

Abstract
Insulin and insulin-like growth factor 1 (IGF-1) receptor signaling pathways differentially modulate cardiac growth under resting conditions and following exercise training. These effects are mediated by insulin receptor substrate 1 (IRS1) and IRS2, which also differentially regulate resting cardiac mass. To determine the role of IRS isoforms in mediating the hypertrophic and metabolic adaptations of the heart to exercise training, we subjected mice with cardiomyocyte-specific deletion of either IRS1 (CIRS1 knockout [CIRS1KO] mice) or IRS2 (CIRS2KO mice) to swim training. CIRS1KO hearts were reduced in size under basal conditions, whereas CIRS2KO hearts exhibited hypertrophy. Following exercise swim training in CIRS1KO and CIRS2KO hearts, the hypertrophic response was equivalently attenuated, phosphoinositol 3-kinase (PI3K) activation was blunted, and prohypertrophic signaling intermediates, such as Akt and glycogen synthase kinase 3β (GSK3β), were dephosphorylated potentially on the basis of reduced Janus kinase-mediated inhibition of protein phosphatase 2a (PP2A). Exercise training increased peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) protein content, mitochondrial capacity, fatty acid oxidation, and glycogen synthesis in wild-type (WT) controls but not in IRS1- and IRS2-deficient hearts. PGC-1α protein content remained unchanged in CIRS1KO but decreased in CIRS2KO hearts. These results indicate that although IRS isoforms play divergent roles in the developmental regulation of cardiac size, these isoforms exhibit nonredundant roles in mediating the hypertrophic and metabolic response of the heart to exercise.
INTRODUCTION
Cardiac hypertrophy is the growth response of the heart to increased workload and has been categorized as physiological or pathological hypertrophy. Cardiac hypertrophy is considered pathological if contractile dysfunction occurs after an initial phase of compensation, which ultimately results in heart failure. Common causes for pathological hypertrophy are valvular disease and hypertension. In contrast, physiological hypertrophy is characterized by adaptive myocyte growth with a new steady state and preserved contractile function, as exemplified by the response to chronic exercise training, also known as “athlete's heart” (1). The differences between physiological and pathological cardiac hypertrophy have been attributed in part to differences in intracellular signaling pathways. For example, insulin receptor- and insulin-like growth factor 1 (IGF-1) receptor-mediated signaling to phosphatidylinositol 3-kinase (PI3K) and Akt1 have been implicated in physiological cardiac hypertrophy, whereas activation of G protein-coupled pathways, such as angiotensin II and adrenergic signaling, has been associated with pathological hypertrophy (2, 3).
We previously reported that cardiomyocyte-selective deletion of the insulin receptor (CIRKO) reduced heart size by 20 to 30% (4), whereas under basal conditions IGF-1 receptor deletion was without effect (5). CIRKO hearts exhibit increased glycolysis and decreased fatty acid oxidation rates (4) and impaired maturation of cardiac mitochondrial oxidative capacity (6). Cardiac IGF-1 production is increased in professional athletes in parallel with physiological cardiac hypertrophy (7), and IGF-1 mRNA levels were increased in hearts obtained from chronically exercise-trained rodents (8). Using a mouse model with cardiomyocyte-specific deletion of the IGF-1 receptor, we showed that IGF-1 signaling mediated the hypertrophic adaptation of the heart to physiological stimuli such as exercise, despite negligible effects at baseline (5).
An obligate requirement for both PI3K and Akt1 signaling in mediating cardiomyocyte hypertrophy in response to chronic exercise training has been demonstrated (9–11). We subsequently showed that PI3K signaling mediated the mitochondrial bioenergetic adaptation to chronic endurance training via Akt-independent signaling pathways (12). Whereas signaling from insulin and IGF-1 receptors to PI3K is mediated by insulin receptor substrate 1 (IRS1) and IRS2, their roles in hypertrophic signaling in the heart are incompletely understood. Studies of germ line knockouts for IRS1 and IRS2 suggest a predominant role for IRS1 in the regulation of somatic growth and for IRS2 in the regulation of metabolism (13–15). We therefore sought to test the hypothesis that IRS isoforms might differentially modulate the hypertrophic and metabolic adaptations of the heart to exercise training. Thus, we generated mice with cardiomyocyte-specific deletion (CIRS knockout [KO]) of either IRS1 (CIRS1KO) or IRS2 (CIRS2KO) using cre/loxP recombination and subjected them to chronic swim training.
We found that resting CIRS1KO hearts were reduced in size, whereas CIRS2KO hearts exhibited baseline cardiac hypertrophy. Following chronic exercise training, hypertrophy developed as expected in wild-type (WT) but not in CIRS1KO or CIRS2KO hearts. In parallel, mitochondrial capacity and substrate oxidation rates increased in WT controls but not in IRS-deficient hearts. Together, these results indicate that although IRS isoforms play divergent roles in the developmental regulation of cardiac size, both isoforms are required for the hypertrophic and bioenergetic response of the heart to chronic exercise training.
MATERIALS AND METHODS
Animals.
CIRS1KO mice (αMHC-Cre+/−:IRS1lox/lox) were generated by cross-breeding mice harboring the homozygous IRS1 allele flanked by loxP sites (IRS1lox/lox) with mice expressing the enzyme Cre recombinase driven by the α-myosin heavy chain promoter (αMHC-Cre+/−) (16). CIRS2KO (αMHC-Cre+/−:IRS2lox/lox) mice were generated similarly. The generation of IRS1lox/lox and IRS2lox/lox mice has been described before (17, 18), and genotyping was performed as previously described (16–18). IRS2lox/lox mice were used as wild-type controls in all experiments. All studies were performed under random feeding conditions in male mice unless otherwise indicated. All mice were back-crossed to the C57/BL6 genetic background for four generations and maintained on the same C57/BL6/129Sv mixed genetic background. Animals were housed with a 12-h light/12-h dark cycle at 22°C with free access to food and water. All experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Utah and the Carver College of Medicine of the University of Iowa.
Exercise training.
Six- to 7-week-old mice were subjected to swim training. Mice underwent two training sessions a day, starting with a 10-min duration per mouse on the first day. The duration of each swimming session increased by 10 min/day until a maximum duration of 90 min twice a day was achieved. The sessions were separated by at least a 4-h interval. Mice were trained for 28 additional days (for a total training period of 36 days) (5). Mice were euthanized and tissues harvested 1 h after the final swimming session.
Immunoblotting.
Western blot analysis was performed as described in detail in the supplemental material (19).
WGA stains and stereological quantification.
Myocardial fragments were embedded in paraffin, portioned into 5-μm-thick sections, stained with wheat germ agglutinin (WGA)-Alexa Fluor 488 conjugate (Invitrogen Corporation, Carlsbad, CA), and covered with SlowFade Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Images were obtained and processed at the University of Utah Fluorescence Microscopy Core Facility. From each sample, left ventricular myocardium was identified in 2 chamber cross sections, and 20 microscopic fields were analyzed at random, the stage of the microscope being moved blindly. For stereological analysis, cross-sectioned left ventricular cardiomyocytes with complete WGA staining of the cellular membrane and central nuclear localization were selected. The cardiomyocyte diameter was determined using the Image-Pro Plus software package (Media Cybernetics, Bethesda, MD).
Hemodynamic measurements.
Mice were anesthetized (400 mg chloral hydrate/kg body weight) and placed in the supine position on a heating pad. A 1.4-F micromanometer-tipped pressure catheter (Millar Instruments, Houston, TX) was retrogradely introduced into the left ventricle via the right carotid artery. Hemodynamic measurements were obtained and analyzed as previously described using LabChart7 Pro software (ADInstruments, Colorado Springs, CO) (20, 21).
Mitochondrial function in saponin-permeabilized cardiac fibers.
Mitochondrial oxygen consumption and ATP production were determined in subendocardial muscle fibers (19). See the supplemental materials for details.
Measurement of PI3K activity.
Cardiac lysates were immunoprecipitated with anti-PI3K p85 antibody (Millipore, Billerica, MA), and PI3K enzymatic activity was measured using a competitive enzyme-linked immunosorbent assay (ELISA) kit (Echelon, Salt Lake City, UT). Briefly, about 25 mg of tissue was homogenized in ice-cold homogenization buffer (20 mmol/liter Tris-HCl, 1 mmol/liter CaCl2, 137 mmol/liter NaCl, 1% Triton X-100, 1.5 mmol/liter MgCl2, pH 7.4) supplemented with the HALT protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL) using a hand-driven homogenizer, incubated on ice for 20 min, and centrifuged at 12,000 × g for 10 min. Supernatants were transferred to new reaction tubes, and protein concentration was determined. Samples were divided for measurement of PI3K p85 subunit protein expression or PI3K enzymatic activity.
For measurement of PI3K enzymatic activity, homogenization buffer was added to homogenate containing 500 μg of protein to a total reaction volume of 1 ml. Five micrograms of anti-PI3K p85 antibody (Millipore, Billerica, MA) was then added, and the mixture was incubated overnight at 4°C with constant rotation. Next, protein A-agarose beads (Millipore, Billerica, MA) were added, the mixture was incubated for 1 h, and immunoprecipitated complexes were collected by centrifugation. The supernatant was discarded, the pellet was washed, and 30 μl of KBZ buffer and 30 μl of 10 μM phosphatidylinositol (4,5)-bisphosphate (PIP2) substrate were added followed by incubation for 3 h at 37°C according to the manufacturer's protocol. The amount of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) formed by PI3K activity was detected using a competitive ELISA, developed using the colorimetric reagent 3,3′,5,5′-tetramethylbenzidine (TMB), and read using a Synergy HT multidetection reader (Bio-Tek Instruments, Winooski, VT) at a wavelength of 450 nm. Enzymatic activity was expressed as amounts of PIP3 produced, normalized either to total protein or to anti-PI3K p85 protein expression.
Quantification of mtDNA content.
DNA was extracted and purified using the DNeasy tissue kit (Invitrogen, Carlsbad, CA), and real-time PCR was performed with an ABI Prism instrument (Applied Biosystems, Foster City, CA) with SYBR green chemistry (Invitrogen). The mitochondrial DNA (mtDNA) content relative to nuclear DNA content was determined using the following primers: nuclear DNA-encoded NADH dehydrogenase flavoprotein 1, forward, CTTCCCCACTGGCCTCAAG, and reverse, CCAAAACCCAGTGATCCAGC; mtDNA-encoded CO1, forward, TGCTAGCCGCAGGCATTAC, and reverse, GGGTGCCCAAAGAATCAGAAC (22).
Mitochondrial enzyme activity assays.
Citrate synthase (CS) and hydroxyacyl-coenzyme A (hydroxyacyl-CoA) dehydrogenase (HADH) enzyme activities were determined as previously described (21). For measurement of skeletal muscle CS activity, absorbance was monitored at 412 nm using an Ultrospec 3000 spectrophotometer (Amersham Pharmacia Biotech, NJ) with a total reaction volume of 1 ml. CS and HADH activities in heart homogenates were measured using a Synergy HT multidetection reader (Bio-Tek Instruments, Winooski, VT) with a total reaction volume of 200 μl as previously described (21).
Isolated working hearts.
Hearts were perfused with Krebs-Henseleit buffer (118.5 mmol/liter NaCl, 25 mmol/liter NaHCO3, 4.7 mmol/liter KCl, 1.2 mmol/liter MgSO4, 1.2 mmol/liter KH2PO4, 2.5 mmol/liter CaCl2, 0.5 mmol/liter EDTA, pH 7.4) containing 5 mmol/liter glucose and 0.4 mmol/liter palmitate using a sealed perfusion apparatus. Glucose oxidation and glycolysis were measured in one set of hearts, and palmitate oxidation rates were measured in a second set of experiments. Glucose oxidation was determined by measuring 14CO2 released by the metabolism of [U-14C]glucose (specific activity, 296 MBq/mol). Glycolytic flux was assessed by the amount of 3H2O released from the metabolism of exogenous [5-3H]glucose (specific activity, 177 MBq/mol). Palmitate oxidation was calculated from the amount of 3H2O released from the metabolism of exogenous [9,10-3H]palmitate (specific activity, 42 GBq/mol).
Measurements of flow and pressure (Millar pressure catheter; Millar Instruments, Houston, TX) were obtained every 20 min throughout the 60-min perfusion. The oxygen content of freshly oxygenated buffer (arterial partial pressure of oxygen [PaO2]) and oxygen concentration in the pulmonary artery effluent, which was collected using a capillary tube (venous partial pressure of oxygen [PvO2]), were measured using a fiber optic oxygen sensor (Ocean Optics, Orlando, FL). The following formulae were used to determine myocardial O2 consumption (MVO2), cardiac hydraulic work (CHW), and cardiac efficiency (CE).
Myocardial O2 consumption, measured in ml · min−1 · g−1 wet heart weight (WHW), was determined as follows: MVO2 = [(PaO2 − PvO2)/100] · (coronary flow/WHW) · (725/760) · (1,000 · C), where PaO2 is the arterial partial pressure of oxygen in mm Hg, PvO2 is the venous partial pressure of oxygen in mm Hg, 725 and 760 are the atmospheric pressures in mm Hg at the University of Utah and at sea level, respectively, and C is the Bunsen coefficient for plasma, i.e., 0.0212.
Cardiac hydraulic work, measured in J · min−1 · g−1 WHW, was determined as follows: CHW = CO · DevP · 1.33 · 10−4/g WHW, where CO is the cardiac output in ml/min and DevP is the developed pressure in mm Hg.
Cardiac efficiency, as a percentage, was determined as follows. CE = hydraulic work/MVO2 · 100. MVO2 (ml/min) was converted to μmol/min by multiplying by the conversion factor 0.0393 and then to joules per min (J/min) using the conversion formula of 1 μmol O2 = 0.4478 J, as previously described (19).
Glucose uptake in isolated cardiomyocytes.
Myocytes were isolated from hearts obtained from 8-week-old female mice. Glucose uptake was measured in the presence or absence of 1 nM insulin as previously described (4).
Measurement of cardiac glycogen content.
Snap-frozen tissue was homogenized in 0.3 mol/liter perchloric acid and assayed in a buffer containing 50 mmol/liter Na acetate and 0.02% bovine serum albumin with or without amyloglucosidase (Sigma, St. Louis, MO). Absorption was monitored at 340 nm using an Ultrospec 3000 spectrophotometer (Amersham Pharmacia Biotech, NJ) and compared with a glucose standard of 0 to 80 μmol. Data are presented as glucose released from glycogen corrected to tissue weight (23).
Statistical analysis.
All data are expressed as means ± standard errors of the means (SEM). Unpaired Student's t test was used to analyze differences relative to sedentary controls with the same genotype. One-way analysis of variance (ANOVA) was performed when analyzing differences between 3 genotypes (WT, CIRS1KO, and CIRS2KO) under basal conditions (glucose tolerance tests and serum insulin levels) followed by Fisher's protected least significant difference (PLSD) test. Two-way ANOVA was performed to analyze differences by genotype and exercise swim training or insulin stimulation when comparing 6 groups (WT, CIRS1KO, CIRS2KO with or without swim training or insulin stimulation), followed by Fisher's protected least significant difference test. Statistical calculations were performed using the GraphPad Prism software (GraphPad, San Diego, CA). For all analyses, a probability value of <0.05 was considered significantly different.
RESULTS
IRS isoforms play redundant roles in the regulation of myocardial insulin signaling.
CIRS1KO and CIRS2KO mice were born at the expected Mendelian distribution and survived to adulthood (data not shown). Cardiomyocyte-specific deletion of IRS1 or IRS2 was confirmed by immunoblotting (Fig. 1a and b) and had no effect on glucose tolerance and serum insulin levels (see Fig. S1 in the supplemental material). WT and IRS-deficient hearts were perfused with insulin, and Akt, AS160, S6K, and S6 phosphorylation were determined. The fold increase in insulin-mediated Akt phosphorylation at Ser473 was not impacted by loss of IRS1 or IRS2 individually. The activation of Akt on Thr308 was also not altered by IRS2 deficiency, although it was reduced by 50% in IRS1-deficient hearts. Similarly, absolute levels of AS160 and phosphorylated AS160 were also reduced in IRS1-deficient hearts (see Fig. S2 in the supplemental material). Despite these changes, insulin stimulation led to an equivalent fold increase in AS160, S6K, and S6 phosphorylation in IRS-deficient hearts relative to WT controls (Fig. 1c to h), and insulin-stimulated glucose uptake levels in isolated cardiomyocytes obtained from WT, CIRS1KO, and CIRS2KO hearts were similar (Fig. 1i). These data indicate redundant roles for IRS isoforms in myocardial insulin signaling and insulin-stimulated glucose uptake.
FIG 1.

Preserved insulin-mediated signaling in CIRS1KO and CIRS2KO hearts. (a and b) Representative immunoblots for IRS1 and IRS2 in homogenates of various tissues from 8-week-old CIRS1KO and CIRS2KO mice and ventricle homogenates from mice with genotypes as indicated. (c to h) Representative immunoblots (c) and quantification of densitometry (d to h) in protein lysates obtained from isolated working hearts perfused for 1 h with Krebs-Henseleit buffer containing 5 mM glucose and 0.4 mM palmitate in the presence or absence of 1 nM insulin (n = 4). (i) Glucose uptake in isolated cardiomyocytes obtained from genotypes as indicated in the presence or absence of 1 nM insulin. Data are presented as fold changes relative to the same genotype, no insulin (n = 7 to 9). Two-way ANOVA was performed to analyze differences by insulin stimulation and genotype followed by Fisher's PLSD post hoc analysis. Significance: phosphorylation of Akt at Ser473, P < 0.05 for insulin (d) and Thr308, P < 0.05 for insulin, genotype, and their interaction (e); phosphorylation of AS160 at Thr642 (f), phosphorylation of p70 S6K at Thr389 (g), phosphorylation of S6 at Ser235/236 (h), and glucose uptake in isolated cardiomyocytes (i), P < 0.05 for insulin. *, P < 0.05 versus WT (same insulin concentration); †, P < 0.05 versus same genotype (0 nM insulin); ‡, P < 0.05 versus CIRS1KO (same insulin concentration).
Expression of both IRS isoforms is required for the hypertrophic response to exercise training.
Citrate synthase (CS) activity levels in skeletal muscle homogenates obtained from WT, CIRS1KO, and CIRS2KO mice following exercise training were equivalently increased, indicating similar degrees of exercise training across all genotypes (Fig. 2a). At 12 weeks of age, heart weight-to-tibia length ratios (HW/TL) were reduced by 12.8% (P < 0.05) in CIRS1KO mice compared to WT controls under sedentary conditions. In contrast, HW/TL was increased in CIRS2KO mice by 8.4% (P < 0.05) at the same age. WT mice increased HW/TL by 10.2% (P < 0.05) following 5 weeks of exercise swim training, but we observed no increase in CIRS1KO and CIRS2KO mice, respectively (Fig. 2b and c; see also Table S1 in the supplemental material). These results were confirmed by stereological quantification of cardiomyocyte diameter using wheat germ agglutinin (WGA) staining (Fig. 2d and e). Left ventricular catheterization revealed a mild impairment in contractile function in CIRS1KO hearts following exercise training, and maximum dP/dt was significantly lower in CIRS1KO mice than in CIRS2KO mice (Fig. 2f to h; see also Table S2 in the supplemental material). However, mRNAs of heart failure markers were not further induced in IRS-deficient hearts by exercise training (see Fig. S3 in the supplemental material). Thus, IRS proteins play redundant roles in insulin-mediated signaling in the heart but are both required for the hypertrophic response following exercise training despite exhibiting divergent roles in the developmental regulation of cardiac size.
FIG 2.

IRS proteins modulate the hypertrophic response to exercise training. Two-way ANOVA was performed to analyze differences by swim training and genotype followed by Fisher's PLSD post hoc analysis. (a) Citrate synthase activity in mixed gastrocnemius muscle homogenates (n = 6; P < 0.05 for swim training); (b) heart weights (HW; P < 0.05 for genotype, P < 0.05 for the interaction between swim training and genotype); (c) heart weights normalized to tibia lengths (HW/TL) under sedentary conditions and following swim training (n = 10; P < 0.05 for swim training, genotype, and their interaction). (d and e) Representative pictures and quantification of WGA staining (n = 6 per group; scale bar, 20 μm; P < 0.05 for genotype, P < 0.05 for the interaction between swim training and genotype). The intensities of each panel were adjusted individually to allow visualization of cell size. (f to h) In vivo, left ventricular hemodynamic parameters (maximum [Max] or minimum [Min] rates of change of left ventricle [LV] contraction [dP/dt] and LV developed pressure [Dev P]) under sedentary conditions and following exercise training (n = 9 to 12; Max dP/dt and LV Dev P, P < 0.05 for genotype, respectively). *, P < 0.05 versus WT (same treatment); †, P < 0.05 versus sedentary (same genotype); ‡, P < 0.05 versus CIRS1KO (same treatment).
Activation of PI3K in response to exercise is prevented, and downstream signaling targets are dephosphorylated, in IRS-deficient hearts following exercise training.
To determine the underlying mechanisms for the attenuated hypertrophic response in IRS-deficient hearts, we investigated the activation of PI3K/Akt signaling pathways, which may mediate physiological cardiac hypertrophy (5, 12). Exercise increased PI3K enzymatic activity normalized to PI3K p85 subunit protein expression by ∼50% in WT but not in IRS-deficient hearts, with no changes in PI3K p85 subunit protein content across all groups (Fig. 3a to c). Exercise increased Akt phosphorylation 2-fold at Thr308 and by about ∼ 50% at Ser473 in WT hearts (Fig. 3d to f). Similar to PI3K activity, the increase in Thr308 phosphorylation of Akt was prevented in IRS1- and IRS2-deficient hearts (Fig. 3e). Unexpectedly, we observed a striking dephosphorylation of Akt (Ser473), glycogen synthase kinase 3β (GSK3β), and 4E-BP1, in IRS-deficient hearts following exercise training relative to sedentary controls (Fig. 3f to h). These signaling changes would be predicted to antagonize the hypertrophic response (24, 25).
FIG 3.

Attenuated PI3K activation and increased dephosphorylation of prohypertrophic signaling intermediates in CIRS1KO and CIRS2KO hearts following exercise training. Two-way ANOVA was performed to analyze differences by swim training and genotype followed by Fisher's PLSD post hoc analysis. (a) PI3 kinase enzymatic activity (P < 0.05 for genotype, P < 0.05 for the interaction between swim training and genotype); (b) densitometric quantification of PI3 kinase p85 subunit protein; (c) PI3 kinase enzymatic activity normalized to p85 protein (n = 6; P < 0.05 for the interaction between swim training and genotype). (d to k) Representative Western blots (d) and densitometric quantification of P AKT Thr308/Total AKT (P < 0.05 for genotype, P < 0.05 for the interaction between swim training and genotype) (e), P AKT Ser473/Total AKT (P < 0.05 for genotype, P < 0.05 for the interaction between swim training and genotype) (f), P GSK3b Ser9/Total GSK3b (P < 0.05 for swim training, P < 0.05 for the interaction between swim training and genotype) (g), 4E-BP1 γ/GAPDH (P < 0.05 for swim training, P < 0.05 for the interaction between swim training and genotype) (h), P PP2A Tyr307/Total PP2A (P < 0.05 for swim training, genotype, and their interaction) (i), P JAK2 Tyr221/Total JAK2 (P < 0.05 for swim training, P < 0.05 for the interaction between swim training and genotype) (j), and P STAT3 Tyr705/Total STAT3 (P < 0.05 for swim training, P < 0.05 for the interaction between swim training and genotype) (n = 8) (k). *, P < 0.05 versus WT (same treatment); †, P < 0.05 versus sedentary (same genotype); ‡, P < 0.05 versus CIRS1KO (same treatment). n.s., no significant difference observed.
We examined potential mechanisms for kinase dephosphorylation and focused on the protein phosphatase 2a (PP2A) by examining the phosphorylation of PP2A at Tyr307, which decreases the phosphatase activity of PP2A (26–28). Exercise training was associated with reduced phosphorylation of PP2A at Tyr307 in IRS-deficient hearts (Fig. 3i). We then examined the phosphorylation of Janus kinase 2 (JAK2) at Tyr221, which increases its kinase activity to phosphorylate PP2A at Tyr307 (29). Consistent with reduced PP2A phosphorylation, JAK2 phosphorylation at Tyr221 was reduced in IRS1- and IRS2-deficient hearts following exercise (Fig. 3j). Impaired JAK2 signaling in IRS-deficient hearts (30) was corroborated by the observation that phosphorylation of the JAK2 target, STAT3 at Tyr705 (31), was increased in WT controls following swim training but was unchanged in IRS1- or IRS2-deficient hearts (Fig. 3k). The loss of PP2A phosphorylation in IRS mutant animals was specific for exercise training, as PP2A phosphorylation was not altered in WT or mutant hearts following insulin stimulation (see Fig. S4 in the supplemental material).
IRS proteins mediate the mitochondrial bioenergetic response following exercise training.
The mitochondrial adaptations to exercise training are dependent upon an induction of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), in addition to an Akt-independent role of PI3K signaling (12). Thus, we hypothesized that reduced PI3K activation would impair the mitochondrial bioenergetic response to exercise training. ADP-stimulated mitochondrial oxygen consumption (VADP) and ATP synthesis measured in saponin-permeabilized cardiac fibers supplemented with pyruvate or palmitoyl-carnitine as the substrates were increased in WT hearts following swim training (+25.5% to 35.1%, P < 0.05 each). In contrast, no increase was observed in CIRS1KO and CIRS2KO hearts following exercise training (Fig. 4a to d). Similarly, activity of the mitochondrial enzymes citrate synthase and hydroxyacyl-coenzyme A dehydrogenase (HADH) increased in WT hearts following swim training (+33.5% to 72.5%, P < 0.05 each) but not in IRS-deficient hearts (Fig. 4e and f). Furthermore, expression of mitochondrial proteins involved in fatty acid oxidation and oxidative phosphorylation and mitochondrial DNA content increased in WT but not in IRS-deficient hearts in response to exercise training (Fig. 4g to m). PGC-1α protein levels increased with exercise training in WT hearts; however, this effect was absent in CIRS1KO hearts, and PGC-1α protein levels actually decreased with exercise in CIRS2KO hearts (Fig. 4n). The induction of PGC-1α mRNA levels and PGC-1α downstream targets was significantly repressed in exercise-trained IRS-deficient hearts relative to WT controls (see Fig. S5 in the supplemental material). Importantly, phosphorylation of AMPK (Thr172) and its downstream target eEF2 (Thr56) were not increased in IRS-deficient hearts following swim training (see Fig. S6 in the supplemental material). Together, these data indicate that expression of each IRS isoform is required for the mitochondrial adaptations that accompany physiological cardiac hypertrophy via synergistic effects on PI3K and PGC-1α.
FIG 4.

IRS proteins mediate mitochondrial bioenergetics and the increase in PGC-1α protein in response to exercise training. Two-way ANOVA was performed to analyze differences by swim training and genotype followed by Fisher's PLSD post hoc analysis. (a to d) Mitochondrial capacity as measured by maximum ADP-stimulated mitochondrial respiration (VADP) and ATP synthesis in saponin-permeabilized cardiac fibers. VADP and ATP synthesis (P < 0.05 for swim training, genotype, and their interaction) with pyruvate (a and b) and VADP (P < 0.05 for swim training, genotype, and their interaction) (c) and ATP synthesis (P < 0.05 for swim training, P < 0.05 for genotype) (d) with palmitoyl-carnitine as the substrates each combined with malate as indicated (n = 4 to 6). (e and f) Citrate synthase (P < 0.05 for swim training, genotype, and their interaction) (e) and hydroxyacyl-CoA dehydrogenase (HADH) enzymatic activity (P < 0.05 for swim training, P < 0.05 for genotype) (f) (n = 8). (g to l) Representative Western blots (g) and densitometric analysis of CPT1b and CPT2 (P < 0.05 for swim training, P < 0.05 for the interaction between swim training and genotype) (h and i) and electron transport chains subunit complex I NDUFA9 (P < 0.05 for genotype) (j), complex II 30 kDa subunit (P < 0.05 for swim training, genotype, and their interaction) (k), and complex V subunit α (P < 0.05 for swim training, P < 0.05 for genotype) (l), each normalized to Coomassie blue stains. (m and n) Mitochondrial DNA content measured by RT-PCR (P < 0.05 for swim training, genotype, and their interaction) (m) and protein levels of the transcriptional coactivator PGC-1α normalized to GAPDH (P < 0.05 for genotype, P < 0.05 for the interaction between swim training and genotype; n = 3 to 8) (n). PGC-1α knockout heart homogenate was used as a negative control for PGC-1α immunoblotting (38). *, P < 0.05 versus WT (same treatment); †, P < 0.05 versus sedentary (same genotype); ‡, P < 0.05 versus CIRS1KO (same treatment).
IRS proteins mediate the metabolic response to exercise training.
The increase in mitochondrial capacity following exercise training correlates with increased fatty acid oxidation and glucose oxidation rates (32). We hypothesized that the impaired mitochondrial bioenergetic response in IRS-deficient hearts would impair the metabolic adaptations following swim training. In isolated perfused working hearts, palmitate oxidation rates increased in WT hearts following swim training (+95.0%, P < 0.05), while CIRS1KO and CIRS2KO hearts showed no difference relative to sedentary controls of the same genotype. Similarly, glucose oxidation and glycolysis rates increased ∼2-fold in WT hearts and by ∼55% in CIRS1KO hearts following exercise (P < 0.05 each), and this effect was absent in CIRS2KO hearts. Absolute values for glucose oxidation were significantly lower in CIRS2KO hearts than in CIRS1KO hearts following swim training (Fig. 5a to c; see also Table S3 in the supplemental material). Cardiac efficiency was significantly reduced in resting IRS1- and IRS2-deficient hearts, individually. Although exercise training increased cardiac efficiency in all genotypes, cardiac efficiency remained low in exercise-trained IRS1- or IRS2-deficient hearts. Cardiac tissue glycogen content increased only in WT hearts compared to sedentary controls (+56.2%, P < 0.05) (Fig. 5d; see also Table S4 in the supplemental material). Together, these data indicate that normal expression of both IRS isoforms is required for the metabolic response to chronic exercise training.
FIG 5.
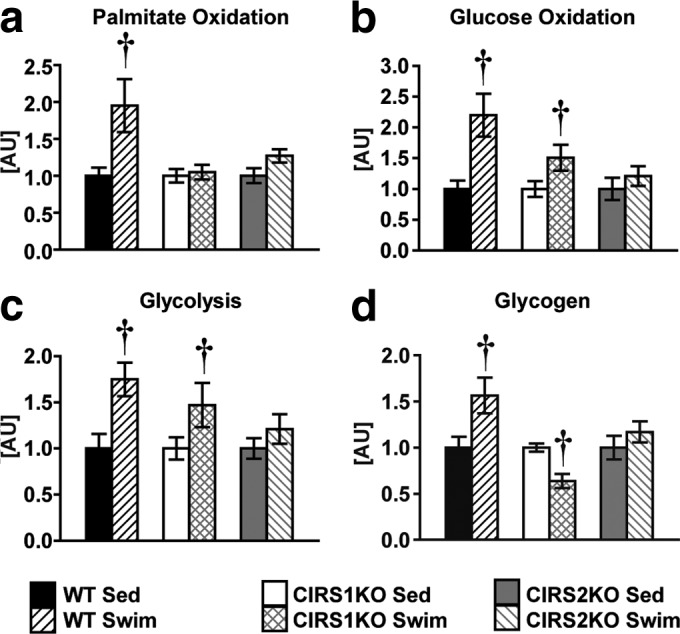
IRS proteins mediate the metabolic response to exercise training. (a to c) Palmitate oxidation, glucose oxidation, and glycolysis rates in isolated working WT, CIRS1KO, and CIRS2KO hearts under sedentary conditions and following chronic exercise training (n = 6 to 10). Hearts were perfused with 5 mmol/liter glucose and 0.4 mmol/liter palmitate. (d) Cardiac glycogen content as indicated (n = 7 to 8). Data are presented as fold changes compared to sedentary controls (same genotype); †, P < 0.05 versus sedentary (same genotype).
DISCUSSION
Here, we show that although IRS isoforms play divergent roles in the developmental regulation of cardiac size, expression of both IRS isoforms is required for the hypertrophic and bioenergetic response of the heart to chronic exercise training.
We previously reported that embryonic deletion of the IGF-1 receptor has no effect on heart size (5), whereas deletion of the insulin receptor results in reduced size of the heart (4, 6). While insulin and IGF-1 receptor signaling is transduced by IRS proteins, the present study revealed that CIRS1KO hearts are smaller. These data are consistent with data obtained from mice with germ line deletion of IRS proteins, indicating a predominant role for IRS1 on the regulation of somatic growth (13–15). Thus, IRS1 signaling plays a predominant role in the developmental regulation of cardiac growth, and this supports the hypothesis that insulin receptor-mediated cardiac growth might be predominantly regulated by IRS1 in the nonstressed heart. In contrast, basal cardiac size was increased in IRS2-deficient hearts relative to controls, raising the possibility that signaling pathways that are transduced via IRS2 negatively regulate developmental cardiac size via mechanisms that remain to be determined.
Previous reports indicate that Akt1 and PI3 kinase signaling is required for physiological cardiac hypertrophy (9, 10, 12, 33, 34). Furthermore, we also reported that mice with cardiomyocyte-specific deletion of the IGF-1 receptor exhibited an attenuated hypertrophic response following chronic swim training (5, 35). Unexpectedly, Akt phosphorylation was not decreased in IGF-1 receptor knockout hearts following swim training relative to WT controls. We observed increased 5′ adenosine monophosphate-activated protein kinase (AMPK) phosphorylation in IGF-1 receptor KO hearts, which is indicative of energetic stress. AMPK has been reported to inhibit hypertrophic growth by phosphorylating eukaryotic elongation factor 2 (eEF2) (36). Therefore, we concluded that there was a predominant contribution of energetic stress in the attenuated hypertrophic response of IGF-1 receptor knockout hearts. This contrasts with observations in IRS-deficient hearts following exercise training. Despite mitochondrial dysfunction in CIRS1KO and CIRS2KO hearts, AMPK phosphorylation (Thr172) was not increased following swim training, and eEF2 phosphorylation (Thr56) was unchanged between the groups. However, we observed decreased Akt phosphorylation in CIRS1KO and CIRS2KO hearts following swim training, relative to untrained mice of all genotypes and to exercise-trained WT controls. This suggested that additional mechanisms beyond blunted activation of PI3K-mediated Akt signaling could contribute to the impaired cardiac hypertrophic response following exercise training in IRS-deficient hearts. To further understand the underlying mechanisms for this reduction in Akt phosphorylation, we examined PP2A, which has been shown to dephosphorylate Akt (28). PP2A phosphorylation at Tyr307 was decreased in IRS-deficient hearts following swim training. This would be predicted to increase PP2A activity, which, by reducing Akt phosphorylation, could provide a plausible mechanism for the blunted hypertrophic response. Insulin stimulation had no effect on PP2A phosphorylation on Tyr307 in the heart. Thus, we focused our attention on signaling mechanisms that are independent of insulin/IGF-1 receptor signaling, which could influence PP2A phosphorylation. We observed decreased JAK2 phosphorylation at Tyr221 and impaired JAK2-mediated signaling in IRS-deficient hearts following chronic exercise training relative to wild-type controls. The mechanisms by which IRS1 or IRS2 deficiency impairs exercise-mediated activation of the JAK-STAT pathway remain to be elucidated. Previous studies showed that phosphorylation at Tyr221 increases the kinase activity of JAK2 (30) and JAK2 phosphorylates PP2A at Tyr307 (29), thereby decreasing its phosphatase activity (26, 27). Thus, impaired JAK2-mediated inhibition of PP2A represents a potential mechanism for increased Akt dephosphorylation, which we posit attenuates the hypertrophic response of IRS-deficient hearts to exercise training.
We observed preserved insulin-mediated signaling in CIRS1KO and CIRS2KO hearts both under basal conditions and following insulin stimulation compared to WT controls. Thus, expression of one IRS isoform is sufficient for insulin-mediated Akt activation, while expression of both IRS isoforms is required for mediating the hypertrophic response following chronic exercise training. These observations raise the possibility that IRS1 and IRS2 exist in distinct spatiotemporal domains within cardiomyocytes, leading to distinct signaling complexes that are entrained by divergent upstream signals such as hormonal activation of insulin or IGF-1 on one hand or by exercise on the other. Moreover, the nonredundant roles of IRS1 and IRS2 in exercise-induced cardiac hypertrophy raise the possibility that these proteins directly interact to mediate the downstream signals.
We previously reported that cardiomyocyte-specific deletion of insulin receptors (CIRKO) impairs mitochondrial bioenergetics and reduced the expression of proteins involved in the citric acid cycle and fatty acid oxidation (6). We also observed mitochondrial dysfunction in IRS-deficient hearts under basal conditions. Furthermore, we previously described a central role for a PI3K-dependent but Akt-independent signaling pathway that promotes the mitochondrial adaptations to exercise training (12). Interestingly, PI3K activity was impaired in IRS-deficient hearts compared to WT controls following exercise training. Taken together with our previous studies, these data imply that impaired PI3K signaling could contribute to the attenuated mitochondrial bioenergetic response in IRS-deficient hearts following exercise training.
What might account for the near-complete inhibition of exercise-induced PI3K activity when one IRS isoform is absent? Although our studies do not completely answer this question, it is important to note that protein levels of the PI3K p85 regulatory subunit protein were unchanged between the groups. It is recognized that the stoichiometry of p85 and IRS proteins governs IRS-associated activation of PI3K, so that increased binding of p85 to IRS proteins could reduce PI3K activation (37). Thus, the possibility exists that deletion of one IRS isoform would increase the stoichiometric binding of p85 to the remaining IRS protein, thereby reducing binding to the p110 catalytic subunit. This would limit PI3K activation and reduce the expected increase in mitochondrial energetics. Why such a mechanism could apply to exercise-induced signaling but not to insulin signaling is not known and will be the subject of future investigations. In addition, we also observed either unchanged or decreased PGC-1α protein levels following exercise training in IRS-deficient hearts, in the absence of a similar decline in mRNA. Thus, IRS signaling might also regulate either the translation or the stability of the PGC-1α protein. Although the mechanisms linking IRS signaling to PGC-1α protein levels remain to be elucidated, the reduction in PGC-1α protein clearly represents a second mechanism that contributes to the attenuated mitochondrial bioenergetic response to exercise training following loss of one IRS isoform.
Substrate oxidation rates and cardiac efficiency increased in exercise-trained WT hearts compared to sedentary controls, and this effect was attenuated following deletion of either IRS isoform. Similarly, glycogen content increased only in WT hearts. In addition to orchestrating the mitochondrial adaptations following exercise training, PGC-1α plays an important role for postexercise glycogen resynthesis as shown in skeletal muscle (23). Together, impaired induction of PGC-1α expression and PI3K activity provides potential mechanisms for the attenuated metabolic response and decreased tissue glycogen content in IRS-deficient hearts following exercise training.
In summary, the present study has identified an essential role for IRS signaling in cardiac growth. IRS isoforms play divergent roles in the developmental regulation of cardiac size and may play redundant roles in insulin signaling (see Fig. S7 in the supplemental material). IRS1 or IRS2 deficiency is sufficient to block the hypertrophic and mitochondrial adaptations to exercise training. We believe that a unifying mechanism for the blunted hypertrophic and mitochondrial adaptations might be impaired activation of PI3K and increased dephosphorylation of Akt. Some differences in the responses of IRS1- and IRS2-deficient hearts to exercise, such as PGC-1α levels, glycogen content, and the fold change in glucose utilization were also noted and are summarized in Fig. 6. The mechanisms for these differences between isoform-specific mutants will be the subject of future investigation. However, our data indicate nonredundant roles of IRS isoforms in mediating the hypertrophic, mitochondrial, and metabolic responses of the heart to exercise training (Fig. 6).
FIG 6.
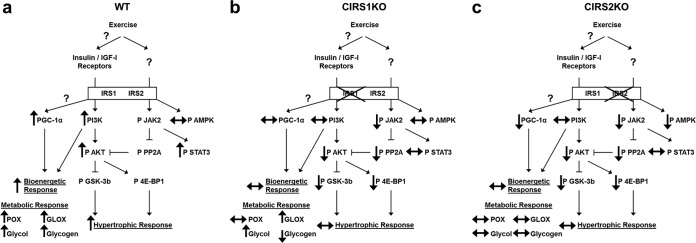
Proposed model for attenuated hypertrophic and bioenergetic response in IRS-deficient hearts to exercise training. Changes in exercise-trained WT (a), CIRS1KO (b), and CIRS2KO (c) hearts relative to sedentary controls of the same genotype are shown. POX, palmitate oxidation; GLOX, glucose oxidation; Glycol, glycolysis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants RO1DK092065, RO1HL070070, and RO1HL108379 UO1HL087947 to E.D.A., who is an established investigator of the American Heart Association. C.R. was supported by a postdoctoral fellowship from the German Research Foundation (DFG), A.R.W. was supported by an advanced postdoctoral fellowship from the JDRF (10-2009-672) and by grant K99 HL111322 from the NIH, and R.O.P. was supported by a postdoctoral fellowship from the AHA (Western Affiliates) and by grant T32 HL007576 from the NIH. R.M. and M.R. were supported by fellowships from Maastricht University and the Netherlands Heart Foundation.
Footnotes
Published ahead of print 7 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00426-14.
REFERENCES
- 1.Abel ED, Doenst T. 2011. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 90:234–242. 10.1093/cvr/cvr015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dorn GW., II 2007. The fuzzy logic of physiological cardiac hypertrophy. Hypertension 49:962–970. 10.1161/HYPERTENSIONAHA.106.079426 [DOI] [PubMed] [Google Scholar]
- 3.Shiojima I, Walsh K. 2006. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 20:3347–3365. 10.1101/gad.1492806 [DOI] [PubMed] [Google Scholar]
- 4.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. 2002. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Invest. 109:629–639. 10.1172/JCI13946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, LeRoith D, Abel ED. 2008. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 22:2531–2543. 10.1210/me.2008-0265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudina S, Bugger H, Sena S, O'Neill BT, Zaha VG, Ilkun O, Wright JJ, Mazumder PK, Palfreyman E, Tidwell TJ, Theobald H, Khalimonchuk O, Wayment B, Sheng X, Rodnick KJ, Centini R, Chen D, Litwin SE, Weimer BE, Abel ED. 2009. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 119:1272–1283. 10.1161/CIRCULATIONAHA.108.792101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neri Serneri GG, Boddi M, Modesti PA, Cecioni I, Coppo M, Padeletti L, Michelucci A, Colella A, Galanti G. 2001. Increased cardiac sympathetic activity and insulin-like growth factor-I formation are associated with physiological hypertrophy in athletes. Circ. Res. 89:977–982. 10.1161/hh2301.100982 [DOI] [PubMed] [Google Scholar]
- 8.Scheinowitz M, Kessler-Icekson G, Freimann S, Zimmermann R, Schaper W, Golomb E, Savion N, Eldar M. 2003. Short- and long-term swimming exercise training increases myocardial insulin-like growth factor-I gene expression. Growth Horm. IGF Res. 13:19–25. 10.1016/S1096-6374(02)00137-5 [DOI] [PubMed] [Google Scholar]
- 9.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. 2006. Akt1 is required for physiological cardiac growth. Circulation 113:2097–2104. 10.1161/CIRCULATIONAHA.105.595231 [DOI] [PubMed] [Google Scholar]
- 10.McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. 2003. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 100:12355–12360. 10.1073/pnas.1934654100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, Riggi L, Kang PM, Izumo S. 2004. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J. Biol. Chem. 279:4782–4793. 10.1074/jbc.M310405200 [DOI] [PubMed] [Google Scholar]
- 12.O'Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, Guo A, Zaha VG, Davis DK, Schell JC, Boudina S, Wayment B, Litwin SE, Shioi T, Izumo S, Birnbaum MJ, Abel ED. 2007. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 6:294–306. 10.1016/j.cmet.2007.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Araki E, Lipes MA, Patti ME, Bruning JC, Haag B, III, Johnson RS, Kahn CR. 1994. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 372:186–190. 10.1038/372186a0 [DOI] [PubMed] [Google Scholar]
- 14.Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Kaburagi Y, Satoh S, Sekihara H, Yoshioka S, Horikoshi H, Furuta Y, Ikawa Y, Kasuga M, Yazaki Y, Aizawa S. 1994. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 372:182–186. 10.1038/372182a0 [DOI] [PubMed] [Google Scholar]
- 15.Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF. 1998. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391:900–904. 10.1038/36116 [DOI] [PubMed] [Google Scholar]
- 16.Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste-Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB. 1999. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J. Clin. Invest. 104:1703–1714. 10.1172/JCI7605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, White MF. 2008. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 8:65–76. 10.1016/j.cmet.2008.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin X, Taguchi A, Park S, Kushner JA, Li F, Li Y, White MF. 2004. Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes. J. Clin. Invest. 114:908–916. 10.1172/JCI22217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, Bugger H, Buchanan J, Wang X, Moreira AB, Doenst T, Medina-Gomez G, Litwin SE, Lelliott CJ, Vidal-Puig A, Abel ED. 2011. PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ. Res. 109:783–793. 10.1161/CIRCRESAHA.111.243964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sena S, Rasmussen IR, Wende AR, McQueen AP, Theobald HA, Wilde N, Pereira RO, Litwin SE, Berger JP, Abel ED. 2007. Cardiac hypertrophy caused by peroxisome proliferator-activated receptor-gamma agonist treatment occurs independently of changes in myocardial insulin signaling. Endocrinology 148:6047–6053. 10.1210/en.2006-1559 [DOI] [PubMed] [Google Scholar]
- 21.Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, Bugger H, Frank D, Bevins J, Chen D, Perry CN, Dong XC, Valdez S, Rech Sheng MX, Weimer BC, Gottlieb RA, White MF, Abel ED. 2013. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J. Clin. Invest. 123:5319–5333. 10.1172/JCI71171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R. 2011. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 17:1076–1085. 10.1038/nm.2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wende AR, Schaeffer PJ, Parker GJ, Zechner C, Han DH, Chen MM, Hancock CR, Lehman JJ, Huss JM, McClain DA, Holloszy JO, Kelly DP. 2007. A role for the transcriptional coactivator PGC-1alpha in muscle refueling. J. Biol. Chem. 282:36642–36651. 10.1074/jbc.M707006200 [DOI] [PubMed] [Google Scholar]
- 24.Hardt SE, Sadoshima J. 2002. Glycogen synthase kinase-3beta: a novel regulator of cardiac hypertrophy and development. Circ. Res. 90:1055–1063. 10.1161/01.RES.0000018952.70505.F1 [DOI] [PubMed] [Google Scholar]
- 25.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. 1999. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13:1422–1437. 10.1101/gad.13.11.1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Parsons S, Brautigan DL. 1994. Tyrosine phosphorylation of protein phosphatase 2A in response to growth stimulation and v-src transformation of fibroblasts. J. Biol. Chem. 269:7957–7962 [PubMed] [Google Scholar]
- 27.Chen J, Martin BL, Brautigan DL. 1992. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 257:1261–1264. 10.1126/science.1325671 [DOI] [PubMed] [Google Scholar]
- 28.Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, Obata T, Ebina Y, Kashiwagi A, Olefsky JM. 2004. Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 24:8778–8789. 10.1128/MCB.24.19.8778-8789.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokoyama N, Reich NC, Miller WT. 2003. Determinants for the interaction between Janus kinase 2 and protein phosphatase 2A. Arch. Biochem. Biophys. 417:87–95. 10.1016/S0003-9861(03)00333-3 [DOI] [PubMed] [Google Scholar]
- 30.Argetsinger LS, Kouadio JL, Steen H, Stensballe A, Jensen ON, Carter-Su C. 2004. Autophosphorylation of JAK2 on tyrosines 221 and 570 regulates its activity. Mol. Cell. Biol. 24:4955–4967. 10.1128/MCB.24.11.4955-4967.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH, Guan KL. 2000. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science 290:144–147. 10.1126/science.290.5489.144 [DOI] [PubMed] [Google Scholar]
- 32.Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, Bonen A, Keller A, Dunaway GA, Popov KM, Hochachka PW, Allard MF. 2004. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 287:H1055–H1063. 10.1152/ajpheart.00925.2003 [DOI] [PubMed] [Google Scholar]
- 33.Luo J, McMullen JR, Sobkiw CL, Zhang L, Dorfman AL, Sherwood MC, Logsdon MN, Horner JW, DePinho RA, Izumo S, Cantley LC. 2005. Class IA phosphoinositide 3-kinase regulates heart size and physiological cardiac hypertrophy. Mol. Cell. Biol. 25:9491–9502. 10.1128/MCB.25.21.9491-9502.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, Cantley LC, Izumo S. 2000. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19:2537–2548. 10.1093/emboj/19.11.2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikeda H, Shiojima I, Ozasa Y, Yoshida M, Holzenberger M, Kahn CR, Walsh K, Igarashi T, Abel ED, Komuro I. 2009. Interaction of myocardial insulin receptor and IGF receptor signaling in exercise-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 47:664–675. 10.1016/j.yjmcc.2009.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. 2004. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J. Biol. Chem. 279:32771–32779. 10.1074/jbc.M403528200 [DOI] [PubMed] [Google Scholar]
- 37.Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ, Iannacone M, Accili D, Cantley LC, Kahn CR. 2002. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J. Clin. Invest. 109:141–149. 10.1172/JCI13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. 2005. PGC-1alpha deficiency causes multisystem energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3:e101. 10.1371/journal.pbio.0030101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.