

Abstract
Variable-number tandem repeats (VNTRs) mutate rapidly and can be useful markers for genotyping. While multilocus VNTR analysis (MLVA) is increasingly used in the detection and investigation of food-borne outbreaks caused by Salmonella enterica serovar Typhimurium (S. Typhimurium) and other bacterial pathogens, MLVA data analysis usually relies on simple clustering approaches that may lead to incorrect interpretations. Here, we estimated the rates of copy number change at each of the five loci commonly used for S. Typhimurium MLVA, during in vitro and in vivo passage. We found that loci STTR5, STTR6, and STTR10 changed during passage but STTR3 and STTR9 did not. Relative rates of change were consistent across in vitro and in vivo growth and could be accurately estimated from diversity measures of natural variation observed during large outbreaks. Using a set of 203 isolates from a series of linked outbreaks and whole-genome sequencing of 12 representative isolates, we assessed the accuracy and utility of several alternative methods for analyzing and interpreting S. Typhimurium MLVA data. We show that eBURST analysis was accurate and informative. For construction of MLVA-based trees, a novel distance metric, based on the geometric model of VNTR evolution coupled with locus-specific weights, performed better than the commonly used simple or categorical distance metrics. The data suggest that, for the purpose of identifying potential transmission clusters for further investigation, isolates whose profiles differ at one of the rapidly changing STTR5, STTR6, and STTR10 loci should be collapsed into the same cluster.
INTRODUCTION
Genetic subtyping of bacterial isolates is critical in the investigation of food-borne infections. Multilocus variable-number tandem-repeat (VNTR) analysis (MLVA) is a high-resolution typing method that has become widespread among public health laboratories for the investigation of Salmonella and other food-borne outbreaks (1–3) (see Fig. S1 in the supplemental material). A range of methods are available for subtyping of Salmonella (4), but MLVA is particularly well suited to outbreak investigation as it provides high resolution, is efficient and robust, and generates repeatable results suitable for sharing between laboratories using the same standardized techniques (1, 5–7). An MLVA profile is usually expressed as a string of numbers of length N, which represents the number of copies of repeated sequences at each of a set of N loci under analysis. For example, the profile 3-4-3-10-12 indicates 3 repeat copies at locus 1, 4 repeat copies at locus 2, and so on. Where repeat copies differ in length, allele codes can be assigned to distinguish specific types of repeat combinations (8).
Salmonella enterica, in particular, serovar Typhimurium (S. Typhimurium), is among the most common causes of food-borne outbreaks globally. MLVA was first proposed for the analysis of S. Typhimurium in 2003 by Lindstedt and colleagues (9), who then developed a multiplex assay targeting the five most variable loci (STTR3, STTR5, STTR6, STTR9, and STTR10) (10). The 5-locus multiplex scheme has since been widely adopted in Europe, Australia, and elsewhere (3, 8, 11). Since repeat lengths in STTR3 can differ, this locus is sometimes expressed using allele codes as presented in reference 8 or as the total size in base pairs. Similar schemes are also in use or development for other S. enterica serovars, including Enteritidis, Gallinarum, Heidelberg, Dublin, and Typhi (12–18), and almost a hundred papers reported the use of MLVA for Salmonella analysis between 2004 and 2013 (see the supplemental material).
Analysis of MLVA profiles typically relies on distance-based methods for constructing trees or dendrograms to infer the relationships between isolates with different profiles (see, e.g., the PulseNet MLVA protocols [http://www.pulsenetinternational.org/protocols/mlva/] and the supplemental material) or for identifying clusters of closely related isolates (see, e.g., references 11 and 19). Dendrogram or tree construction typically uses either “simple” distance (the number of loci that differ) or “categorical” distance (the number of repeat differences summed across loci) (see, e.g., Table S1 in the supplemental material, which summarizes the MLVA results reported in a cross section of papers from 2012). Cluster identification typically starts by identifying groups of isolates in a given spatial and temporal range that share identical MLVA profiles (11, 19) and sometimes expands to include isolates that differ slightly in their profiles. However, these simplistic approaches share two key problems. First, they treat all loci equally; e.g., a single-copy change at locus 1 is considered equivalent to a single-copy change at locus 2. Yet it is clear from the variations in locus-specific diversity metrics for many organisms, including Salmonella (20), and from in vitro passage experiments in other organisms (21, 22) that different VNTR loci mutate at different rates. Second, these approaches do not account for the fact that multiple copies of a tandem-repeat sequence can be gained or lost in multiple or single events; e.g., a decrease of 2 copies could be the result of a single deletion event in which 2 copies were lost or the result of 2 separate deletion events, with each affecting 1 copy. Yet in vitro passage studies in Yersinia pestis and Escherichia coli have shown that multicopy changes can occur and that the probability of a change of a given size occurring can be approximated using a geometric model for VNTR evolution (GMVE) (22). The eBURST approach to multilocus analysis (23), developed for multilocus sequence typing (MLST), has also been applied to MLVA data. This method links profiles that share loci, creating chains or complexes of related profiles, but does not attempt to infer relationships beyond this.
It is generally assumed that MLVA profiles (i.e., repeat copy numbers) change rapidly enough to distinguish bacterial clones from one another—e.g., to distinguish outbreak strains from other circulating strains—but not so rapidly that clonal relationships are masked by changes arising during the course of an outbreak. However, the rates at which MLVA profiles change have not been directly investigated for Salmonella; hence, it is unclear how S. Typhimurium MLVA profiles should be interpreted in the context of outbreak detection and investigation. Parallel serial-passage experiments (PSPE) have been used to directly estimate the rate of VNTR mutations, and the relative rates of single-copy changes, among several pathogenic bacteria, including Y. pestis, E. coli O157:H7, and Burkholderia pseudomallei (21, 22, 24). This allows GMVE model-based analysis and interpretation of MLVA data for these organisms (22, 24). Some public health laboratories give more weight to selected loci when interpreting S. Typhimurium MLVA profiles (personal communications, Microbiological Diagnostic Unit [MDU] staff members); however, this is not based on direct measurement of mutation rates and has not been formalized. The persistence and VNTR stability of S. Typhimurium DT41 were recently studied through a series of experiments in chicken flocks in Denmark (25). The study detected a few changes, at loci STTR5, STTR6, and STTR10 but not STTR3 or STTR9, suggesting that the VNTRs used for S. Typhimurium MLVA are quite stable but that the levels of stability differ among loci. Similar patterns were also reported during in vitro passage of a range of S. Typhimurium phage types and for MLVA profiles from Belgian isolates (26). However, mutation rates were not estimated directly in either of these studies and they did not provide enough data to construct a GMVE model for S. Typhimurium.
In the present study, we aimed to estimate the different rates of change in copy number at the five loci targeted by the S. Typhimurium MLVA scheme, during in vitro and in vivo passage of a clinical isolate. We then used these rates to assess alternative methods for analyzing S. Typhimurium MLVA profiles, including tree construction and cluster identification, using whole-genome sequencing of a small number of representative strains to determine the true underlying relationships.
MATERIALS AND METHODS
Bacterial isolates and sequencing.
All S. Typhimurium isolates in this study shared a phage type that is a variant of definitive type 135 (DT135). This variant does not yet have an official phage type designation but is commonly referred to as 135a or 135@ and is among the most common phage types detected in Australian food-borne outbreaks (11, 27–29). All isolates were sourced from the Microbiological Diagnostic Unit Public Health Laboratory, Victoria. STm5, used in the passage experiments, was isolated from a human gastroenteritis patient during a 2005 outbreak in Tasmania, and its MLVA profiles are 2-11-10-10-212 using the European scheme (8) and 3-13-11-11-523 using the Australian nomenclature for the same data. MLVA was performed on a total of 201 outbreak-linked S. Typhimurium 135@ isolates and on 2 isolates from sporadic cases in Tasmania, as summarized in Table 1 and Fig. 1. STm5 and 11 other Tasmanian isolates (see Table 1 and Fig. 2) were subjected to whole-genome sequencing on an Illumina HiSeq platform; details of sequencing and analysis are reported elsewhere (30).
TABLE 1.
Summary of 203 S. Typhimurium isolates analyzed in this study
| State | Yr | n | Outbreaka | Sequenced isolate(s) |
|---|---|---|---|---|
| Tasmania | 2005 | 8 | 2005 (OB1) | STm8 |
| 2005 | 79 | 2005 (OB2) | STm1, STm2, STm3, STm5b | |
| 2005 | 42 | 2005 (OB3) | STm11 | |
| 2007 | 10 | 2007 (OB6) | STm9 | |
| 2008 | 45 | 2008 (OB7) | STm6, STm7, STm12 | |
| 2005 | 1 | Sporadic | STm10 | |
| 2007 | 1 | Sporadic | STm4 | |
| Victoria | 2002 | 2 | 2002 (1) | |
| 2002 | 3 | 2002 (2) | ||
| 2007 | 3 | 2007 | ||
| 2008 | 5 | 2008 (1) | ||
| 2008 | 4 | 2008 (2) |
OB, outbreak.
Passaged isolate.
FIG 1.
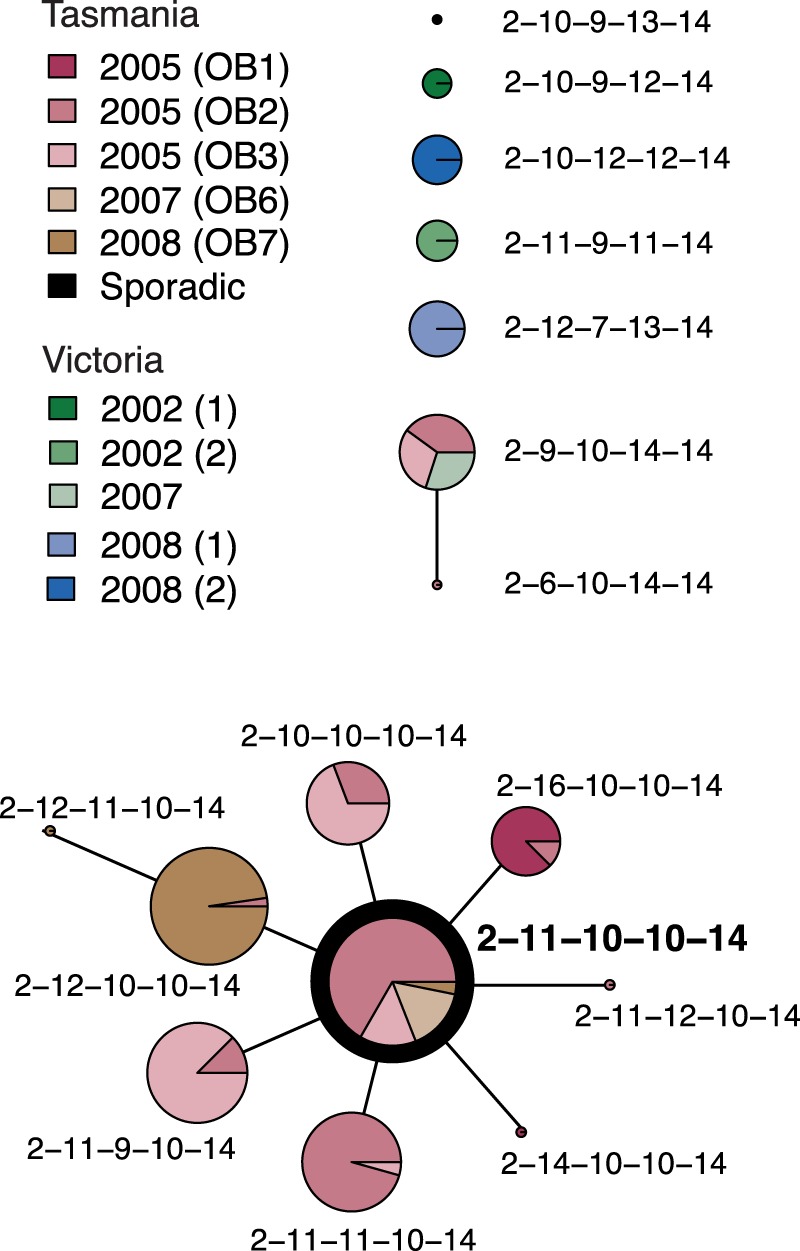
eBURST analysis of 203 S. Typhimurium 135@ isolates. Each MLVA profile is shown as a pie chart whose size represents the number of isolates with that profile, and colored slices indicate the outbreaks (OB) to which these isolates were linked during epidemiological investigations conducted at the time. The inferred founder profile is highlighted in bold.
FIG 2.

Whole-genome phylogeny of 12 S. Typhimurium 135@ isolates from Tasmania. Numbers on branches indicate the number of SNPs (base changes) on each branch; divergence dates were estimated previously from SNP data using BEAST (30). Red branches indicate the outbreak clone. Each isolate is labeled with respect to its STm number (within the icon indicating specimen type), its MLVA profile, the outbreak from which it derives (OB [as described for Fig. 1]), and the proportion of isolates from that outbreak that share this profile. *, most-common MLVA profile, inferred to be ancestral to the outbreak clone. Copy number changes relative to this profile are highlighted in yellow and green. (Adapted from BMC Genomics [30] with additional annotation.)
In vitro passage.
Parallel serial-passage experiments (PSPE), modeled on those described in references 21, 22, and 31, were performed using isolate STm5. Briefly, the PSPE procedure involved 100 independent clonal lineages subcultured on Luria-Bertani (LB) media (Difco) plates at 24-h intervals for a total of 10 passages. At each stage of each passage, one colony was picked at random and split into two; one half-colony was subcultured to continue the passage, and the other half-colony was suspended in LB–15% glycerol and stored at −80°C for later MLVA. DNA was extracted from all 100 lineages at the final time point (i.e., after 10 passages) and subjected to MLVA testing. Where mutations were identified at the final time point, the half-colonies stored from earlier time points from the same clonal lineage were tested to determine the precise stage of passage during which each mutation arose and to determine whether the observed change in copy number was the result of a single mutational events or multiple mutational events.
The number of generations per 24-h subculture was estimated by picking at random three replicate colonies following 24 h of growth of STm5 on LB media plates, suspending each colony in 1 ml phosphate-buffered saline (Media Preparation Unit, Department of Microbiology and Immunology, University of Melbourne), plating the bacteria using serial dilution, and counting CFU, as described in reference 21. The total number of generations during 24-h in vitro passage was therefore equal to the average number of generations per colony × 100 lineages × 10 passages; specifically, 28.6 × 100 × 10 = 28,600 generations.
In vivo passage.
Animal experiments were approved by the Animal Ethics Committee of the University of Melbourne and were conducted in accordance with the relevant Australian legislation. Salmonella-susceptible C57BL/6 mice and Salmonella-resistant B6.Nramp1R/R mice were generated by backcrossing the Nramp1R/R gene from CBA mice onto the C57BL/6 background through 13 backcrosses, followed by brother × sister mating to generate homozygous B6.NrampR/R mice. The genotype of B6.Nramp1R/R mice was confirmed using allele-specific single nucleotide polymorphism (SNP) PCR (32). Initial pilot experiments were performed by inoculating five mice of each strain with 3 × 103 CFU of a fresh subculture of isolate STm5 (18 h static at 37°C; optical density at 600 nm [OD600] = 0.6) from unpassaged stocks. Mice were inoculated intravenously and monitored for weight loss and other signs of illness and then euthanized when moribund in accordance with animal ethics and defined humane endpoints. Susceptible C57BL/6 mice became moribund after 5 to 7 days, but resistant mice did not become ill. However, between 102 and 106 CFU of Salmonella was cultured from the spleen of all inoculated mice, indicating that the STm5 isolate could establish an infection in mice at this dose (clinical infection in susceptible mice, subclinical infection in resistant mice). Five rounds of serial in vivo passage of STm5 were then performed in the Salmonella-susceptible C57BL/6 mouse strain; three rounds of passage were done in Salmonella-resistant mouse strain B6.Nramp1R/R. The C57BL/6 mice were inoculated orally with 2 × 107 to 3 × 107 CFU of STm5 bacteria (n = 5 mice for passages 1 to 3; n = 10 mice for passages 4 and 5) and euthanized when moribund (approximately 6 to 7 days postinfection). The B6.NrampR/R mice were infected orally with 2 × 107 to 3 × 107 CFU of STm5 bacteria (n = 5 mice for passage 1 and 2; n = 9 mice for passage 3), and all mice were euthanized at 13 to 14 days postinfection. Spleens were harvested from euthanized mice, homogenized, and cultured on xylose lysine deoxycholate (XLD) media (Oxoid) for selective isolation of Salmonella. Following overnight culture, three half-colonies were randomly sampled from each homogenate and subjected to DNA extraction and MLVA profiling. One of the three remaining half-colonies was randomly selected and subcultured overnight and was used to inoculate a further five mice in the next round of passage.
Multilocus VNTR analysis.
Genomic DNA was extracted from colonies or half-colonies using a QIAextractor instrument with a DX reagent kit (Qiagen). DNA was subjected to MLVA profiling using a multiplex assay targeting five VNTR loci (10). MLVA profiles were expressed as repeat copy numbers for loci STTR9, STTR5, STTR6, and STTR10, followed by an allele code for STTR3, using the methods and nomenclature described in reference 8. For example, profile 02-11-10-10-212 indicates 2 repeat copies for locus STTR9, 11 repeat copies for STTR5, 10 repeat copies for STTR6, 10 repeat copies for STTR10, and allele 212 at locus STTR3. For cluster analysis, which relies on analysis of repeat copy numbers, STTR3 was expressed as the number of repeat copies estimated from the total amplicon size, according to reference 8.
Statistical analysis.
All statistical analyses were performed in R (33). Distance-based analysis of MLVA profiles involved the calculation of a pairwise distance matrix representing the distance between pairs of strains according to the specified distance metric, followed by tree inference by clustering of the distance matrix using the upgma function of the phangorn R package. All distances were calculated with and without locus weights, with wk representing weight for locus k defined as 1/rk, where rk represents the mutation rate for locus k (determined by in vitro passage in this study). Simple pairwise distance determinations were based on the total number of loci at which two MLVA profiles differ, defined as dist , where k represents the locus index and ni,k represents the copy number at locus k in strain i. Categorical distance incorporates the number of VNTR copies at which two MLVA profiles differ, defined as dist . Finally, we introduce a novel GMVE-based distance metric, which handles copy number changes not in a linear fashion as in the categorical distance but in a scaled fashion according to the GMVE model, defined as dist , where f(ni,k, nj,k) = 0 if ni,k = nj,k and f(ni,k, nj,k) = 1 − P × P̂(| ni,k − ni,k| − 1) and P is the rate of single-copy mutational changes as a proportion of all changes (as observed during in vitro passage).
An alternative method, using a combination of maximum parsimony (MP) to infer possible phylogenies and calculation of likelihoods of each possible phylogeny under a GMVE model, was performed as previously described (22) but with a modification to include locus weights. Briefly, maximum-parsimony trees were inferred using the Paup program and the Wagner parsimony measure for discrete characters. The likelihood for each tree was calculated by inferring all the copy number changes that are required to explain the tree (using the chglist command in Paup) and calculating the product of the probabilities of each change, weighted by locus weights, such that , where nx = the copy number in change x and wx = the weight of the locus affected by change x, as defined above.
Scripts implementing these methods are available from the authors on request. eBURST analysis was performed using the eBURST website (http://eburst.mlst.net/) (23).
RESULTS
Estimation of VNTR mutation rates at S. Typhimurium MLVA loci during experimental passage.
In order to determine the rates of copy number change at the five VNTR loci targeted by the European S. Typhimurium scheme (8, 10), we performed a series of 100 parallel serial-passage experiments (PSPE) using a representative clinical isolate of S. Typhimurium phage type 135@, STm5. Each clonal lineage was passaged 10 times (see Materials and Methods), resulting in a total of 1,000 24-h subcultures. We calculated the mean number of generations in a colony during 24 h of passage to be 28.6 generations (95% confidence interval [CI], 28.2 to 29.0), similar to those estimated previously for Salmonella (25 generations) (34), E. coli O157:H7 (28 generations) (21), and Y. pestis (25 generations) (22). Hence, a total of 28,600 (95% CI, 28,200 to 29,000) generations of STm5 growth were observed during 10 rounds of in vitro passage in 100 parallel lineages. MLVA profiling of the 100 cultures obtained following 10 rounds of passage identified a total of 10 copy number changes across three of the five loci analyzed, as summarized in Table 2. For each of the passaged lineages in which a change was observed, MLVA of subcolonies stored from passages 1 to 9 confirmed that each change was the result of a single mutational event.
TABLE 2.
Estimated mutation rates at MLVA loci in Salmonella Typhimuriuma
| Locus | Gene | Gene function | Repeat length(s) (bp) | No. of changes observeda |
Mutation rate |
Diversityb |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In vitro | In vivo | Per generation, in vitro | Per day, in vitro | Per day, in vivo | General (10) | General (20) | General (this study) | Outbreak (20) | Outbreak (this study) | ||||
| STTR9 | Intergenic | Upstream of mannitol dehydrogenase | 9 | 0 | 0 | <3.5E-5 | <1.0E-3 | <2.3E-3 | 5.10E-01 | 3.70E-01 | 3.10E-01 | 8.70E-03 | 0 |
| STTR5 | yohM | Nickel/cobalt efflux protein | 6 | 2 (+1, +1) | 1 (−3) | 7.00E-05 | 2.00E-03 | 2.30E-03 | 8.70E-01 | 9.20E-01 | 9.10E-01 | 4.50E-01 | 6.30E-01 |
| STTR6 | Intergenic | Phage related | 6 | 4 (+1, +1, −1, −2) | 2 (+1, −2) | 1.40E-04 | 4.00E-03 | 4.50E-03 | 9.00E-01 | 9.10E-01 | 9.00E-01 | 7.20E-01 | 4.80E-01 |
| STTR10 | Intergenic | pSLT plasmid | 6 | 4 (+1, +1, +1, −1) | 1 (−2) | 1.40E-04 | 4.00E-03 | 2.30E-03 | 9.20E-01 | 9.10E-01 | 8.40E-01 | 5.50E-01 | 2.10E-01 |
| STTR3 | bigA | Surface-exposed virulence protein | 27/33 | 0 | 0 | <3.5E-5 | <1.0E-3 | <2.3E-3 | 2.50E-01 | 6.80E-01 | 2.80E-01 | 2.80E-02 | 0 |
Total numbers of observed changes are given; sizes and directions of changes are given in parentheses (e.g., “+1” indicates a 1-copy increase).
Diversity measures are Simpson's diversity values; “Outbreak” refers to diversity measures within sets of isolates epidemiologically defined as belonging to the same outbreak; “General” refers to large isolate collections not limited to outbreaks.
We also investigated MLVA mutation rates during in vivo passage of STm5 in a murine infection model (see Materials and Methods). We observed a total of four changes in the VNTR copy number at three loci, with patterns similar to in vitro change patterns (see Table 2). As we were unable to estimate the number of bacterial generations involved during the course of each murine infection, we compared the in vitro and in vivo mutation rates on a temporal scale (per-day mutation rates in Table 2). The per-day mutation rates were similar during in vivo passage and in vitro passage (see Table 2), suggesting that the VNTR loci may be under similar mutational pressures in the in vivo and in vitro environments.
The mutation rates calculated from in vitro passage were inversely correlated with published measures of Simpson's diversity for the five VNTR loci (10, 20). The evidence for correlation was strongest using Simpson's diversity estimates obtained from clonal sets of isolates linked to individual outbreaks, i.e., for estimates of diversity that reflect short-term evolution (R2 = −0.97 and P = 0.005 versus R2 = −0.88 and P = 0.05). This suggests that locus-specific mutation rates per generation could be accurately estimated from the corresponding locus-specific Simpson's diversity (D) measures, using the following formula: rate = 1 × 10−5/(1 − D). Hence, locus-specific relative mutation rates, or weights (w), could be estimated simply as w = 1/(1 − D). We hypothesize that this relationship is likely to apply to other bacteria and thus provides a rational basis for estimating relative locus-specific weights in the absence of experimental data on VNTR mutation rates.
Case study: understanding MLVA profiles during a real outbreak series.
S. Typhimurium 135@ was responsible for a series of outbreaks in Tasmania, Australia, between 2005 and 2008 (27, 28). A total of 184 S. Typhimurium 135@ isolates were collected during these outbreaks and profiled using MLVA. The results of an eBURST analysis of these profiles, plus results of analyses of a further 2 sporadic cases and 17 isolates from outbreaks in the nearby state of Victoria (total n = 203; Table 1), are shown in Fig. 1. The MLVA profiles of Tasmanian outbreak isolates displayed very little variation. The most common profile, 02-11-10-10-14, was isolated from outbreaks in 2005, 2007, and 2008, and each outbreak was dominated by 1 to 2 other MLVA profiles that represented single-locus variants of 02-11-10-10-14. This suggests that the Tasmanian outbreaks were linked but distinct transmission events, potentially stemming from an ancestral strain that possessed the MLVA profile 02-11-10-10-14. In contrast, the MLVA profiles of Victorian isolates were distinct from those of this group, with no indication of a direct transmission link between these and the Tasmanian outbreaks. To determine the underlying phylogenetic relationships among the Tasmanian S. Typhimurium 135@ outbreaks, we previously sequenced the genomes of the STm5 passaged isolate, a further nine Tasmanian outbreak-related isolates, and isolates from two sporadic cases in Tasmania (Table 1) (30). The outbreak isolates were selected for sequencing on the basis of their MLVA profiles and outbreak origins to include the most common MLVA profiles isolated during each outbreak. The two sporadic isolates sequenced were those with the MLVA profiles closest to the outbreak-linked 02-11-10-10-14 profile of the isolates collected in Tasmania during the period of the outbreaks. Epidemiological investigations conducted at the time showed that the outbreak-related isolates, but not the sporadic isolates, could be traced back to a single farm at which the bacterium was also detected (27, 28). The whole-genome sequence (WGS) phylogeny (reproduced in Fig. 2) confirmed that the outbreak isolates belong to a single clonal group that includes two isolates from the farm (the “outbreak clone”) and is genetically distant from the sporadic cases isolated in the same area (>75 SNPs; Fig. 1) (30). The analysis also indicated that in this population of S. Typhimurium, SNPs accumulated in the chromosome at a rate of 3 to 5 per year (30).
According to the WGS phylogeny, the most recent common ancestor (mrca) of the isolates with profile 02-11-10-10-14 is close to the mrca of the outbreak clone (red circle in Fig. 2), consistent with this being the ancestral MLVA profile, as was inferred from MLVA data alone via eBURST analysis. Isolates from the same outbreak were nearly identical at the genome level (0 to 5 SNPs), but each outbreak represented a distinct sublineage of the outbreak clone (separated from other outbreaks by ≥10 SNPs; Fig. 2), again consistent with the observation from MLVA data that each outbreak was associated with a single-locus variant of a common MLVA profile and thus that the outbreaks likely represented distinct chains of transmission from the farm. The only deviations from the ancestral MLVA profile to be identified among members of the outbreak clone involved single-locus changes at either STTR5 or STTR6 (yellow in Fig. 2), two of the most rapidly changing loci in our in vitro and in vivo passage analyses (Table 2). This suggests that single locus changes at the rapidly changing VNTR loci STTR5 and STTR6 are roughly equivalent, as a measure of evolutionary change, to 0 to 10 chromosomal SNPs (i.e., the number of SNPs occurring in 0 to 3 years). The more distantly related sporadic isolates STm4 and STm10 differed further at locus STTR10 (green in Fig. 2), the only other VNTR locus at which mutations were detected during experimental passage. Hence, within our small set of sequenced isolates (n = 12), the “real world” VNTR mutation rates observed were consistent with those observed during in vitro and in vivo passage (with some indication that STTR10 may actually mutate slower than STTR5 or STTR6 in natural populations). The data suggest that changes at 2 to 3 of these VNTR loci (STTR5, STTR6, and STTR10) are indicative of a substantially longer evolutionary time than a single locus change would be and that the changes are equivalent to 50 to 100 chromosomal SNPs (i.e., the number of SNPS occurring in 10 to 30 years).
Locus-specific mutation rates and the GMVE model for tree construction from MLVA data.
We compared the abilities of several alternative analysis methods to recover the underlying phylogenetic relationships depicted in Fig. 2 directly from MLVA profiles of the full set of 203 S. Typhimurium 135@ isolates from Tasmania and Victoria (Table 1). The eBURST analysis of the data has already been shown (Fig. 1), and the additional methods considered were as follows: (i) maximum-parsimony reconstruction coupled with GMVE likelihood estimation, as proposed in references 22, 24, and 35, and (ii) unweighted-pair group method using average linkages (UPGMA) clustering using (a) a novel distance metric based on the GMVE model, (b) simple clustering, and (c) categorical clustering. Each method was considered using both unweighted analysis and locus-specific weights based on the locus-specific mutation rates estimated from the passage experiments (Table 2) (see Materials and Methods). The resulting tree topologies were assessed with respect to their ability to (i) differentiate the Tasmanian outbreak clone from other isolates (including sporadic Tasmanian cases and Victorian outbreaks) and (ii) differentiate the Tasmanian outbreak clone isolates from different outbreaks into separate clusters.
Maximum-parsimony (MP) analysis of the MLVA profiles identified 156 MP tree topologies, of which 18 alternative tree topologies shared maximum likelihood under the GMVE model. The majority-rules consensus of these 18 trees did not differentiate the outbreak clone from the Victorian isolates; however, incorporating locus-specific mutation rates into the GMVE likelihood calculations produced better results, resulting in six maximum-likelihood topologies that separated the outbreak clone from the Victorian outbreaks (Fig. 3A).
FIG 3.

Tree building with MLVA data. Analysis of 203 STm135@ isolates associated with Tasmanian and Victorian outbreaks. Pie graphs indicate MLVA profiles; circle size indicates the number of isolates with that profile, and colored segments indicate proportions of isolates from different outbreaks (colored according to the inset legend). Isolates belonging to the outbreak clone are highlighted with a gray background. GMVE, geometric model of VNTR evolution; MP, maximum-parsimony inference. Otherwise, trees are the result of UPGMA clustering of the specified distance metric.
Clustering using GMVE distances and locus-specific weights correctly separated the outbreak clone from other isolates and also clustered profiles from the same outbreaks (Fig. 3B). This method performed better than unweighted simple or categorical clustering (Fig. 3C and D), which broadly grouped the outbreak clone members but did not cluster profiles from the same outbreak. The inclusion of locus-specific weights into simple or categorical clustering did not improve the performance of these metrics. Hence, the data suggest that improvements in the construction of trees from MLVA profiles can be most easily obtained using the GMVE distance coupled with locus-specific weights.
Identifying clusters from MLVA profiles.
Some public health and surveillance laboratories use regular reviews of S. Typhimurium MLVA profiles to detect potential transmission clusters that may warrant further investigation. Cluster definitions in use include (i) identical profiles, (ii) profiles with ≤1 or a specified number of locus differences, and (iii) more-complex rules about which VNTR loci may differ and the scale of differences. The WGS tree (Fig. 2) shows that a cluster definition based on identical profiles would fail to cluster isolates that are otherwise genetically identical and clearly part of the same transmission chain. For example, STm3 and STm5, food and case isolates from the second 2005 outbreak that differ by a single SNP and were collected 6 days apart, differed by one repeat copy at locus STTR6. A more relaxed approach to cluster identification which allows copy number change at one of the three most rapidly changing loci (STTR5, STTR6, and STTR10; see Table 2) would identify clusters consistent with the WGS data. In contrast, in our WGS data set, copy number changes at >1 of the rapidly mutating loci were associated with the much more distant phylogenetic relationships (>75 SNPs, ∼15 years of independent evolution).
To further explore the effects of different cluster identification rules, we compared retrospective cluster definitions based on MLVA data for the 203 135@ isolates from Australian outbreaks to outbreak definitions based on epidemiological investigations conducted at the time (Table 1). Using a cluster definition based on identical profiles, only 68% of isolates would have been correctly included in their epidemiologically determined outbreak clusters. Allowing one (or two) differences at one of the three rapidly mutating loci would result in 91% (or 98%) of outbreak isolates being correctly assigned to epidemiologically determined clusters.
DISCUSSION
The stability of the MLVA loci through 1,000 rounds of in vitro passage suggests that the likelihood of mutations being introduced during regular laboratory manipulation (usually only 1 to 2 subcultures) is very low. That is, MLVA assays as currently performed can be expected to give an accurate indication of the true VNTR status of the bacteria at the point of isolation, with very low risk of observed differences being the result of mutations arising in the laboratory. Furthermore, the similarity in relative rates of change observed between loci during in vitro passage and in vivo passage, as well as real-world variation during outbreaks, is consistent with the hypothesis that VNTR loci evolve at similar rates under a variety of growth conditions.
These results are also consistent with recent studies of VNTR stability that examined a range of S. Typhimurium strains, albeit through fewer passages. In S. Typhimurium DT41, occasional changes were identified during passage experiments performed in vivo (in chickens) and in vitro (8 passage chains with daily subculture on blood agar for 100 to 200 days) (25). The changes occurred mostly in STTR6, followed by STTR10 and STTR5. A second study performed 50 12-h passages in LB broth, using 31 S. Typhimurium isolates with different MLVA profiles and a range of phage types (DT104, DT193, DT120, DT195, U302, and DT12) (26). Changes were observed in STTR6 (n = 6 changes), STTR10 (n = 4), and STTR5 (n = 2), and a single change was also detected in STTR9. It was not possible to calculate rates per generation in the reported studies, which did not directly track clonal lineages. However, there is clear agreement across all three studies regarding the order of instability among the S. Typhimurium MLVA loci, namely, STTR6 > STTR10 > STTR5 ≫ STTR9 and STTR3. This suggests that our results and the mutation rates are likely to be broadly applicable to different S. Typhimurium phage types and strains. However, neither the current study nor the others reported to date were sufficiently powered to detect and quantify subtle differences in per-unit-of-time mutation rates under different growth conditions, and a larger study incorporating much longer total passage times (and ideally incorporating a wider range of in vitro and in vivo growth conditions) would be required to prove the absolute equivalence of mutation rates under different conditions or in different genetic backgrounds.
The present report reveals clear trends that have immediate utility for deciding how best to handle MLVA data analysis of S. Typhimurium and bacterial pathogens more generally. Using WGS data and detailed epidemiological trace-back methods to determine the true underlying genetic relationships behind a series of outbreaks, our analysis shows that accurate conclusions about this case study could be drawn from MLVA data alone. Similarly, a recent study comparing MLVA and WGS for analysis of Clostridium difficile transmission showed that MLVA was as informative as WGS, which is considered the gold standard for transmission tracking (36).
With respect to cluster detection for identifying outbreaks and ruling out unrelated strains, our data indicate that the best cluster definition is one that includes isolates with variation in none or one of the rapidly changing loci STTR5, STTR6, and STTR10 but that excludes strains with more than one difference or with any difference in locus STTR3 or STTR9. This definition is supported by the only other available study of VNTR stability analysis in S. Typhimurium, which also did not observe changes at STTR3 or STTR9 during environmental or in vivo passage (25), and by Simpson's diversity measures for the five VNTR loci calculated among the members of our outbreak set and an independent collection of S. Typhimurium outbreak isolates, as summarized in Table 2 (20).
Finally, our data indicate that, for the purpose of tree construction based on MLVA profiles, a weighted GMVE model is superior to the simple or categorical distances that have been most frequently used in published studies of MLVA data. This is consistent with the expectation that a distance based on the GMVE that attempts to account for the mutational processes which generate variation in MLVA profiles will lead to more-accurate inferences of genetic relationships between isolates. Standard eBURST analysis, which has rarely been used in published studies reporting MLVA data, provided an accurate, if incomplete, representation of the relationships between profiles and does not require prior knowledge of any mutational parameters.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an Early Career Researcher grant from the University of Melbourne, a grant from the government of Victoria, Australia, and a Victorian Life Sciences Computation Initiative (VLSCI) (no. VR0082). K.E.H. and O.L.C.W. were supported by fellowships from the National Health and Medical Research Council (NHMRC) of Australia (no. 628930 and no. 454774) and H.C. by an NHMRC program grant (no. 606788). The Microbiological Diagnostic Unit (MDU) Public Health Laboratory is funded by the Department of Health Victoria.
We thank the MDU staff members and the staff members of all laboratories who sent isolates for typing.
Footnotes
Published ahead of print 23 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01820-14.
REFERENCES
- 1.van Belkum A. 2007. Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA). FEMS Immunol. Med. Microbiol. 49:22–27. 10.1111/j.1574-695X.2006.00173.x [DOI] [PubMed] [Google Scholar]
- 2.Paranthaman K, Haroon S, Latif S, Vinnyey N, de Souza V, Welfare W, Tahir M, Cooke E, Stone K, Lane C, Peters T, Puleston R. 2013. Emergence of a multidrug-resistant (ASSuTTm) strain of Salmonella enterica serovar Typhimurium DT120 in England in 2011 and the use of multiple-locus variable-number tandem-repeat analysis in supporting outbreak investigations. Foodborne Pathog. Dis. 10:850–855. 10.1089/fpd.2013.1513 [DOI] [PubMed] [Google Scholar]
- 3.Lindstedt BA, Torpdahl M, Vergnaud G, Le Hello S, Weill FX, Tietze E, Malorny B, Prendergast DM, Ni Ghallchoir E, Lista RF, Schouls LM, Soderlund R, Borjesson S, Akerstrom S. 2013. Use of multilocus variable-number tandem repeat analysis (MLVA) in eight European countries, 2012. Euro Surveill. 18:20385 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20385 [DOI] [PubMed] [Google Scholar]
- 4.Wattiau P, Boland C, Bertrand S. 2011. Methodologies for Salmonella enterica subsp. enterica subtyping: gold standards and alternatives. Appl. Environ. Microbiol. 77:7877–7885. 10.1128/AEM.05527-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foley SL, Lynne AM, Nayak R. 2009. Molecular typing methodologies for microbial source tracking and epidemiological investigations of Gram-negative bacterial foodborne pathogens. Infect. Genet. Evol. 9:430–440. 10.1016/j.meegid.2009.03.004 [DOI] [PubMed] [Google Scholar]
- 6.Grissa I, Bouchon P, Pourcel C, Vergnaud G. 2008. On-line resources for bacterial micro-evolution studies using MLVA or CRISPR typing. Biochimie 90:660–668. 10.1016/j.biochi.2007.07.014 [DOI] [PubMed] [Google Scholar]
- 7.Larsson JT, Torpdahl M, MLVA working group. Møller Nielsen E. 2013. Proof-of-concept study for successful inter-laboratory comparison of MLVA results. Euro Surveill. 18:20566 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20566 [DOI] [PubMed] [Google Scholar]
- 8.Larsson JT, Torpdahl M, Petersen RF, Sorensen G, Lindstedt BA, Nielsen EM. 2009. Development of a new nomenclature for Salmonella typhimurium multilocus variable number of tandem repeats analysis (MLVA). Euro Surveill. 14:19174 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19174 [PubMed] [Google Scholar]
- 9.Lindstedt BA, Heir E, Gjernes E, Kapperud G. 2003. DNA fingerprinting of Salmonella enterica subsp. enterica serovar Typhimurium with emphasis on phage type DT104 based on variable number of tandem repeat loci. J. Clin. Microbiol. 41:1469–1479. 10.1128/JCM.41.4.1469-1479.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindstedt BA, Vardund T, Aas L, Kapperud G. 2004. Multiple-locus variable-number tandem-repeats analysis of Salmonella enterica subsp. enterica serovar Typhimurium using PCR multiplexing and multicolor capillary electrophoresis. J. Microbiol. Methods 59:163–172. 10.1016/j.mimet.2004.06.014 [DOI] [PubMed] [Google Scholar]
- 11.Sintchenko V, Wang Q, Howard P, Ha CW, Kardamanidis K, Musto J, Gilbert GL. 2012. Improving resolution of public health surveillance for human Salmonella enterica serovar Typhimurium infection: 3 years of prospective multiple-locus variable-number tandem-repeat analysis (MLVA). BMC Infect. Dis. 12:78. 10.1186/1471-2334-12-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dewaele I, Rasschaert G, Bertrand S, Wildemauwe C, Wattiau P, Imberechts H, Herman L, Ducatelle R, De Reu K, Heyndrickx M. 2012. Molecular characterization of Salmonella Enteritidis: comparison of an optimized multi-locus variable-number of tandem repeat analysis (MLVA) and pulsed-field gel electrophoresis. Foodborne Pathog. Dis. 9:885–895. 10.1089/fpd.2012.1199 [DOI] [PubMed] [Google Scholar]
- 13.Hopkins KL, Peters TM, de Pinna E, Wain J. 2011. Standardisation of multilocus variable-number tandem-repeat analysis (MLVA) for subtyping of Salmonella enterica serovar Enteritidis. Euro Surveill. 16:19942 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19942 [PubMed] [Google Scholar]
- 14.Kang MS, Kwon YK, Oh JY, Call DR, An BK, Song EA, Kim JY, Shin EG, Kim MJ, Kwon JH, Chung GS. 2011. Multilocus variable-number tandem-repeat analysis for subtyping Salmonella enterica serovar Gallinarum. Avian Pathol. 40:559–564. 10.1080/03079457.2011.613915 [DOI] [PubMed] [Google Scholar]
- 15.Young CC, Ross IL, Heuzenroeder MW. 2012. A new methodology for differentiation and typing of closely related Salmonella enterica serovar Heidelberg isolates. Curr. Microbiol. 65:481–487. 10.1007/s00284-012-0179-3 [DOI] [PubMed] [Google Scholar]
- 16.Octavia S, Lan R. 2009. Multiple-locus variable-number tandem-repeat analysis of Salmonella enterica serovar Typhi. J. Clin. Microbiol. 47:2369–2376. 10.1128/JCM.00223-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tien YY, Ushijima H, Mizuguchi M, Liang SY, Chiou CS. 2012. Use of multilocus variable-number tandem repeat analysis in molecular subtyping of Salmonella enterica serovar Typhi isolates. J. Med. Microbiol. 61:223–232. 10.1099/jmm.0.037291-0 [DOI] [PubMed] [Google Scholar]
- 18.Kjeldsen MK, Torpdahl M, Campos J, Pedersen K, Nielsen EM. 2014. Multiple-locus variable-number tandem repeat analysis of Salmonella enterica subsp. enterica serovar Dublin. J. Appl. Microbiol. 116:1044–1054. 10.1111/jam.12441 [DOI] [PubMed] [Google Scholar]
- 19.Torpdahl M, Sorensen G, Lindstedt BA, Nielsen EM. 2007. Tandem repeat analysis for surveillance of human Salmonella Typhimurium infections. Emerg. Infect. Dis. 13:388–395. 10.3201/eid1303.060460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiou CS, Hung CS, Torpdahl M, Watanabe H, Tung SK, Terajima J, Liang SY, Wang YW. 2010. Development and evaluation of multilocus variable number tandem repeat analysis for fine typing and phylogenetic analysis of Salmonella enterica serovar Typhimurium. Int. J. Food Microbiol. 142:67–73. 10.1016/j.ijfoodmicro.2010.06.001 [DOI] [PubMed] [Google Scholar]
- 21.Vogler AJ, Keys C, Nemoto Y, Colman RE, Jay Z, Keim P. 2006. Effect of repeat copy number on variable-number tandem repeat mutations in Escherichia coli O157:H7. J. Bacteriol. 188:4253–4263. 10.1128/JB.00001-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogler AJ, Keys CE, Allender C, Bailey I, Girard J, Pearson T, Smith KL, Wagner DM, Keim P. 2007. Mutations, mutation rates, and evolution at the hypervariable VNTR loci of Yersinia pestis. Mutat. Res. 616:145–158. 10.1016/j.mrfmmm.2006.11.007 [DOI] [PubMed] [Google Scholar]
- 23.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186:1518–1530. 10.1128/JB.186.5.1518-1530.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price EP, Hornstra HM, Limmathurotsakul D, Max TL, Sarovich DS, Vogler AJ, Dale JL, Ginther JL, Leadem B, Colman RE, Foster JT, Tuanyok A, Wagner DM, Peacock SJ, Pearson T, Keim P. 2010. Within-host evolution of Burkholderia pseudomallei in four cases of acute melioidosis. PLoS Pathog. 6:e1000725. 10.1371/journal.ppat.1000725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barua H, Lindblom IL, Bisgaard M, Christensen JP, Olsen RH, Christensen H. 2013. In vitro and in vivo investigation on genomic stability of Salmonella enterica Typhimurium DT41 obtained from broiler breeders in Denmark. Vet. Microbiol. 166:607–616. 10.1016/j.vetmic.2013.06.035 [DOI] [PubMed] [Google Scholar]
- 26.Wuyts V, Mattheus W, De Laminne de Bex G, Wildemauwe C, Roosens NH, Marchal K, De Keersmaecker SC, Bertrand S. 2013. MLVA as a tool for public health surveillance of human Salmonella Typhimurium: prospective study in Belgium and evaluation of MLVA loci stability. PLoS One 8:e84055. 10.1371/journal.pone.0084055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephens N, Coleman D, Shaw K. 2008. Recurring outbreaks of Salmonella typhimurium phage type 135 associated with the consumption of products containing raw egg in Tasmania. Commun. Dis. Intell. Q. Rep. 32:466–468 [PubMed] [Google Scholar]
- 28.Stephens N, Sault C, Firestone SM, Lightfoot D, Bell C. 2007. Large outbreaks of Salmonella Typhimurium phage type 135 infections associated with the consumption of products containing raw egg in Tasmania. Commun. Dis. Intell. Q. Rep. 31:118–124 [PubMed] [Google Scholar]
- 29.OzFoodNet Working Group. 2009. Monitoring the incidence and causes of diseases potentially transmitted by food in Australia: annual report of the OzFoodNet Network, 2008. Commun. Dis. Intell. Q. Rep. 33:389–413 [PubMed] [Google Scholar]
- 30.Hawkey J, Edwards DJ, Dimovski K, Hiley L, Billman-Jacobe H, Hogg G, Holt KE. 2013. Evidence of microevolution of Salmonella Typhimurium during a series of egg-associated outbreaks linked to a single chicken farm. BMC Genomics 14:800. 10.1186/1471-2164-14-800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Girard JM, Wagner DM, Vogler AJ, Keys C, Allender CJ, Drickamer LC, Keim P. 2004. Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local, regional, and global scales. Proc. Natl. Acad. Sci. U. S. A. 101:8408–8413. 10.1073/pnas.0401561101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lovelace MD, Yap ML, Yip J, Muller W, Wijburg O, Jackson DE. 2013. Absence of platelet endothelial cell adhesion molecule 1, PECAM-1/CD31, in vivo increases resistance to Salmonella enterica serovar Typhimurium in mice. Infect. Immun. 81:1952–1963. 10.1128/IAI.01295-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornick K. 5 April 2014. R FAQ. http://cran.r-project.org/doc/FAQ/R-FAQ.html
- 34.Lind PA, Andersson DI. 2008. Whole-genome mutational biases in bacteria. Proc. Natl. Acad. Sci. U. S. A. 105:17878–17883. 10.1073/pnas.0804445105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colman RE, Vogler AJ, Lowell JL, Gage KL, Morway C, Reynolds PJ, Ettestad P, Keim P, Kosoy MY, Wagner DM. 2009. Fine-scale identification of the most likely source of a human plague infection. Emerg. Infect. Dis. 15:1623–1625. 10.3201/eid1510.090188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eyre DW, Fawley WN, Best EL, Griffiths D, Stoesser NE, Crook DW, Peto T, Walker AS, Wilcox MH. 2013. Comparison of multilocus variable-number tandem-repeat analysis and whole-genome sequencing for investigation of Clostridium difficile transmission. J. Clin. Microbiol. 51:4141–4149. 10.1128/JCM.01095-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.