

Abstract
Extracellular signal-regulated kinase 3 (ERK3) is an atypical member of the mitogen-activated protein kinase (MAPK) family whose function is largely unknown. Given the central role of MAPKs in T cell development, we hypothesized that ERK3 may regulate thymocyte development. Here we have shown that ERK3 deficiency leads to a 50% reduction in CD4+ CD8+ (DP) thymocyte number. Analysis of hematopoietic chimeras revealed that the reduction in DP thymocytes is intrinsic to hematopoietic cells. We found that early thymic progenitors seed the Erk3−/− thymus and can properly differentiate and proliferate to generate DP thymocytes. However, ERK3 deficiency results in a decrease in the DP thymocyte half-life, associated with a higher level of apoptosis. As a consequence, ERK3-deficient DP thymocytes are impaired in their ability to make successful secondary T cell receptor alpha (TCRα) gene rearrangement. Introduction of an already rearranged TCR transgene restores thymic cell number. We further show that knock-in of a catalytically inactive allele of Erk3 fails to rescue the loss of DP thymocytes. Our results uncover a unique role for ERK3, dependent on its kinase activity, during T cell development and show that this atypical MAPK is essential to sustain DP survival during RAG-mediated rearrangements.
INTRODUCTION
Mitogen-activated protein kinases (MAPKs) are evolutionarily conserved serine/threonine kinases that play key roles in transducing extracellular signals into a wide variety of cellular responses (1). Extracellular signal-regulated kinase 3 (ERK3) (Mapk6 gene product) is an atypical member of the MAPK family which displays ∼45% homology to the classical MAPKs ERK1/ERK2 in the kinase domain (2). Despite the similarity of their catalytic cores, several structural and functional properties of ERK3 set it apart from ERK1/ERK2 and other classical MAPKs (3). Unlike classical MAPKs, ERK3 is constitutively phosphorylated in the activation loop even in unstimulated cells (4). This suggests that upstream regulation of ERK3 and downstream targeting of substrates are likely to involve mechanisms different from those for other family members. Consistent with this idea, ERK3 is phosphorylated and activated by group I p21-activated kinases (5, 6).
Little is known about the effector pathways of ERK3. ERK3 was shown to interact with the MAPK-activated protein kinase 5 (MK5), leading to its phosphorylation and activation (7, 8). Moreover, this interaction stabilizes ERK3 and excludes both ERK3 and MK5 from the nucleus (7, 8). ERK3 ablation reduces MK5 activity by 50% in cells (8). The remaining MK5 activity is due to the closely related MAPK, ERK4, which is also able to activate MK5 (9, 10). The cellular and molecular functions of MK5 are poorly understood (11, 12), but it was recently shown to regulate Rag transcription during B cell differentiation (13), raising the possibility that the ERK3-MK5 axis also influences T cell differentiation.
The exact physiological functions of ERK3 remain to be established, but accumulating evidence points to a role in differentiation. For example, ERK3 transcripts are upregulated during differentiation of P19 embryonal carcinoma cells into neuronal or muscle cells (2). The ERK3 protein also markedly accumulates during differentiation of C2C12 myoblasts into muscle cells (14). Notably, overexpression of ERK3 in fibroblasts causes G1 cell cycle arrest, suggesting a possible role in cell cycle exit (14). In mice, ERK3 deficiency is associated with a defect in type II pneumocyte differentiation, leading to early neonatal death (15). Recent work has also uncovered a role for ERK3 in neuronal morphogenesis (16).
T cell generation in the thymus is a complex biological process that combines differentiation, proliferation, death, selection, and lineage commitment. Early thymic progenitors (ETPs) seed the thymus, where they commit to the T lineage to generate αβ and γδ T cells (17). The most immature thymocytes are double-negative (DN) thymocytes that lack CD4 and CD8 expression. The first two developmental stages (DN1 and DN2) involve proliferation to expand the rare committed T cell progenitors. Thymocyte proliferation stops at the DN3 stage to allow T cell receptor β (TCRβ) rearrangement since RAG2 is unstable in proliferating cells (18, 19). If TCRβ rearrangement is successful, DN3 cells express the pre-TCR that will transmit concomitant survival, proliferation, and differentiation signals (β-selection). This generates actively dividing DN4 cells that further differentiate into double-positive CD4+ CD8+ (DP) thymocytes (20). At this stage, proliferation stops during TCRα locus rearrangement, and conventional αβ TCR expression starts. Since TCR rearrangement creates sequence diversity, DP thymocytes undergo an educational process to allow the survival of cells expressing a useful TCR restricted to self-major histocompatibility complex (MHC) molecules (positive selection), while thymocytes expressing an autoreactive TCR are eliminated (negative selection) (21, 22). These DP cells further differentiate into CD4+ or CD8+ single-positive (SP) thymocytes (CD4SP and CD8SP thymocytes), depending on their MHC specificity, and exit as naive T cells into the peripheral lymphoid organs (21, 22).
DP thymocyte differentiation can be divided into three steps based on the TCR expression level. Newly generated DP thymocytes do not express the TCR (TCRlo) and are actively rearranging the TCRα locus to generate a functional TCRα chain. At this developmental stage, DNA double-strand breaks (DSBs) are generated by RAG1 and RAG2. These DNA DSBs have to be repaired correctly by nonhomologous end joining (NHEJ) to generate the TCRα chain coding sequence (23, 24) and to maintain genomic integrity (25). If DP thymocytes successfully rearrange the TCRα locus, they express intermediate TCR levels (TCRint), allowing them to test TCR specificity. Positive selection will occur if the DP thymocytes express a useful self-MHC-restricted TCR. This event will lead to the upregulation of TCR expression (TCRhi) and to the maturation of the DP thymocyte into an SP thymocyte. If the DP thymocyte fails to make a functional TCRα rearrangement or if the expressed TCR does not allow the DP thymocyte to be positively selected, TCRα locus rearrangement will continue. These successive rounds of secondary TCRα gene rearrangement will proceed from proximal to more distal V-J segments and require prolonged DP thymocyte survival (26). This survival is dependent on the expression of the antiapoptotic molecule Bcl-xL (26–28). Several transcription factors (c-Myb, RORγt, TCF-1, and HEB) are important for the proper expression of Bcl-xL in DP thymocytes (26, 29–32). The ablation of their expression in genetically modified mouse models leads to impaired DP thymocyte survival and reduced TCRα secondary gene rearrangements (26, 29–32).
Classical MAPKs are known to play crucial signaling roles at different steps of T cell differentiation. For example, ERK1/2, the closest homologs of ERK3, are essential for β-selection and positive selection during thymocyte differentiation (33). Given the importance of other MAPK family members in T cell development and the postulated role of ERK3 in differentiation, we hypothesized that ERK3 may be involved in the control of thymocyte proliferation and/or differentiation. We showed that in the absence of ERK3, the number of DP thymocytes is decreased by 2-fold. Analysis of hematopoietic chimeras revealed that the reduction in DP thymocyte number is intrinsic to hematopoietic cells. We also demonstrated that the reduction in DP thymocyte number is a consequence of their decreased survival. The number of DP thymocytes is also reduced in knock-in mice expressing a catalytically inactive form of ERK3, highlighting the importance of ERK3 kinase activity in a physiological differentiation event. Our results show that ERK3 controls unique thymocyte differentiation events distinct from those of conventional MAPKs.
MATERIALS AND METHODS
Mice.
ERK3-deficient and ERK4-deficient mice have been described (15, 34). Erk3+/− 129/SvEv and Erk3+/− C57BL/6 mice were bred under specific-pathogen-free conditions at the Maisonneuve-Rosemont Hospital Research Center in accordance with the Canadian Council of Animal Care. Erk3+/− mice were intercrossed to generate Erk3−/− embryos and newborn mice. Knock-in mice expressing a kinase-dead allele of ERK3 were generated by introducing the double mutation K49A/K50A (KA) in exon 2 of the Erk3 gene (characterization of these mice will be reported elsewhere; the KA mutation in subdomain II of the kinase domain abolishes ERK3 catalytic activity in vitro [4]). We have confirmed that the ERK3 wild type and ERK3KA mutant are expressed at similar levels in mouse tissues, including the thymus. RAG1-deficient OT-II TCR transgenic mice were obtained from Taconic. These mice were bred to Erk3+/− mice and intercrossed to generate ERK3-deficient OT-II/RAG1−/− mice. C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA).
Antibodies and flow cytometry.
Anti-CD4 (L3/T4, CT-CD4), anti-CD25 (CL8925B, CL8925F, and PC615.3), anti-CD8 (CL168, CL169B, and CT-CD8a), anti-TCRβ (CL7200B, H57-597), anti-CD3 (CT-CD3), and anti-NK (DX5) antibodies were purchased from Cedarlane Laboratories (Hornby, Ontario, Canada). Anti-CD8 (53-6.7), anti-CD44 (IM7), antibromodeoxyuridine (anti-BrdU) (B44), anti-c-Kit, and lineage cocktail antibodies were purchased from BD Biosciences (San Jose, CA). Anti-TCRγδ (UC7-13D5) was purchased from eBioscience. Anti-phospho-H2AX was purchased from Cell Signaling Technology. Anti-Bcl-xL (7B2.5) was purchased from Abcam. Annexin V staining was performed according to the manufacturer's protocol (Biolegend, San Diego, CA). Stainings of cell surface markers were done as previously described (35). Samples were analyzed on a BD FACSCalibur or BD FACSCanto I flow cytometer.
FDG staining.
Fluorescein di(β-D-galactopyranoside) (FDG) staining was performed as described previously (36). Briefly, cells were surface stained and resuspended in phosphate-buffered saline (PBS). Warmed FDG (7.5 mM) was added to cells while gently vortexing. The reaction was stopped by adding ice-cold PBS, and cells were kept on ice for 5 min. After centrifugation, cells were resuspended in PBS supplemented with 10% horse serum and incubated at 15°C for 15 to 20 min to enhance β-galactosidase activity before flow cytometry analysis.
BrdU incorporation.
Mice at 18.5 days of gestation were injected intraperitoneally (i.p.) with 1 mg BrdU (Sigma Chemicals Co., St. Louis, MO). One hour later, thymocytes were stained for cell surface markers, followed by fixation and permeabilization using a BD Cytofix/Cytoperm kit (BD Biosciences) and staining with anti-BrdU or isotype control antibody.
In vitro differentiation of ETPs.
Fetal thymus 14.5-day ETPs were sorted as lineage cocktail-negative (lin−), c-Kithi, and CD25− and seeded at 103 cells/well in 24-well tissue culture plates containing OP9-DL1 cells (37) supplemented with 1 ng/ml interleukin 7 (IL-7) and 5 ng/ml Flt3 ligand. Cells were cultured for 12 days before analysis.
Immunoblot analysis.
Thymocytes were lysed in 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 50 mM NaF, 5 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μM leupeptin, 1 μM pepstatin A, and 1% (vol/vol) Triton X-100 for 1 min at 4°C. Lysate proteins were resolved on 7.5% SDS acrylamide gels and transferred to Hybond-ECL membranes (Amersham). Blots were probed with polyclonal anti-ERK3 (Cell Signaling Technology) and anti-β-actin (LabVision, CA) antibodies and subsequently with horseradish peroxidase-conjugated anti-rabbit IgG. Immunoreactive bands were detected by enhanced chemiluminescence (PerkinElmer LAB Inc., MA).
FTOC.
Embryonic-day-14.5 (E14.5) fetal thymic lobes were placed on the surfaces of 0.8-μm-pore-size Nuclepore Track-Etch membranes (Fisher Scientific, CA), cultured on 2 ml of complete Iscove's modified Dulbecco's medium (Life Technologies), and incubated for 5 days at 37°C and 7.5% CO2.
mRNA quantification.
DP thymocytes from E18.5 mice were sorted, and RNA was extracted using TRIzol (Life Technologies). RNA was reverse transcribed using Superscript II with oligo(dT) (Life Technologies), and real-time PCRs were carried out on an ABI Prism 7500 instrument (Life Technologies) using Power SYBR green (Life Technologies). The primer sequences are listed in Table 1. All reactions were carried out in triplicate, and average values were used for quantification. Hprt was used as an endogenous control. Relative expression was calculated as described previously (38).
TABLE 1.
Primers for mRNA quantification by qPCR
| Gene name | Primer sequence |
|
|---|---|---|
| Forward (5′–3′) | Reverse (5′–3′) | |
| Tcra | AAAGAGACCAACGCCACCTAC | GAGGATTCGGAGTCCCATAAC |
| Pmaip1 | CCACCTGAGTTCGCAGCTCAA | GTTGAGCACACTCGTCCTTCAA |
| Mcl1 | ACGGGACTGGCTTGTCAAACAAAG | GCACATTTCTGATGCCGCCTTCTA |
| Bcl2l11 | CGGATCGGAGACGAGTTCA | TTCAGCCTCGCGGTAATCA |
| Rorc | CAAGCGGCTTTCAGGCTTCAT | GACTGTGTGGTTGTTGGCATTGTAG |
| Bcl2a1 | CAAGAGCAGATTGCCCTGGATGTA | AAGCCATCTTCCCAACCTCCATTC |
| Rag1 | GCTATCTCTGTGGCATCGAGTG | GGTGTTGAATTTCATCGGGTG |
| Rag2 | TGCCGAGTTTAATTCCTGGCTTGG | GTGTACTTCCTGCTTGTGGATGTG |
| Hprt | CTCCTCAGACCGCTTTTTGC | TAACCTGGTTCATCATCGCTAATC |
Quantification of DNA DSBs at Jα gene segments.
Jα DNA DSBs were quantified using ligation-mediated PCR, as previously described (39), with modifications. Primer and linker sequences are provided in Table 2. Briefly, 1.5 μg of genomic DNA (gDNA) from sorted DP thymocytes was ligated to 20 pmol of asymmetrical linker (linkers 1 and 2) using T4 DNA ligase (New England BioLabs), following the manufacturer's instructions. Quantitative PCRs were performed with ligated gDNA using combinations of the linker primer and Jα61, Jα49, or Jα27 primer, respectively. PCRs were run in triplicate using Power SYBR green (Life Technologies) on an ABI 7500 real-time PCR system. Cd14 quantitative PCR (qPCR) was used to normalize the amount of gDNA. The ΔCT value for each sample was determined by calculating the difference between the threshold cycle (CT) value of the target and the CT value of the reference gene (Cd14). Then, the ΔΔCT value for each sample was determined by subtracting the mean of the ΔCT value of the sample from the ΔCT value of a reference sample composed of ligated gDNA from mouse thymocytes. The relative level of target gene expression was calculated using 2−ΔΔCT.
TABLE 2.
Primers for quantification of DNA DSBs at Jα gene segments
| Target | Primer sequence |
|
|---|---|---|
| Forward (5′–3′) | Reverse (5′–3′) | |
| Linker 1 | GCGGTGACCCGGGAGATCTGAATTC | |
| Linker 2 | GAATTCAGATC | |
| Linker primer | CCGGGAGATCTGAATTCCAC | |
| Jα27 | ATGGCAGATAGAATGGAGCGG | |
| Jα49 | AGGGAAAGTGACAACCAGGC | |
| Jα61 | TCCAAAAGAGGAAAGGAAGGCAGTC | |
| CD14 | GCTCAAACTTTCAGAATCTACCGAC | CAGAAGCAACAGCAACAA |
Quantification of TCRα secondary gene rearrangement.
Erk3+/+ and Erk3−/− thymocytes were harvested and immediately immersed in TRIzol for RNA extraction, according to the manufacturer's protocol (Life Technologies). Two micrograms of RNA was reverse transcribed to cDNA using Superscript II with oligo(dT) (Life Technologies). The cDNA obtained was serially diluted 7 times, and each dilution was amplified with different primer combination (Table 3) to evaluate TCRα gene segment usage. PCR products were analyzed in 2% agarose gel. Pictures were taken using an Alpha imager. The quantification of band intensity (nonsaturating and in the linear range) was measured using ImageJ software. Quantitative PCR was performed as described above with cDNA diluted 1/10 with the primer combinations described in Table 4, except that Tcra (primer located in the constant region) was used as the endogenous control.
TABLE 3.
Primers for measurement of TCRα secondary gene rearrangement by semiquantitative RT-PCR
| Target | Primer sequence |
|
|---|---|---|
| Forward (5′–3′) | Reverse (5′–3′) | |
| Vα3-Cα | CCCAGTGGTTCAAGGAGTGA | TTCAGCAGGAGGATTCGGAG |
| Vα6-Cα | CTGACTCATGTCAGCCTGAGAG | TTCAGCAGGAGGATTCGGAG |
| Vα8-Cα | CAACAAGAGGACCGAGCACC | TTCAGCAGGAGGATTCGGAG |
| Vα14-Cα | TGGGAGATACTCAGCAACTCTGG | TTCAGCAGGAGGATTCGGAG |
| Vα19-Cα | CTGCTTCTGACAGAGCTCCAG | TTCAGCAGGAGGATTCGGAG |
| Vα3-Jα2 | CCCAGTGGTTCAAGGAGTGA | ACCACTTAGTCCTCCAGTATTC |
| Vα3-Jα18 | CCCAGTGGTTCAAGGAGTGA | CAGGTATGACAATCAGCTGAGTCC |
| Vα3-Jα30 | CCCAGTGGTTCAAGGAGTGA | AGATGTGTCCCTTTTCCAAAGATG |
| Tcra constant region (Cα) | TTCAAAGAGACCAACGCCAC | TTCAGCAGGAGGATTCGGAG |
TABLE 4.
Primers for quantification of TCRα secondary gene rearrangement by qPCR
| Target | Primer sequence |
|
|---|---|---|
| Forward (5′–3′) | Reverse (5′–3′) | |
| Vα3-Jα30 | CCCAGTGGTTCAAGGAGTGA | AGATGTGTCCCTTTTCCAAAGATG |
| Vα6-Cα | ACCACTGTATCCTGAGAGTAG | CTGGTACACAGCAGGTTC |
| Vα19-Cα | CTGCTTCTGACAGAGCTCCAG | CTGGTACACAGCAGGTTC |
| Vα3-Jα2 | CCCAGTGGTTCAAGGAGTGA | ACCACTTAGTCCTCCAGTATTC |
| Tcra constant region (Cα) | TTCAAAGAGACCAACGCCAC | TTCAGCAGGAGGATTCGGAG |
Hematopoietic chimeras.
Fetal liver cells (2 × 106) from E14.5 embryos were injected intravenously (i.v.) into lethally irradiated (12 Gy) syngeneic mice (5 to 7 weeks old). Mice were analyzed 8 weeks postreconstitution.
Statistical analysis.
Statistical analyses for differences between groups were performed using Student's t test except for the TCRα secondary rearrangement measurement, where an analysis of variance (ANOVA) with a Bonferroni posttest was performed. Welch's correction was applied for unequal variances when required. Data are presented as means ± standard errors of the means (SEM). All tests were two sided, and a P value of <0.05 was considered statistically significant.
RESULTS
ERK3 expression during thymic differentiation.
To evaluate the role of ERK3 during thymic differentiation, we first characterized its expression profile in thymocyte subsets using ERK3 mutant mice in which the Erk3 coding sequence is replaced by the LacZ reporter (15), allowing the identification of cells actively transcribing Erk3 by measuring β-galactosidase activity. Erk3 transcription is apparent at the DN1 stage and increases up to the DN4 stage of thymocyte differentiation (Fig. 1A). The transcription of the Erk3 gene at the DP stage is lower than in the DN subsets and is further decreased in SP thymocytes (Fig. 1A). Since the ERK3 protein has a very short half-life in proliferating cells (14), we confirmed that Erk3 transcription leads to a detectable level of the ERK3 protein in thymocyte extracts (Fig. 1B).
FIG 1.

Reduced thymocyte number and defective CD4SP thymocyte differentiation in Erk3−/− newborn thymus. (A) Representative overlay histograms of ERK3 expression during thymocyte differentiation. Thymocytes from Erk3+/− (thick line) and Erk3+/+ (thin line) mice were cell surface stained, permeabilized, and exposed to FDG. Numbers indicate the percentages of FDG+ cells. (B) ERK3 protein expression in Erk3+/+ (WT) and Erk3−/− (KO) thymus. Lysates of newborn thymocytes from Erk3+/+ and Erk3−/− mice were immunoblotted with an anti-ERK3 antibody. Murine embryonic fibroblasts (MEF) were used as controls. (C) Representative thymic profile of newborn Erk3+/+ and Erk3−/− mice. The percentages of cells in the different thymocyte subsets are indicated on the dot plot. (D) Cell numbers for the different thymocyte populations are shown for Erk3+/+ and Erk3−/− newborn mice. The bars represent the mean values; each dot corresponds to one mouse. Data are representative of at least 4 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed Student's t test.
Reduced thymic cell number and lack of CD4SP thymocytes in the absence of ERK3.
We next evaluated the impact of ERK3 ablation on thymocyte differentiation. Erk3−/− mice die within 24 h after birth (15), limiting analysis to thymi from newborn mice. We first confirmed the absence of the ERK3 protein in Erk3−/− thymus (Fig. 1B) and then analyzed the effect of ERK3 deficiency on T cell development. DN and DP thymocytes were present in similar percentages in Erk3+/+ and Erk3−/− thymi, but CD4SP thymocyte differentiation was almost abrogated in Erk3−/− thymus (Fig. 1C). At birth, CD8SP thymocytes are not apparent in wild-type thymus, precluding analysis of ERK3 function in these cells (Fig. 1C). Interestingly, thymic cell number was reduced by 50% in the absence of ERK3 (Fig. 1D). This is apparent in all thymocyte subsets but not statistically significant for DN (Fig. 1D). These results show that ERK3 loss decreases thymocyte number, particularly from the DP stage onwards.
Defective thymocyte differentiation in FTOCs.
To rule out the possibility that defective thymocyte differentiation in Erk3−/− newborn mice was due to their smaller size or their moribund status (15), we analyzed T cell differentiation in fetal thymic organ cultures (FTOCs). Fetal thymic lobes from E14.5 embryos are similar in size (not shown) and cell number (Fig. 2A). After 5 days of culture, DP thymocytes were present in lower percentages in Erk3−/− FTOCs (Fig. 2B). As observed in newborn thymus, Erk3−/− FTOCs exhibit a 50% reduction in cell number (Fig. 2C). The decrease was statistically significant for DP and CD4SP thymocytes (Fig. 2C). As in newborn mice, CD8SP differentiation is not apparent after 5 days of FTOC differentiation. All of the CD8+ thymocytes in the FTOCs are TCR negative and as such represent intermediate single-positive (ISP) thymocytes, the differentiation step between DN4 and DP. These results indicate that defective thymocyte differentiation in the absence of ERK3 is due to a thymus-intrinsic effect and not to the health status of Erk3−/− mice.
FIG 2.

Defective thymocyte development in the absence of ERK3 is intrinsic to the thymus. (A) Cell numbers of Erk3+/+ and Erk3−/− E14.5 fetal thymus. (B) Representative dot plot of Erk3+/+ and Erk3−/− FTOCs cultured for 5 days. The percentages of cells in the different thymocyte subsets are indicated on the dot plot. (C) Cell numbers per thymic lobe for the different thymocyte populations are shown for Erk3+/+ and Erk3−/− FTOCs. Data are representative of 4 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed Student's t test.
Reduced thymic cell number is a hematopoietic cell-dependent phenotype.
In order to circumvent the embryonic lethality of ERK3-deficient mice and to study adult T cell differentiation in the presence of a wild-type epithelium (Erk3 is transcribed in thymic epithelial cells; data not shown), fetal liver cells from Erk3+/+ and Erk3−/− E14.5 embryos were transferred into lethally irradiated mice. Thymocyte differentiation was normal in hematopoietic chimeras lacking ERK3 (Fig. 3A). However, thymocyte number was reduced by 50% in these hematopoietic chimeras. This is apparent for the DP, CD4SP, and CD8SP subsets but statistically significant only for DP and CD8SP thymocytes (Fig. 3B). In comparison to the results obtained for Erk3−/− mice (see Fig. 1 and 2), the decrease in CD4SP thymocytes is less obvious and seems to be a consequence of the reduction in the number of DP thymocytes rather than differentiation. These findings suggest that the reduced thymocyte number phenotype in Erk3−/− mice is intrinsic to hematopoietic cells and does not require ERK3 ablation in the thymic epithelium. Thus, results from three different experimental approaches (Fig. 1 to 3) reproducibly demonstrate a reduction in DP thymocytes in Erk3−/− mice.
FIG 3.

Reduced thymocyte number in ERK3-deficient hematopoietic chimeras. (A) T cell differentiation in Erk3+/+ and Erk3−/− hematopoietic chimeras. CD4/CD8 profiles are shown for Erk3+/+ and Erk3−/− hematopoietic chimeras 8 weeks postreconstitution. (B) Compilation of the cell numbers of total thymocytes and of the different thymocyte populations in Erk3+/+ and Erk3−/− hematopoietic chimeras. Each dot corresponds to one mouse, and the bars represent the mean values. (C) Normal distribution of DN1 to DN4 thymocytes in Erk3-deficient hematopoietic chimeras. Data are representative of 3 or 4 independent experiments. *, P < 0.05; ***, P < 0.001; two-tailed Student's t test.
Loss of ERK3 does not affect the early steps of thymocyte differentiation.
The decreased number of cells, particularly the DP subset, in Erk3−/− mice prompted us to evaluate if the early steps of thymocyte differentiation occurred normally in the absence of ERK3. These differentiation steps were not affected by the absence of ERK3, as indicated by the normal distribution of the DN1 (CD44+ CD25−), DN2 (CD44+ CD25+), DN3 (CD44− CD25+), and DN4 (CD44− CD25−) populations in thymus from newborn mice (Fig. 4A and B). In addition, while DN1 number showed a statistically significant decrease in Erk3−/− mice (Fig. 4B), ETP (lin− c-Kit+ CD25−) numbers were similar in Erk3+/+ and Erk3−/− mice (Fig. 4C). Analysis of DN1 to DN4 populations in Erk3+/+ and Erk3−/− hematopoietic chimeras gave similar results (see Fig. 3C). Altogether, these results suggest that the reduced number of DP thymocytes in the absence of ERK3 is not a consequence of defective differentiation of ETPs into DP thymocytes.
FIG 4.

Distribution of DN thymocyte subsets in Erk3+/+ and Erk3−/− mice. (A) CD44/CD25 dot plots gated on lineage-negative cells are shown for Erk3+/+ and Erk3−/− newborn thymus. The percentages of DN1 to DN4 cells are indicated on the profile (DN1, CD44+ CD25−; DN2, CD44+ CD25+; DN3, CD44− CD25+; DN4, CD44− CD25−). (B) Compilation of DN1 to DN4 cell numbers in Erk3+/+ and Erk3−/− newborn mice. Cell numbers for the different thymocyte populations are shown. The bars represents the mean values; each dot corresponds to one mouse. Data are representative of 8 independent experiments. (C) CD25/c-Kit profiles gated on lineage-negative cells are shown for Erk3+/+ and Erk3−/− newborn thymus. Numbers indicate ETP percentages in the left panel; ETP cell numbers are shown in the right panel. Each dot represents one mouse, and the bars are the mean values. *, P < 0.05, two-tailed Student's t test.
Normal progenitor-to-DP transition in Erk3−/− mice.
The decreased DP thymocyte number in ERK3-deficient mice could result from reduced proliferation of DN thymocytes during their differentiation into DP thymocytes. We therefore measured the proliferation rate of DN thymocytes. We injected BrdU into female mice at day 18.5 of gestation and collected the embryos 1 h later to measure the number of cells in S phase. In the absence of ERK3, there was no difference in BrdU incorporation in the different DN thymocyte subsets (Fig. 5A). Furthermore, a similar proliferation rate was observed in intermediate-single-positive (ISP) thymocytes (Fig. 5B), which represent cells at the intermediate differentiation step between DN4 and DP thymocytes. Therefore, the reduction in DP thymocyte number in Erk3−/− mice cannot be explained by decreased proliferation after β-selection, since DN3, DN4, and ISP thymocytes proliferate at similar rates. We observed an increased percentage of BrdU+ cells within the DP thymocyte subset in the absence of ERK3 (Fig. 5B). However, the numbers of BrdU+ DP thymocytes are similar in Erk3+/+ and Erk3−/− mice (Fig. 5C). In such a short period of BrdU labeling, BrdU+ DP thymocytes represent newly generated cells that have incorporated BrdU during the DN-to-DP transition, and therefore, the increased proportion of BrdU+ DP thymocytes from Erk3−/− mice reflects the loss of older DP thymocytes. Thus, these results indicate that the generation of DP from DN thymocytes is not affected by ERK3 deficiency. To further demonstrate that the generation of DP thymocytes occurs normally in the absence of ERK3, we asked whether sorted ETPs from Erk3+/+ and Erk3−/− mice were able to differentiate as efficiently into DP thymocytes in vitro (37). Sorted ETPs plated on OP9-DL1 cells generated similar percentages and numbers of DP thymocytes whether they were isolated from Erk3+/+ or Erk3−/− mice (Fig. 5D). Altogether, this indicates that the decreased DP thymocyte number does not result from impaired proliferation or generation from ETPs.
FIG 5.

Normal rate of DN thymocyte proliferation and differentiation into DP thymocytes in the absence of ERK3. (A and B) Proliferation of the different thymocyte subsets. Thymocytes were surface stained, followed by intracellular staining with anti-BrdU antibody (thick line) or isotype control (thin line). The overlay histograms show BrdU incorporation by the different subsets of thymocytes from Erk3+/+ and Erk3−/− mice. Numbers indicate the percentages of BrdU+ cells. (C) Quantification of DP thymocyte proliferation. The numbers of BrdU+ DP thymocytes are shown. The bars represents the mean values; each dot represents one mouse. (D) ETPs were cultured for 12 days on OP9-DL1 cells. CD4/CD8 profiles are shown for Erk3+/+ and Erk3−/− cultures. Cell recovery and the percentage of DP thymocytes are indicated on the dot plot. Data are representative of at least 3 independent experiments, except for panel D, which represents one experiment with 2 mice per group.
Reduced TCR expression by Erk3−/− DP thymocytes.
The normal rate of DP thymocyte generation suggests that the reduced DP thymocyte number is due to events occurring at the DP stage. At this step of differentiation, DP thymocyte survival is critical to allow sufficient time to test different TCRα gene rearrangements. DP thymocytes can be separated into three subsets based on their TCR expression level. TCRlo cells represent thymocytes that are undergoing TCRα gene rearrangement, TCRint cells are cells that have successfully rearranged the TCRα chain and are in the process of testing their TCR specificity, and TCRhi cells are DP thymocytes that have received a positive selection signal and will further differentiate into SP thymocytes. To understand the molecular basis of the decrease in DP thymocyte number, we have quantified the TCR cell surface expression level on DP thymocytes and have noticed a marked decrease in the proportion and number of cells expressing intermediate (TCRint) and high (TCRhi) TCR levels (Fig. 6A and B). This reduced TCR expression does not result from defective TCRβ chain production, since Erk3−/− thymocytes express the same amount of intracellular TCRβ chain (not shown). The decreased expression of the TCR was associated with a reduction in Tcra transcription (Fig. 6C), suggesting that fewer DP thymocytes were able to make a successful rearrangement at the Tcra locus.
FIG 6.

Reduced TCR expression in Erk3−/− DP thymocytes. (A) Cell surface expression of TCRβ in Erk3−/− DP thymocytes from E18.5 embryos. The representative overlay shows TCRβ cell surface expression by Erk3+/+ (thin line) and Erk3−/− (thick line) DP thymocytes. (B) Quantification of the percentages (top) and numbers (bottom) of TCRlo, TCRint, and TCRhi DP thymocytes. The bar represents the mean value, and each dot corresponds to one mouse. (C) Real-time PCR analysis of Tcra gene expression on sorted DP thymocytes from Erk3+/+ and Erk3−/− mice. (D) Overlay histograms show phospho-H2AX staining in Erk3+/+ (thin line) or Erk3−/− (thick line) DP (TCRlo, TCRint, and TCRhi) thymocytes from E18.5 embryos. Numbers on the graph indicate the means ± SEM of the percentages of phospho-H2AX-positive cells. (E) The numbers of phospho-H2AX-positive cells in total DP thymocytes are shown for Erk3+/+ and Erk3−/− newborn mice. (F) Quantification of DNA DSBs at different Jα gene segments. Genomic DNA isolated from DP thymocytes of Erk3+/+ and Erk3−/− mice was subjected to ligation-mediated qPCR. (G) mRNA quantification of Rag1 and Rag2 on sorted DP thymocytes from Erk3+/+ and Erk3−/− mice. Data are representative of at least 3 independent experiments. *, P < 0.05; ***, P < 0.01; two-tailed Student's t test.
The reduction in TCR expression by ERK3-deficient DP thymocytes could be a direct consequence of defective Tcra gene segment rearrangement. This could occur via defective DSB repair or via a reduced efficiency of Tcra gene rearrangement. To test the first possibility, we assayed the presence of phosphorylated histone H2AX, a marker of DNA DSBs (40–42). The percentage of phosphorylated H2AX (p-H2AX) was increased in DP thymocytes expressing low, intermediate, and high TCR levels (Fig. 6D). However, these results did not translate into increased numbers of p-H2AX DP thymocytes (Fig. 6E), possibly due to a decrease in the total number of DP thymocytes in Erk3−/− mice. Since phosphorylation of H2AX can occur during apoptosis (43), we directly measured DNA DSB accumulation by ligation-mediated PCR (39). We found that DNA DSBs did not accumulate in Erk3−/− DP thymocytes, and this was true for distal, intermediate, and proximal Jα segments (Fig. 6F). These results suggest that the reduction in Erk3−/− DP thymocyte number is not a consequence of DNA DSB-mediated cell death. However, a trend (P = 0.09) toward a reduction in DNA DSBs at Jα49 and Jα27 gene segments (Fig. 6F) suggests that ERK3-deficient thymocytes may be less able to perform successive TCRα gene rearrangements. In light of the recent report of Chow et al. (13) showing that MK5 (a binding partner and substrate of ERK3) regulates Rag transcription, we evaluated whether ERK3 deficiency leads to a reduction in Rag1 or Rag2 expression. Loss of ERK3 did not affect Rag1 and Rag2 transcription (Fig. 6G). These results suggest that the reduction in the proportion of DP thymocytes expressing the TCR is not due to defective expression of Rag1 and Rag2, nor is it a consequence of the accumulation of DNA DSBs at the Tcra locus.
Decreased Erk3−/− DP thymocyte survival is responsible for reduced TCR expression and DP thymocyte number.
The trend in the reduction of DNA DSBs at the distal Jα27 gene segment and the reduced TCR expression suggest that Erk3−/− DP thymocytes may not survive long enough to make successive secondary TCRα gene rearrangements. To directly test this possibility, we evaluated the capacity of Erk3−/− DP thymocytes to form distal rearrangements at the TCRα locus. Indeed, the usage of more distal Vα (Vα14 and -19) and Jα (Jα2) gene segments was reduced while proximal rearrangement (Vα6) was not affected by ERK3 deficiency in DP thymocytes (Fig. 7A and B). These semiquantitative results were confirmed by qPCR for some of the distal (Vα19-Cα and Vα3-Jα2) and proximal (Vα6-Cα and Vα3-Jα30) rearrangements (Fig. 7C). This reduction in distal TCRα gene rearrangement has been observed in several mouse models where DP thymocyte survival is compromised (26, 29–32), since DP thymocytes need to survive long enough to make successive rounds of secondary TCRα gene rearrangement. To test if ERK3 loss reduces DP thymocyte survival, we measured the expression of pro- and antiapoptotic molecules known to affect the survival of DP thymocytes. Erk3−/− DP thymocytes showed a decrease in mRNA expression of the antiapoptotic gene Mcl1 and an increase in expression of Noxa (encoded by the Pmaip1 gene), a proapoptotic BH3-only member of the Bcl-2 family (Fig. 8A). No differences in the expression of the Bcl2l11 (encoding Bim), Bcl2a1, and Rorc (encoding Rorγt) genes were observed (Fig. 8B). Moreover, similar levels of the Bcl-xL protein were detected in Erk3+/+ and Erk3−/− DP thymocytes (Fig. 8C). We next investigated whether DP thymocytes lacking ERK3 were more susceptible to cell death. As shown in Fig. 8D, a 2-fold increase in cell death (annexin V+) was observed for Erk3−/− DP thymocytes at day 5 of FTOCs. Furthermore, ERK3-deficient DP TCRint and DP TCRhi thymocytes but not DP TCRlo thymocyes showed a significant increase in cell death (Fig. 8D). These results suggest that ERK3 controls DP thymocyte survival to ensure successive waves of TCRα gene rearrangement. If ERK3 is necessary to promote prolonged DP thymocyte survival to allow sufficient time for the expression of a positively selectable and useful TCR, the introduction of an already rearranged TCR should restore DP thymocyte number in Erk3−/− mice. Strikingly, introduction of the OT-II TCR into ERK3-deficient thymocytes completely rescued DP thymocyte cell number (Fig. 9). Altogether, these results suggest that the reduction of DP thymocyte number in the absence of ERK3 is a consequence of reduced DP thymocyte survival, which then affects distal TCRα gene rearrangement and TCR expression.
FIG 7.

Reduced usage of distal Tcra gene segments in ERK3-deficient mice. (A) Semiquantitative reverse transcription-PCR (RT-PCR) of different rearranged segments is shown for Erk3+/+ (left) and Erk3−/− (right) mice. The first panel represents different pairs, from the most proximal Vα segment (Vα6; top) to the most distal Vα segment (Vα19; bottom). The second panel represents different Jα and Vα3 combinations. The third panel uses a combination of both distal Vα14 and Jα18 segments. The fourth panel represents the amplification of the Tcra constant region (Cα) and serves as the endogenous control. The top triangles indicate the cDNA dilution. (B) Compilation of cDNA band intensity measurement (intensity score) normalized to the intensity of the Cα band. In each graph, more proximal to more distal rearrangements are shown from left to right. Results are presented as a ratio of Erk3+/+ to Erk3−/− intensity scores. The mean ± SEM are shown for each pair segment. (C) Quantitative PCR analysis of TCRα secondary gene rearrangements. Data are representative of at least 2 independent experiments. *, P < 0.05; ANOVA with Bonferroni posttest was used for panel B, and a two-tailed Student's t test was used for panel C.
FIG 8.

Decreased survival of DP thymocytes in ERK3-deficient mice. (A) mRNA quantification of the antiapoptotic Mcl1 and proapoptotic Pmaip1 genes in sorted DP thymocytes from Erk3+/+ and Erk3−/− mice. (B) mRNA quantification of the Bcl2l11, Bcl2a1, and Rorc genes in sorted DP thymocytes from Erk3+/+ and Erk3−/− mice. (C) Bcl-xL representative overlay histogram gated on DP thymocytes from Erk3+/+ and Erk3−/− mice. Fluorescence minus one (FMO) was used as a negative control. (D) Quantification of DP thymocyte cell death. Annexin V-positive (AnnV+) cells among total, TCRlo, TCRint, and TCRhi DP thymocyte populations after 5 days of culture in FTOCs are shown. Data are representative of at least 2 independent experiments. *, P < 0.05, two-tailed Student's t test.
FIG 9.
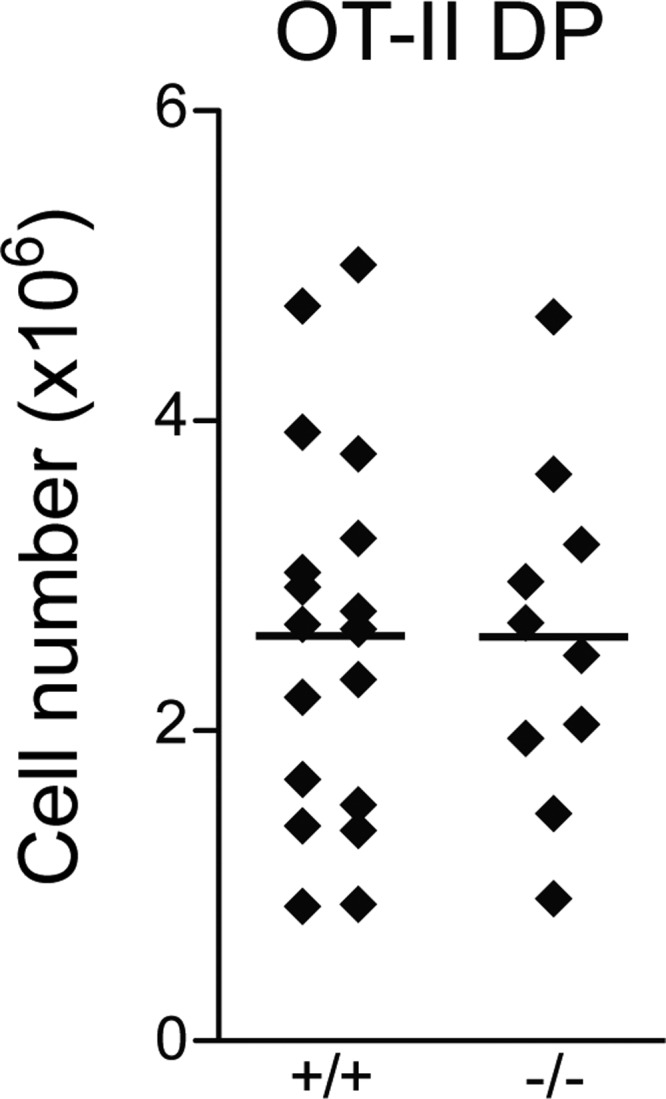
Introduction of a TCR transgene restores DP thymocyte number in Erk3−/− mice. Numbers of DP thymocytes in newborn OT-II Rag1−/− Erk3−/− and OT-II Rag1−/− Erk3+/+ mice are shown. Each dot represents one mouse, and the bars represent mean values.
The kinase activity of ERK3 is required for normal DP thymocyte differentiation.
To get further insight into the mechanism by which ERK3 regulates DP thymocyte survival, we evaluated T cell differentiation in Erk3KA/KA knock-in mice, which express a catalytically inactive form of ERK3. Expression of a kinase-dead form of ERK3 recapitulated the phenotype observed in ERK3 null mice, as evidenced by a reduction of DP and CD4SP thymocyte numbers and decreased TCR expression by DP thymocytes (Fig. 10). These results demonstrate that the kinase activity of ERK3 is absolutely required for normal thymocyte differentiation.
FIG 10.

The kinase activity of ERK3 is required for normal thymocyte differentiation. (A) Representative thymic profiles of Erk3+/+ and Erk3KA/KA E18.5 mice. The percentages of cells in the different thymocyte subsets are indicated on the dot plot. (B) Cell numbers for the different thymocyte populations are shown for Erk3+/+ and Erk3KA/KA E18.5 mice. The bars represent the mean values; each dot corresponds to one mouse. (C) Reduced cell surface expression of the TCR by Erk3KA/KA DP thymocytes. The representative overlay shows TCRβ cell surface expression by Erk3+/+ (thin line) and Erk3KA/KA (thick line) DP thymocytes. (D) Compilation of the percentages of TCRlo, TCRint, and TCRhi DP thymocytes in Erk3+/+ and Erk3KA/KA mice. Each dot represents one mouse, and the bars indicate the mean values. Data are representative of 2 independent experiments. ***, P < 0.001, two-tailed Student's t test.
DISCUSSION
Our results identify the atypical MAPK ERK3 as a new regulator of T cell development. In mice lacking ERK3, DP thymocyte number is reduced 2-fold in a hematopoietic cell-autonomous fashion. This reduction is not due to a decreased rate of generation from ETPs or to defective proliferation after β-selection, indicating that ERK3 is not necessary for the generation of DP thymocytes. Analysis of the maturation state of DP thymocytes using TCR expression levels has shed light on the role of ERK3. Indeed, we observed a reduced proportion of TCR-expressing DP cells in Erk3−/− mice. This defective expression of the TCR suggests that TCR gene rearrangement is affected by ERK3 deficiency. The lack of DNA DSB accumulation at the TCR Jα gene segments ruled out that the Erk3−/− DP thymocyte number was reduced as a consequence of cell death mediated by genotoxic stress. It also indicates that ERK3 expression is not necessary for proper repair of DNA DSBs occurring at the TCRα locus during rearrangement. Furthermore, in the absence of ERK3, we have observed a reduction of DNA DSBs at Jα gene segments in DP thymocytes, which is more important for distal Jα segments. This suggests that Erk3−/− DP thymocytes make fewer secondary TCRα gene rearrangements. This hypothesis is further supported by the reduced usage of distal V (5′ in the locus) and J (3′ in the locus) segments that we observed in ERK3-deficient DP thymocytes. The decrease rate of secondary TCRα gene rearrangement is not the consequence of reduced Rag expression. The similar levels of Rag1 and Rag2 transcripts suggest that ERK3 does not work in concert with MK5 to control Rag transcription in thymocytes.
Reduced secondary TCRα gene rearrangement is observed in several experimental systems where DP thymocyte survival is compromised (26, 29–32), since a certain time window is required for thymocytes to perform these successive TCRα gene rearrangements. Accordingly, ERK3-deficient DP thymocytes show an increased rate of apoptosis, which suggests that they have a shorter life span, explaining the reduced usage of distal TCRα locus rearrangement. Unexpectedly, the reduced half-life of Erk3−/− DP thymocytes is not the consequence of decreased Bcl-xL expression, which is thought to be the major regulator of DP thymocyte survival in other genetically modified mouse models with reduced secondary TCRα gene rearrangement (26, 29–32). In these models, the deficiency in the transcription factors RORγt, TCF-1, and c-Myb all affect DP thymocyte survival by reducing the transcription of Bcl-xL (26, 29–32). The fact that we observed normal expression of Bcl-xL suggests that ERK3 is not involved in the regulation of these transcription factors or of other regulators of Bcl-xL expression. Indeed, we did not observe any difference in the level of transcription of Rorc between Erk3+/+ and Erk3−/− DP thymocytes. Instead of a decrease in Bcl-xL expression, ERK3 deficiency in DP thymocytes leads to an increase in the mRNA level of the proapoptotic gene Pmaip1 (Noxa) and a decrease in the mRNA level of the antiapoptotic gene Mcl1, which may tip the balance between pro- and antiapoptotic factors toward apoptosis. Of interest, these two molecules have been previously shown to also influence DP thymocyte survival (44, 45). Thus, our results further highlight that Bcl-xL is not the only member of the pro- and antiapoptotic Bcl-2 family controlling DP thymocyte survival in vivo.
The selective role for ERK3 in promoting survival of thymocytes only during TCRα chain rearrangement probably reflects the fact that in DN thymocytes every successful TCRβ chain rearrangement is permissive to β-selection and further differentiation.
Importantly, we have demonstrated that the catalytic activity of ERK3 is essential for proper differentiation of T cells in the thymus. Indeed, introduction of a kinase-dead form of ERK3 at the Erk3 locus recapitulates the phenotype that is observed in the absence of ERK3. To our knowledge, this is the first demonstration of a role for the kinase activity of ERK3 in vivo. This also suggests that the long C-terminal extension of ERK3 is dispensable for thymic differentiation. Our ERK3 mouse mutants offer unique models to identify the key in vivo substrates of ERK3.
The partial thymocyte differentiation phenotype observed in the absence of ERK3 suggests that other signaling effectors have redundant functions during thymocyte differentiation. One possible candidate is the paralogous MAPK ERK4, which shares high homology with ERK3 (3). However, we did not observe any defect in thymocyte differentiation in Erk4−/− mice (not shown) or any aggravation of the phenotype in ERK3/ERK4 double-deficient mice (not shown). Further studies are required to identify the network of signaling proteins instructing thymocyte differentiation in the absence of ERK3.
Another important and unique role of ERK3 that was unveiled by our studies is the control of CD4SP thymocyte differentiation. Lack of ERK3 expression only in thymocytes is not sufficient to block CD4SP thymocyte development, since these cells mature normally when ERK3 is inactivated in hematopoietic cells. This suggests that the absence of ERK3 in the thymic epithelium may be responsible for the phenotype observed in newborn mice. It is possible that ERK3-deficient thymocytes lack expression of molecules that are involved in the thymocyte-thymic epithelial cell cross talk that controls maturation and maintenance of thymic epithelial cells (46–48). Even though CD4SP thymocyte differentiation occurs in ERK3-deficient hematopoietic chimeras, their reduced usage of distal V and J segments during TCRα chain rearrangement will impact the diversity of the TCR repertoire, which may then influence the T cell response to certain antigens.
Our results further highlight the unique physiological functions of ERK3 among MAPK family members. ERK3 has no redundant functions with its closest relatives, ERK1/2, during T cell development (33) and controls unique thymocyte differentiation events, suggesting that this atypical MAPK has evolved to assume functions different from those of conventional MAPKs.
In conclusion, we have identified two novel and unique functions for ERK3 during T cell development. First, ERK3 expression in thymocytes is necessary to promote DP thymocyte survival to allow successive TCRα gene rearrangements. Second, ERK3 expression by thymic epithelial cells is essential for the differentiation of CD4+ T cells. The identification of the role of ERK3 in these processes may help in designing better strategies for immune reconstitution in immunocompromised patients.
ACKNOWLEDGMENTS
We thank members of the laboratory for helpful discussion. We acknowledge C. Perreault, S. Lesage, and H. Melichar for critical reading of the manuscript. We thank N. Henley and M. Dupuis for cell sorting, C. Beauchamp, J. Rooney, M. Saba El Leil, and K. Lévesque for technical assistance, M. Chagnon for statistical analysis, and the staff of the Animal Facility for mouse care.
This work was supported by grants from the Natural Sciences and Engineering Council of Canada (NSERC) (grant number 262146-2009) to N.L. and the Canadian Institutes of Health Research (CIHR) (MOP-93729) to S. Meloche. M.M. was supported by the Cole Foundation and the University of Montreal. S.B. was supported by a CIHR Fellowship, and K.C. was supported by an NSERC summer studentship. S. Meloche holds the Canada Research Chair in Cellular Signaling. N.L. was supported by a CIHR New Investigator Award and a Senior Scholarship of the Fonds de la Recherche en Santé du Québec.
Footnotes
Published ahead of print 7 July 2014
REFERENCES
- 1.Pearson G, Robinson F, Gibson TB, Xu BE, Karandikar M, Berman K, Cobb MH. 2001. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22:153–183. 10.1210/edrv.22.2.0428 [DOI] [PubMed] [Google Scholar]
- 2.Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. 1991. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65:663–675. 10.1016/0092-8674(91)90098-J [DOI] [PubMed] [Google Scholar]
- 3.Coulombe P, Meloche S. 2007. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim. Biophys. Acta 1773:1376–1387. 10.1016/j.bbamcr.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 4.Deleris P, Rousseau J, Coulombe P, Rodier G, Tanguay PL, Meloche S. 2008. Activation loop phosphorylation of the atypical MAP kinases ERK3 and ERK4 is required for binding, activation and cytoplasmic relocalization of MK5. J. Cell. Physiol. 217:778–788. 10.1002/jcp.21560 [DOI] [PubMed] [Google Scholar]
- 5.Deleris P, Trost M, Topisirovic I, Tanguay PL, Borden KL, Thibault P, Meloche S. 2011. Activation loop phosphorylation of ERK3/ERK4 by group I p21-activated kinases (PAKs) defines a novel PAK-ERK3/4-MAPK-activated protein kinase 5 signaling pathway. J. Biol. Chem. 286:6470–6478. 10.1074/jbc.M110.181529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De la Mota-Peynado A, Chernoff J, Beeser A. 2011. Identification of the atypical MAPK Erk3 as a novel substrate for p21-activated kinase (Pak) activity. J. Biol. Chem. 286:13603–13611. 10.1074/jbc.M110.181743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schumacher S, Laass K, Kant S, Shi Y, Visel A, Gruber AD, Kotlyarov A, Gaestel M. 2004. Scaffolding by ERK3 regulates MK5 in development. EMBO J. 23:4770–4779. 10.1038/sj.emboj.7600467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seternes OM, Mikalsen T, Johansen B, Michaelsen E, Armstrong CG, Morrice NA, Turgeon B, Meloche S, Moens U, Keyse SM. 2004. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 23:4780–4791. 10.1038/sj.emboj.7600489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aberg E, Perander M, Johansen B, Julien C, Meloche S, Keyse SM, Seternes OM. 2006. Regulation of MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAPK ERK4/MAPK4. J. Biol. Chem. 281:35499–35510. 10.1074/jbc.M606225200 [DOI] [PubMed] [Google Scholar]
- 10.Kant S, Schumacher S, Singh MK, Kispert A, Kotlyarov A, Gaestel M. 2006. Characterization of the atypical MAPK ERK4 and its activation of the MAPK-activated protein kinase MK5. J. Biol. Chem. 281:35511–35519. 10.1074/jbc.M606693200 [DOI] [PubMed] [Google Scholar]
- 11.Gaestel M. 2006. MAPKAP kinases—MKs —two's company, three's a crowd. Nat. Rev. Mol. Cell Biol. 7:120–130. 10.1038/nrm1834 [DOI] [PubMed] [Google Scholar]
- 12.Perander M, Keyse SM, Seternes OM. 2008. Does MK5 reconcile classical and atypical MAP kinases? Front. Biosci. 13:4617–4624. 10.2741/3027 [DOI] [PubMed] [Google Scholar]
- 13.Chow KT, Timblin GA, McWhirter SM, Schlissel MS. 2013. MK5 activates Rag transcription via Foxo1 in developing B cells. J. Exp. Med. 210:1621–1634. 10.1084/jem.20130498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coulombe P, Rodier G, Pelletier S, Pellerin J, Meloche S. 2003. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol. Cell. Biol. 23:4542–4558. 10.1128/MCB.23.13.4542-4558.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klinger S, Turgeon B, Levesque K, Wood GA, Aagaard-Tillery KM, Meloche S. 2009. Loss of Erk3 function in mice leads to intrauterine growth restriction, pulmonary immaturity, and neonatal lethality. Proc. Natl. Acad. Sci. U. S. A. 106:16710–16715. 10.1073/pnas.0900919106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brand F, Schumacher S, Kant S, Menon MB, Simon R, Turgeon B, Britsch S, Meloche S, Gaestel M, Kotlyarov A. 2012. The extracellular signal-regulated kinase 3 (mitogen-activated protein kinase 6 [MAPK6])-MAPK-activated protein kinase 5 signaling complex regulates septin function and dendrite morphology. Mol. Cell. Biol. 32:2467–2478. 10.1128/MCB.06633-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhandoola A, von Boehmer H, Petrie HT, Zuniga-Pflucker JC. 2007. Commitment and developmental potential of extrathymic and intrathymic T cell precursors: plenty to choose from. Immunity 26:678–689. 10.1016/j.immuni.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 18.Hoffman ES, Passoni L, Crompton T, Leu TM, Schatz DG, Koff A, Owen MJ, Hayday AC. 1996. Productive T-cell receptor beta-chain gene rearrangement: coincident regulation of cell cycle and clonality during development in vivo. Genes Dev. 10:948–962. 10.1101/gad.10.8.948 [DOI] [PubMed] [Google Scholar]
- 19.Lin WC, Desiderio S. 1994. Cell cycle regulation of V(D)J recombination-activating protein RAG-2. Proc. Natl. Acad. Sci. U. S. A. 91:2733–2737. 10.1073/pnas.91.7.2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aifantis I, Mandal M, Sawai K, Ferrando A, Vilimas T. 2006. Regulation of T-cell progenitor survival and cell-cycle entry by the pre-T-cell receptor. Immunol. Rev. 209:159–169. 10.1111/j.0105-2896.2006.00343.x [DOI] [PubMed] [Google Scholar]
- 21.Starr TK, Jameson SC, Hogquist KA. 2003. Positive and negative selection of T cells. Annu. Rev. Immunol. 21:139–176. 10.1146/annurev.immunol.21.120601.141107 [DOI] [PubMed] [Google Scholar]
- 22.Labrecque N, Baldwin T, Lesage S. 2011. Molecular and genetic parameters defining T-cell clonal selection. Immunol. Cell Biol. 89:16–26. 10.1038/icb.2010.119 [DOI] [PubMed] [Google Scholar]
- 23.Bassing CH, Alt FW. 2004. The cellular response to general and programmed DNA double strand breaks. DNA Repair 3:781–796. 10.1016/j.dnarep.2004.06.001 [DOI] [PubMed] [Google Scholar]
- 24.Gellert M. 2002. V(D)J. recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 71:101–132. 10.1146/annurev.biochem.71.090501.150203 [DOI] [PubMed] [Google Scholar]
- 25.Rooney S, Chaudhuri J, Alt FW. 2004. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol. Rev. 200:115–131. 10.1111/j.0105-2896.2004.00165.x [DOI] [PubMed] [Google Scholar]
- 26.Guo J, Hawwari A, Li H, Sun Z, Mahanta SK, Littman DR, Krangel MS, He YW. 2002. Regulation of the TCRalpha repertoire by the survival window of CD4(+)CD8(+) thymocytes. Nat. Immunol. 3:469–476. 10.1038/ni791 [DOI] [PubMed] [Google Scholar]
- 27.Grillot DA, Merino R, Nunez G. 1995. Bcl-XL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J. Exp. Med. 182:1973–1983. 10.1084/jem.182.6.1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW, Thompson CB. 1995. Bclx regulates the survival of double-positive thymocytes. Proc. Natl. Acad. Sci. U. S. A. 92:4763–4767. 10.1073/pnas.92.11.4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D'Cruz LM, Knell J, Fujimoto JK, Goldrath AW. 2010. An essential role for the transcription factor HEB in thymocyte survival, Tcra rearrangement and the development of natural killer T cells. Nat. Immunol. 11:240–249. 10.1038/ni.1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu T, Simmons A, Yuan J, Bender TP, Alberola-Ila J. 2010. The transcription factor c-Myb primes CD4+CD8+ immature thymocytes for selection into the iNKT lineage. Nat. Immunol. 11:435–441. 10.1038/ni.1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan J, Crittenden RB, Bender TP. 2010. c-Myb promotes the survival of CD4+CD8+ double-positive thymocytes through upregulation of Bcl-xL. J. Immunol. 184:2793–2804. 10.4049/jimmunol.0902846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang R, Xie H, Huang Z, Ma J, Fang X, Ding Y, Sun Z. 2011. T cell factor 1 regulates thymocyte survival via a RORgammat-dependent pathway. J. Immunol. 187:5964–5973. 10.4049/jimmunol.1101205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer AM, Katayama CD, Pages G, Pouyssegur J, Hedrick SM. 2005. The role of erk1 and erk2 in multiple stages of T cell development. Immunity 23:431–443. 10.1016/j.immuni.2005.08.013 [DOI] [PubMed] [Google Scholar]
- 34.Rousseau J, Klinger S, Rachalski A, Turgeon B, Deleris P, Vigneault E, Poirier-Heon JF, Davoli MA, Mechawar N, El Mestikawy S, Cermakian N, Meloche S. 2010. Targeted inactivation of Mapk4 in mice reveals specific nonredundant functions of Erk3/Erk4 subfamily mitogen-activated protein kinases. Mol. Cell. Biol. 30:5752–5763. 10.1128/MCB.01147-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lacombe MH, Hardy MP, Rooney J, Labrecque N. 2005. IL-7 receptor expression levels do not identify CD8+ memory T lymphocyte precursors following peptide immunization. J. Immunol. 175:4400–4407. 10.4049/jimmunol.175.7.4400 [DOI] [PubMed] [Google Scholar]
- 36.Chan S, Correia-Neves M, Dierich A, Benoist C, Mathis D. 1998. Visualization of CD4/CD8 T cell commitment. J. Exp. Med. 188:2321–2333. 10.1084/jem.188.12.2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitt TM, Zuniga-Pflucker JC. 2002. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 17:749–756. 10.1016/S1074-7613(02)00474-0 [DOI] [PubMed] [Google Scholar]
- 38.Mathieu M, Cotta-Grand N, Daudelin JF, Boulet S, Lapointe R, Labrecque N. 2012. CD40-activated B cells can efficiently prime antigen-specific naive CD8 T cells to generate effector but not memory T cells. PLoS One 7:e30139. 10.1371/journal.pone.0030139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McMurry MT, Hernandez-Munain C, Lauzurica P, Krangel MS. 1997. Enhancer control of local accessibility to V(D)J. recombinase. Mol. Cell. Biol. 17:4553–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ismail IH, Hendzel MJ. 2008. The gamma-H2A.X: is it just a surrogate marker of double-strand breaks or much more? Environ. Mol. Mutagen. 49:73–82. 10.1002/em.20358 [DOI] [PubMed] [Google Scholar]
- 41.Rogakou EP, Boon C, Redon C, Bonner WM. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146:905–916. 10.1083/jcb.146.5.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858–5868. 10.1074/jbc.273.10.5858 [DOI] [PubMed] [Google Scholar]
- 43.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. 2000. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 275:9390–9395. 10.1074/jbc.275.13.9390 [DOI] [PubMed] [Google Scholar]
- 44.Yelamos J, Monreal Y, Saenz L, Aguado E, Schreiber V, Mota R, Fuente T, Minguela A, Parrilla P, de Murcia G, Almarza E, Aparicio P, Menissier-de Murcia J. 2006. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J. 25:4350–4360. 10.1038/sj.emboj.7601301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dzhagalov I, Dunkle A, He YW. 2008. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J. Immunol. 181:521–528. 10.4049/jimmunol.181.1.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boehm T, Scheu S, Pfeffer K, Bleul CC. 2003. Thymic medullary epithelial cell differentiation, thymocyte emigration, and the control of autoimmunity require lympho-epithelial cross talk via LTbetaR. J. Exp. Med. 198:757–769. 10.1084/jem.20030794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shores EW, Van Ewijk W, Singer A. 1991. Disorganization and restoration of thymic medullary epithelial cells in T cell receptor-negative scid mice: evidence that receptor-bearing lymphocytes influence maturation of the thymic microenvironment. Eur. J. Immunol. 21:1657–1661. 10.1002/eji.1830210711 [DOI] [PubMed] [Google Scholar]
- 48.van Ewijk W, Shores EW, Singer A. 1994. Crosstalk in the mouse thymus. Immunol. Today 15:214–217. 10.1016/0167-5699(94)90246-1 [DOI] [PubMed] [Google Scholar]