAP1 with the MT fraction. HPAEC were stimulated with HGF (50 ng/ml); this was followed by isolation of the MT-enriched fractionation. Western blotting was used to detect IQGAP1 in the MT fraction, normalized to tubulin content, and in total lysates isolated from control and HGF-treated EC. (A) Time course analysis of HGF-induced IQGAP1 association with the MT fraction. (B) Effect of c-Met inhibitor (inh; 50 nM carboxamide, 30 min) on HGF-induced IQGAP1 association with the MT fraction. (C) Effect of siRNA-induced Rac1 knockdown (50 nM, 48 h) on HGF-induced IQGAP1 association with the MT fraction. Rac1 protein depletion was confirmed by Western blot analysis of total cell lysates with a Rac1 antibody. Results are representative of three to five independent experiments.
HGF stimulates the association of EB1, IQGAP1, and cortactin.
The results described above suggest that HGF-induced actin cytoskeletal remodeling is dependent on IQGAP1 and involves cortactin accumulation at the cell periphery. In addition, HGF stimulates IQGAP1 association with MT. We next tested if HGF induces interactions among IQGAP1, cortactin, and MT proteins. Myc-IQGAP1 was coexpressed with GFP-cortactin or GFP-EB1 in HeLa cells. Immunoprecipitation with an anti-Myc antibody, followed by immunoblotting with a GFP antibody, revealed that both cortactin and EB-1 coimmunoprecipitated with IQGAP1 (Fig. 5A). GFP, which served as a negative control, was not found in a complex with Myc-IQGAP1.
FIG 5.
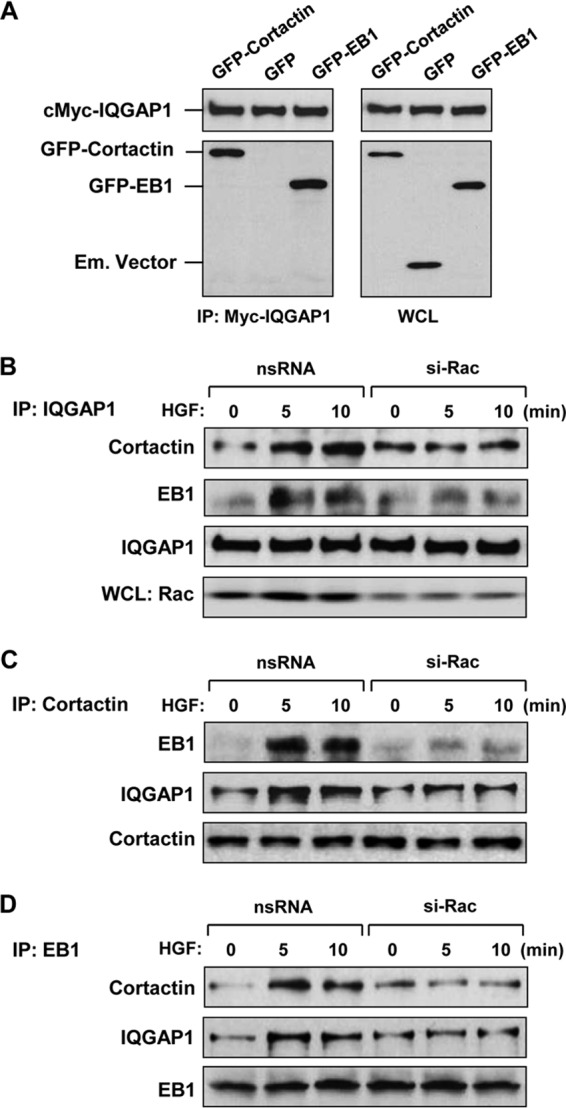
HGF induces Rac1-dependent association of IQGAP1, EB1, and cortactin. (A) Recombinant Myc-IQGAP1 and GFP-cortactin or GFP-EB1 was coexpressed in HeLa cells. Control transfections were performed with the empty (Em.) GFP vector. After immunoprecipitation (IP) with an anti-Myc tag antibody, the presence of overexpressed proteins in the immune complex was tested by Western blot analysis with anti-Myc tag and anti-GFP tag antibodies (left panel). Expression of recombinant proteins in HeLa cells was confirmed by Western blot analysis of whole-cell lysates (WCL) (right panel). (B to D) HPAEC transfected with nonspecific RNA (nsRNA) or Rac1-specific siRNA (si-Rac) were stimulated with HGF (50 ng/ml), and endogenous IQGAP1 (B), cortactin (C), and EB1 (D) proteins were immunoprecipitated under nondenaturing conditions with the appropriate antibodies. The presence of protein partners in immune complexes was tested by Western blotting. Rac1 protein depletion was confirmed by Western blot analysis of total cell lysates with a Rac1 antibody. Results are representative of three to six independent experiments.
Interactions among endogenous IQGAP1, cortactin, and EB1 in the human pulmonary endothelium were evaluated by using coimmunoprecipitation assays. HGF induced the association of IQGAP1 with cortactin and EB-1, which was abolished in EC with Rac1 knockdown (Fig. 5B). A series of reciprocal immunoprecipitation assays with antibodies to cortactin and EB1 confirmed the specificity of the interactions of endogenous IQGAP1, cortactin, and EB1 induced by HGF (Fig. 5C and D). These interactions were also dependent on Rac1.
To examine whether the cortactin-EB-1 interaction is mediated by IQGAP1, endogenous IQGAP1 was knocked down with siRNA and coimmunoprecipitation assays with EB1 or cortactin antibodies were performed with control and HGF-stimulated cells. The results show that IQGAP1 knockdown blocked HGF-induced complex formation between EB1 and cortactin (Fig. 6A and B). Quantitative analyses of these immunoprecipitation studies are presented in the bar graphs in Fig. 6A and B. Imaging studies performed with human pulmonary endothelium revealed increased EB1-cortactin colocalization at the cell periphery upon HGF stimulation (Fig. 6C). These effects were inhibited in IQGAP1 knockdown cells. Collectively, these results suggest that IQGAP1 serves as a scaffold for the recruitment of cortactin and EB1 in response to HGF treatment.
FIG 6.

HGF-induced EB1 and cortactin interactions are IQGAP1 dependent. (A, B) HPAEC treated with nonspecific RNA (nsRNA) or IQGAP1-specific siRNA (si-IQGAP1) were used for reciprocal coimmunoprecipitation (IP) assays with EB1 (A) and cortactin (B) antibodies, followed by Western blot assay detection of cortactin and EB1. Bar graphs depict quantitative densitometry analysis of immunoblotting data. Results are presented as mean ± SD; *, P < 0.05; n = 4. RDU, relative density units. (C) HPAEC grown on glass coverslips were stimulated with HGF (50 ng/ml); this was followed by double immunofluorescence staining for cortactin (red) and EB1 (green). The insets are shown at the bottom as higher-magnification images of the peripheral cell areas marked by quadrangles in the upper panels and depict cortactin and EB1 colocalization, which is yellow. Shown are representative results of three independent experiments. Bars = 5 μm.
IQGAP1ΔC mutant protein impairs HGF-induced peripheral MT growth and attenuates cortactin-EB1 association and EC barrier enhancement.
The IQGAP1 domain structure defines multiple interactions with its binding partners. The C-terminal domain has been implicated in IQGAP1 targeting to MT (18). We used a truncated mutant IQGAP1 protein with a preserved GRD (involved in interaction with Cdc42/Rac1 and responsible for IQGAP1 activation) but lacking amino acids 1502 to 1657 at the C terminus (termed IQGAP1ΔC) to test the role of the C terminus in EB-1-cortactin interactions and MT growth. In comparison with the expression of wild-type IQGAP1 in HPAEC, the expression of IQGAP1ΔC attenuated HGF-induced MT growth (Fig. 7). The bar graph in Fig. 7 presents a quantitative image analysis of peripheral MT density in EC transfected with the wild-type IQGAP1 and mutant IQGAP1ΔC proteins.
FIG 7.

Effect of the IQGAP1ΔC mutant protein on HGF-induced stimulation of peripheral MT network formation. Cells grown on coverslips were transfected with wild-type IQGAP1 (IQGAP1-WT) or IQGAP1ΔC and stimulated with HGF (50 ng/ml, 10 min). The MT network was visualized by immunofluorescence staining of methanol-fixed cells with an antibody against β-tubulin. Transfected cells were visualized by staining with an anti-Myc tag antibody. The insets are shown as magnified images on the right with details of MT structure in nontransfected (inset 1) and IQGAP1ΔC-transfected (inset 2) cells. Shown are representative results of three independent experiments. Bar = 5 μm. The bar graph presents a quantitative analysis of peripheral MT density in EC transfected with IQGAP1-WT or IQGAP1ΔC. Results are presented as mean ± SD; *, P < 0.05; n = 5 independent experiments, 10 cells per condition.
A coimmunoprecipitation assay with a cortactin antibody showed that expression of IQGAP1ΔC reduced the HGF-induced interaction between cortactin and EB1 (Fig. 8A). Expression of IQGAP1ΔC also attenuated HGF-induced cortactin phosphorylation (Fig. 8B), which was previously shown to be mediated by Rac1-dependent mechanisms (30). In addition, expression of IQGAP1ΔC both attenuated HGF-induced peripheral actin polymerization (Fig. 8C) and suppressed the EC barrier-enhancing response to HGF as monitored by elevation of TER in HGF-stimulated pulmonary EC monolayers (Fig. 8D).
FIG 8.

IQGAP1ΔC attenuates HGF-induced EB1-cortactin interactions, cortactin phosphorylation, and EC barrier enhancement. (A) HPAEC transfected with full-length IQGAP1 (IQGAP1-WT) or IQGAP1ΔC were stimulated with HGF (50 ng/ml, 10 min) and used in a coimmunoprecipitation (IP) assay with a cortactin antibody, followed by a Western blot assay for EB1 and cortactin. The bar graph represents a quantitative analysis of the Western blot assay data; *, P < 0.05; n = 4. (B) HGF-induced cortactin phosphorylation in EC expressing full-length IQGAP1 or IQGAP1ΔC was evaluated by a Western blot assay with a phospho-Y421-cortactin antibody. Equal protein loading was confirmed by probing with a cortactin antibody. (C) Expression of IQGAP1ΔC abrogates HGF-induced activation of cortical actin cytoskeleton and lamellipodium formation. F-actin was visualized by staining with Texas Red-phalloidin. IQGAP1ΔC-expressing cells were visualized by immunofluorescence staining with an anti-Myc tag antibody. The insets are shown as magnified images at the bottom with details of peripheral F-actin staining in nontransfected (inset 1) and IQGAP1ΔC-transfected (inset 2) cells. Shown are representative results of three independent experiments. Bar = 10 μm. (D) Permeability measurements. EC transfected with IQGAP1ΔC or wild-type IQGAP1 (IQGAP1-WT) were stimulated with HGF (50 ng/ml), and TER was monitored for 1.5 h. The Western blot assay at the upper right shows the expression of recombinant IQGAP1-WT and IQGAP1ΔC in HPAEC. The bar graph below it depicts EC permeability changes at the time point corresponding to the maximal TER increase. Results are represented as mean ± SD; *, P < 0.05; n = 5.
DISCUSSION
This study demonstrates a novel role for IQGAP1 in the mechanisms of HGF-induced barrier enhancement in vascular endothelium. Stimulation of EC with HGF promoted peripheral MT growth and protrusion of EB1-positive MT plus ends to the EC peripheral submembrane area which was dependent on IQGAP1. These effects were coupled to increased cortactin localization to the EC cortical layer and increased cortical actin polymerization, leading to decreased basal permeability of the EC monolayer. Experiments with IQGAP1 knockdown show that HGF-induced cortactin phosphorylation and peripheral accumulation, leading to peripheral actin cytoskeleton remodeling, is dependent on the presence of IQGAP1 and strongly depends on the ability of IQGAP1 to form a functional complex with cortactin and EB1. IQGAP1 knockdown also attenuated actin cortical remodeling, peripheral MT growth, and enhancement of the EC barrier function in response to HGF. Our unpublished studies show a similar effect of IQGAP1 inhibition on the attenuation of EC permeability in response to the barrier-enhancing phospholipid oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (A. Birukova and K. Birukov, unpublished data). These results indicate that Rac-mediated IQGAP1 activation and IQGAP1-dependent regulation of peripheral cytoskeletal dynamics may be a universal mechanism that regulates EC barrier function with barrier-protective agonists.
Cross talk between MT and the actin cytoskeleton has been shown to be associated with increased endothelial permeability in response to barrier-disruptive agonists such as thrombin, TGF-β, TNF-α, and LPS or direct MT disruption by colchicine or nocodazole (11, 13, 24, 31–33). The mechanism of this cross talk included partial dissolution of the MT network, release and activation of MT-associated Rho-specific guanine nucleotide exchange factor GEF-H1, stimulation of Rho-dependent MLC phosphorylation, actin stress fiber formation, cell contraction, and formation of intercellular gaps (11, 31).
In contrast to cross talk by barrier-disruptive agonists, HGF stimulation promoted Rac1-dependent association of IQGAP1 with MT and the formation of an EB1-IQGAP1-cortactin complex. The formation of this functional complex further stimulated local Rac1 signaling, as indicated by increased cortactin phosphorylation and subcortical accumulation (Fig. 2). The presence of EB1, an MT plus-end binding protein, in the coimmunoprecipitation complex with IQGAP1 and cortactin supports a role for this complex in capturing the MT tips in the actin cytoskeletal compartment at the cell periphery.
The presence of Rac1 activity was critical for the previously described formation of an IQGAP1–CLIP-170–Rac tripartite complex, which captured MT at the leading edge and promoted directional lamellipodium formation in epithelial cells (34). In agreement with these findings, our data strongly suggest that MT capture by IQGAP1 determines cortical actin dynamics mediated by a Rac mechanism. Although precise regulation of leading-edge dynamics by MT-bound IQGAP1 awaits further investigation, we speculate that IQGAP1-directed functional interaction of MT with actin filaments at the cell periphery recruits additional activators of Rac1 signaling and thus contributes to local Rac1 regulation at the leading edge of the cell. This regulation plays a key role in the reestablishment of cell monolayer integrity and the EC barrier.
Mapping of IQGAP1 protein interaction sites shows interactions of MT-associated partners CLASP-2 and CLIP-170 with the IQGAP1 C terminus (17, 34). On the other hand, the interaction of IQGAP1 with the actin-binding protein N-WASP has been mapped to the N-terminal CHD or the C-terminal domain and shown to promote Rac-dependent actin polymerization and branched actin filament assembly (35, 36). Our results show that IQGAP1 is essential for the formation of a functional complex between EB-1 and cortactin. These data represent a novel mechanism of MT anchoring to the peripheral actin cytoskeleton, which induces an EC barrier-enhancing response.
Expression of a deletion mutant form of IQGAP1 (termed IQGAP1ΔC) that lacks the C terminus involved in the interaction with MT but retains the GRD (involved in the interaction with Cdc42/Rac1 and responsible for IQGAP1 activation) abolished the HGF-induced formation of an IQGAP1-EB1-cortactin complex. Expression of IQGAP1ΔC also inhibited HGF-induced enrichment of MT at the cell periphery and partially suppressed cortical actin polymerization.
Our data show that Rac1 is required for the IQGAP1-dependent formation of an EB1-cortactin complex. This complex promotes MT capture at the cell periphery, leading to activated cortical actin polymerization and endothelial barrier enhancement. This complex may be disrupted by the expression of IQGAP1ΔC, which lacks the MT-binding domain. Thus, the C-terminal IQGAP1 domain (comprising amino acids 1502 to 1657) is critical for the formation of a tripartite IQGAP1-EB1-cortactin complex.
On the basis of our findings, we propose a model of IQGAP1-dependent control of vascular endothelial integrity via MT-actin cross talk (Fig. 9). Increased IQGAP1-MT interaction induced by barrier-protective stimuli such as HGF promotes Rac-dependent expansion of MT growth to the cell periphery. EB1 at the MT growing ends interacts with IQGAP1. The resultant IQGAP1-EB1 complex captures cortactin, leading to local activation of actin polymerization and enhancement of the peripheral F-actin rim. This actin cytoskeleton remodeling tightens the intercellular gaps and increases the endothelial barrier function. HGF-induced formation of the EB1-IQGAP1-cortactin tripartite complex may further promote local Rac signaling at the cell periphery, which is essential for the maintenance of EC barrier integrity; this possibility requires further investigation.
FIG 9.
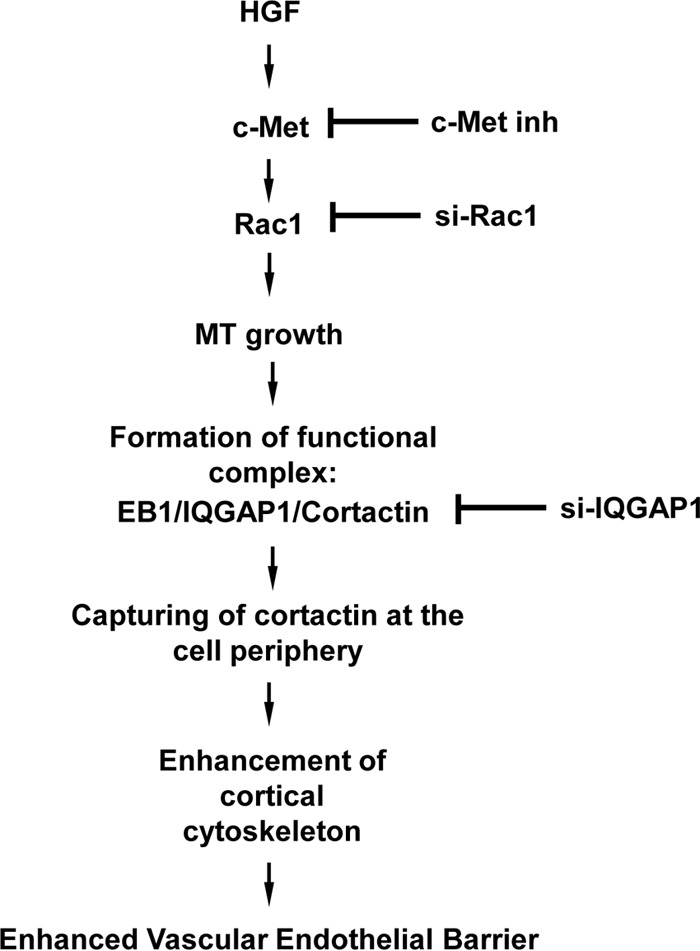
Scheme of how IQGAP1 mediates HGF-induced vascular endothelial barrier enhancement effects via stimulation of MT-actin cross talk. HGF-induced activation of Rac1 mediated by the c-Met receptor stimulates MT peripheral growth, and MT plus-end tips reaching the cell cortical region are captured on the cortical actin cytoskeleton via the formation of a tripartite complex of cortactin, IQGAP1, and MT. The formation of this complex coordinates cortical actin dynamics and supports local Rac1 activity levels, leading to enhanced cortical actin polymerization and endothelial barrier enhancement. inh, inhibitor; si-Rac1, Rac-1-specific siRNA; si-IQGAP1, IQGAP1-specific siRNA.
ACKNOWLEDGMENTS
This work was supported by the grants from the National Heart, Lung, and Blood Institute (HL89257 and HL107920 to A.A.B.) and by the Intramural Program of the National Institutes of Health (to D.B.S.).
Footnotes
Published ahead of print 14 July 2014
REFERENCES
- 1.Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. 2007. HGF attenuates thrombin-induced permeability in the human pulmonary endothelial cells by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the Rho pathway. FASEB J. 21:2776–2786. 10.1096/fj.06-7660com [DOI] [PubMed] [Google Scholar]
- 2.Liu F, Schaphorst KL, Verin AD, Jacobs K, Birukova A, Day RM, Bogatcheva N, Bottaro DP, Garcia JG. 2002. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. FASEB J. 16:950–962. 10.1096/fj.01-0870com [DOI] [PubMed] [Google Scholar]
- 3.Rosen EM, Goldberg ID. 1995. Scatter factor and angiogenesis. Adv. Cancer Res. 67:257–279. 10.1016/S0065-230X(08)60715-0 [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Himi T, Morita I, Murota S. 2000. Hepatocyte growth factor protects cultured rat cerebellar granule neurons from apoptosis via the phosphatidylinositol-3 kinase/Akt pathway. J. Neurosci. Res. 59:489–496. [DOI] [PubMed] [Google Scholar]
- 5.Ware LB, Matthay MA. 2002. Keratinocyte and hepatocyte growth factors in the lung: roles in lung development, inflammation, and repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L924–L940 [DOI] [PubMed] [Google Scholar]
- 6.Date I, Takagi N, Takagi K, Kago T, Matsumoto K, Nakamura T, Takeo S. 2004. Hepatocyte growth factor attenuates cerebral ischemia-induced learning dysfunction. Biochem. Biophys. Res. Commun. 319:1152–1158. 10.1016/j.bbrc.2004.05.100 [DOI] [PubMed] [Google Scholar]
- 7.Singleton PA, Salgia R, Moreno-Vinasco L, Moitra J, Sammani S, Mirzapoiazova T, Garcia JG. 2007. CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J. Biol. Chem. 282:30643–30657. 10.1074/jbc.M702573200 [DOI] [PubMed] [Google Scholar]
- 8.Dudek SM, Garcia JG. 2001. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 91:1487–1500 [DOI] [PubMed] [Google Scholar]
- 9.Birukov KG, Zebda N, Birukova AA. 2013. Barrier enhancing signals in pulmonary edema. Compr. Physiol. 3:429–484. 10.1002/cphy.c100066 [DOI] [PubMed] [Google Scholar]
- 10.Krendel M, Zenke FT, Bokoch GM. 2002. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 4:294–301. 10.1038/ncb773 [DOI] [PubMed] [Google Scholar]
- 11.Birukova AA, Birukov KG, Adyshev D, Usatyuk P, Natarajan V, Garcia JG, Verin AD. 2005. Involvement of microtubules and Rho pathway in TGF-beta1-induced lung vascular barrier dysfunction. J. Cell. Physiol. 204:934–947. 10.1002/jcp.20359 [DOI] [PubMed] [Google Scholar]
- 12.Birukova AA, Fu P, Xing J, Yakubov B, Cokic I, Birukov KG. 2010. Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 298:L837–L848. 10.1152/ajplung.00263.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kratzer E, Tian Y, Sarich N, Wu T, Meliton A, Leff A, Birukova AA. 2012. Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am. J. Respir. Cell Mol. Biol. 47:688–697. 10.1165/rcmb.2012-0161OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. 2003. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am. J. Respir. Cell Mol. Biol. 28:574–581. 10.1165/rcmb.2002-0075OC [DOI] [PubMed] [Google Scholar]
- 15.Tian X, Tian Y, Sarich N, Wu T, Birukova AA. 2012. Novel role of stathmin in microtubule-dependent control of endothelial permeability. FASEB J. 26:3862–3874. 10.1096/fj.12-207746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown MD, Sacks DB. 2006. IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 16:242–249. 10.1016/j.tcb.2006.03.002 [DOI] [PubMed] [Google Scholar]
- 17.Watanabe T, Noritake J, Kakeno M, Matsui T, Harada T, Wang S, Itoh N, Sato K, Matsuzawa K, Iwamatsu A, Galjart N, Kaibuchi K. 2009. Phosphorylation of CLASP2 by GSK-3beta regulates its interaction with IQGAP1, EB1 and microtubules. J. Cell Sci. 122:2969–2979. 10.1242/jcs.046649 [DOI] [PubMed] [Google Scholar]
- 18.White CD, Erdemir HH, Sacks DB. 2012. IQGAP1 and its binding proteins control diverse biological functions. Cell Signal. 24:826–834. 10.1016/j.cellsig.2011.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaibuchi K, Kuroda S, Fukata M, Nakagawa M. 1999. Regulation of cadherin-mediated cell-cell adhesion by the Rho family GTPases. Curr. Opin. Cell Biol. 11:591–596. 10.1016/S0955-0674(99)00014-9 [DOI] [PubMed] [Google Scholar]
- 20.Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K. 2005. IQGAP1: a key regulator of adhesion and migration. J. Cell Sci. 118:2085–2092. 10.1242/jcs.02379 [DOI] [PubMed] [Google Scholar]
- 21.Swart-Mataraza JM, Li Z, Sacks DB. 2002. IQGAP1 is a component of Cdc42 signaling to the cytoskeleton. J. Biol. Chem. 277:24753–24763. 10.1074/jbc.M111165200 [DOI] [PubMed] [Google Scholar]
- 22.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. 2009. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ. Res. 104:978–986. 10.1161/CIRCRESAHA.108.193367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Briggs MW, Li Z, Sacks DB. 2002. IQGAP1-mediated stimulation of transcriptional co-activation by beta-catenin is modulated by calmodulin. J. Biol. Chem. 277:7453–7465. 10.1074/jbc.M104315200 [DOI] [PubMed] [Google Scholar]
- 24.Birukova AA, Birukov KG, Smurova K, Adyshev DM, Kaibuchi K, Alieva I, Garcia JG, Verin AD. 2004. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 18:1879–1890. 10.1096/fj.04-2328com [DOI] [PubMed] [Google Scholar]
- 25.Dubrovskyi O, Birukova AA, Birukov KG. 2013. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab. Invest. 93:254–263. 10.1038/labinvest.2012.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birukov KG, Bochkov VN, Birukova AA, Kawkitinarong K, Rios A, Leitner A, Verin AD, Bokoch GM, Leitinger N, Garcia JG. 2004. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ. Res. 95:892–901. 10.1161/01.RES.0000147310.18962.06 [DOI] [PubMed] [Google Scholar]
- 27.Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JG, Verin AD. 2004. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc. Res. 67:64–77. 10.1016/j.mvr.2003.09.007 [DOI] [PubMed] [Google Scholar]
- 28.Komarova Y, De Groot CO, Grigoriev I, Gouveia SM, Munteanu EL, Schober JM, Honnappa S, Buey RM, Hoogenraad CC, Dogterom M, Borisy GG, Steinmetz MO, Akhmanova A. 2009. Mammalian end binding proteins control persistent microtubule growth. J. Cell Biol. 184:691–706. 10.1083/jcb.200807179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zebda N, Tian Y, Tian X, Gawlak G, Higginbotham K, Reynolds AB, Birukova AA, Birukov KG. 2013. Interaction of p190RhoGAP with C-terminal domain of p120-catenin modulates endothelial cytoskeleton and permeability. J. Biol. Chem. 288:18290–18299. 10.1074/jbc.M112.432757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Head JA, Jiang D, Li M, Zorn LJ, Schaefer EM, Parsons JT, Weed SA. 2003. Cortactin tyrosine phosphorylation requires Rac1 activity and association with the cortical actin cytoskeleton. Mol. Biol. Cell 14:3216–3229. 10.1091/mbc.E02-11-0753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kakiashvili E, Speight P, Waheed F, Seth R, Lodyga M, Tanimura S, Kohno M, Rotstein OD, Kapus A, Szaszi K. 2009. GEF-H1 mediates tumor necrosis factor-alpha-induced Rho activation and myosin phosphorylation: role in the regulation of tubular paracellular permeability. J. Biol. Chem. 284:11454–11466. 10.1074/jbc.M805933200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu BP, Chrzanowska-Wodnicka M, Burridge K. 1998. Microtubule depolymerization induces stress fibers, focal adhesions, and DNA synthesis via the GTP-binding protein Rho. Cell Adhes. Commun. 5:249–255. 10.3109/15419069809040295 [DOI] [PubMed] [Google Scholar]
- 33.Zhang Q, Magnusson MK, Mosher DF. 1997. Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol. Biol. Cell 8:1415–1425. 10.1091/mbc.8.8.1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukata M, Watanabe T, Noritake J, Nakagawa M, Yamaga M, Kuroda S, Matsuura Y, Iwamatsu A, Perez F, Kaibuchi K. 2002. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 109:873–885. 10.1016/S0092-8674(02)00800-0 [DOI] [PubMed] [Google Scholar]
- 35.Benseñor LB, Kan HM, Wang N, Wallrabe H, Davidson LA, Cai Y, Schafer DA, Bloom GS. 2007. IQGAP1 regulates cell motility by linking growth factor signaling to actin assembly. J. Cell Sci. 120:658–669. 10.1242/jcs.03376 [DOI] [PubMed] [Google Scholar]
- 36.Le Clainche C, Schlaepfer D, Ferrari A, Klingauf M, Grohmanova K, Veligodskiy A, Didry D, Le D, Egile C, Carlier MF, Kroschewski R. 2007. IQGAP1 stimulates actin assembly through the N-WASP-Arp2/3 pathway. J. Biol. Chem. 282:426–435. 10.1074/jbc.M607711200 [DOI] [PubMed] [Google Scholar]