

Abstract
Escherichia coli displays O antigens on the outer membrane that play an important role in bacterial interactions with the environment. The O antigens of enterohemorrhagic E. coli O104 and O5 contain a Galβ1-3GalNAc disaccharide at the reducing end of the repeating unit. Several other O antigens contain this disaccharide, which is identical to the mammalian O-glycan core 1 or the cancer-associated Thomsen-Friedenreich (TF) antigen. We identified the wbwC genes responsible for the synthesis of the disaccharide in E. coli serotypes O104 and O5. To functionally characterize WbwC, an acceptor substrate analog, GalNAcα-diphosphate-phenylundecyl, was synthesized. WbwC reaction products were isolated by high-pressure liquid chromatography and analyzed by mass spectrometry, nuclear magnetic resonance, galactosidase and O-glycanase digestion, and anti-TF antibody. The results clearly showed that the Galβ1-3GalNAcα linkage was synthesized, confirming WbwCECO104 and WbwCECO5 as UDP-Gal:GalNAcα-diphosphate-lipid β1,3-Gal-transferases. Sequence analysis revealed a conserved DxDD motif, and mutagenesis showed the importance of these Asp residues in catalysis. The purified enzymes require divalent cations (Mn2+) for activity and are specific for UDP-Gal and GalNAc-diphosphate lipid substrates. WbwC was inhibited by bis-imidazolium salts having aliphatic chains of 18 to 22 carbons. This work will help to elucidate mechanisms of polysaccharide synthesis in pathogenic bacteria and provide technology for vaccine synthesis.
INTRODUCTION
The O antigens of lipopolysaccharides (LPS) in Gram-negative bacteria consist of many repeats of a specific oligosaccharide unit and are a major contributor to the antigenic variability of the bacterial cell surface, conferring defense to the bacteria against host killing mechanisms (1–5).
The O antigen gene clusters contain the corresponding glycosyltransferase (GT) genes, most of which have not yet been biochemically characterized. These GTs are likely to be responsible for completing the synthesis of the O antigen repeating unit by adding sugars to the nonreducing end of the oligosaccharide (6, 7). The repeating units are thought to be preassembled on the cytosolic face of the inner membrane, where the catalytic domains of GTs are directed into the cytoplasm with access to the nucleotide sugar donor substrate pools, as well as to the membrane-bound undecaprenol-phosphate (P-Und)-linked acceptor substrates (8, 9).
E. coli strains O104 and O5 produce Shiga toxins (10), which are known to cause severe bloody diarrhea in humans, sometimes leading to hemolytic uremic syndrome and death (11, 12). Shiga toxin-producing E. coli O104 (STEC) has acquired a second gene encoding a Shiga toxin variant (13). Several outbreaks of O104 infections have been reported worldwide (12); for example, an outbreak occurred in 2011 in Germany, in which thousands were infected, 53 people died, and many still suffer from the consequences of infections. In light of growing worldwide antibiotic resistance, alternative therapies such as immune therapies become more important. E. coli O104 synthesizes an O antigen with a tetrasaccharide repeating-unit structure of (-4-d-Galα1-4Neu5,7,9Ac3α2-3-d-Galβ1-3-d-GalNAcβ1-)n (14), which could be the basis for a vaccine. The O5 antigen has the repeating-unit structure (4-d-Quip3NAcβ1-3-d-Ribfβ1-4-d-Galpβ1-3-d-GalNAcα1-)n (15). Thus, the two O antigens share the Galβ1-3GalNAcα disaccharide sequence, which is identical to O-glycan core 1 of mammalian glycoproteins and the cancer-associated Thomsen-Friedenreich (TF) antigen (16, 17). The genes of the O104 antigen gene cluster include three putative glycosyltransferase genes, wbwA, wbwB, and wbwC, in addition to CMP-NeuNAc synthesis genes, the O antigen polymerase wzy gene, and a flippase wzx gene (18). This suggests that O104 antigen synthesis follows the Wzy-dependent pathway. Since the wbwC gene from E. coli O104 has 44% identity to the orf11 gene from E. coli O5, we propose that these genes encode orthologous enzymes that synthesize the Galβ1-3GalNAcα linkage.
The assembly of the O104 and O5 antigen repeating units is thought to be initiated by the transfer of GalNAcα-phosphate to undecaprenol-phosphate (P-Und) by the membrane-bound enzyme GlcNAc/GalNAc-phosphotransferase WecA (see Fig. S1 in the supplemental material). Alternatively, GlcNAcα-phosphate may be transferred, followed by 4-epimerization of GlcNAc (19) to form the acceptor substrate for WbwC, GalNAcα-PP-Und. Membrane-associated GTs then transfer sugar residues from nucleotide sugars to the PP-Und-linked acceptor substrates to complete the synthesis of the repeating unit. The lipid-linked saccharide is flipped across the inner membrane, and repeating units are polymerized in the periplasmic space. The completed O antigen is then ligated to the lipid A-core moiety to synthesize LPS, which is transported to the outer membrane by the Lpt complex (20, 21).
Most of the bacterial GTs are still putative and require biochemical proof of activity. We have characterized several Gal and Glc transferases that catalyze the second step in O antigen repeating-unit synthesis (22–25). The synthesis of a natural substrate analog, GlcNAcα-diphosphate phenylundecyl [GlcNAcα-O-PO3-PO3-(CH2)11-O-Ph, GlcNAc-PP-PhU], numbered 14 in Fig. 1, was instrumental in these studies (26). We recently reported that the enzyme encoded by orf11 from E. coli O5 was a Gal-transferase that utilized GalNAcα-diphosphate phenylundecyl (GalNAc-PP-PhU [compound 8 in Fig. 1]) as the acceptor, as well as the fluorescent acceptor GalNAcα-diphosphate (anthracen-9-ylmethoxy)undecyl, allowing both radioactive and fluorescence-based activity assays (27). However, the enzyme had not been fully characterized. In this work, we report the thorough characterization of two WbwC Gal-transferases, the second enzymes in the assembly of repeating units from E. coli O104 and O5. We improved the syntheses of GalNAc-PP-PhU and GlcNAc-PP-PhU to achieve higher yields and showed that WbwC enzymes synthesize the mammalian core 1/TF antigen and can be inhibited by imidazolium salts of specific structures. The enzymes are targets for the development of antibacterial compounds and are useful for the chemoenzymatic synthesis of glycan-based vaccines that may help to prevent tumor growth.
FIG 1.

Synthesis of acceptor substrates 8 and 14. Reagents and conditions were as follows: PhOH, K2CO3, KI, acetone, reflux temperature (a); POCl3, Et3N, hexanes, 0°C → rt (b); Ac2O, pyridine, rt (c); CF3SO3Si(CH3)3, 1,2-dichloroethane, 50 to 60°C (d); dibenzyl phosphate, 1,2-dichloroethane, rt (e); H2, Pd/C, MeOH, rt (f); compound 2, diisopropylamine, 1,1′-carbonyldiimidazole, THF, rt (g); NaOMe, MeOH, rt, Amberlite IRA-120 resin (Na+ form) (h).
MATERIALS AND METHODS
Materials.
Reagents and materials were obtained from Sigma-Aldrich unless otherwise stated. GalNAc and GlcNAc derivatives were synthesized as described below, in the supplemental material, and in previous publications (26, 28–30). The synthesis of the undecyl (U)-containing fluorescent substrate P1-[11-(anthracen-9-ylmethoxy)undecyl]-P2-(2-acetamido-2-deoxy-α-d-galactopyranosyl) diphosphate (GalNAc-PP-AnthrU) was reported previously (27, 30). Glycopeptides and 4-deoxy-GalNAcα-Bn were synthesized by Hans Paulsen, University Hamburg, Hamburg, Germany. GalNAcβ1-4GlcNAcβ-Bn was synthesized by Khushi Matta at the Roswell Park Cancer Institute, Buffalo, NY. Inhibitors were synthesized as described previously (28, 30, 31).
Chemical synthesis.
The syntheses of acceptor substrates 8 and 14 are shown in Fig. 1. The ether-alcohol (compound 1) was synthesized from 11-bromoundecanol by treatment with phenol-potassium carbonate-potassium iodide in acetone, following a similar procedure by Visscher et al. (32). The phosphate (compound 2) was synthesized by the treatment of alcohol 1 with phosphorus oxychloride in triethylamine-hexanes, by a modification of the procedure by Bernardes et al. (33) for the formation of citronellylphosphate. The acetylated compound 3 was synthesized by the treatment of d-galactosamine hydrochloride with acetic anhydride in pyridine. Treatment of compound 3 with trimethylsilyl triflate in 1,2-dichloroethane gave the oxazoline (compound 4), following a procedure similar to that published by Nakabayashi et al. (34). The dibenzyl phosphate (compound 5) was prepared by the treatment of oxazoline (compound 4) with dibenzyl phosphate in 1,2-dichloroethane, following essentially the procedure of Bernardes et al. (33). Hydrogenolysis of compound 5 in methanol (MeOH) using Pd/C as a catalyst gave the debenzylated sugar-phosphate (compound 6), following essentially the procedure of Montoya-Peleaz et al. (26). The coupling of the in situ-generated diisopropylammonium salt of phosphate 2 and sugar-phosphate 6 using 1,1′-carbonyldiimidazole in tetrahydrofuran to form compound 7, followed by deprotection of compound 7 using sodium methoxide in MeOH, gave the final acceptor substrate 8; the procedure followed was essentially that of Montoya et al. (26), but using the d-galactosamine derivative instead of the d-glucosamine derivative. The purification procedure was altered, and the enzyme-substrate reaction product was isolated and characterized in the disodium salt form. A similar synthetic strategy starting from d-glucosamine hydrochloride was followed to produce the GlcNAc-containing acceptor substrate 14, also isolated and characterized in the disodium salt form. All procedures and purification methods were modified appropriately from the existing referenced procedures. Further characterizing data, and nuclear magnetic resonance (NMR) spectroscopic data obtained in alternative solvent systems, have been obtained. These data and detailed synthetic procedures are documented in the supplemental material.
Bacterial growth, plasmids, and protein expression.
The putative glycosyltransferase genes wbwC from E. coli O104 (strain G1629) and orf11 from E. coli O5 (strain G1675) (see Fig. S2 in the supplemental material) were amplified by PCR, using primer pairs wl-49644/wl-49645 (5-CGGGATCCATGAAATTTAGCGTTCTGTT-3/5-CCGCTCGAGTTATTTCCGCAATATATT-3) and wl-11103/wl-11104 (5-CGGGATCCATGAATGATAGTTCAAGATCAT-3/5-CCGCTCGAGTTAATTCTTAGTCCTTGAAATAC-3), respectively. A total of 30 cycles were performed under the following conditions: denaturation at 95°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 1 min, in a final volume of 50 μl. The amplified genes were then cloned into expression plasmid vector pET28a (KanR), containing a cleavable His tag-encoded sequence at the C terminus, and the presence of the inserts was confirmed by sequencing using an ABI 3730 sequencer. Plasmid constructs were transformed into E. coli BL21 for protein expression (26). For the induction of plasmid-derived enzyme, bacteria were grown overnight at 37°C in 5 ml of Luria broth containing 50 μg/ml of kanamycin with constant shaking at 150 rpm. The bacterial suspension (5 ml) was transferred to 125 ml of Luria broth containing kanamycin, and the mixture was incubated at 37°C. Isopropyl 1-thio-β-d-galactopyranoside (IPTG) was added to a final concentration of 1 mM when the suspension reached an optical density at 600 nm of 0.8. Cells were grown for an additional 4 h at 37°C and were then harvested by centrifugation for 15 min at 2,200 × g. Pellets were washed in phosphate-buffered saline (PBS). A total of 10 ml of PBS containing 10% glycerol was added, and aliquots of bacteria were stored at −20°C for enzyme assays.
Enzyme purification.
WbwCECO104 and WbwCECO5 proteins contained a His6 tag at the C terminus. After IPTG induction, bacteria were harvested by centrifugation, washed with PBS (pH 7.4), resuspended into the same buffer, and sonicated in four cycles for 30 s with 2-min intervals on ice (30). The homogenate was then centrifuged at 8,000 × g in a Centronics M-1200 centrifuge (Boehringer-Mannheim) for 30 min. The WbwC fusion proteins in the supernatant were purified using a HisPur Ni2+-nitrilotriacetic acid (-NTA) Sepharose column (Thermo). Bound proteins were eluted with a gradient of 200 to 400 mM imidazole buffer in PBS. Each fraction was analyzed by sodium dodecyl-polyacrylamide gel electrophoresis (SDS-PAGE) (12% gel). The desired fractions that contained fusion protein were pooled and stored at −80°C. Western blot analysis was performed with rabbit antibody against the His6 tag as the primary antibody (Thermo) and horseradish peroxidase-conjugated goat anti-rabbit IgG as the secondary antibody (Santa Cruz Biotechnology). Purified enzymes and cell lysates of the wild type or mutants (0.5 to 0.7 μg protein/μl) were diluted 3:1 in 4× SDS loading buffer consisting of 40 mM Tris-HCl (pH 8.0), 4% SDS, 8% glycerol, 4% β-mercaptoethanol, and bromophenol blue and run on SDS-PAGE gels (12% gel). Proteins were electrophoretically transferred onto nitrocellulose and then probed sequentially with rabbit anti-His tag antibody (1:1,000) and anti-rabbit IgG (1:10,000). Alternatively, mouse anti-His antibody and goat anti-mouse antibody (1:5,000) (Cell Biolabs Inc.) were used. Labeling was visualized with WEST-one spray (iNtRON) or Western lighting ECL Pro (Perkin-Elmer) and exposure of fluorescence on film. Prestained protein standards (GeneDirex) were used to calibrate the gels. The relative protein amounts were determined by densitometric analysis of the Western blot film, performed by using ImageJ1.43 software. The enzyme activities of purified protein were normalized according to the relative amounts of protein detected on Western blots. Protein contents were determined by measuring absorbance at 280 nm in a microvolume spectrophotometer (Nanodrop 2000; Thermo). Purified enzyme (see Fig. S3 and S4 in the supplemental material) was used for enzyme characterization.
Galactosyltransferase assays.
For Gal-transferase assays, bacterial homogenates were prepared by sonication as described before (23), or purified enzyme was used as indicated. The standard assay mixtures contained, in a total of 40 μl, 0.1 mM acceptor substrate GalNAc-PP-PhU (compound 8 in Fig. 1), 5 mM MnCl2, 2.5 mM dithiothreitol (DTT), 0.125 M 2-(N-morpholino) ethanesulfonic acid (MES) buffer, pH 7, 10 μl purified enzyme homogenate (5 to 6 μg protein), and 0.44 mM UDP-[3H]Gal (2,000 to 3,500 cpm/nmol). Control assays lacked the acceptor. All assays were carried out in at least duplicate determinations, and results were confirmed by repeating the experiments at least once. Mixtures were incubated for 10 min at 37°C, and reactions were quenched by the addition of 700 μl of ice-cold water and freezing. Enzyme reaction product was isolated using Sep-Pak C18 columns, eluted first in water and then in MeOH, and quantified by scintillation counting as described previously (22, 25). High-pressure liquid chromatography (HPLC) separations were carried out as described previously (24), using a C18 column and acetonitrile-water as the mobile phase. Peaks were monitored by measuring absorbance at 195 nm and scintillation counting of fractions. Kinetic parameters were determined using the program GraphPad Prism. In assays using potential hydrophobic inhibitors, mixtures contained 10% MeOH or ethyl alcohol (EtOH) in the assay.
Enzyme reaction product identification by HPLC and MS.
The enzyme reaction products were prepared in large-scale radioactive and nonradioactive forms. The separation of substrates and enzyme reaction products was achieved by HPLC using a C18 column at a flow rate of 1 ml/min and acetonitrile-water mixtures (27/73) as the mobile phase, monitored by absorbance at 195 nm. Product peaks were dried and analyzed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry (MS) or electrospray ionization (ESI)-MS in the negative-ion mode, as described previously (30).
Identification of enzyme reaction product structure by NMR.
To prepare large amounts of nonradioactive enzyme reaction product for nuclear magnetic resonance (NMR), WbwC assays were carried out as follows. The assay mixture (20 ml) contained 4 μmol GalNAc-PP-PhU (compound 8), 30 μmol UDP-Gal, 5 ml bacterial cell homogenate in 50 mM sucrose, 0.1 mmol MnCl2, and 2.5 mmol MES buffer, pH 7.0. After incubation for 60 min at 37°C, 10 ml of cold water was added to stop the reaction. The reaction mixture was then applied in aliquots to 50 C18 Sep-Pak columns, and the reaction products were eluted with MeOH. Following flash evaporation and lyophilization of the eluates, the reaction product was resuspended in water and further purified by reversed-phase HPLC (24) using standard compounds. Pooled fractions containing product were flash evaporated and lyophilized. Dried product was dissolved in D2O and analyzed by 600-MHz NMR. Spectra were collected in one-dimensional (1D) and 2D experiments using a Bruker spectrometer as described previously (22).
Linkage confirmation using galactosidases or O-glycanase.
The anomeric configuration of the linkage formed in the radioactive WbwC reaction product was confirmed by incubation with specific galactosidases: green coffee bean α-galactosidase (100 U/ml), E. coli (β1-4-specific) β-galactosidase (100 U/ml), bovine testicular (β1-3-, -4-, and -6-specific) β-galactosidase (100 U/ml), or O-glycanase (Glyko, 1.25 U/ml). Aliquots of radioactive reaction product (2,000 cpm) were treated in a total volume of 100 μl with 25 μl MacIlvaine buffer (0.1 M citric acid–0.2 M Na-phosphate, pH 4.3), 10 μl 0.1% bovine serum albumin, and galactosidase or O-glycanase. The activities of galactosidases were confirmed with Galα- and Galβ-p-nitrophenyl as the substrates. Control incubations lacked the enzymes. Mixtures were incubated for 30 min at 37°C, diluted with 800 μl of water, and applied to columns of 0.4 ml AG1x8 (Cl− form). Released radioactivity was eluted with 2.8 ml water and quantified, while unreacted negatively charged WbwC reaction products stayed bound to the AG1x8 columns.
Test for TF antigen cross-reactivity.
To test whether the enzyme reaction products represented the cancer-associated TF antigen, JAAFII mouse monoclonal antibody against the TF antigen was used as the primary antibody (kindly donated by K. Rittenhouse, Roswell Park Cancer Institute, Buffalo, NY, USA) and horseradish peroxidase-conjugated goat anti-mouse IgG as the secondary antibody (Santa Cruz Biotechnology). A 50-nmol amount of either WbwCECO104 or WbwCECO5 reaction product Galβ1-3GalNAcα-PP-PhU, WbdN reaction product Glcβ1-3GlcNAcα-PP-PhU (23), compound 8 or its GlcNAc derivative 14, or Galβ1-3GalNAcα-Bn as the positive TF antigen control was incubated with 0.5 ml 1:100 TF antigen-specific antibody (4 mg/ml in Tris-buffered saline) overnight at 4°C. This was followed by incubation with 200 ng secondary antibody (goat anti-mouse IgG, 1:1,000) for 2 h at room temperature (rt). A 2-μl volume was saved as a positive control for antibody, and the remainder was slowly loaded onto C18 Sep-Pak columns. Columns were washed first with 5 ml water and then with 5 ml MeOH. MeOH fractions were dried by rotary evaporation and dissolved in 500 μl H2O. Samples were then transferred to small tubes, lyophilized, and dissolved in 10 μl H2O. Samples were spotted onto nitrocellulose membrane and dried. Labeling was visualized with WEST-one spray (iNtRON) and exposure of fluorescence on film.
Effect of amino acid-specific reagents.
To evaluate the role of amino acids in Gal-transferase activity, the purified enzyme preparations were incubated with amino acid-specific reagents prior to the assays for 10 min at rt in reaction mixtures lacking UDP-Gal. The reaction was initiated by the addition of UDP-Gal. p-Hydroxyphenylglyoxal (HPG; reacts with Arg), 5,5′-dithio-bis-2-nitrobenzoic acid (DTNB; reacts with Cys), and N-ethylmaleimide (NEM; reacts with Cys) were used at 0.2 mM concentration in the assay. Disulfide bond-reducing agent dithiothreitol was used at 0.1 to 4 mM in the assay and β-mercaptoethanol (beta-ME) at 1 to 18 mM.
Construction of WbwCECO104 mutants.
The QuikChange site-directed mutagenesis kit (Agilent, Santa Clara, CA) was used to construct mutants D91A and D125A of WbwCECO104. Primers were designed using the Quikchange Primer Design program. For the D91A mutant, the forward primer was 5′CAATGAACTGATTTTTAGGATGGCTACTGATGATATTTGTTTGCCTG3′ and the reverse primer was 5′CAGGCAAACAAATATCATCAGTAGCCATCC AAAAATCAGTTCATTG3′. For the D125A mutant, the forward primer was 5′GGGAAGTGTCATTGAAGAATTTGCTAATACAATGAAAATTAGGCAAG3′ and the reverse primer was 5′CTTGCCTAATTTTCATTGTATTAGCAAATTCTTCAATGGCACTTCCC3′. Amplification was performed with a DNA thermal cycler (model Eppendorf Mastercycler Gradient) by using a single denaturation step at 95°C for 1 min followed by an 18-cycle program consisting of denaturation at 95°C for 1 min, annealing at 55°C for 1.5 min, and extension at 68°C for 6 min. A total of 10 μl of each reaction mixture was electrophoresed on a 1% agarose gel in 45 mM Tris-borate buffer–1 mM EDTA, pH 8.3, at 150 V for 1 h and stained with ethidium bromide. The PCR mixture was transformed into E. coli Top10 cells. Bacterial colonies harboring mutated genes were isolated, and plasmids were isolated and sequenced (Robarts Research Institute, London, ON, Canada) to confirm the correct mutations. Mutants D45A, D46A, D93A, D94A, C21A, C82A, and C96A of WbwCECO104 and mutants D96A, D98A, and D99A of WbwCECO5 were constructed by Mutagenex (Hillsborough, NJ). Bacterial growth, enzyme activity assays, SDS-PAGE, and Western blotting were carried out as described above.
RESULTS
Sequence comparisons of WbwCECO104 and WbwCECO5.
A protein-protein BLAST comparison revealed that the amino acid sequences of WbwCECO104 and WbwCECO5 have 47% identity (see Fig. S2 in the supplemental material). WbwC from E. coli O104 shares 42% identity with uncharacterized WbwC from E. coli O81, 99% identity with WbwC from Escherichia fergusonii (35), and 79% identity with WfaM from E. coli O24 (36). Homologs of WbwC are also found in a number of other bacterial strains. None of these enzymes have been characterized. The other putative glycosyltransferases (WbwA and WbwB) encoded by genes located in the O104 antigen gene cluster (18) share only 13% and 11% identity, respectively, with WbwCECO104. WbiP from E. coli O127 (37), which also synthesizes a Galβ1-3GalNAc linkage, shows only 22% and 25% sequence identity with WbwCECO104 and WbwCECO5 (see Fig. S2 in the supplemental material). WbbD from E. coli O7 (26) and WbgO from E. coli O55 (38) both form a Galβ1-3GlcNAc linkage. WbbD shares 37% and 35% sequence identity with WbwCECO104 and WbwCECO5, respectively, whereas WbgO shares 23% and 20% sequence identity with WbwCECO104 and WbwCECO5. Human core 1 Gal-transferase (C1GalT1), which also forms a Galβ1-3GalNAc linkage, shows very low sequence identity to WbwCECO104 (12%) and to WbwCECO5 (11%). WbwC is predicted to form a GT-A glycosyltransferase fold and has been classified in the CAZy GT2 family, which includes many other bacterial and mammalian inverting β-glycosyltransferases (22–24, 37–39). All of the WbwC homologs have a DxD sequence that might be involved in catalysis (40).
Properties of WbwCECO104 and WbwCECO5.
Based on the amino acid composition, the molecular masses of His6-tagged WbwCECO104 and WbwCECO5 were calculated to be 31.2 and 31.4 kDa, respectively (protein molecular mass calculator; Science Gateway, www.sciencegateway.org). The two recombinant proteins from the bacterial lysate were induced by 1 mM IPTG in a 4-h incubation at 37°C and showed intense bands on SDS-PAGE at an apparent molecular mass of 30 to 32 kDa. Induction at rt, overnight or after 6 h at 0.5 or 1 mM IPTG did not improve the level of expression. The His6-tagged WbwCECO104 and WbwCECO5 were purified by NTA Sepharose affinity chromatography (see Fig. S3 and S4 in the supplemental material). Western blotting showed a major protein band at 30 kDa for WbwCECO104 for both the cell lysate and the purified enzyme at about 90% purity. WbwCECO5 was expressed and purified similarly but was slightly larger (32 kDa). A single elution with 150 mM imidazole was optimal for eluting the protein from the NTA column (about 50% purity). The specific activities of purified WbwCECO104 and WbwCECO5 were enriched 49-fold and 14-fold, respectively. The final yield of purified WbwCECO104 and WbwCECO5 proteins obtained from 150 ml starting culture was 5 to 7 mg (0.6 mg/ml). Under standard conditions, the transfer of Gal to GalNAcα-O-PO3-PO3-(CH2)11-O-Ph was linear for up to 10 min of incubation time and up to 6 μg protein per assay for purified WbwCECO104 and WbwCECO5. Gal-transferase activities in the bacterial homogenates were stable for several days at rt for at least 2 weeks at 4°C and for at least 4 months at −20°C.
The presence of the conserved DxD motif (DTDD in the WbwCECO104 and DSDD in the WbwCECO5 sequence) suggested the involvement of divalent metal cations as cofactors in Gal-transferase catalysis (40, 41). No activity was observed in the presence of 5 mM EDTA. At 5 mM concentration, Mn2+ was the most efficient cofactor among the divalent metal ions (set to 100% activity), but Mg2+ also stimulated the activity (22% for WbwCECO104 and 15% for WbwCECO5). The optimal concentration of Mn2+ in the WbwC assays was between 2.5 and 5 mM (data not shown). None of the other metal ions tested at 5 mM concentration (Co2+, Pb2+, Ca2+, Zn2+, and Cu2+) were effective in stimulating WbwC Gal-transferase activity. Both WbwCECO104 and WbwCECO5 activities were shown to have a broad pH optimum between 7 and 8 (data not shown).
Effects of detergents on WbwCECO104 and WbwCECO5 activity.
The hydrophobicity plots of both WbwC enzymes did not reveal any transmembrane domains, but in vivo, the enzymes could be loosely associated with the bacterial membrane. Membrane components or detergents could also affect the availability of the sugar-diphosphate-lipid acceptor substrate. We therefore determined whether the purified WbwCECO104 enzyme activity was affected by the presence of 0.125 to 0.5% Triton X-100, NP-40 (nonyl phenoxylpolyethoxylethanol), or n-octyl-β-d-glucoside (Octylglucoside). There was no effect with NP-40 treatment, but the presence of the other detergents reduced WbwCECO104 Gal-transferase activities to various degrees (data not shown). Interestingly, the effect of detergents on WbwCECO5 enzyme activity differed from the effect on WbwCECO104, and WbwCECO5 activity was reduced by all detergents tested. SDS at 0.005% in the reaction mixtures completely abolished both WbwCECO104 and WbwCECO5 activities.
Acceptor substrate specificities of WbwCECO104 and WbwCECO5.
The acceptor substrate specificities of purified WbwCECO104 and WbwCECO5 were tested with a series of synthetic acceptor substrate analogs, including compounds 8 and 14, GalNAcα-glycopeptides, GalNAc α and β linked to a number of different hydrophobic aglycone groups, and GlcNAc derivatives (Table 1). The only two compounds in this series that could serve as acceptor substrates were GalNAc-PP-PhU (compound 8) and a fluorescent acceptor (GalNAc-PP-AnthrU), but the corresponding GlcNAc analog GlcNAc-PP-PhU or any other GalNAc or GlcNAc derivative could not. This suggested that the presence of a diphosphate bridge in the acceptor as well as the axial 4-hydroxyl of the acceptor sugar was required for the activity of both WbwCECO104 and WbwCECO5. The kinetic parameters for purified WbwCECO104 and WbwCECO5 were similar, with apparent Km values of 0.12 mM and 0.10 mM, respectively, for GalNAc-PP-PhU, and apparent maximum rate of metabolism (Vmax) values of 1.57 μmol/h/mg for WbwCECO104 and 1.42 μmol/h/mg for WbwCECO5 (Fig. 2). For UDP-Gal, the apparent Km values for WbwCECO104 and WbwCECO5 were 0.73 mM and 1.20 mM, respectively, with apparent Vmax values of 0.87 μmol/h/mg and 2.67 μmol/h/mg, respectively. At high acceptor concentration, substrate inhibition was observed. These values were similar to those obtained with crude enzymes.
TABLE 1.
Acceptor substrate specificities of WbwCECO104 and WbwCECO5a
| Compound | Concn(mM) | Relative activity |
|
|---|---|---|---|
| WbwCECO104 | WbwCECO5 | ||
| GalNAcα-PO3-PO3-(CH2)11-O-Ph (compound 8) | 0.1 | 100 | 100 |
| GalNAcα-PO3-PO3-AnthrUb | 0.1 | 49.0 | 46.1 |
| GlcNAcα-PO3-PO3-(CH2)11-O-Ph (compound 14) | 1 | <1 | <1 |
| GalNAcα-Bn | 1 | <1 | <1 |
| 4-Deoxy-GalNAcα-Bn | 1 | <1 | <1 |
| A-(GalNAcα)T | 1 | <1 | <1 |
| N-Ac-V-(GalNAcα)TP-NH2 | 1 | <1 | <1 |
| GalNAc | 1 | <1 | <1 |
| AHGVT-(GalNAcα)SAPDTRPAPGSTAPPA | 1 | <1 | <1 |
| GalNAcβ1–4GlcNAcβ-Bn | 1 | <1 | <1 |
FIG 2.

Kinetics of WbwCECO104 and WbwCECO5 reactions. The standard assay as described in Materials and Methods was used to measure Gal transfer by purified WbwC enzymes. (A) WbwCECO104 reaction with acceptor 8 (0.25 mM) as a function of UDP-Gal concentration. The apparent Km for UDP-Gal was 0.73 mM with an apparent Vmax of 0.87 μmol/h/mg protein. (B) WbwCECO104 reaction as a function of acceptor 8 concentration. UDP-Gal concentration was 2.2 mM. The apparent Km for 8 was 0.12 mM with an apparent Vmax of 1.57 μmol/h/mg protein. (C) WbwCECO5 reaction with 0.25 mM acceptor 8 as a function of UDP-Gal concentration. The apparent Km for UDP-Gal was 1.20 mM with an apparent Vmax of 2.67 μmol/h/mg protein. (D) WbwCECO5 reaction as a function of acceptor 8. UDP-Gal donor concentration was 1.09 mM. The apparent Km for acceptor 8 was 0.10 mM with an apparent Vmax of 1.42 μmol/h/mg protein. Substrate inhibition was apparent at high acceptor concentration (not shown). All results were analyzed by regression analysis with GraphPad Prism.
Donor specificities of WbwCECO104 and WbwCECO5.
The donor specificities of purified WbwCECO104 and WbwCECO5 were examined by replacing 0.435 mM UDP-[3H]Gal with a number of other nucleotide sugars in the assay at the same concentration. No activities were observed with UDP-[3H]Glc, UDP-[3H]GalNAc, UDP-[3H]GlcNAc, CMP-[3H]sialic acid, or GDP-[3H]Man. This was indicative of a strict specificity of WbwCECO104 and WbwCECO5 for UDP-Gal as the donor substrate.
Analysis of WbwCECO104 and WbwCECO5 reaction products.
To determine the structures of WbwCECO104 and WbwCECO5 reaction products, aliquots of assay mixtures were applied to Sep-Pak C18 columns and enzyme reaction product was eluted with MeOH. MALDI-MS analysis of the MeOH eluates showed the presence of substrate (m/z 626) and enzyme product (m/z 788) (data not shown). The Sep-Pak step was required for the efficient HPLC separation of the reaction product. On reversed-phase HPLC, using 24% acetonitrile–76% water as the mobile phase, the product Gal-GalNAc-PP-PhU eluted between 35 and 45 min, well separated from substrate. The radioactive product served as a standard for HPLC. Pooled fractions containing purified reaction products showed m/z 788 for Gal-GalNAc-PP-PhU by MALDI-MS. This demonstrated that one Gal residue (m/z 162) had been added to the substrate by both WbwCECO104 and WbwCECO5.
NMR analysis of WbwC reaction products.
The 1H-NMR spectrum of the WbwCECO104 product showed a new doublet at 4.41 ppm for H-1 of Gal with a coupling constant of J1,2 = 8.3 Hz, which is indicative of Gal in β-linkage (Table 2 and Fig. 3). By comparison, the spectrum of the enzyme reaction product of β3-GalT WbbD (Galβ1-3GlcNAc-PP-PhU) showed a similar chemical shift for Gal H-1 at 4.35 ppm (24). To determine whether Gal was β1-3, β1-4, or β1-6 linked to GalNAc, we conducted two-dimensional NMR experiments, including rotating-frame nuclear Overhauser effect correlation spectroscopy (ROESY) (Fig. 4) and heteronuclear single quantum coherence (HSQC), which identified the carbon and proton chemical shifts of the Gal and GalNAc residues (Table 2). The GalNAc H-3 signal of substrate 8 (3.78 to 3.88 ppm) shifted to 4.00 in the WbwC reaction product. A large difference between substrate and product in the 13C signals was also seen for the GalNAc C-3 signal, which was 66.9 ppm in the substrate and 77.1 ppm in the product. However, the GalNAc H-4 signal also shifted from 3.90 to 3.94 ppm to 4.19 ppm in the product, but neither H-6 nor C-4 or C-6 signals showed large differences between substrate and product. A strong coupling between Gal H-1 and GalNAc H-3 was seen in the ROESY spectrum (Fig. 4), showing that Gal was indeed linked to position 3 of GalNAc. The NMR spectra of WbwCECO5 reaction product (data not shown) were virtually identical to those from WbwCECO104. This identifies the product structures as Galβ1-3GalNAcα-PP-PhU and proves that WbwC is a UDP-Gal:GalNAc-diphosphate-R β1,3-Gal-transferase.
TABLE 2.
1H and 13C NMR parameters of WbwCECO104 enzyme substrate 8 and WbwCECO104 reaction product Galβ1-3GalNAcα-PO3-PO3-(CH2)11-O-Pha
| Residue | 1H (ppm) | 13C (ppm) |
|---|---|---|
| WbwC reaction product Galβ1-3GalNAcα-PO3-PO3-(CH2)11-O-Ph | ||
| Gal-1 | 4.41 J1,2 = 8.3 Hz | 104.5 |
| Gal-2 | 3.45 | 70.7 |
| Gal-3 | 3.55 | 72.1 |
| Gal-4 | 3.82 | 68.3 |
| Gal-5 | 3.57 | 74.5 |
| Gal-6 | 3.65 | 60.2 |
| GalNAc-1 | 5.45 J1,2 = 7.2 Hz, 3.7 Hz | 94.7 |
| GalNAc-2 | 4.32 | 48.2 |
| GalNAc-3 | 4.00 | 77.1 |
| GalNAc-4 | 4.19 | 68.4 |
| GalNAc-5 | 4.13 | 71.6 |
| GalNAc-6 | 3.67 | 60.6 |
| GalNAc-N-acetyl | 1.97 | |
| Substrate 8, GalNAcα-PO3-PO3-(CH2)11-O-Ph | ||
| GalNAc-1 | 5.39 J1,2 = 7.0 Hz, 3.4 Hz | 94.6 |
| GalNAc-2 | 4.13 | 49.8 |
| GalNAc-3 | 3.78–3.88 | 66.9 |
| GalNAc-4 | 3.90–3.94 | 67.3 |
| GalNAc-5 | 4.07 | 72.1 |
| GalNAc-6 | 3.58, 3.69 | 61.1 |
| GalNAc-N-acetyl | 2.00 | |
The substrate for the WbwCECO104 reaction was GalNAcα-PO3-PO3-(CH2)11-O-Ph (compound 8 in Fig. 1) as described in Materials and Methods. Enzyme reaction product was purified by Sep-Pak C18 column and reversed-phase HPLC; 600-MHz NMR spectra were collected in 1D and 2D experiments in D2O. Assignments were made by HSQC and ROESY spectra. The coupling constants were difficult to determine with certainty due to crowding of signals. The spectra for the WbwCECO5 reaction product were virtually identical to those shown for the WbwCECO104 reaction product.
FIG 3.

One-dimensional 600-MHz 1H-NMR spectra of WbwCECO104 Gal-transferase reaction product measured in D2O. WbwCECO104 and WbwCECO5 reaction products have virtually identical spectra. Sample preparation is described in Materials and Methods.
FIG 4.

Two-dimensional 600-MHz ROESY spectrum of WbwCECO104 Gal-transferase reaction product measured in D2O. Experiments were performed at rt. There is a clear correlation between the Gal H-1 and GalNAc H-3 signals indicating the Gal1-3GalNAc linkage.
Glycosidase digestions of WbwC enzyme reaction products.
Galactosidase digestions were used to confirm the anomeric linkage between Gal and GalNAc in the [3H]Gal-GalNAc-PP-PhU enzyme reaction products synthesized by WbwCECO104 and WbwCECO5. The released free [3H]Gal was separated by AG1x8 columns that bound the uncleaved reaction product. WbwCECO104 and WbwCECO5 reaction products were resistant to green coffee bean α-galactosidase and to Jack bean (β1-4-specific) β-galactosidase, indicating that Gal was not α linked or in β1-4 linkage (Fig. 5). In contrast, treatment with bovine testicular β-galactosidase, which cleaves Gal in β1-3, β1-4, and β1-6 linkages, released 41% and 36% of the radioactivity from WbwCECO104 and WbwCECO5 reaction products, respectively. O-glycanase is an enzyme that cleaves O-glycan core 1, Galβ1-3GalNAcα, from glycopeptides and core 1-linked hydrophobic compounds, but it was not known if the enzyme also acts on diphosphate-linked core 1 structures. Indeed, O-glycanase released 29% and 32% of the radioactivity from the disaccharide reaction products of WbwCECO104 and WbwCECO5. This enzyme therefore acts on a variety of core 1-containing glycoconjugates, including those with negatively charged diphosphates attached to core 1. These results confirm that the WbwC reaction products have the structure Galβ1-3GalNAcα-PP-PhU.
FIG 5.
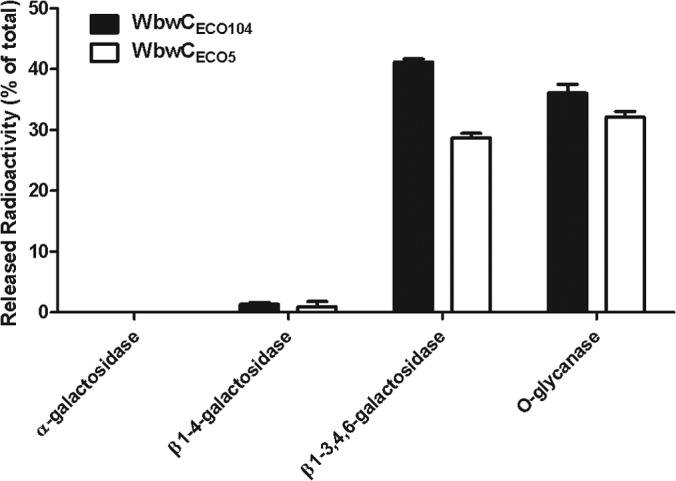
Glycosidase digestion of WbwC reaction products. Product linkage confirmation using glycosidase digestion on WbwC reaction products. The anomeric configuration of the linkage formed in the radioactive enzyme product was determined by incubation with specific galactosidases or O-glycanase. Mixtures were incubated for 30 min at 37°C and applied to AG1x8 columns. Released radioactivity was eluted and quantified. The percentages of release of radioactive Gal (or Gal-GalNAc for O-glycanase) are shown relative to the total radioactivity of WbwC reaction products in the assay. The bars indicate variations between duplicates.
Cross-reactivity of WbwC enzyme reaction products with TF antigen-specific antibody.
To test whether the enzyme reaction products represented the TF antigen in spite of the presence of the diphosphate group, aliquots of reaction products (50 nmol) were incubated with TF antigen-specific antibody as the primary antibody and horseradish peroxidase-conjugated anti-mouse IgG as the secondary antibody. The complexes were then separated by C18 Sep-Pak, which binds the hydrophobic phenylundecyl moiety. As a positive control, core 1–benzyl was used, and the substrate GalNAc-PP-PhU, the corresponding GlcNAc-PP-PhU, and the WbdN reaction product Glcβ1-3GalNAcα-PP-PhU (23) were used as negative controls. After washing with water, MeOH elution fractions were concentrated and applied to nitrocellulose membrane and stained for peroxidase activity. The results showed that after separation by C18 Sep-Pak, only the positive control and WbwCECO104 and WbwCECO5 reaction products stained positively for the TF antigen (see Fig. S5 in the supplemental material). It is interesting that Gal-GalNAc-PP-PhU bound to C18 Sep-Pak in the presence of antibodies and that the core 1 structure was clearly recognized by the antibody as the TF antigen, in spite of being linked to a charged diphosphate-lipid group. This confirms that both WbwCECO104 and WbwCECO5 synthesized the TF antigen.
Role of specific amino acids in WbwCECO104 and WbwCECO5 activities.
There are many positively charged amino acid residues in WbwCECO104 and WbwCECO5 that may possibly be involved in the binding of the negatively charged substrates, and we previously identified an essential Lys residue in Gal-transferase WfeD (25). The inclusion of 0.2 mM HPG in the assay mixture (without DTT) for purified WbwC enzymes showed 22% inhibition of WbwCECO104 activity and minimal inhibition of WbwCECO5 activity (Fig. 6). This suggested a potential role of Arg in the protein structure or catalysis of WbwCECO104.
FIG 6.
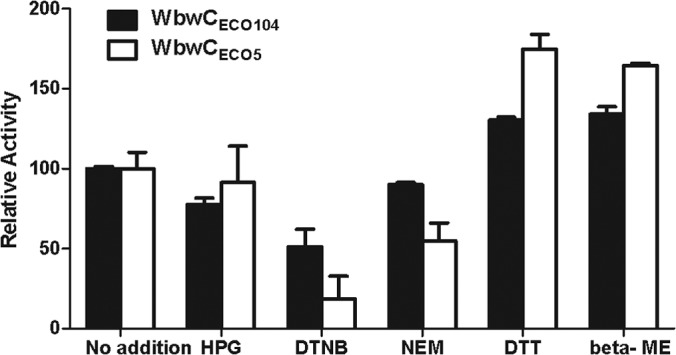
Effects of amino acid-specific reagents on activity of WbwC. Amino acid-specific reagents were preincubated with purified enzyme for 10 min, prior to the assays as described in Materials and Methods. The activities are shown relative to the positive control set to 100 (no addition of amino acid reagent). The bars indicate variations between duplicates. HPG, 0.2 mM hydroxyphenylglyoxal; DTNB, 0.2 mM 5,5′-dithio-bis-2-nitrobenzoic acid; NEM, 0.2 mM N-ethylmaleimide; DTT, 0.2 mM dithiothreitol; beta-ME, 18 mM beta-mercaptoethanol.
There are a number of conserved Cys residues in WbwCECO104. In the absence of DTT, DTNB (0.2 mM), which reacts with reduced SH groups, inhibited WbwCECO104 and WbwCECO5 activities by 49% and 81%, respectively, while 0.2 mM NEM showed 10% and 45% inhibition, respectively. In contrast, DTT (0.2 mM), which reduces disulfide bonds, stimulated WbwCECO104 and WbwCECO5 activities. β-Mercaptoethanol (18 mM) also increased the activities of WbwCECO104 and WbwCECO5 by 30% and 75%, respectively (Fig. 6). This suggested that enzymes may have formed disulfide bonds upon storage that interfered with activity, although these cytosolic enzymes are expected to be naturally in the reduced state. All of the Cys mutants of WbwCECO104 (C21A, C82A, and C96A) were fully active, indicating that these Cys residues are not required for activity. Reagents that covalently bind to Cys-SH groups may thus lead to less active enzyme by blocking the access of substrates.
Importance of a novel DxDD motif for WbwCECO104 and WbwCECO5 activity.
Most inverting glycosyltransferases have a DxD motif, which can contain an Asp or Glu residue representing a catalytic base (40). The WbwC sequences contain a DxDD motif that had not yet been recognized and is conserved mainly in β3-glycosyltransferases within the GT2 family. To evaluate the roles of these acidic amino acids, Asp residues were mutated to the neutral Ala in WbwCECO104 and WbwCECO5. Mutations of any of the Asp residues within the DTDD sequence in WbwCECO104 and the DSDD sequence in WbwCECO5 were found to drastically reduce the activities of both WbwCECO104 (residues D91, D93, and D94) and WbwCECO5 (residues D96, D98, and D99). No activity was detected with mutants lacking the first Asp residue of the DxDD motif (D91A mutant of WbwCECO104 and D96A mutant of WbwCECO5) (Fig. 7). In contrast, none of the other conserved Asp residues in the WbwCECO104 enzyme appeared to contribute to catalysis, and D45A, D46A, and D125A mutants had activities comparable to that of the wild-type enzyme. All mutants were well expressed, as shown by SDS-PAGE and Western blots (data not shown). This suggests that the WbwC enzyme family has a novel DxDD motif in which the first Asp residue is essential and the remaining Asp residues are important for catalysis.
FIG 7.
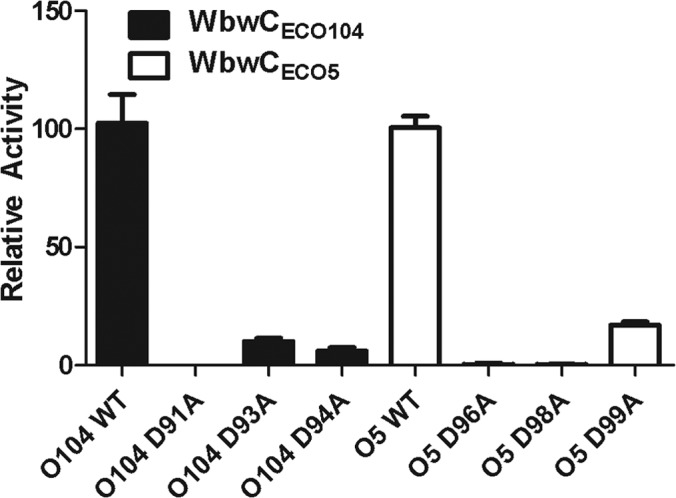
Relative activities of site-specific mutants of the DxDD motif of WbwCECO104 and WbwCECO5. Mutants of WbwCECO104 and WbwCECO5 were produced by replacing Asp with Ala as indicated and assayed as described in Materials and Methods. WT, wild type (unaltered controls of WbwCECO104 and WbwCECO5 set to 100). Relative activities are shown with error bars indicating differences in activities between multiple assays. Western blots showed that all mutants were well expressed.
Inhibition of WbwCECO104 and WbwCECO5 activity.
A number of diimidazolium salts, i.e., those having two positively charged imidazolium groups linked by aliphatic chains of 20 to 22 carbons, strongly inhibited WbwCECO104 and WbwCECO5 activities in the absence of DTT in the assay (Table 3). The solvents alone, without inhibitors (up to 10% in the assay) showed no effect. Especially compound QT 149 [1,22-bis-(3-methyl-1H-imidazolium-1-yl)docosane dimesylate], having an aliphatic chain of 22 carbons in length, effectively inhibited purified WbwCECO104 and WbwCECO5 with 50% inhibitory concentrations (IC50s) of 8.0 μM and 17.6 μM, respectively. In contrast, inhibitors of mammalian β1,4-Gal-transferase (N-butyryl-glucosamine-β-1-thio-2-naphthyl) (29) and core 1 β1,3-Gal-transferase (N-butyryl-galactosamineα-Bn) (39) showed no inhibition.
TABLE 3.
Inhibition of WbwCECO104 and WbwCECO5 activitiesa
| Compound | Concn (mM) | % Inhibition of activity over control |
|
|---|---|---|---|
| WbwCECO104 | WbwCECO5 | ||
| N-butyryl-glucosamineβ-thio-2-naphthyl | 0.5 | <1 | <1 |
| N-butyryl-galactosamineα-benzyl | 5.0 | <1 | <1 |
| QT137, 1,4-bis-(3-methyl-1H-imidazolium-1-yl)butane dichloride | 0.5 | 9.0 | <1 |
| QT139, 1,6-bis-(3-methyl-1H-imidazolium-1-yl)hexane dichloride | 0.5 | 23.0 | <1 |
| QT136, 1,18-bis-(3-methyl-1H-imidazolium-1-yl)octadecane dichloride | 0.5 | 24.1 | 55.2 |
| QT135, 1,20-bis-(3-methyl-1H-imidazolium-1-yl)eicosane dichloride | 0.5 | 87.8 | 97.5 |
| QT148, 1,20-bis-(3-methyl-1H-imidazolium-1-yl)eicosane dimesylate | 0.5 | 79.8 | 93.1 |
| QT149, 1,22-bis-(3-methyl-1H-imidazolium-1-yl)docosane dimesylate | 0.5 | 99.2 | 99.7 |
WbwC assays were performed in duplicate determinations using purified enzymes as described in Materials and Methods. The acceptor concentration was 0.1 mM substrate 8.
DISCUSSION
Two similar enzymes, WbwCECO104 and WbwCECO5, have been identified as UDP-Gal:GalNAcα-diphosphate-lipid β1,3-d-Gal-transferases that catalyze the second step of the O antigen repeating-unit assembly in E. coli serotypes O104 and O5, respectively. The enzymes have the same function in synthesizing the Galβ1-3GalNAc linkage and have very similar properties, although the amino acid sequences show only 47% identity.
Analyses of the structures of the WbwCECO104 and WbwCECO5 enzyme reaction products by HPLC, MS, and NMR spectroscopy, by digestions with galactosidases and O-glycanase, and by Western blots using TF antigen-specific antibody clearly showed that both WbwC enzymes add one Gal residue in β1-3 linkage to GalNAc-PP-PhU, an analog of the natural undecaprenyl-linked substrate. The reaction product structure corresponds to the disaccharide at the reducing end of the O104 and O5 antigen repeating unit and establishes WbwCECO104 and WbwCECO5 as the second enzymes in the respective O104 and O5 antigen repeating-unit biosynthesis pathways.
There are several bacterial strains that have the internal Galβ1-3GalNAc linkage in their O antigen structures (14, 15, 42). The β1,3-Gal-transferase WbiP has been characterized from E. coli O127, which contains the internal Galβ1-3GalNAcα structure in the O antigen (37). WbiP synthesizes the Galβ1-3GalNAcα linkage but does not have a requirement for the diphosphate group in the acceptor and can act on free GalNAc, with a preference for the GalNAcα linkage. Thus, a number of simple oligosaccharides serve as the WbiP substrate, and these assays do not require extensive chemical synthesis of complex sugar-phosphates. We describe improved syntheses of GalNAc-PP-PhU and GlcNAc-PP-PhU in this paper, which should enhance the characterization of other bacterial enzymes with yet-unknown function that may require the diphosphate group in the acceptor substrate.
Like other characterized members of the GT2 family, WbwC enzymes are typical inverting transferases, requiring divalent metal ion for activity and having DxD motifs (40, 41). We have shown in this study that all three Asp residues of the DxDD motif are required for full activity in both WbwCECO104 and WbwCECO5, with the first Asp being essential for activity and likely representing the catalytic base, and the other two Asp residues likely contributing to the acidic properties of the first Asp residue. Thus, we identified a new DxDD motif that is found in several members of the GT2 family. These enzymes have a predicted GT-A fold with the conserved DxDD motif in the N-terminal half of the protein (between amino acids 85 and 102). Most of these enzymes are inverting β1,3-glycosyltransferases, including β3Gal-transferases WbwC, WbbD, WbiP, WbgO, CgtB, LsgF, WbnJ, and Eps6 and the β1,3-Glc-transferases WbdN, WfaP, WfgD. In β3Gal-transferase WbiP from E. coli, the first and second Asp residues of the DxDD motif (Asp88 and Asp90) were mutated and suggested to be involved in UDP-Gal binding (37). Our study is the first to examine all three Asp residues of the DxDD motif. Interestingly, other enzymes of the GT2 family that are not β3-glycosyltransferases, e.g., β1,4-Gal- or β1,4-Glc-transferases, do not possess the DxDD motif. This suggests a functional importance of DxDD in the transfer of a Gal or Glc residue to position 3 of either GalNAc- or GlcNAc-containing acceptor substrates. We are awaiting the crystal structure of one of these enzymes in order to confirm the role of the DxDD motif in catalysis.
Another class of enzymes, the terpenoid cyclases, also have a conserved DxDD motif (43, 44). In these enzymes, the Asp residues are thought to cooperatively act as catalytic acids. The multiple Asp residues may thus enhance the ability of the catalytic Asp to protonate and in addition, in the terpenoid cyclases, are involved in binding the inhibitory metal ion Mg2+.
The TF (or T) antigen has been recognized as an important tumor antigen that can be targeted for the clinical development of carbohydrate-based anticancer vaccines (16, 45). Various chemical strategies have been developed to synthesize TF antigen-containing glycopeptides (46). CgtB, a β1,3-Gal-transferase involved in the synthesis of the ganglioside-like Galβ1-3GalNAcβ linkage found in the lipooligosaccharides of Campylobacter jejuni (47), prefers the β linkage in the GalNAc-R acceptor but also acts on GalNAcα-R substrates and can synthesize the linkage of the TF antigen structure. WbiP also synthesizes the TF antigen, but direct proof using anti TF antibody has not been provided, although it is likely that anti-TF antigen antibodies bind the disaccharide Galβ1-3GalNAcα linked to a variety of structures, including oligosaccharides, peptides, and hydrophobic aglycones. We showed here that the TF antigen can also be linked to a diphosphate group. Thus, WbwC can be regarded as a T synthase (16, 17, 39) and may be useful for a chemoenzymatic synthesis of the TF antigen. The diphosphate group can easily be removed with pyrophosphatases and phosphatases to yield the reducing TF disaccharide that could further be linked to an aglycone group suitable for vaccine synthesis.
Human T synthase (core 1 β1,3-Gal-transferase, classified in the GT31 family of inverting glycosyltransferases), which synthesizes the TF antigen in mucin-type O-glycans, has minimal sequence identity with WbwC but may have a similar architecture at the catalytic site, which remains to be elucidated. In spite of similar properties of mammalian and bacterial Gal-transferases, it is interesting that human core 1 β1,3-Gal-transferase, which transfers Gal to GalNAcα-benzyl, does not accept GalNAcα-PP-PhU as a substrate and prefers peptide aglycones (39). In contrast, WbwC uses GalNAcα-PP-PhU as the acceptor but not GalNAcα-benzyl or GalNAcα-peptides. Another difference between the human and bacterial enzymes is the inhibition by bis-imidazolium salts that inhibit WbwC but not the human enzyme. In contrast, N-butyryl-galactosamine α-Bn inhibits only human T synthase and not WbwC.
Both WbwCECO104 and WbwCECO5 are specific for the axial 4-hydroxyl of the GalNAc and the diphosphate in the acceptor substrate. The requirement for the diphosphate linked to GalNAc or GlcNAc in the acceptors appears to be a characteristic of the second enzymes in the O antigen assembly pathways, which include WbwC (22–25, 48, 49). It is not known, however, if the α linkage of GalNAc is required and if both phosphate groups are necessary for high activity. Based on our experience, we expect that at least one phosphate residue directly linked to GalNAcα is an absolute requirement for WbwC, but this remains to be shown upon synthesis of the appropriate acceptor analogs (26, 50, 51). Previous studies showed that different lipid chains in the acceptor were compatible with high activities of Gal- and Glc-transferases (22, 24, 48). In the current study, we used two acceptors that differed in the terminal ring structure in the aglycone, suggesting that the structure of the lipid moiety may be of minor importance for in vitro activity. The acceptors may form micelles where GalNAc-PP is exposed while the lipid moiety is enclosed; this may mimic the in vivo Und-linked substrates that are embedded in the inner membrane.
There are two other putative glycosyltransferase genes in the E. coli O104-antigen gene cluster (wbwA and wbwB) that may encode the α2,3-NeuNAc-transferase and α1,4-Gal-transferase necessary to sequentially complete the assembly of the O104 repeating unit (18). WbwC can be used to synthesize the substrate for the next step in O104 antigen synthesis and to determine the functions of the enzymes encoded by the wbwA and wbwB genes. The wzx and wzy genes in the O104 gene cluster are expected to encode a flippase and polymerase, respectively (2, 18, 52, 53), which indicates that the O104 antigen biosynthesis is governed by the Wzy-polymerase-dependent pathway (8, 9, 52).
All of the bacterial enzymes characterized to date that assemble O antigens appeared to lack a transmembrane domain and probably reside close to the cytosolic face of the inner membrane with access to both the soluble nucleotide sugar pools and the membrane-bound Und substrates. Enzymes may be tethered to the membrane by a yet-unknown mechanism, possibly by an enzyme complex formation that maintains their high activity. In our in vitro assays of recombinant enzymes, this organization is lacking, explaining why these glycosyltransferases often show relatively low activity in the purified form. The acceptor substrate has detergent-like properties and may form micelles together with added detergents in vitro. The fact that the two WbwC enzymes differ in their response to detergents may be due to a characteristic recognition of substrates by the enzymes. The amino acid sequences of WbwCECO104 and WbwCECO104 differ by 47%, and different hydrophobic sequences may be exposed. However, the enzymes have very similar characteristics; thus, the amino acids essential for substrate binding and catalysis and the dimensions of the active sites may be identical in the two enzymes, which remains to be shown by protein structure analyses.
As the second enzyme in the O104 biosynthesis pathway, WbwCECO104 is a suitable target for blocking O104 antigen synthesis and thus reducing a virulence factor. The bis-imidazolium inhibitors are interesting drug candidates. Specific bis-imidazolium salts inhibit selected glycosyltransferases (8, 17) with IC50s between 20 and 250 μM, while WbwC showed relatively low IC50s of 8 to 18.6 μM. These inhibitors probably bind properly spaced negatively charged and hydrophobic patches on the enzyme and may distort protein structure or block either substrate binding or catalysis. Since bis-imidazolium salts inhibit only selected Gal-transferases, the mechanism of inhibition may not involve binding to negatively charged donor or acceptor substrates. Current investigations are aimed at elucidating the mode of inhibition in vitro and in bacteria.
Our present study shows the thorough characterization of two bacterial enzymes that synthesize a humanlike antigen. This work provides a possible biotechnology approach for the synthesis of functionally important bacterial and mammalian oligosaccharides.
Supplementary Material
ACKNOWLEDGMENTS
We thank Francoise Sauriol (Queen's University) for carrying out the NMR analyses and Jiaxi Wang for the mass spectra analyses (Queen's University). We thank Vladimir V. Veselovsky and his team at the Zelinsky Institute, Russian Academy of Sciences, Moscow, Russia, for the synthesis of the fluorescent substrate. We thank Kate Rittenhouse, Roswell Park Cancer Institute, Buffalo, NY, USA, for the anti-TF antibody and John Schutzbach for critically reading the manuscript.
This work was funded in part by a Discovery grant from the Natural Science and Engineering Research Council of Canada (to I.B.) and a grant from the Canadian Institutes of Health Research (to I.B. and W.A.S.), by a National 973 Program of China grant (2012CB721101), a National Natural Science Foundation of China (NSFC) Key Program grant (31030002), and the National Key Program for Infectious Diseases of China (2013ZX10004216-001-001) (to L.F.).
Footnotes
Published ahead of print 23 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01698-14.
REFERENCES
- 1.Ho TD, Waldor MK. 2007. Enterohemorrhagic Escherichia coli O157:H7 Gal mutants are sensitive to bacteriophage P1 and defective in intestinal colonization. Infect. Immun. 75:1661–1666. 10.1128/IAI.01342-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635–700. 10.1146/annurev.biochem.71.110601.135414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheng H, Lim JY, Watkins MK, Minnich SA, Hovde CJ. 2008. Characterization of an Escherichia coli O157:H7 O-antigen deletion mutant and effect of the deletion on bacterial persistence in the mouse intestine and colonization at the bovine terminal rectal mucosa. Appl. Environ. Microbiol. 74:5015–5022. 10.1128/AEM.00743-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strauss J, Burnham NA, Camesano TA. 2009. Atomic force microscopy study of the role of LPS O-antigen on adhesion of E. coli. J. Mol. Recognit. 22:347–355. 10.1002/jmr.955 [DOI] [PubMed] [Google Scholar]
- 5.Zhang L, Radziejewska-Lebrecht J, Krajewska-Pietrasik D, Toivanen P, Skurnik M. 1997. Molecular and chemical characterization of the lipopolysaccharide O-antigen and its role in the virulence of Yersinia enterocolitica serotype O8. Mol. Microbiol. 23:63–76. 10.1046/j.1365-2958.1997.1871558.x [DOI] [PubMed] [Google Scholar]
- 6.Reeves PR, Hobbs M, Valvano MA, Skurnik M, Whitfield C, Coplin D, Kido N, Klena J, Maskell D, Raetz CR. 1996. Bacterial polysaccharide synthesis and gene nomenclature. Trends Microbiol. 4:495–503. 10.1016/S0966-842X(97)82912-5 [DOI] [PubMed] [Google Scholar]
- 7.Shimizu T, Yamasaki S, Tsukamoto T, Takeda Y. 1999. Analysis of the genes responsible for the O-antigen synthesis in enterohaemorrhagic Escherichia coli O157. Microb. Pathog. 26:235–247. 10.1006/mpat.1998.0253 [DOI] [PubMed] [Google Scholar]
- 8.Reeves PR. 1994. Biosynthesis and assembly of lipopolysaccharide. New Comp. Biochem. 27:281–314. 10.1016/S0167-7306(08)60416-0 [DOI] [Google Scholar]
- 9.Whitfield C. 1995. Biosynthesis of lipopolysaccharide O antigens. Trends Microbiol. 3:178–185. 10.1016/S0966-842X(00)88917-9 [DOI] [PubMed] [Google Scholar]
- 10.Qin J, Cui Y, Zhao X, Rohde H, Liang T, Wolters M, Li D, Belmar Campos C, Christner M, Song Y, Yang R. 2011. Identification of the Shiga toxin-producing Escherichia coli O104:H4 strain responsible for a food poisoning outbreak in Germany by PCR. J. Clin. Microbiol. 49:3439–3440. 10.1128/JCM.01312-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Creydt VP, Nuñez P, Boccoli J, Silberstein C, Zotta E, Goldstein J, Ibarra C. 2006. Role of the Shiga toxin in the hemolytic uremic syndrome. Medicina (B. Aires) 66(Suppl 3):S11–S15 (In Spanish.) [PubMed] [Google Scholar]
- 12.Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, Bernard H, Fruth A, Prager R, Spode A, Wadl M, Zoufaly A, Jordan S, Kemper MJ, Follin P, Müller L, King LA, Rosner B, Buchholz U, Stark K, Krause G, Investigation Team HUS 2011. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 365:1771–1780. 10.1056/NEJMoa1106483 [DOI] [PubMed] [Google Scholar]
- 13.Muniesa M, Hammerl JA, Hertwig S, Appel B, Brüssow H. 2012. Shiga toxin-producing Escherichia coli O104:H4: a new challenge for microbiology. Appl. Environ. Microbiol. 78:4065–4073. 10.1128/AEM.00217-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gamian A, Romanowska E, Ulrich J, Defaye J. 1992. The structure of the sialic acid-containing Escherichia coli O104 O-specific polysaccharide and its linkage to the core region in lipopolysaccharide. Carbohydr. Res. 236:195–208. 10.1016/0008-6215(92)85016-S [DOI] [PubMed] [Google Scholar]
- 15.Urbina F, Nordmark EL, Yang Z, Weintraub A, Scheutz F, Widmalm G. 2005. Structural elucidation of the O-antigenic polysaccharide from the enteroaggregative Escherichia coli strain 180/C3 and its immunochemical relationship with E. coli O5 and O65. Carbohydr. Res. 340:645–650. 10.1016/j.carres.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 16.Brockhausen I, Gao Y. 2012. Structural glycobiology, p 177–214 In Yuriev E, Ramsland PA. (ed), Structural glycobiology: applications in cancer research. CRC Press, Taylor & Francis Group, Boca Raton, FL [Google Scholar]
- 17.Springer GF. 1997. Immunoreactive T and Tn epitopes in cancer diagnosis, prognosis, and immunotherapy. J. Mol. Med. 75:594–602. 10.1007/s001090050144 [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Briggs CE, Rothemund D, Fratamico P, Luchansky JB, Reeves PR. 2001. Sequence of the E. coli O104 antigen gene cluster and identification of O104 specific genes. Gene 270:231–236. 10.1016/S0378-1119(01)00471-1 [DOI] [PubMed] [Google Scholar]
- 19.Rush JS, Alaimo C, Robbiani R, Wacker M, Waechter CJ. 2010. A novel epimerase that converts GlcNAc-P-P-undecaprenol to GalNAc-P-P-undecaprenol in Escherichia coli O157. J. Biol. Chem. 285:1671–1680. 10.1074/jbc.M109.061630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freinkman E, Chng SS, Kahne D. 2011. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc. Natl. Acad. Sci. U. S. A. 108:2486–2491. 10.1073/pnas.1015617108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Reeves PR. 1998. Organization of Escherichia coli O157 O antigen gene cluster and identification of its specific genes. Infect. Immun. 66:3545–3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brockhausen I, Hu B, Liu B, Lau K, Szarek WA, Wang L, Feng L. 2008. Characterization of two beta-1,3-glucosyltransferases from Escherichia coli serotypes O56 and O152. J. Bacteriol. 190:4922–4932. 10.1128/JB.00160-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao Y, Liu B, Strum S, Schutzbach JS, Druzhinina TN, Utkina NS, Torgov VI, Danilov L, Veselovsky VV, Vlahakis JZ, Szarek WA, Wang L, Brockhausen I. 2012. Biochemical characterization of WbdN, a beta1,3-glucosyltransferase involved in O-antigen synthesis in enterohemorrhagic Escherichia coli O157. Glycobiology 22:1092–1102. 10.1093/glycob/cws081 [DOI] [PubMed] [Google Scholar]
- 24.Riley JG, Menggad M, Montoya-Peleaz PJ, Szarek WA, Marolda CL, Valvano MA, Schutzbach JS, Brockhausen I. 2005. The wbbD gene of E. coli strain VW187 (O7:K1) encodes a UDP-Gal: GlcNAc{alpha}-pyrophosphate-R {beta}1,3-galactosyltransferase involved in the biosynthesis of O7-specific lipopolysaccharide. Glycobiology 15:605–613 [DOI] [PubMed] [Google Scholar]
- 25.Xu C, Liu B, Hu B, Han Y, Feng L, Allingham JS, Szarek WA, Wang L, Brockhausen I. 2011. Biochemical characterization of UDP-Gal:GlcNAc-pyrophosphate-lipid β-1,4-Galactosyltransferase WfeD, a new enzyme from Shigella boydii type 14 that catalyzes the second step in O-antigen repeating-unit synthesis. J. Bacteriol. 193:449–459. 10.1128/JB.00737-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montoya-Peleaz PJ, Riley JG, Szarek WA, Valvano MA, Schutzbach JS, Brockhausen I. 2005. Identification of a UDP-Gal:GlcNAc-R galactosyltransferase activity in Escherichia coli VW187. Bioorg. Med. Chem. Lett. 15:1205–1211. 10.1016/j.bmcl.2004.11.077 [DOI] [PubMed] [Google Scholar]
- 27.Vinnikova AN, Druzhinina TN, Danilov LL, Utkina NS, Torgov VI, Veselovsky VV, Wang S, Liu B, Wang L, Brockhausen I. 2013. Synthesis of a fluorescent acceptor substrate for glycosyltransferases involved in the assembly of O-antigens of enterohemorrhagic Escherichia coli O157 and O5. Carbohydr. Res. 366:17–24. 10.1016/j.carres.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 28.Brockhausen I, Benn M, Bhat S, Marone S, Riley JG, Montoya-Peleaz P, Vlahakis JZ, Paulsen H, Schutzbach JS, Szarek WA. 2006. UDP-Gal: GlcNAc-R beta1,4-galactosyltransferase—a target enzyme for drug design. Acceptor specificity and inhibition of the enzyme. Glycoconj. J. 23:525–541 [DOI] [PubMed] [Google Scholar]
- 29.Gao Y, Lazar C, Szarek WA, Brockhausen I. 2010. Specificity of β1,4-galactosyltransferase inhibition by 2-naphthyl 2-butanamido-2-deoxy-1-thio-β-d-glucopyranoside. Glycoconj. J. 27:673–684. 10.1007/s10719-010-9312-3 [DOI] [PubMed] [Google Scholar]
- 30.Gao Y, Vinnikova A, Brockhausen I. 2013. Functional identification of bacterial glucosyltransferase WbdN. Glycosyltransferases: methods and protocols in method molecular biology. Humana Press, Springer Science and Business Media, New York, NY: [DOI] [PubMed] [Google Scholar]
- 31.Gao Y, Vlahakis JZ, Szarek WA, Brockhausen I. 2013. Selective inhibition of glycosyltransferases by bivalent imidazolium salts. Bioorg. Med. Chem. 21:1305–1311. 10.1016/j.bmc.2012.12.034 [DOI] [PubMed] [Google Scholar]
- 32.Visscher I, Stuart MC, Engberts JB. 2006. The influence of phenyl and phenoxy modification in the hydrophobic tails of di-n-alkyl phosphate amphiphiles on aggregate morphology. Org. Biomol. Chem. 4:707–712. 10.1039/b514285g [DOI] [PubMed] [Google Scholar]
- 33.Bernardes GJL, Kikkeri R, Maglinao M, Laurino P, Collot M, Hong SY, Lepenies B, Seeberger PH. 2010. Design, synthesis and biological evaluation of carbohydrate-functionalized cyclodextrins and liposomes for hepatocyte-specific targeting. Org. Biomol. Chem. 8:4987–4996. 10.1039/c0ob00372g [DOI] [PubMed] [Google Scholar]
- 34.Nakabayashi S, Warren CD, Jeanloz RW. 1986. A new procedure for the preparation of oligosaccharide oxazolines. Carbohydr. Res. 150:C7–C10. 10.1016/0008-6215(86)80028-3 [DOI] [PubMed] [Google Scholar]
- 35.Fegan N, Barlow RS, Gobius KS. 2006. Escherichia coli O157 somatic antigen is present in an isolate of E. fergusonii. Curr. Microbiol. 52:482–486. 10.1007/s00284-005-0447-6 [DOI] [PubMed] [Google Scholar]
- 36.Cheng J, Wang Q, Wang W, Wang Y, Wang L, Feng L. 2006. Characterization of E. coli O24 and O56 O antigen gene clusters reveals a complex evolutionary history of the O24 gene cluster. Curr. Microbiol. 53:470–476. 10.1007/s00284-006-0032-7 [DOI] [PubMed] [Google Scholar]
- 37.Yi W, Perali RS, Eguchi H, Motari E, Woodward R, Wang PG. 2008. Characterization of a bacterial β-1,3-galactosyltransferase with application in the synthesis of tumor-associated T-antigen mimics. Biochemistry 47:1241–1248. 10.1021/bi7020712 [DOI] [PubMed] [Google Scholar]
- 38.Liu XW, Xia C, Li L, Guan WY, Pettit N, Zhang HC, Chen M, Wang PG. 2009. Characterization and synthetic application of a novel beta1,3-galactosyltransferase from Escherichia coli O55:H7. Bioorg. Med. Chem. 17:4910–4915. 10.1016/j.bmc.2009.06.005 [DOI] [PubMed] [Google Scholar]
- 39.Gao Y, Aryal RP, Ju T, Cummings Gahlay RDG, Jarvis DL, Matta KL, Vlahakis JZ, Szarek WA, Brockhausen I. 2013. Acceptor specificities and selective inhibition of recombinant human Gal- and GlcNAc-transferases that synthesize core structures 1, 2, 3 and 4 of O-glycans. Biochim. Biophys. Acta 1830:4274–4281. 10.1016/j.bbagen.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breton C, Snajdrová L, Jeanneau C, Koca J, Imberty A. 2006. Structures and mechanisms of glycosyltransferases. Glycobiology 16:29R–37R. 10.1093/glycob/cwj016 [DOI] [PubMed] [Google Scholar]
- 41.Ünligil UM, Rini JM. 2000. Glycosyltransferase structure and mechanism. Curr. Opin. Struct. Biol. 10:510–517. 10.1016/S0959-440X(00)00124-X [DOI] [PubMed] [Google Scholar]
- 42.Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Wang Q, Reeves PR, Wang L. 2008. Structure and genetics of Shigella O antigens. FEMS Microbiol. Rev. 32:627–653. 10.1111/j.1574-6976.2008.00114.x [DOI] [PubMed] [Google Scholar]
- 43.Mann FM, Prisic S, Davenport EK, Determan MK, Coates RM, Peters RJ. 2010. A single residue switch for Mg(2+)-dependent inhibition characterizes plant class II diterpene cyclases from primary and secondary metabolism. J. Biol. Chem. 285:20558–20563. 10.1074/jbc.M110.123307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peters RJ, Croteau RB. 2002. Abietadiene synthase catalysis: conserved residues involved in protonation-initiated cyclization of geranylgeranyl diphosphate to (+)-copalyl diphosphate. Biochemistry 41:1836–1842. 10.1021/bi011879d [DOI] [PubMed] [Google Scholar]
- 45.Karsten U, Papsdorf G, Pauly A, Vojtesek B, Moll R, Lane EB, Clausen H, Stosiek P, Kasper M. 1993. Subtypes of non-transformed human mammary epithelial cells cultured in vitro: histo-blood group antigen H type 2 defines basal cell-derived cells. Differentiation 54:55–56. 10.1111/j.1432-0436.1993.tb01588.x [DOI] [PubMed] [Google Scholar]
- 46.Brocke C, Kunz H. 2002. Synthesis of tumor-associated glycopeptide antigens. Bioorg. Med. Chem. 10:3085–3112. 10.1016/S0968-0896(02)00135-9 [DOI] [PubMed] [Google Scholar]
- 47.Bernatchez S, Gilbert M, Blanchard M-C, Karwaski M-F, Li J, DeFrees S, Wakarchuk WW. 2007. Variants of the β1,3-galactosyltransferase CgtB from the bacterium Campylobacter jejuni have distinct acceptor specificities. Glycobiology 17:1333–1343. 10.1093/glycob/cwm090 [DOI] [PubMed] [Google Scholar]
- 48.Brockhausen I, Riley JG, Joynt M, Yang X, Szarek WA. 2008. Acceptor substrate specificity of UDP-Gal:GlcNAc-R β1,3-galactosyltransferase (WbbD) from Escherichia coli O7:K1. Glycoconj. J. 25:663–673. 10.1007/s10719-008-9127-7 [DOI] [PubMed] [Google Scholar]
- 49.Yi W, Yao Q, Zhang Y, Motari E, Lin S, Wang PG. 2006. The wbnH gene of Escherichia coli O86:H2 encodes an α-1,3-N-acetylgalactosaminyl transferase involved in the O-repeating unit biosynthesis. Biochem. Biophys. Res. Commun. 344:631–639. 10.1016/j.bbrc.2006.03.181 [DOI] [PubMed] [Google Scholar]
- 50.Brockhausen I, Larsson EA, Hindsgaul O. 2008. A very simple synthesis of GlcNAc-alpha-pyrophosphoryl-decanol: a substrate for the assay of a bacterial galactosyltransferase. Bioorg. Med. Chem. Lett. 18:804–807. 10.1016/j.bmcl.2007.11.031 [DOI] [PubMed] [Google Scholar]
- 51.Riley JG, Xu C, Brockhausen I. 2010. Synthesis of acceptor substrate analogs for the study of glycosyltransferases involved in the second step of the biosynthesis of O-antigen repeating units. Carbohydr. Res. 345:586–597. 10.1016/j.carres.2009.12.022 [DOI] [PubMed] [Google Scholar]
- 52.Samuel G, Reeves P. 2003. Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr. Res. 338:2503–2519. 10.1016/j.carres.2003.07.009 [DOI] [PubMed] [Google Scholar]
- 53.Valvano MA. 2003. Export of O-specific lipopolysaccharide. Front. Biosci. 8:s452–s471. 10.2741/1079 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.