

Abstract
Wnt5a can activate β-catenin-independent pathways for regulation of various cellular functions, such as migration, that play critical roles in wound repair. Investigation of Wnt5a signaling may help identify therapeutic targets for enhancing corneal endothelial wound healing that could provide an alternative to corneal transplantation in patients with blindness from endothelial dysfunction. However, Wnt5a signaling in corneal endothelial cells (CECs) has not been well characterized. In this study, we show transient induction of Wnt5a by interleukin-1β (IL-1β) stimulation proceeds through NF-κB in human CECs. This leads to binding of Fzd5 to Ror2, resulting in activation of disheveled protein (Dvl) and subsequently disheveled-associated activator of morphogenesis 1 (DAAM1). This leads to activation of Cdc42 and subsequent inhibition of RhoA. Inhibition of RhoA leads to parallel dephosphorylation and inactivation of LIM domain kinase 2 along with dephosphorylation and activation of slingshot 1, resulting in dephosphorylation and activation of cofilin and leading to enhanced cell migration. These findings suggest that Wnt5a enhances cell migration through activation of Cdc42 and inactivation of RhoA in human CECs.
INTRODUCTION
The cornea is the anterior transparent tissue of the eye and is composed of epithelial, stromal, and endothelial layers. The corneal endothelium consists of a monolayer of differentiated cells that play a critical role in regulating through their pump function corneal hydration, which is essential for maintenance of corneal transparency and sharp vision. Adult human corneal endothelial cells (CECs) are arrested in the G1 phase of the cell in vivo even when injured (1, 2). Because of this, there is an age-related decline in CEC density that can be accelerated by inflammation or injury. A decrease in CEC density below a critical threshold leads to vision loss from decreased transparency secondary to corneal edema. Vision loss from corneal edema is a major indication for corneal transplantation in developed nations. Approximately 50,000 corneal transplants are performed in the United States annually, and there fortunately is still an excess of donor tissue in the United States. However, there are over 10 million patients globally who cannot be transplanted due to a regional lack of donor tissue (3).
Unlike other tissues where cell proliferation and migration are important components of injury repair, cell migration and enlargement play critical roles in corneal endothelial wound repair. The etiology of this peculiar repair mechanism is still unclear but may be related to the need for maintaining transparency which is critical for sharp vision. A variety of soluble factors have previously been shown to enhance cell migration, including interleukin 1β (IL-1β), a major mediator of inflammation and wound healing in the cornea (4–6). We previously reported that IL-1β stimulation induces fibroblast growth factor 2 (FGF2) expression through NF-κB and AP-1 pathways, leading to enhanced migration of human and rabbit CECs (7–9). However, IL-1β and FGF2 have also been shown to be important mediators of the endothelial-mesenchymal transition (EMT) that leads to retrocorneal fibrous membrane (RCFM) formation (4, 8, 10), which represents end-stage pathology for the eye, where restoration of vision is no longer possible. We therefore investigated other downstream targets of IL-1β that could enhance corneal wound healing without leading to RCFM formation.
The mammalian Wnt family of secreted glycoprotein ligands consists of 19 members, and they play essential roles in development and cellular homeostasis (11, 12). Binding of Wnt proteins to the frizzled (Fzd) family of receptors initiates a complex signaling cascade that can be broadly divided into canonical and noncanonical pathways. In the canonical pathway, binding of the Wnt ligand to the Fzd-LRP5/6 receptor complex leads to cytoplasmic stabilization and nuclear transport of β-catenin (12, 13). Canonical Wnt signaling regulates cell proliferation, cell fate specification, and body axis patterning during development (14, 15). During development, local repression of posteriorizing signals, including canonical Wnt proteins, is critical in specifying forebrain structures, including the eye (16–18). In zebrafish, local repression of canonical Wnt signaling is required for formation of the eye field (19). During corneal development, local repression of canonical Wnt signaling is also critical for development of the corneal epithelium (20). Noncanonical Wnt pathways, such as planar cell polarity (PCP) and calcium pathways (21, 22), do not signal through β-catenin and have not been studied as extensively as the canonical pathway. Noncanonical Wnt proteins, such as Wnt5a, bind to different frizzled (Fzd) family members, including Fzd5 (23–25) and Fzd7 (21, 26), and coreceptors such as Ror2, Ryk, and PTK7 (27, 28). In the PCP pathway, binding of Wnt ligand and its receptor complex leads to assembly of disheveled protein (Dvl) and its effector proteins, including disheveled-associated activator of morphogenesis 1 (DAAM1) (29–31). They then regulate cytoskeleton organization and cell migration through Rho GTPases such as RhoA, Rac1, and Cdc42 (30–33). Although the function of noncanonical Wnt signaling in the cornea is unclear, robust expression of Wnt5a can be observed in fetal but not adult human corneal endothelium (34).
Rho, Rac, and Cdc42 of the Rho GTPase family play a role in regulation of assembly and organization of the actin cytoskeleton in eukaryotic cells. Cdc42 and Rac play essential roles in cell polarity during migration by regulating pseudopodium and lamellipodium formation at the leading edge, while Rho plays a role in substrate adhesion that is necessary for generating contractile force during cell movement (35–38). The activation of Rho GTPases via noncanonical Wnt signaling has been shown to lead to regulation of LIM domain kinases (LIMK), slingshot (SSH), and cofilin, resulting in modulation of actin turnover in cytoskeleton assembly during cell movement (39–42). Despite extensive investigation of Rho GTPases, interactions among individual Rho GTPase proteins, with the exception of WGEF in activation of RhoA during Xenopus laevis gastrulation (43), remain largely unclear.
It has been reported that IL-1β can induce Wnt5a expression through NF-κB in chondrocytes (44). Moreover, NF-κB has been reported to bind and activate the human Wnt5a promoter (45). In light of previously reported functions of noncanonical Wnt proteins, we hypothesized that they play an important role in IL-1β-induced migration of human CECs. In this report, we show that brief stimulation of human CECs with IL-1β induces Wnt5a expression through NF-κB, which ultimately leads to enhanced cell migration. Binding of Wnt5a to Fzd5 and Ror2 leads to activation of Dvl and subsequent binding between DAAM1 and Cdc42. Activation of Cdc42 results in inhibition of RhoA which, in turn, leads to enhanced cell migration through regulation of cofilin in human CECs. Prior to this study, the effects of noncanonical Wnt signaling on human CEC migration were not known. We provide evidence that noncanonical Wnt signaling enhances cell migration and we show that there are regulatory interactions among Rho GTPases in human CECs.
MATERIALS AND METHODS
Materials.
FGF2 and antibodies against Wnt5a, RhoA, cofilin, and phospho-cofilin were purchased from Cell Signaling Technology (Danvers, MA). Anti-Fzd5 antibody was purchased from Fitzgerald Industries International (Acton, MA). Sulfasalazine, Wnt antagonist III, Box5, Dvl-PDZ domain inhibitor, Y27632, NSC23766, and SB203580 were obtained from Calbiochem (San Diego, CA). IL-1β, anti-β-actin, α-tubulin, and peroxidase-conjugated secondary antibodies were obtained from Sigma-Aldrich (St. Louis, MO). Recombinant human interleukin-1 receptor antagonist (IL-1ra), Wnt5a, and Wnt3a were obtained from R&D Systems (Minneapolis, MN). ML141 was purchased from Tocris Bioscience (Minneapolis, MN). XAV939 and AZD4547 were purchased from Selleckchem (Houston, TX). RhoA activator was obtained from Cytoskeleton (Denver, CO). Antibodies against Ror2, β-catenin, LIMK2, and phospho-LIMK2 were obtained from GeneTex (Irvine, CA). Antibodies against DAAM1, Cdc42, and lamin B were purchased from Santa Cruz Biotechnology (Dallas, TX). Antibodies against SSH1 and phospho-SSH1 were purchased from Bethyl Laboratory (Montgomery, TX) and ECM Biosciences (Versailles, KY), respectively.
Cell culture.
Immortalized human CEC line hCEC-B4G12 (DSMZ, Braunschweig, Germany) was cultured as previously described (7). Briefly, hCEC-B4G12 cells were cultured in human endothelial serum-free medium supplemented with 10 ng/ml human recombinant FGF2 without antibiotics. Cells were grown in a humidified atmosphere containing 5% CO2 at 37°C. For subculture, confluent cultures were treated with 0.05% trypsin and 5 mM EDTA in phosphate-buffered saline (PBS) for 5 min. Cells were plated at a concentration of 1 × 106 cells in 100-mm tissue culture dishes coated with 10 mg/ml chondroitin-6-sulfate and 10 μg/ml laminin. Second-passage human CECs were used for all experiments. Culture medium was changed twice a week. In some experiments, pharmacologic inhibitors were used in the presence of IL-1β (5 ng/ml), Wnt5a (300 ng/ml), or Wnt3a (50 ng/ml) stimulation: IL-1ra (50 ng/ml), AZD4547 (FGF receptor inhibitor; 0.1 μM), SB203580 (p38 inhibitor; 20 μM), sulfasalazine (inhibitor for IκB degradation; 2 mM), Wnt antagonist III, Box5 (3 μg/ml), Dvl-PDZ domain inhibitor II (25 μM), Y27632 (Rho-associated coil kinase [ROCK] inhibitor; 10 μM), ML141 (Cdc42 inhibitor; 10 μM), or NSC23766 (Rac1 inhibitor; 100 μM).
Scratch-induced directional migration assay.
Cell migration assay was performed using our previously published protocols with some modifications (7, 46). Briefly, human CECs were plated in 24-well tissue culture dishes at a concentration of 4 × 104 cells and maintained until the cells reached greater than 95% confluence. At this time, cells were FGF2 starved for 24 h. A micropipette tip was then used to wound the monolayer, creating a linear, cross-stripe of scrapes, 2 mm apart. Cells were then washed with PBS to remove floating cellular debris and refed for an additional 16 h with the indicated medium. Wound closure or cell migration was photographed at 16 h postwounding by using an inverted EVOS microscope equipped with a digital camera (Advanced Microscopy Group, Bothell, WA). The individual gaps were measured under each culture condition and at various time points by using the SPOT program (version 2.1.2; Diagnostic Instruments, Inc.). The residual gap between the migrating cells from the opposing wound edge under experimental conditions was expressed as the percentage of migrating cells under control conditions. All experiments were conducted in the presence of 5 μg/ml mitomycin C to inhibit cell proliferation (47).
Protein preparation, protein assay, SDS-PAGE, Western blotting, coimmunoprecipitation, and nuclear fractionation.
All assays were performed following previously reported protocols (7, 8, 48, 49). The following gel concentrations were used to separate proteins: 15% polyacrylamide gel for Cdc42, Rac1, RhoA, cofilin, and p-cofilin; 10% polyacrylamide gel for Wnt5a, lamin B, α-tubulin, and β-actin; 8% polyacrylamide gel for Fzd 5, β-catenin, LIMK2, p-LIMK2; 6% polyacrylamide gel for Ror2, DAAM1, SSH1, and p-SSH1. For coimmunoprecipitation of Fzd5 and Ror2, the protein cross-linking procedures were performed using dithiobis succinimidyl propionate (DTSP), a substance that has been shown to cross-link cell membranes (50). Briefly, cells were harvested using enzyme-free cell dissociation buffer (EMD Millipore, Billerica, MA). The harvested cells were combined with 2 mM DTSP, vigorously shaken, and placed on ice for 1 h. The cells were then rinsed with 2 mM glycine in PBS to block the DTSP activity and lysed with RIPA lysis and extraction buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 0.5% Nonidet P-40, and protease inhibitors). Concentration of lysates was assessed with the Bradford protein assay system (Bio-Rad Laboratories, Hercules, CA) and then used for coimmunoprecipitation.
ChIP assay.
FGF2-starved human CECs were pretreated with sulfasalazine for 2 h, maintained with or without IL-1β for 10 min, and then maintained for 16 h. Chromatin immunoprecipitation (ChIP) assays were performed as previously described (7). After isolating purified protein-DNA complexes using anti-NF-κB (p65) antibody, transcription factor binding to Wnt5a promoter was assessed by PCR using the following primers: 5′-ATCAGCGTCTGGAAGCAGACG-3′ (N1, −638) and 5′-CCACAGTTGAGTAGTGGTACA-3′ (N2, −486); 5′-TGTACCACTACTCAACTGTGG-3′ (C1, −464), 5′-CCGTTTCCAACGTCCATCAGC-3′ (C2, −337), and 5′-CGCAGGCAACTGTTCCACGGA-3′ (C3, −308). PCR conditions were as follows: 5 min at 94°C followed by 33 cycles of 30 s at 94°C, 30 s at 55°C, and 30 s at 72°C, and a final extension for 2 min at 72°C. PCR products were separated by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining.
siRNA knockdown.
Corneal endothelial cells seeded on 6-well plates were transfected at 70% confluence with 0.5, 1, or 1.5 μM Accell SMARTpool small interfering RNA (siRNA) targeting p65 (RelA), Jun, or Dvl2 via Accell delivery medium per the manufacturer's instructions (Dharmacon, Pittsburgh, PA). At 42 h after transfection, the medium was changed to the indicated culture medium and maintained for 6 more hours. The knockdown of p65, Jun, and Dvl2 was analyzed by immunoblotting. Accell nontargeting pool siRNA was used as a negative control, and transfection efficiency was measured with Accel Red cyclophilin B control siRNA.
Ex vivo human cornea.
Discarded rims from human donor corneas were obtained following corneal transplantation and placed endothelial side up in wells of a 24-well tissue culture plate. The tissues were serum starved for 24 h and then were incubated with IL-1β (5 ng/ml) or experimental medium for 6 h. After the 6-hour incubation, the endothelium-Descemet's membrane complex was carefully stripped from the underlying corneal stroma and incubated in RIPA lysis and extraction buffer. Concentrations of the resultant lysates were assessed with the Bradford protein assay system (Bio-Rad Laboratories, Inc.) and then Western blot assays were performed.
Cdc42 and Rac1 GTPase activation assay.
Cdc42 and Rac activities were quantified using Cdc42 and Rac activation assay kits (EMD Millipore) with the p21 binding domain of human PAK-1 as bait according to the manufacturer's instructions. Briefly, CECs were cultured under the designated culture conditions and then lysed with Mg2+ lysis buffer. A total of 9 mg of cleared cell lysate was split into three equal aliquots. As negative and positive controls for the pulldown assay, two of the aliquots were added to 1 mM GDP or 100 μM GTPγS, respectively, and incubated for 15 min at 30°C with agitation to deplete or enrich Cdc42/Rac-GTP. The third aliquot remained untreated and was kept on ice while the controls were loaded. Glutathione S-transferase (GST)–PAK–PBD beads were added to each aliquot, and the reaction mixtures were incubated for 1 h at 4°C with gentle agitation. After the pulldown reaction, the supernatants were removed by brief centrifugation, and the precipitated proteins bound to the beads were subjected to immunoblot analysis with monoclonal antibody to Cdc42 or to Rac1. The total amount of each small G protein was also determined by Western blot analysis using total cell lysates and then used to normalize the protein concentration of each lane.
RhoA GTPase activation assay.
RhoA activation was quantified by measuring the amounts of RhoA-GTP via an enzyme-linked immunosorbent assay (ELISA)-based G-LISA RhoA Activation Biochem assay kit (Cytoskeleton). The total cellular fractions from the cells maintained under each culture condition were used to measure the RhoA activity. Active RhoA-GTP from lysates is bound to the Rho-GTP binding domain of a Rho effector immobilized in the 96-well plate, whereas inactive Rho-GDP is removed during the washing step. RhoA-GTP is subsequently detected by primary anti-RhoA antibody and horseradish peroxidase (HRP)-conjugated secondary antibody. The HRP reaction signal was quantified by measuring absorbance at 490 nm. Results were normalized to the absorbance of lysate from control cells and expressed as the percentage of this value for comparison of RhoA activity (level of GTP-bound RhoA) in different cell lysates.
Statistical analysis.
Analysis of variance (ANOVA) was performed to compare means within groups, and post hoc Tukey's honest significant difference tests were done to perform pairwise comparisons between means in a group.
RESULTS
IL-1β-enhanced cell migration and Wnt5a expression in human CECs.
A one-way ANOVA test showed that the mean migration rates were significantly different between controls and groups treated with IL-1β, IL-1β plus AZD4547 (AZD), FGF2, and FGF2 plus AZD: F(4, 80) = 502, P < 0.00001. Tukey's post hoc test showed that stimulation of human CECs with IL-1β (P < 0.01) or FGF2 (P < 0.01) resulted in an approximately 2-fold enhancement of migration (Fig. 1A). Cotreatment with AZD, an antagonist to FGFR1, FGFR2, and FGFR3, completely inhibited FGF2-enhanced cell migration (P < 0.01) but not IL-1β-enhanced cell migration (P > 0.05), suggesting a role for another signaling pathway downstream of IL-1β for mediating cell migration. IL-1β stimulation resulted in expression of Wnt5a in human CECs that, in turn, was antagonized by cotreatment with IL-1 receptor antagonist (Fig. 1B). Cotreatment with sulfasalazine, an NF-κB antagonist, also inhibited Wnt5a expression in human CECs, but SB203580, an AP-1 antagonist, did not affect Wnt5a expression. Increasing amounts of p65 siRNA were able to decrease the amount of IL-1β-induced Wnt5a expression, while Jun siRNA had no effect on Wnt5a expression (Fig. 1C). There was a dose-dependent knockdown of p65 and Jun protein levels.
FIG 1.

Effect IL-1β stimulation on human corneal endothelial cells. (A) Treatment of human CECs with of IL-1β (*, P < 0.01) and FGF2 (+, P < 0.01) resulted in enhanced cell migration as measured in a scratch-induced directional migration assay. Cotreatment with AZD4547 (AZD), an antagonist against FGF receptors 1 to 3, abolished the FGF2-enhanced (++, P < 0.01) but not the IL-1β-enhanced (P > 0.05) migration in human CECs. One-way ANOVA: F(4, 80) = 512, P < 0.00001, n = 17 per sample. Tukey's post hoc test, HSD[0.05] = 10.9; HSD[0.01] = 13.1. (B) Treatment of human CECs with IL-1β resulted in expression of Wnt5a. IL-1β-induced Wnt5a expression in human CECs was inhibited by cotreatment with interleukin 1 receptor antagonist (IL-1ra) and sulfasalazine (Sul) but not with SB203580 (SB). (C) Transfection with p65 siRNA but not Jun siRNA resulted in a dose-dependent decrease in Wnt5a expression in human corneal endothelial cells. There was a dose-dependent knockdown in p65 and Jun protein levels in cells transfected with p65 siRNA and Jun siRNA, respectively. C, control.
Expression of Fzd5 and Ror2 in human CECs.
To further investigate the role of Wnt5a signaling in human CECs, we examined whether Frizzled 5 (Fzd5) and the tyrosine-protein kinase transmembrane receptor Ror2, known components of the Wnt5a pathway, were expressed in human CECs (23, 27). Constitutive expression of Fzd5 and Ror2 was observed under control and IL-1β-stimulated culture conditions, and cotreatment with IL-1 receptor antagonist did not have an effect on the expression of the receptors in human CECs (Fig. 2A). Fzd5 and Ror2 did not bind under control conditions, but treatment with IL-1β or Wnt5a resulted in binding of Fzd5 to Ror2 (Fig. 2B). IL-1β-mediated binding was inhibited by cotreatment with IL-1ra or sulfasalazine (Fig. 2B).
FIG 2.

IL-1β-induced binding of Fzd5 and Ror2. (A) Constitutive expression of Frizzled 5 (Fzd5) and tyrosine-protein kinase transmembrane receptor Ror2 was detected in human CECs under all culture conditions tested. (B) Immunoprecipitation (IP) and immunoblotting (IB) with anti-Fzd5 and anti-Ror2 antibodies showed binding between Fzd5 and Ror2 in the presence Wnt5a and IL-1β. IL-1β-induced binding was inhibited by cotreatment with IL-1ra and sulfasalazine (Sul). C, control.
Binding of NF-κB to the Wnt5a promoter in human CECs.
The human Wnt5a promoter contains both conserved and putative NF-κB binding sites. To determine whether those binding sites are functional, we performed ChIP assays in human CECs with a combination of PCR primers spanning the NF-κB binding sites under various conditions (Fig. 3A). Treatment of human CECs with IL-1β resulted in pulldown of DNA fragments containing both NF-κB binding sites in the Wnt5a promoter, generating the expected 150-, 173-, 180-, 301-, and 330-bp PCR fragments (Fig. 3B). Human CECs cultured under control conditions or cotreated with IL-1β and sulfasalazine failed to protect the NF-κB binding sites.
FIG 3.

Binding of NF-κB to the Wnt5a promoter in human CECs. (A) Schematic illustration of putative NF-κB binding sites within the human Wnt5a promoter. The location of each primer used in the ChIP assays is indicated. (B) The expected PCR product sizes for the primer pairs were as follows: N1-C1, 173 bp; N1-C2, 301 bp; N1-C3, 330 bp; N2-C2, 150 bp; N2-C3, 180 bp. Stimulation with IL-1β resulted in NF-κB binding to the Wn5a promoter in human CECs. Cotreatment with sulfasalazine (Sul) inhibited NF-κB binding to the promoter. Anti-histone H3 antibody was used for the positive control (+), and normal rabbit IgG was used for the negative control (−) in the ChIP assays.
Wnt5a-enhanced cell migration proceeds via a β-catenin-independent pathway.
A one-way ANOVA test showed that the mean migration rates were significantly different between the control and groups treated with Wnt5a, Wnt5a plus Wnt antagonist III (Box5), Wnt5a plus disheveled-PDZ domain inhibitor (DI), and Wnt5a plus XAV939: F(4, 80) = 415, P < 0.00001. Tukey's post hoc test showed Wnt5a stimulation of human CECs resulted in a 2-fold increase in the migration rate over control CECs (P < 0.01) (Fig. 4A). Cotreatment of Wnt5a-stimulated human CECs with Box5 (P < 0.01) or DI (P < 0.01) inhibited Wnt5a-enhanced migration, while cotreatment with XAV939, an axin stabilizer, had no effect on Wnt5a-enhanced migration (P > 0.05) (Fig. 4A). β-Catenin was detected in the cytoplasmic fraction but not in the nuclear fraction of human CECs treated with Wnt5a (Fig. 4B). β-Catenin was detected in both cytoplasmic and nuclear fractions of cells treated with Wnt3a, a known canonical Wnt protein. β-Catenin was detected in the cytoplasmic fraction but not in the nuclear fraction of control human CECs (Fig. 4B).
FIG 4.

Wnt5a enhances human CEC migration through the β-catenin-independent pathway. (A) Wnt5a stimulation (+) resulted in a 2-fold increase in migration rates (*, P < 0.01). Wnt5a-induced cell migration was completely blocked by treatment with Wnt antagonist III (Box5) (+, P < 0.01) or DI (++, P < 0.01) but not by treatment with XAV939 (XAV) (P > 0.05). One-way ANOVA: F(4, 80) = 415, P < 0.00001, n = 17 per sample. Tukey's post hoc test: HSD[0.05] = 11.1; HSD[0.01] = 13.4. (B) β-Catenin was detected in the nuclear fraction of Wnt3a-treated cells but not in control (C) or Wnt5a-treated cells. Lamin B and α-tubulin were used as nuclear and cytoplasmic controls, respectively.
Wnt5a-induced binding of DAAM1 and Cdc42.
Because binding of DAAM1 to Rho GTPases has previously been shown in noncanonical Wnt signal transduction in other cell types (30, 31, 51, 52), we asked whether this was also the case in human CECs. Stimulation with Wnt5a resulted in binding of DAAM1 and Cdc42, while cotreatment with Wnt antagonist III (Box5) or disheveled-PDZ domain inhibitor (DI) abolished Wnt5a-mediated binding of DAAM1 and Cdc42 (Fig. 5). Stimulation of human CECs with FGF2, which has been shown to activate Cdc42 (46) in the absence of Wnt5a stimulation, did not result in binding of DAAM1 and Cdc42.
FIG 5.

Wnt5a-induced binding of DAAM1 and Cdc42. Binding between DAAM1 and Cdc42 was induced by Wnt5a stimulation (+) in human CECs. However, inhibition of Wnt5a signaling by cotreatment with Wnt antagonist III (Box5) or DI inhibited Wnt5a-induced binding between Cdc42 and DAAM1. Treatment with FGF2 had no effect on binding between DAAM1 and Cdc42 in the absence of Wnt5a. C, control.
Regulation of RhoA, Rac1, and Cdc42 by Wnt5a.
We focused next on Rho GTPases because they have previously been reported to regulate cell migration and planar cell polarity through the β-catenin-independent pathway (32, 33). Activated Cdc42 (Cdc42-GTP) was detected in Wnt5a-stimulated but not in control human CECs (Fig. 6A). Cdc42 activation was inhibited by cotreatment with Wnt antagonist III (Box5), disheveled-PDZ domain inhibitor (DI), or Cdc42 inhibitor ML141 (ML), while cotreatment with RhoA activator (RA), the Rho kinase inhibitor Y27632 (Y), or the Rac1 inhibitor Nsc23766 (NSC) had no effect (Fig. 6A). Activated Rac1 (Rac1-GTP) was detected in Wnt5a-stimulated but not control human CECs (Fig. 6B). Rac1 activation was inhibited by cotreatment with Box5, DI, or NSC, while cotreatment with RA, Y, or ML had no effect (Fig. 6B). A one-way ANOVA test showed a significant difference in RhoA activities among the experimental group as a whole: F(8, 45) = 470, P < 0.00001. Tukey's post hoc test showed that RhoA activity was dramatically inhibited in Wnt5a-stimulated human CECs compared to control CECs (P < 0.01) (Fig. 6C). Wnt5a-mediated inhibition of RhoA activity was reversed by cotreatment with Box5 (P < 0.01), DI (P < 0.01), ML (P < 0.01), or RA (P < 0.01). Cotreatment with NSC (P < 0.05) or Y (P > 0.05) had no effect on Wnt5a-mediated inhibition of RhoA activity (Fig. 6C). A one-way ANOVA test showed that the mean migration rates were significantly different among the experimental group as a whole: F(5, 102) = 419, P < 0.00001. Tukey's post hoc test showed that Wnt5a stimulation resulted in a 2-fold increase in the migration rate in human CECs compared to controls (P < 0.01), and this effect was further enhanced by cotreatment with Y (P < 0.01) (Fig. 6D). Cotreatment with ML led to an inhibition of Wnt5a-enhanced migration (P < 0.01), whereas cotreatment with NSC led to a mild attenuation (P < 0.01) (Fig. 6D). Cotreatment with a combination of Y and ML resulted in reversal of ML-mediated migration inhibition in Wnt5a-stimulated cells (P < 0.01) (Fig. 6D). Transfection of human CECs with Dvl2 siRNA (Dvl si) resulted in knockdown of Dvl2 protein levels and decreased amounts of Cdc42-GTP in response to Wnt5a stimulation (Fig. 7A). The nontargeting control siRNA had no effect on Dvl2 protein levels or Cdc42-GTP levels. Wnt antagonist III (Box5) and disheveled-PDZ domain inhibitor (DI) had no effect on Dvl2 protein levels but inhibited Wnt5a-mediated activation of Cdc42. Dvl2 siRNA also reversed Wnt5a-mediated inhibition of RhoA (Fig. 7B). A one-way ANOVA test showed a significant difference in RhoA activities among the various groups: F(5, 18) = 196, P < 0.00001. Tukey's post hoc test showed that RhoA activity was significantly inhibited by Wnt5a stimulation compared to the control (P < 0.01) (Fig. 7B). Wnt5a-mediated inhibition of RhoA activity was reversed by transfection with Dvl2 siRNA (P < 0.01) but not by the nontargeting control siRNA. Cotreatment with Box5 (P < 0.01) or DI (P < 0.01) also reversed Wnt5a-mediated inhibition of RhoA activity (Fig. 7B).
FIG 6.

Regulation of RhoA, Rac1, and Cdc42 by Wnt5a in human CECs. (A) GTP-bound Cdc42 was detected in Wnt5a-treated (+) cells. Cdc42 activation was blocked by cotreatment with Wnt antagonist III (Box5), DI, or Cdc42 inhibitor ML141 (ML). Cotreatment with RhoA activator (RA), Y27632 (Y), or Nsc23766 (Nsc) had no effect on activation of Cdc42. (B) GTP-bound Rac1 was detected in Wnt5a-treated (+) cells. Rac1 activation was inhibited by cotreatment with Box5, DI, or Nsc but not by cotreatment with ML, RA, or Y. (C) Wnt5a stimulation led to decreased RhoA activity (*, P < 0.01). This was reversed by cotreatment with Box5 (**, P < 0.01), DI (***, P < 0.01), ML (+, P < 0.01), or RA (†, P < 0.01), but cotreatment with Nsc (++, P > 0.05) or Y (+++, P > 0.05) had no effect. Treatment with RA in the absence of Wnt5a stimulation resulted in higher RhoA activity than RA treatment in the presence of Wnt5a (††, P < 0.01). One-way ANOVA: F(8, 45) = 470, P < 0.00001, n = 6 per sample. Tukey's post hoc test: HSD[0.05] = 10.8; HSD[0.01] = 12.7. (D) Treatment with Wnt5a (+) increased the rate of migration by 2-fold (*, P < 0.01). Wnt5a-enhanced migration was further increased by cotreatment with Y (**, P < 0.01). Cotreatment with ML (***, P < 0.01), and to a much lesser extent with Nsc (+, P < 0.01), inhibited Wnt5a-enhanced migration. Cotreatment with Y and ML led to reversal of migration inhibition by ML in Wnt5a-stimulated cells (++, P < 0.01). One-way ANOVA: F(4, 102) = 419, P < 0.00001, n = 18 per sample. Tukey's post hoc test: HSD[0.05] = 12.4; HSD[0.01] = 14.7. C, control.
FIG 7.

Regulation of CDC42 and RhoA by Wnt5a in human CECs. (A) Cdc42-GTP was detected in Wnt5a-treated (+) cells. Wnt5a-mediated activation of Cdc42 was inhibited by transfection with Dvl2 siRNA (Dvl si) but not nontargeting control siRNA (NT). Cotreatment with Wnt antagonist III (Box5) or DI also inhibited Wnt5a-mediated activation of Cdc42. Transfection with Dvl si but not NT resulted in knockdown of Dvl2 levels. (B) Wnt5a stimulation (+) led to an inhibition of relative RhoA activity (*, P < 0.01). Transfection with Dvl si (**, P < 0.01) but not NT (P > 0.05) resulted in a reversal of Wnt5a-mediated inhibition of RhoA. Cotreatment with Box5 (+, P < 0.01) and DI (++, P < 0.01) also resulted in reversal of Wnt5a-mediated inhibition of RhoA. One-way ANOVA: F(5, 18) = 196, P < 0.00001, n = 4 per sample. Tukey's post hoc test: HSD[0.05] = 7.1; HSD[0.01] = 8.9. C, control.
Regulation of LIMK2, SSH1, and cofilin by Wnt5a.
We next turned our attention to signaling targets downstream of Rho GTPases in the Wnt5a-enhanced migration, with particular focus on the role of RhoA in actin reorganization. RhoA was attractive because Cdc42 appeared to act mainly through RhoA inhibition in human CEC migration (Fig. 5D), and recent studies have reported that LIMK2 and SSH 1 to 3, which are regulated by Rho GTPases, modulate actin rearrangement by regulating cofilin (39–42). Wnt5a stimulation of human CECs led to dephosphorylation of LIMK2 and SSH1, resulting in dephosphorylation of cofilin (Fig. 8). Cotreatment of Wnt5a-stimulated human CECs with Box5, DI, ML, or RA prevented Wnt5a-induced dephosphorylation of LIMK2 and SSH1, resulting in phosphorylation of cofilin (Fig. 8). Cotreatment of Wnt5a-stimulated human CECs with the Rho kinase inhibitor Y27632 resulted in dephosphorylation of LIMK2 and SSH1, with the resulting dephosphorylation of cofilin (Fig. 8). Interestingly, simultaneous cotreatment with Y and ML in Wnt5a-stimulated human CECs led to reversal of ML-mediated phosphorylation of LIMK2 and SSH1 and led to dephosphorylation of cofilin (Fig. 8).
FIG 8.
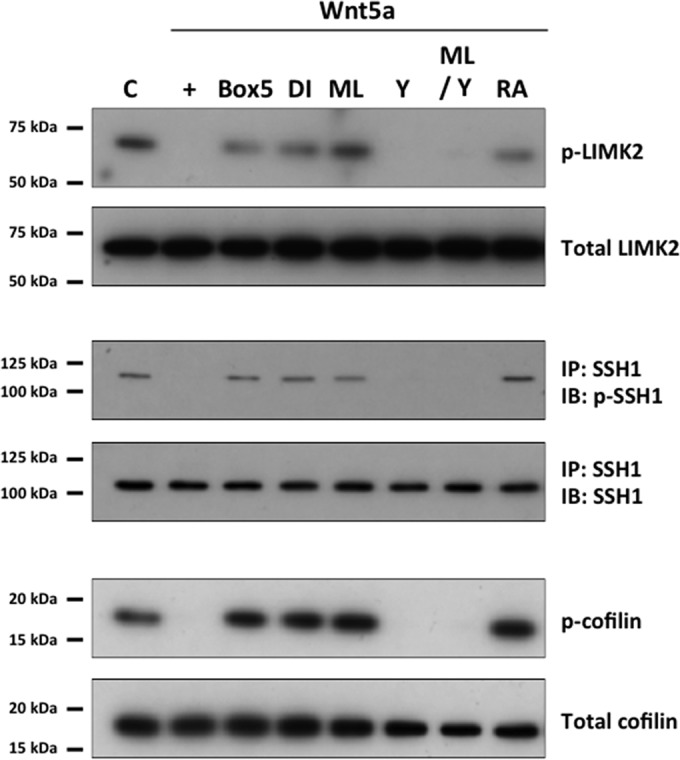
Regulation of LIMK2, SSH1, and cofilin by Wnt5a in human CECs. LIMK2 and SSH1 were observed to be in phosphorylated states, indicating active LIMK2 and inactive SSH1, under control conditions (C). Stimulation with Wnt5a (+) resulted in dephosphorylation of both proteins, leading to LIMK2 inactivation and SSH1 activation. Cotreatment of Wnt5a-stimulated cells with Wnt antagonist III (Box5), DI, ML, or RA inhibited Wnt5a-mediated dephosphorylation of both proteins. Cotreatment with Y27632 (Y) had no effect on Wnt5a-mediated dephosphorylation of LIMK2 and SSH1. Simultaneous treatment with ML and Y reversed ML-mediated phosphorylation of LIMK2 and SSH1 in Wnt5a-stimulated cells. Cofilin was phosphorylated when LIMK2 and SSH1 were phosphorylated and dephosphorylated when LIMK2 and SSH1 were dephosphorylated. C, control.
IL-1β-induced Wnt5a expression in ex vivo human corneal endothelium.
To address the possibility that IL-1β-induced Wnt5a expression in human CECs was an artifact of cell culture conditions, we asked whether IL-1β could also induce Wnt5a expression in ex vivo human corneal endothelium. Stimulation of ex vivo human corneal endothelium with IL-1β resulted in significant amounts of Wnt5a being expressed at 6 h poststimulation, and this was abolished by cotreatment with IL-1 receptor antagonist or sulfasalazine but not with SB203580 (Fig. 9A). Fzd5 and Ror2 were also expressed constitutively, regardless of culture conditions in IL-1β-stimulated ex vivo human corneal endothelium (Fig. 9B).
FIG 9.
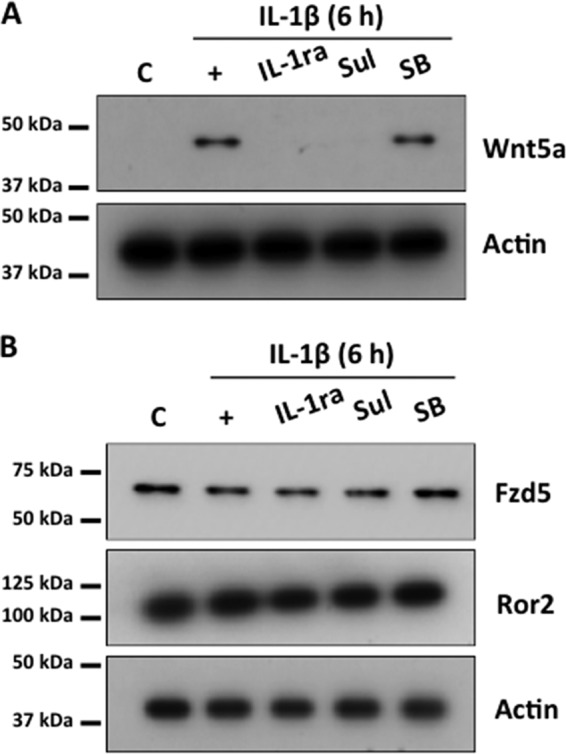
Expression of Wnt5a in human ex vivo corneal endothelium. (A) Wnt5a expression was detected in ex vivo human corneal endothelium 6 h after treatment with IL-1β. This was inhibited by cotreatment with IL-1ra or sulfasalazine (Sul) but not by SB203580 (SB). (B) Constitutive expression of Fzd5 and Ror2 was observed in ex vivo human corneal endothelium. C, control.
DISCUSSION
The demand for donor corneas in developed nations is expected to rise due to increases in the age of the general population and the number of people undergoing intraocular surgery. This, in addition to the 10 million corneal blindness patients worldwide not treated due to a lack of donor tissue currently, will impart a greater sense of urgency to the development of pharmacologic therapies for vision loss secondary to corneal endothelial dysfunction. The mechanism of corneal wound healing is unusual in that cell proliferation does not play an important role; rather, cell migration and enlargement are critical in wound repair. Corneal endothelial cells that have been severely injured can undergo endothelial-mesenchymal transition (EMT), where they show increased migration, proliferation, and fibrosis, which lead to formation of retrocorneal fibrous membranes (RCFM) and make visual rehabilitation no longer possible. Although RCFM formation is undesirable, modulation of individual components of EMT may provide a means of enhancing corneal endothelial wound healing without RCFM formation. FGF2 induced by IL-1β has been shown to enhance migration of human CECs; unfortunately, it has also been implicated as a major mediator of EMT in corneal endothelial cells (7, 10, 46, 50, 53). This led us to investigate other downstream targets of IL-1β signaling in human CECs that could enhance cell migration.
We previously reported that IL-1β and FGF2 treatment enhanced human CEC migration and that this could be completely abolished by SU5402 (7). SU5402, unfortunately, also inhibited other signaling pathways, including the noncanonical Wnt pathway (J. G. Lee and M. Heur, unpublished data). Attenuation but not complete inhibition of IL-1β-enhanced migration by AZD4547 (Fig. 1A) strongly suggested the presence of another signaling pathway downstream of IL-1β that could enhance migration in human CECs. Several reports linking IL-1β with Wnt5a signaling led us to investigate its potential as a candidate (44, 54, 55). Expression of Wnt5a has been reported in fetal but not adult corneal endothelium in humans (34), and this is consistent with our Western blot analysis results, where Wnt5a could not be detected in differentiated human CECs in vitro (Fig. 1B) or adult corneal endothelium ex vivo (Fig. 9) without IL-1β stimulation. Constitutive expression of Fzd5 and Ror2 (Fig. 2A) strongly suggested that human CECs are primed to respond to noncanonical Wnt signaling, and as expected, IL-1β and Wnt5a stimulation led to binding between Fzd5 and Ror2 (Fig. 2B). IL-1β-induced Wnt5a expression in human CECs is mediated through NF-κB, as shown by inhibition of Wnt5a expression by sulfasalazine (Fig. 1B) and the dose-dependent decrease with p65 siRNA knockdown (Fig. 1C). This was further supported by pulldown of NF-κB binding sites in the human Wnt5a promoter in chromatin immunoprecipitation experiments (Fig. 3). These data strongly suggest that Wnt5a expression in human CECs proceeds through previously described pathways.
As in other cell types, Wnt5a signaling through the β-catenin-independent pathway in human CECs was strongly suggested when XAV939, an axin stabilizer, did not inhibit Wnt5a-enhanced migration (Fig, 4A), and this was confirmed by the absence of nuclear translocation of β-catenin in Wnt5a-stimulated human CECs (Fig. 4B). Wnt5a stimulation also led to binding of DAAM1 and Cdc42, indicating that human CECs also utilize previously described components of noncanonical Wnt signaling (Fig. 5). Interestingly, Wnt5a stimulation led to inhibition of RhoA activity in human CECs, which in turn was reversed by ML141, a Cdc42 antagonist (Fig. 6C). This, along with reversal of ML141-mediated inhibition of cell migration in Wnt5a-stimulated cells by simultaneous treatment with ML141 and Y27632, indicated that Cdc42 acts upstream of RhoA in regulating migration of human CECs (Fig. 6D). Furthermore, knockdown of Dvl2 by using siRNA resulted in decreased levels of Cdc42-GTP and reversal of Wnt5a-mediated activation of RhoA (Fig. 7). Rac1 appears to play a very minor role in Wnt5a-enhanced cell migration, as cotreatment with Nsc23766 resulted in only a mild attenuation of cell migration (Fig. 6D). This is in contrast to other cell types, where Rac1-mediated lamellipodium formation is important in cell migration (56–58). RhoA-mediated stress fiber formation is critical in stationary cells and inhibits cell migration (39, 59–61), and this is consistent with the results of our migration assays in human CECs, where Y27632 enhanced migration.
Downstream of Rho kinase, LIMK2, SSH1, and cofilin appear to play a role in Wnt5a-enhanced migration in human CECs. Under conditions where cell migration was enhanced, e.g., with Wnt5a and Y27632 treatment, LIMK2, SSH1, and cofilin were dephosphorylated, indicating that LIMK2 was inactive and that SSH1 and cofilin were active. Also, under conditions where cell migration was inhibited, e.g., Wnt antagonist III, disheveled-PDZ domain inhibitor, Cdc42 antagonist, and RhoA activator treatments, LIMK2, SSH1, and cofilin were phosphorylated, indicating that LIMK2 was active and that SSH1 and cofilin were inactive. This is broadly consistent with previous reports demonstrating the roles of LIMK2, SSH1, and cofilin in regulation of actin cytoskeleton dynamics and cell migration. LIM kinases have been shown to be the target of various kinases, including Rho kinase, p21-activated protein kinases, and myotonic dystrophy kinase-related Cdc42 binding kinase-α, which are downstream of RhoA, Rac1, and Cdc42 respectively (39, 62, 63). However, simultaneous treatment with ML141 and Y27632, leading to dephosphorylation (Fig. 7), suggested that Rho kinase is the main regulator of LIMK2 in human CECs. Regulation of SSH1 and cofilin in human CECs appears to be consistent with previous reports, where phosphorylation of SSH1 by protein kinase D1 (PKD1), which itself is activated downstream of RhoA, results in its binding to 14-3-3 proteins and sequestration from F-actin, thereby leading to inhibition of its phosphatase activity (62, 64). IL-1β-stimulated expression of Wnt5a and constitutive expression of Fzd5 and Ror2 in human CECs did not appear to be cell culture-related artifacts, given that ex vivo human corneal endothelium responded in the same manner (Fig. 9).
There is still much that warrants further investigation of noncanonical Wnt signaling and Rho GTPases in pathogenic mechanisms of human disease, in particular, those of the eye. The role of Wnt5a in human eye diseases has not been thoroughly investigated, but Wnt5a has been implicated in regulation of the mesenchymal transition of epithelial cells in various forms of cancer. The evidence suggests that it can either inhibit or promote the mesenchymal transition, depending on the tissue, such as the colon or pancreas, and either inhibit or promote metastasis (65–67). Rho kinase inhibitors also show therapeutic promise for several eye diseases (68–70), indicating further investigation of the signaling pathways could prove to be valuable. In conclusion, IL-1β induces Wnt5a expression through NF-κB in human CECs. Wnt5a then enhances migration via noncanonical Wnt signaling that involves Fzd5, Ror2, Dvl, and DAAM1 and subsequent regulation of Rho GTPases Cdc42 and RhoA, leading to inhibition of LIMK2 and activation of SSH1 (Fig. 10).
FIG 10.

The role of noncanonical Wnt signaling in human CEC migration. Stimulation by IL-1β leads to expression of Wnt5a mediated through NF-κB. Wnt5a then induces binding of Fzd5 and Ror2, resulting in activation of Dvl. This leads to binding of DAAM1 and activated Cdc42, which in turn results in inhibition of RhoA. Inhibition of RhoA then leads to dephosphorylation of LIMK2 (inhibition) and SSH1 (activation), thereby resulting in dephosphorylation and activation of cofilin and leading to enhanced migration of human CECs.
ACKNOWLEDGMENTS
This study was supported by EY021485 from the National Eye Institute (M.H.) and a Career Development Award from Research to Prevent Blindness (M.H.).
Footnotes
Published ahead of print 14 July 2014
REFERENCES
- 1.Joyce NC, Navon SE, Roy S, Zieske JD. 1996. Expression of cell cycle-associated proteins in human and rabbit corneal endothelium in situ. Invest. Ophthalmol. Vis. Sci. 37:1566–1575 [PubMed] [Google Scholar]
- 2.Senoo T, Joyce NC. 2000. Cell cycle kinetics in corneal endothelium from old and young donors. Invest. Ophthalmol. Vis. Sci. 41:660–667 [PubMed] [Google Scholar]
- 3.Whitcher JP, Srinivasan M, Upadhyay MP. 2001. Corneal blindness: a global perspective. Bull. World Health Organ. 79:214–221 [PMC free article] [PubMed] [Google Scholar]
- 4.Song JS, Lee JG, Kay EP. 2010. Induction of FGF-2 synthesis by IL-1β in aqueous humor through PI3-kinase and p38 in rabbit corneal endothelium. Invest. Ophthalmol. Vis. Sci. 51:822–829. 10.1167/iovs.09-4240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Djalilian AR, Nagineni CN, Mahesh SP, Smith JA, Nussenblatt RB, Hooks JJ. 2006. Inhibition of inflammatory cytokine production in human corneal cells by dexamethasone, but not cyclosporin. Cornea 25:709–714. 10.1097/01.ico.0000208815.02120.90 [DOI] [PubMed] [Google Scholar]
- 6.Moore JE, McMullen TC, Campbell IL, Rohan R, Kaji Y, Afshari NA, Usui T, Archer DB, Adamis AP. 2002. The inflammatory milieu associated with conjunctivalized cornea and its alteration with IL-1 RA gene therapy. Invest. Ophthalmol. Vis. Sci. 43:2905–2915 [PubMed] [Google Scholar]
- 7.Lee JG, Heur M. 2013. Interleukin-1β enhances cell migration through AP-1 and NF-κB pathway-dependent FGF2 expression in human corneal endothelial cells. Biol. Cell 105:175–189. 10.1111/boc.201200077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JG, Kay EP. 2009. Common and distinct pathways for cellular activities in FGF-2 signaling induced by IL-1β in corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 50:2067–2076. 10.1167/iovs.08-3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JG, Kay EP. 2012. NF-κB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Invest. Ophthalmol. Vis. Sci. 53:1530–1538. 10.1167/iovs.11-9102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee HT, Lee JG, Na M, Kay EP. 2004. FGF-2 induced by interleukin-1 beta through the action of phosphatidylinositol 3-kinase mediates endothelial mesenchymal transformation in corneal endothelial cells. J. Biol. Chem. 279:32325–32332. 10.1074/jbc.M405208200 [DOI] [PubMed] [Google Scholar]
- 11.Nusse R. 2005. Wnt signaling in disease and in development. Cell Res. 15:28–32. 10.1038/sj.cr.7290260 [DOI] [PubMed] [Google Scholar]
- 12.Angers S, Moon RT. 2009. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 10:468–477. 10.1038/nrm2717 [DOI] [PubMed] [Google Scholar]
- 13.MacDonald BT, Tamai K, He X. 2009. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17:9–26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang H, He X. 2008. Wnt/beta-catenin signaling: new (and old) players and new insights. Curr. Opin. Cell Biol. 20:119–125. 10.1016/j.ceb.2008.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grigoryan T, Wend P, Klaus A, Birchmeier W. 2008. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 22:2308–2341. 10.1101/gad.1686208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khokhar S, Sethi HS, Sony P, Sudan R, Soni A. 2002. Pseudophakic pupillary block caused by pupillary capture after phacoemulsification and in-the-bag AcrySof lens implantation. J. Cataract Refract. Surg. 28:1291–1292. 10.1016/S0886-3350(02)01305-6 [DOI] [PubMed] [Google Scholar]
- 17.Kimura C, Yoshinaga K, Tian E, Suzuki M, Aizawa S, Matsuo I. 2000. Visceral endoderm mediates forebrain development by suppressing posteriorizing signals. Dev. Biol. 225:304–321. 10.1006/dbio.2000.9835 [DOI] [PubMed] [Google Scholar]
- 18.Kudoh T, Wilson SW, Dawid IB. 2002. Distinct roles for Fgf, Wnt and retinoic acid in posteriorizing the neural ectoderm. Development 129:4335–4346 [DOI] [PubMed] [Google Scholar]
- 19.Heisenberg CP, Brand M, Jiang YJ, Warga RM, Beuchle D, van Eeden FJ, Furutani-Seiki M, Granato M, Haffter P, Hammerschmidt M, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nusslein-Volhard C. 1996. Genes involved in forebrain development in the zebrafish, Danio rerio. Development 123:191–203 [DOI] [PubMed] [Google Scholar]
- 20.Gage PJ, Qian M, Wu D, Rosenberg KI. 2008. The canonical Wnt signaling antagonist DKK2 is an essential effector of PITX2 function during normal eye development. Dev. Biol. 317:310–324. 10.1016/j.ydbio.2008.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Randall RM, Shao YY, Wang L, Ballock RT. 2012. Activation of Wnt planar cell polarity (PCP) signaling promotes growth plate column formation in vitro. J. Orthop. Res. 30:1906–1914. 10.1002/jor.22152 [DOI] [PubMed] [Google Scholar]
- 22.Dejmek J, Safholm A, Kamp Nielsen C, Andersson T, Leandersson K. 2006. Wnt-5a/Ca2+-induced NFAT activity is counteracted by Wnt-5a/Yes-Cdc42-casein kinase 1α signaling in human mammary epithelial cells. Mol. Cell. Biol. 26:6024–6036. 10.1128/MCB.02354-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM. 2002. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 1:279–288. 10.1016/S1535-6108(02)00045-4 [DOI] [PubMed] [Google Scholar]
- 24.Blumenthal A, Ehlers S, Lauber J, Buer J, Lange C, Goldmann T, Heine H, Brandt E, Reiling N. 2006. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 108:965–973. 10.1182/blood-2005-12-5046 [DOI] [PubMed] [Google Scholar]
- 25.Takada R, Hijikata H, Kondoh H, Takada S. 2005. Analysis of combinatorial effects of Wnts and Frizzleds on beta-catenin/armadillo stabilization and Dishevelled phosphorylation. Genes Cells 10:919–928. 10.1111/j.1365-2443.2005.00889.x [DOI] [PubMed] [Google Scholar]
- 26.Nishita M, Itsukushima S, Nomachi A, Endo M, Wang Z, Inaba D, Qiao S, Takada S, Kikuchi A, Minami Y. 2010. Ror2/Frizzled complex mediates Wnt5a-induced AP-1 activation by regulating Dishevelled polymerization. Mol. Cell. Biol. 30:3610–3619. 10.1128/MCB.00177-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grumolato L, Liu G, Mong P, Mudbhary R, Biswas R, Arroyave R, Vijayakumar S, Economides AN, Aaronson SA. 2010. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev. 24:2517–2530. 10.1101/gad.1957710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishita M, Yoo SK, Nomachi A, Kani S, Sougawa N, Ohta Y, Takada S, Kikuchi A, Minami Y. 2006. Filopodia formation mediated by receptor tyrosine kinase Ror2 is required for Wnt5a-induced cell migration. J. Cell Biol. 175:555–562. 10.1083/jcb.200607127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Habas R, Kato Y, He X. 2001. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell 107:843–854. 10.1016/S0092-8674(01)00614-6 [DOI] [PubMed] [Google Scholar]
- 30.Aspenstrom P, Richnau N, Johansson AS. 2006. The diaphanous-related formin DAAM1 collaborates with the Rho GTPases RhoA and Cdc42, CIP4 and Src in regulating cell morphogenesis and actin dynamics. Exp. Cell Res. 312:2180–2194. 10.1016/j.yexcr.2006.03.013 [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y, Tian Y, Du J, Hu Z, Yang L, Liu J, Gu L. 2012. Dvl2-dependent activation of Daam1 and RhoA regulates Wnt5a-induced breast cancer cell migration. PLoS One 7:e37823. 10.1371/journal.pone.0037823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlessinger K, McManus EJ, Hall A. 2007. Cdc42 and noncanonical Wnt signal transduction pathways cooperate to promote cell polarity. J. Cell Biol. 178:355–361. 10.1083/jcb.200701083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Habas R, Dawid IB, He X. 2003. Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 17:295–309. 10.1101/gad.1022203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Huang K, Nakatsu MN, Xue Z, Deng SX, Fan G. 2013. Identification of novel molecular markers through transcriptomic analysis in human fetal and adult corneal endothelial cells. Hum. Mol. Genet. 22:1271–1279. 10.1093/hmg/dds527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmitz AA, Govek EE, Bottner B, Van Aelst L. 2000. Rho GTPases: signaling, migration, and invasion. Exp. Cell Res. 261:1–12. 10.1006/excr.2000.5049 [DOI] [PubMed] [Google Scholar]
- 36.Nobes CD, Hall A. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244. 10.1083/jcb.144.6.1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ridley AJ. 2001. Rho GTPases and cell migration. J. Cell Sci. 114:2713–2722 [DOI] [PubMed] [Google Scholar]
- 38.Huttenlocher A, Sandborg RR, Horwitz AF. 1995. Adhesion in cell migration. Curr. Opin. Cell Biol. 7:697–706. 10.1016/0955-0674(95)80112-X [DOI] [PubMed] [Google Scholar]
- 39.Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. 1999. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285:895–898. 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- 40.Sumi T, Matsumoto K, Takai Y, Nakamura T. 1999. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 147:1519–1532. 10.1083/jcb.147.7.1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sumi T, Matsumoto K, Nakamura T. 2001. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J. Biol. Chem. 276:670–676. 10.1074/jbc.M007074200 [DOI] [PubMed] [Google Scholar]
- 42.Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, Storz P. 2009. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat. Cell Biol. 11:545–556. 10.1038/ncb1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanegashima K, Zhao H, Dawid IB. 2008. WGEF activates Rho in the Wnt-PCP pathway and controls convergent extension in Xenopus gastrulation. EMBO J. 27:606–617. 10.1038/emboj.2008.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ge XP, Gan YH, Zhang CG, Zhou CY, Ma KT, Meng JH, Ma XC. 2011. Requirement of the NF-κB pathway for induction of Wnt-5A by interleukin-1β in condylar chondrocytes of the temporomandibular joint: functional crosstalk between the Wnt-5a and NF-κB signaling pathways. Osteoarthritis Cartilage 19:111–117. 10.1016/j.joca.2010.10.016 [DOI] [PubMed] [Google Scholar]
- 45.Katula KS, Joyner-Powell NB, Hsu CC, Kuk A. 2012. Differential regulation of the mouse and human Wnt5a alternative promoters A and B. DNA Cell Biol. 31:1585–1597. 10.1089/dna.2012.1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JG, Kay EP. 2006. FGF-2-induced wound healing in corneal endothelial cells requires Cdc42 activation and Rho inactivation through the phosphatidylinositol 3-kinase pathway. Invest. Ophthalmol. Vis. Sci. 47:1376–1386. 10.1167/iovs.05-1223 [DOI] [PubMed] [Google Scholar]
- 47.Garweg JG, Wegmann-Burns M, Goldblum D. 2006. Effects of daunorubicin, mitomycin C, azathioprine and cyclosporin A on human retinal pigmented epithelial, corneal endothelial and conjunctival cell lines. Graefe's Arch. Clin. Exp. Ophthalmol. 244:382–389. 10.1007/s00417-005-0017-4 [DOI] [PubMed] [Google Scholar]
- 48.Lee JG, Kay EP. 2007. Two populations of p27 use differential kinetics to phosphorylate Ser-10 and Thr-187 via phosphatidylinositol 3-kinase in response to fibroblast growth factor-2 stimulation. J. Biol. Chem. 282:6444–6454. 10.1074/jbc.M607808200 [DOI] [PubMed] [Google Scholar]
- 49.Lee JG, Song JS, Smith RE, Kay EP. 2011. Human corneal endothelial cells employ phosphorylation of p27Kip1 at both Ser10 and Thr187 sites for FGF-2-mediated cell proliferation via PI 3-kinase. Invest. Ophthalmol. Vis. Sci. 52:8216–8223. 10.1167/iovs.11-8213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee JG, Kay EP. 2006. Cross-talk among Rho GTPases acting downstream of PI 3-kinase induces mesenchymal transformation of corneal endothelial cells mediated by FGF-2. Invest. Ophthalmol. Vis. Sci. 47:2358–2368. 10.1167/iovs.05-1490 [DOI] [PubMed] [Google Scholar]
- 51.Liebner S, Plate KH. 2010. Differentiation of the brain vasculature: the answer came blowing by the Wnt. J. Angiogenesis Res. 2:1. 10.1186/2040-2384-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ang SF, Zhao ZS, Lim L, Manser E. 2010. DAAM1 is a formin required for centrosome re-orientation during cell migration. PLoS One 5:e13064. 10.1371/journal.pone.0013064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ko MK, Kay EP. 2005. Regulatory role of FGF-2 on type I collagen expression during endothelial mesenchymal transformation. Invest. Ophthalmol. Vis. Sci. 46:4495–4503. 10.1167/iovs.05-0818 [DOI] [PubMed] [Google Scholar]
- 54.Ge X, Ma X, Meng J, Zhang C, Ma K, Zhou C. 2009. Role of Wnt-5A in interleukin-1β-induced matrix metalloproteinase expression in rabbit temporomandibular joint condylar chondrocytes. Arthritis Rheum. 60:2714–2722. 10.1002/art.24779 [DOI] [PubMed] [Google Scholar]
- 55.Sonomoto K, Yamaoka K, Oshita K, Fukuyo S, Zhang X, Nakano K, Okada Y, Tanaka Y. 2012. Interleukin-1β induces differentiation of human mesenchymal stem cells into osteoblasts via the Wnt-5a/receptor tyrosine kinase-like orphan receptor 2 pathway. Arthritis Rheum. 64:3355–3363. 10.1002/art.34555 [DOI] [PubMed] [Google Scholar]
- 56.Connolly JO, Simpson N, Hewlett L, Hall A. 2002. Rac regulates endothelial morphogenesis and capillary assembly. Mol. Biol. Cell 13:2474–2485. 10.1091/mbc.E02-01-0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hall A. 1998. Rho GTPases and the actin cytoskeleton. Science 279:509–514. 10.1126/science.279.5350.509 [DOI] [PubMed] [Google Scholar]
- 58.Yuan XB, Jin M, Xu X, Song YQ, Wu CP, Poo MM, Duan S. 2003. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat. Cell Biol. 5:38–45. 10.1038/ncb895 [DOI] [PubMed] [Google Scholar]
- 59.Couchman JR, Rees DA. 1979. The behaviour of fibroblasts migrating from chick heart explants: changes in adhesion, locomotion and growth, and in the distribution of actomyosin and fibronectin. J. Cell Sci. 39:149–165 [DOI] [PubMed] [Google Scholar]
- 60.Burridge K. 1981. Are stress fibres contractile? Nature 294:691–692. 10.1038/294691a0 [DOI] [PubMed] [Google Scholar]
- 61.Arthur WT, Burridge K. 2001. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol. Biol. Cell 12:2711–2720. 10.1091/mbc.12.9.2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edwards DC, Sanders LC, Bokoch GM, Gill GN. 1999. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1:253–259. 10.1038/12963 [DOI] [PubMed] [Google Scholar]
- 63.Sumi T, Matsumoto K, Shibuya A, Nakamura T. 2001. Activation of LIM kinases by myotonic dystrophy kinase-related Cdc42-binding kinase alpha. J. Biol. Chem. 276:23092–23096. 10.1074/jbc.C100196200 [DOI] [PubMed] [Google Scholar]
- 64.Mullin MJ, Lightfoot K, Marklund U, Cantrell DA. 2006. Differential requirement for RhoA GTPase depending on the cellular localization of protein kinase D. J. Biol. Chem. 281:25089–25096. 10.1074/jbc.M603591200 [DOI] [PubMed] [Google Scholar]
- 65.Bo H, Zhang S, Gao L, Chen Y, Zhang J, Chang X, Zhu M. 2013. Upregulation of Wnt5a promotes epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells. BMC Cancer 13:496. 10.1186/1471-2407-13-496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng R, Sun B, Liu Z, Zhao X, Qi L, Li Y, Gu Q. 24 January 2014. Wnt5a suppresses colon cancer by inhibiting cell proliferation and epithelial-mesenchymal transition. J. Cell. Physiol. 10.1002/jcp.24566 [DOI] [PubMed] [Google Scholar]
- 67.Wei W, Li H, Li N, Sun H, Li Q, Shen X. 2013. WNT5A/JNK signaling regulates pancreatic cancer cells migration by phosphorylating paxillin. Pancreatology 13:384–392. 10.1016/j.pan.2013.05.008 [DOI] [PubMed] [Google Scholar]
- 68.Challa P, Arnold JJ. 2014. Rho-kinase inhibitors offer a new approach in the treatment of glaucoma. Expert Opin. Invest. Drugs 23:81–95. 10.1517/13543784.2013.840288 [DOI] [PubMed] [Google Scholar]
- 69.Koizumi N, Okumura N, Ueno M, Nakagawa H, Hamuro J, Kinoshita S. 2013. Rho-associated kinase inhibitor eye drop treatment as a possible medical treatment for Fuchs corneal dystrophy. Cornea 32:1167–1170. 10.1097/ICO.0b013e318285475d [DOI] [PubMed] [Google Scholar]
- 70.Song H, Gao D. 2011. Fasudil, a Rho-associated protein kinase inhibitor, attenuates retinal ischemia and reperfusion injury in rats. Int. J. Mol. Med. 28:193–198. 10.3892/ijmm.2011.659 [DOI] [PubMed] [Google Scholar]