

Abstract
Reversible lysine acetylation (RLA) is used by cells of all domains of life to modulate protein function. To date, bacterial acetylation/deacetylation systems have been studied in a few bacteria (e.g., Salmonella enterica, Bacillus subtilis, Escherichia coli, Erwinia amylovora, Mycobacterium tuberculosis, and Geobacillus kaustophilus), but little is known about RLA in antibiotic-producing actinomycetes. Here, we identify the Gcn5-like protein acetyltransferase AcuA of Saccharopolyspora erythraea (SacAcuA, SACE_5148) as the enzyme responsible for the acetylation of the AMP-forming acetyl coenzyme A synthetase (SacAcsA, SACE_2375). Acetylated SacAcsA was deacetylated by a sirtuin-type NAD+-dependent consuming deacetylase (SacSrtN, SACE_3798). In vitro acetylation/deacetylation of SacAcsA enzyme was studied by Western blotting, and acetylation of lysine residues Lys237, Lys380, Lys611, and Lys628 was confirmed by mass spectrometry. In a strain devoid of SacAcuA, none of the above-mentioned Lys residues of SacAcsA was acetylated. To our knowledge, the ability of SacAcuA to acetylate multiple Lys residues is unique among AcuA-type acetyltransferases. Results from site-specific mutagenesis experiments showed that the activity of SacAcsA was controlled by lysine acetylation. Lastly, immunoprecipitation data showed that in vivo acetylation of SacAcsA was influenced by glucose and acetate availability. These results suggested that reversible acetylation may also be a conserved regulatory posttranslational modification strategy in antibiotic-producing actinomycetes.
INTRODUCTION
Reversible lysine acetylation (RLA) of proteins is now recognized as a ubiquitous and conserved posttranslational modification in a variety of organisms (1–4). Recent studies have identified over 2,000 acetylated proteins, ranging from transcriptional factors and ribosomal proteins to metabolic enzymes related to glycolysis, gluconeogenesis, the tricarboxylic acid (TCA) cycle, and fatty acid and nitrogen metabolisms. This kind of posttranslational modification (PTM) has emerged as an important metabolic regulatory mechanism in bacteria since the discovery of acetylation of the Salmonella enterica acetyl coenzyme A (Ac-CoA) synthetase in 2002 (5). In the last decade, lysine acetylation of proteins has been reported in other microorganisms, including Escherichia coli, Bacillus subtilis, Streptomyces lividans, Mycobacterium tuberculosis, Erwinia amylovora, Thermus thermophilus, and Geobacillus kaustophilus (6–9).
RLA has been found to modulate protein synthesis, central metabolism, and detoxification metabolism. Yu et al. identified 85 acetylated proteins in E. coli, of which 24 (28%) are involved in protein biosynthesis and 16 (19%) are involved in carbohydrate metabolism (10). Zhang et al. also reported that more than 70% of the 91 acetylated proteins in E. coli are metabolic enzymes (53%) and translation regulators (22%) (11). More recently, Wang et al. identified 235 peptides containing acetylated lysines in a total of 191 proteins in Salmonella enterica and found that enzymes involved in the central metabolism are extensively acetylated and that their acetylation profiles change in response to different carbon sources, concomitantly with changes in cell growth and metabolic flux (2). These results suggest an extensive role of acetylation in the regulation of intracellular metabolism in response to rapidly changing conditions.
The acetate-scavenging, AMP-forming acetyl-CoA synthetase (Acs; EC 6.2.1.1; acetate:CoA ligase) was the first enzyme reported to be regulated by acetylation in prokaryotes and has been extensively investigated in S. enterica (5), B. subtilis (12), E. coli (13), Rhodopseudomonas palustris (14), M. tuberculosis (15), Streptomyces lividans (6), and Saccharomyces cerevisiae (16). AMP-forming acetyl-CoA synthetases belong to the acyl-adenylate-forming superfamily, are ubiquitous enzymes whose activity is central to the metabolism of prokaryotic and eukaryotic cells, and are responsible for the assimilation of acetate. AMP-forming acetyl-CoA synthetases catalyze the reaction acetate + CoA + ATP ↔ acetyl-CoA + AMP + PPi, for the reversible conversion of acetate to acetyl-CoA (Ac-CoA), providing the cell the two-carbon metabolite used in many anabolic and energy generation processes.
In E. coli, it is known that the expression of the acs gene is strictly controlled by complex regulatory systems at a transcriptional level (17, 18). Notably, the activity of Acs is also regulated at the posttranslational level by the RLA system. All RLA systems in prokaryotes consist of protein acetyltransferases (referred to as Gcn5-type N-acetyltransferases, or GNATs) and protein deacetylases. GNATs are conserved in all domains of life and catalyze the transfer of the acetyl from the Ac-CoA donor to a primary amine of small molecules and proteins. The GNAT protein superfamily contains over 50,804 members (pfam00583), which are involved in a wide variety of cellular processes. GNAT enzymes that modulate the acetylation state of histones for facilitating transcription (hyperacetylated histones) or gene silencing (hypoacetylated histones) in eukaryotic cells have been extensively studied. However, so far, only a few GNATs responsible for the acetylation of proteins have been identified in bacteria. In Salmonella enterica, the protein acetyltransferase Pat (SePat) inactivates acetyl-CoA synthetase (SeAcs) through lysine acetylation (19). Similar observations have been reported in other Gram-negative bacteria that synthesize SePat homologues, such as E. coli and Rhodopseudomonas palustris (14, 20). RLA has also been investigated in Gram-positive bacteria such as Bacillus subtilis, whose genome encodes a protein acetyltransferase AcuA (BsAcuA) that modifies the acetyl-CoA synthetase of this bacterium (12).
We are interested in acetylation of protein in high G+C DNA Gram-positive actinomycetes, which are important producers of therapeutic antibiotics. In some pathogenic actinomycetes, e.g., M. tuberculosis and Mycobacterium smegmatis, two protein acetyltransferases, PatA and Pat (MtPatA and MsPat, respectively), were found to acetylate acetyl-CoA synthetase and a universal stress protein (15, 21). The SePat homologue of Streptomyces lividans (SlPatA) has protein acetyltransferase activity that modulates the activity of the acetoacetyl-CoA synthetase enzyme of this bacterium (6). Acetyl-CoA synthetase was also found to be regulated in vivo by acetylation in Streptomyces coelicolor, but the acetyltransferase responsible for this acetylation was not identified (22). The B. subtilis AcuA (211 residues) and Rhodopseudomonas palustris KatA (RpKatA) GNAT enzymes are much smaller than SePat-type acetyltransferases since they contain only the GNAT domain. Of relevance to this work is the fact that all previously reported bacterial acetyltransferases acetylate only one lysine residue (in boldface) within the conserved acylation motif PXXXXGK found in AMP-forming acyl-CoA synthetases.
At present, our understanding of the physiological role of RLA in high G+C-content Gram-positive bacteria (e.g., Streptomyces and Saccharopolyspora erythraea) is limited, the function of protein acetyltransferases responsible for acetylation of specific proteins has not been described, and the systematic screening of Pat substrates has not been investigated. Actinomycete genomes generally encode more than 40 protein acetyltransferases (http://pfam.xfam.org/family/PF00583; http://www.ebi.ac.uk/interpro/entry/IPR000182). To understand the role of acetylation in bacteria, it is imperative to identify the functions and biochemical characterization of these enzymes. Further understanding of the RLA regulation of metabolism in actinomycete species is of interest because of the diversity of natural products synthesized by these organisms.
In this study, we identified the Gcn5-like S. erythraea protein acetyltransferase (SACE_5148; hereinafter referred to as SacAcuA) and sirtuin-type NAD-dependent deacetylase (SACE_3798; hereinafter referred to as SacSrtN) responsible for acetylation/deacetylation of acetyl-CoA synthetase (SACE_2375; hereinafter referred to as SacAcsA) and found that SacAcuA, unlike its B. subtilis homologue, can acetylate multiple lysine sites of its protein substrate. Furthermore, it was also demonstrated that the acetylation status of SacAcsA is modulated by extracellular nutrient availability. These results suggested that reversible acetylation may be a regulatory posttranslational modification strategy in antibiotic-producing actinomycetes.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and reagents.
All strains and plasmids used in this work are listed in Table S1 of the supplemental material. Three Saccharopolyspora erythraea strains, the wile-type NRRL2338 (from DSM 40517) and gene null mutant ΔsrtN and ΔacuA strains, were used in this study. Two gene knockout strains were generated by protoplast transformation under polyethylene glycol (PEG) mediation and chromosome homologous recombination, replacing 771 bp of the SacsrtN and SacacuA open reading frames (ORFs) with a thiostrepton (tsr) resistance cassette (23). Thiostrepton-resistant clones were examined for the presence of the SacsrtN and SacacuA deletion by PCR, taking their genome as the template. The deletion was confirmed by Southern blotting and sequencing. S. erythraea strains were grown in the minimal medium (Evans) containing 25 mM TES [N-(Tris(hydroxymethyl)methyl)-2-aminoethanesulfonic acid sodium salt], 2 mM citric acid, 10 mM KCl, 0.25 mM CaCl2, 1.25 mM MgCl2, 2 mM Na2SO4, 1 mM Na2MoO4, 0.5% trace elements (20 μM MnSO4 · 4H2O, 6 μM ZnSO4 · 7H2O, 20 μM H3BO3, 1 μM KI, 2 μM Na2MoO4 · 2H2O, 50 μM CuSO4 · 5H2O, 50 μM CoCl2 · 6H2O), 2.5% (mass/vol) glucose, 2 mM NaH2PO4, and 10 mM NaNO3 (pH 7.2). S. erythraea strains were also cultured in MM medium (2.5 mM l-asparagine, 1 mM K2HPO4, 0.8 mM MgSO4 · 7H2O, 0.035 mM FeSO4 · 7H2O, pH 7.2) supplemented with 20 mM glucose or 60 mM acetate as the solo carbon resource. Aerobic 100-ml batch cultures were grown in 1-liter flasks at 37°C on a rotary shaker at 250 rpm. Cultures were inoculated to an optical density at 600 nm (OD600) of 0.05 unit with exponentially growing precultures.
Protein A-conjugated agarose beads were from Amersham Biosciences. Acetyl lysine antibody (catalog number ICP0380) and acetylated bovine serum albumin (BSA; catalog number ICP6090) were from ImmuneChem Pharmaceuticals, Inc. (Burnaby, British Columbia, Canada). Trichostatin A (TSA) was purchased from Wako Chemicals.
Overproduction and purification of proteins (SacAcsA, SacAcuA, SacSrtN, SeAcs, BsAcsA, BsAcuA, and SePat).
All genes were amplified by PCR from the genomic DNA of Saccharopolyspora erythraea, Salmonella enterica, and Bacillus subtilis. The primers used in this work are listed in Table S2 in the supplemental material. After restriction digest, the genes coding for SacAcsA (SACE_2375), SacSrtN (SACE_3798), SeAcs (STM4275), and BsAcsA (Bsu29680) were cloned into pET-28a; the genes coding for SacAcuA (SACE_5148), BsAcuA (Bsu29690), and SePat (STM2651) were cloned into pGEX-4T-2. The proteins were expressed using the E. coli BL21(λDE3) strain. A single colony was selected to start a 5-ml overnight culture, which was then used to inoculate 50 ml of lysogeny broth (LB) medium supplemented with 1‰ kanamycin or 1‰ ampicillin. The cells were grown at 37°C to about 0.7 OD600 and then induced overnight with 0.5 mM isopropyl-β-d-thiogalactoside (IPTG) at 37°C for 6 h at 20°C.
Cells were harvested by centrifugation and resuspended in phosphate-buffered saline (PBS) buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KHPO4). The cells were disrupted by sonication, and cell debris was removed by centrifugation at 12,000 × g for 15 min. The resulting supernatant of SacAcsA, SacSrtN, SeAcs, and BsAcsA was loaded onto a 2-ml Ni-nitrilotriacetic acid (NTA)-agarose column (Merck) that was preequilibrated with the binding buffer. After the flowthrough was discarded, the column was washed with 20 ml of wash buffer (50 mM NaH2PO4, 300 mM NaCl, and 20 mM imidazole, pH 8.0), and bound proteins were eluted using a linear gradient from 20 to 250 mM imidazole in 50 mM NaH2PO4 and 300 mM NaCl, pH 8.0. The fractions were analyzed by SDS-PAGE. The SacAcuA, BsAcuA, and SePat proteins were purified from these cell extracts using standard glutathione affinity chromatography. Those containing the desired protein were pooled and dialyzed against buffer A (50 mM Tris and 150 mM NaCl, pH 7.5) and then concentrated using an Amicon Ultra-4 10,000-molecular-weight cutoff centrifugal device (Millipore). The protein concentration was determined by the bicinchoninic acid (BCA) method using bovine serum albumin as the standard.
Site-directed mutagenesis of SacAcsA acetylated-site mutants.
The acetylated-site mutants (K237Q, K380Q, K611Q, and K628Q) were introduced into the pET28a(+)::sacacs plasmid using a QuikChange mutagenesis kit (Stratagene) with the primers listed in Table S3 in the supplemental material. The mutations were confirmed by DNA sequencing.
In vitro acetyl-CoA synthetase (Acs) assays.
The specific activity of acetyl-CoA synthetase was determined at 37°C using a microplate reader (BioTek Instruments, Winooski, VT, USA) in a transparent 384-well microplate at 340 nm. The standard reaction mixture contained 100 mM Tris-HCl (pH 7.7), 10 mM l-malate (pH 7.7), 0.2 mM coenzyme A, 8 mM ATP (pH 7.5), 1 mM NAD+, 10 mM MgCl2, 3 units of malate dehydrogenase, 0.4 unit of citrate synthase, and 200 nM purified SacAcsA. The reaction was started with 100 mM potassium acetate. One unit was defined as the amount of enzyme catalyzing the acetate-dependent formation of 1 mmol of NADH min−1 in the coupled assay (24). For determination of the Km value, the acetate or ATP concentration in the assay was varied while the other components remained constant. The apparent steady-state kinetic parameters were estimated by nonlinear regression to fit the data to Michaelis-Menten kinetics.
In vitro protein acetylation assays.
To determine whether SacAcsA was a substrate for SacAcuA, 0.2 μM purified SacAcuA protein or BSA and 5 μM purified unacetylated SacAcsA protein were added to a reaction mixture (200-μl total volume) containing 0.05 M HEPES buffer (pH 7.5), 200 μM tris(2-carboxyethyl) phosphine (TCEP) hydrochloride, and 20 μM Ac-CoA. Reaction mixtures were incubated at 37°C for 2 h (15). After the reaction, the SacAcsA protein samples were divided into two portions: one portion was analyzed by SDS-PAGE and Western blotting, and the other was used for measurement of the Acs activity. The acetylated SacAcsA (SacAcsAAc) was isolated by SDS-PAGE and then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
SeAcs and BsAcsA (5 μM) were incubated with SacAcuA (0.2 μM) and 20 μM Ac-CoA for 2 h at 37°C using the buffer system described above. The effect of SacAcuA, SePat, and BsAcuA on SacAcsA was measured as described above using the SacAcsA wild-type (SacAcsAWT) and mutant proteins (SacAcs with a K237Q mutation [SacAcsK237Q], SacAcsK380Q, SacAcsK611Q, and SacAcsK628Q); the samples were analyzed by SDS-PAGE and Western blotting. The SeAcs and BsAcsA acetylated by SacAcuA were isolated and analyzed by LC-MS/MS as above.
In vitro deacetylation assays.
Samples containing 5 μM unacetylated SacAcsA were first incubated with 0.2 μM SacAcuA and 20 μM Ac-CoA at 37°C for 2 h. After the acetylation reaction, acetylated SacAcsA was isolated from the reaction mixture by ultrafiltration and affinity chromatography. To examine whether acetylated SacAcsA was deacetylated, the purified acetylated SacAcsA protein was added to 50 mM HEPES (pH 8.5) buffer containing 1 mM MgCl2, 1 mM NAD+, and 0.5 μM SacSrtN. The mixture was incubated at 37°C for 3 h (15). The samples were divided into two portions: one portion was resolved by SDS-PAGE and analyzed by Western blotting, and the other was used for measurement of the acetyl-CoA synthetase activity.
In vitro steady-state kinetic assays.
To measure the kinetic parameters of the deacetylation, the acetylated SacAcsA was obtained after incubating 5 μM SacAcsA with 20 μM Ac-CoA and 0.2 μM SacAcuA at 37°C for 3 h. A typical deacetylation reaction mixture contained 0.1 μM SacSrtN, various amounts of one substrate, and a saturating amount of the other. The reaction was initiated with the addition of SacSrtN. Acs species (0.2 nmol, acetylated and nonacetylated) were withdrawn from the reaction mixture every 30 min and tested for the Acs activity as described above (15). The initial velocities of the deacetylation reaction were calculated based on the amount of active Acs generated in the indicated time. Each data point is the average of three identical assays. For the acetylation, we used a method similar to that described above. The data were fitted into the Michaelis-Menten equation to obtain the Km and kcat values.
Western blot analysis.
The protein concentrations of the samples were determined using a BCA Protein Assay kit (Tiangen) with BSA as the standard. Protein samples were separated by SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane for 30 to 60 min at 100 V. The membrane was blocked at 24°C in 1× TBST (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) containing 5% nonfat dry milk (NFDM) for 2 h. Anti-acetyl-lysine (here, anti-AcK) antibody diluted 1:15,000 in TBST–0.5% NFDM was used. After incubation at 4°C for overnight, the blot was washed with TBST three times. The membrane was incubated with horseradish peroxidase (HRP)-conjugated anti-mouse IgG (1 μg/ml in TBST with 3% BSA) at ambient temperature for 2 h. An enhanced chemiluminescence (ECL) system (Pierce, USA) was used for signal detection according to the manufacturer in conjunction with a luminescent image analyzer (DNR Bio-Imaging Systems, Israel).
Purification of SacAcsA from S. erythraea, IP, and immunoblotting (IB).
Cells of Saccharopolyspora erythraea strains (wild type and SacsrtN and SacacuA deletion mutants) grown on different media for anti-Acs immunoprecipitation (IP) were harvested by centrifugation at 3,000 × g for 30 min and then ground and resuspended in PBS buffer in the presence of protease inhibitors (1 mM phenylmethanesulfonyl fluoride and Complete EDTA-free Protease Inhibitor Cocktail Tablets [Roche]) and histone deacetylase inhibitors (100 μM trichostatin A, 50 mM nicotinamide, and 50 mM sodium butyrate). The resuspended cells were disrupted by sonication, and cell debris was removed by centrifugation at 12,000 × g for 45 min. For immunoprecipitation, lysates (about 300 μg) were incubated with 2 μg of anti-Acs antibody (Abmart, Shanghai, China) at 4°C for 2 h, followed by the addition of 40 μl of protein A-agarose (Santa Cruz Biotechnology, Inc.) overnight. After four washes with PBS washing buffer at 4°C, bound proteins were eluted by boiling in SDS sample buffer, resolved by SDS-PAGE, and then subjected to Western blot analysis. Primary antibodies used were anti-Acs antibody and acetyl lysine antibody HRP conjugate (anti-AcK; Immunechem). Secondary antibodies were purchased from Abmart. Binding was visualized using an ECL Western blotting method. After ECL detection, films were scanned by MF-ChemiBIS software, version 3.2 (DNR Bio-Imaging Systems, Israel), and quantified with ImageJ software. The purified SacAcsA from S. erythraea was then analyzed by LC-MS/MS spectrometry.
Mass spectrometry peptide fingerprinting.
Protein digestion was performed according to the filter-aided sample preparation (FASP) procedure described by Wisniewski et al. (25). Briefly, the protein pellet (about 30 μg) was solubilized in 30 μl of SDT buffer (4% SDS, 100 mM dithiothreitol [DTT], 150 mM Tris-HCl, pH 8.0) at 90°C for 5 min. The detergent, DTT, and other low-molecular-weight components were removed using 200 μl of UA buffer (8 M urea, 150 mM Tris-HCl, pH 8.0) by multiple ultrafiltrations (Microcon units; 30 kDa). Then, 100 μl of 0.05 M iodoacetamide in UA buffer was added to block reduced cysteine residues, and the samples were incubated for 20 min in darkness. The filter was washed with 100 μl of UA buffer three times and then with 100 μl of 25 mM NH4HCO3 twice. Finally, the protein suspension was digested with 2 μg of trypsin (Promega) in 40 μl of 25 mM NH4HCO3 overnight at 37°C, and the resulting peptides were collected as a filtrate.
Tryptic digests (approximately 30 μg of predigested protein) were solid-phase extracted and analyzed by microcapillary LC (mLC)-MS/MS using a Micromass MS (Waters) and Q Exactive mass spectrometer (Thermo Finnigan, San Jose, CA) to locate protein acetylation sites. Chromatography of peptides prior to mass spectral analysis was accomplished using high-performance liquid chromatography (HPLC). Columns were made using lengths of fused silica tubing (0.15-mm outside diameter [o.d.]; 150-mm inside diameter [i.d.]) with pulled tips (1-mm orifice) that were packed with Zorbax 300SB-C18 peptide traps (Agilent Technologies, Wilmington, DE). An Agilent HPLC delivered solvents A (0.1% [vol/vol] formic acid in water) and B (0.1% [vol/vol] formic acid in acetonitrile [84% vol/vol], 0.1% formic acid) at either 1 ml/minute to load sample or at 150 to 200 nl/minute to elute peptides as follows: over a 50-min 4% (vol/vol) B to 50% B gradient; over another 4-min 50% (vol/vol) B to 100% B gradient; and over 6 min in 100% (vol/vol) B. As peptides were eluted from the HPLC column/electrospray source, MS/MS spectra were collected.
MS/MS spectra were searched using the MASCOT engine (version 2.2; Matrix Science, London, United Kingdom) against the Uniprot Saccharopolyspora_NRRL23338 database (7,165 sequences; accessed 1 July 2013). For protein identification, the following options were used: peptide mass tolerance, 20 ppm; MS/MS tolerance, 0.1 Da; enzyme, trypsin; missed cleavage, 2; fixed modification, carbamidomethyl (C); variable modification, oxidation (M) and acetylation (K, N-terminal); decoy database pattern, reverse. All reported data were based on 99% confidence for protein identification as determined by a false discovery rate (FDR) of ≤1%.
RESULTS AND DISCUSSION
The SacAcuA enzyme of Saccharopolyspora erythraea is a novel protein acetyltransferase.
It was reported that the activity of acetyl-CoA synthetase was modulated by acetylation in some bacteria (5, 6, 12, 13). To investigate whether acetyl-CoA synthetase from the erythromycin-producing S. erythraea (SacAcsA) is acetylated in vivo, immunoprecipitation (IP) and immunoblotting (IB) analyses were conducted to directly test the acetylation status of SacAcsA. SacAcsA from S. erythraea cells was immunoprecipitated with an antibody to SacAcsA (encoded by SACE_2375). Acetyl-lysine levels were detected on SacAcsA immunoprecipitates with anti-AcK antibody. As expected, the results demonstrated that SacAcsA was acetylated in vivo (Fig. 1A). The S. erythraea genome contains 42 genes putatively encoding GNAT protein acetyltransferases. Significantly, no SePat homologue was found. We found that antibody against AcK reacted strongly with the SacAcsA enzyme isolated from a wild-type strain; in contrast, the antibody exhibited weak reactivity against SacAcsA isolated from a strain devoid of SacAcuA (ΔSacacuA strain) (Fig. 1A). Because the acetylation level of SacAcsA was significantly reduced in the ΔSacacuA strain, we surmised that the SacAcuA might be involved in the acetylation of SacAcsA. The SacacuA gene encodes a single-domain GNAT protein acetyltransferase (155 amino acids; referred to as SacAcuA), which is 31% identical to the B. subtilis AcuA enzyme (BsAcuA). Furthermore, a phylogenetic analysis with the full-length sequence of SacAcuA and the eight protein acetyltransferases for lysine acetylation showed that SacAcuA clustered with BsAcuA (Fig. 1B) (http://www.phylogeny.fr/version2_cgi/simple_phylogeny.cgi), thus suggesting that SacAcuA could acetylate acetyl-CoA synthetases in S. erythraea as BsAcuA does in B. subtilis.
FIG 1.

SacAcuA enzyme is a novel protein acetyltransferase. (A) Wild-type, ΔSacsrtN, and ΔSacacuA strains of S. erythraea were grown in Evans minimal medium. Total protein extracts were collected, and the acetyl-CoA synthetase SacAcsA was immunoprecipitated with anti-Acs antibody and subjected to Western blot analysis. Immunoblotting was performed with anti-SacAcs and anti-AcK antibodies. The band intensities were quantified by densitometry using ImageJ software. (B) The phylogenetic analysis was conducted (http://www.phylogeny.fr/version2_cgi/simple_phylogeny.cgi) with SacAcuA and the following eight protein acetyltransferases: SePat (STM2651), RpPat (RPA4240), E. coli PatZ (EcPatZ) (b2584), SlPatA, MsPat, MtPatA, BsAcuA, and RpKatA (rpa3031). (C) SDS-PAGE analysis of purified recombinant SacAcuA. Lane 1, molecular mass marker; lane 2, purified glutathione S-transferase (GST)-SacAcuA. (D) The purified SacAcsA was in vitro incubated with or without SacAcuA and Ac-CoA at 37°C for 2 h. After incubation, samples were collected and analyzed by SDS-PAGE, and the acetylation levels were determined by Western blotting using specific anti-AcK antibody.
Additionally, the comparison of amino acid sequences of SacAcuA and BsAcuA revealed that SacAcuA contains four conserved motifs, sequentially labeled C, D, A, and B, found in GNAT family members (see Fig. S1 in the supplemental material). Motif A, as the core of the GNAT domain, is the most highly conserved motif and generally has an R/Q-X-X-G-X-G/A sequence that is important for acetyl-CoA recognition and binding. The RGSGVA sequence (conversed residues in boldface) is indeed found in motif A of SacAcuA while it is not observed in BsAcuA. The SWISS-MODEL server of the Protein Model Portal (PMP [http://www.proteinmodelportal.org/?pid=modeling_interactive]) was used online for model building for the structure of SacAcuA (see Fig. S2 in the supplemental material). Further, we confirmed that SacAcuA directly acetylated SacAcsA in vitro by incubating purified SacAcuA (Fig. 1C) with Ac-CoA and recombinant SacAcsA of S. erythraea. As shown in Fig. 1D, the results showed that SacAcsA was a substrate of SacAcuA.
To test the effect of acetylation on enzyme activity, SacAcsA was incubated with SacAcuA in the presence or absence of Ac-CoA for 2 h. In the presence of both Ac-CoA and SacAcuA, SacAcsA activity was reduced >80%, indicating that SacAcuA lysine acetylation effectively decreased SacAcsA activity (Fig. 2A). Time-dependent inactivation of SacAcsA by SacAcuA acetylation was also investigated. Data presented in Fig. 2B show that SacAcsA gradually lost its activity during acetylation by the SacAcuA enzyme.
FIG 2.
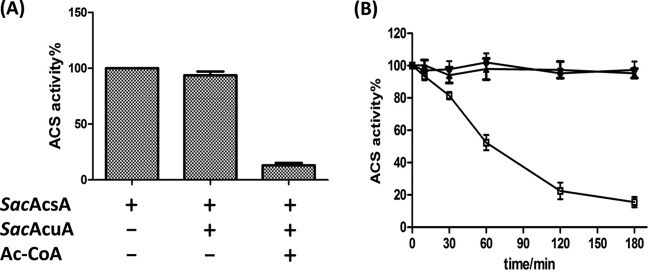
Effects of acetylation on SacAcsA activity. (A) In vitro acetylation affected the activity of SacAcsA. The enzyme activity of SacAcsA after incubation with or without Ac-CoA in the presence of SacAcuA was measured every 30 min. The SacAcsA activity is described as a percentage of the maximum activity determined for SacAcsA before acetylation. Data are expressed as means plus standard deviations of three identical assays. (B) Time-dependent inactivation of SacAcsA by acetylation. SacAcsA activity was measured at different time intervals during incubation with SacAcuA. □, 0.2 μM SacAcuA and 20 μM Ac-CoA; ○, 0.2 μM SacAcuA; △, 5 μM SacAcsA. Each data point represents the average from three independent assays.
To measure the kinetic parameters of the SacAcuA-catalyzed acetylation reaction of SacAcsA, we used a coupled enzymatic assay to monitor the acetylation reaction continuously. The resulting data were fitted using the Michaelis-Menten kinetics model. The kinetic parameters of SacAcuA were compared with those of BsAcuA from Bacillus subtilis and Gcn5 from yeast. The results are shown in Table 1. The Km and kcat values for Ac-CoA are 56 μM and 0.05 s−1, respectively. This is comparable to the intracellular levels reported for acetyl-CoA, which can reach cytoplasmic concentrations of 20 to 600 μM in E. coli (26). Similarly, The Km and kcat values for SacAcsA are 3.9 μM and 0.02 s−1. SacAcuA exhibited a Km value of 56 μM for Ac-CoA substrate, which was slightly higher than that of B. subtilis BsAcuA (Km of 22 μM) (27), which was 22-fold higher than that of the yeast Gcn5 histone acetyltransferase (HAT) (Km of 2.5 μM) (28). These results indicated that SacAcuA was active at high intracellular levels of Ac-CoA. The kcat of SacAcuA for Ac-CoA was 0.05 s−1, a turnover number that was 34-fold lower than the kcat value (1.7 s−1) of the yeast Gcn5 HAT and 6-fold lower than the kcat value (0.3 s−1) of B. subtilis BsAcuA.
TABLE 1.
Kinetic analysis of SacAcuA and SacSrtN
| Enzyme | Substrate | Km (μM) | kcat (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|---|
| SacAcuA | Ac-CoA | 56 ± 1 | 0.049 ± 0.003 | 860 ± 51 |
| AcsA | 3.9 ± 0.7 | 0.019 ± 0.001 | (5.2 ± 0.9) × 103 | |
| BsAcuAa | Ac-CoA | 22 ± 2 | 0.3 | 1.4 × 104 |
| Peptide | 20 ± 2 | 0.3 | 1.5 × 104 | |
| Yeast Gcn5a | Ac-CoA | 2.5 ± 1.4 | 1.7 ± 0.12 | 6.8 × 105 |
| H3 peptide | 490 ± 80 | 1.7 ± 0.12 | 3.5 × 103 | |
| SacSrtN | Ac-AcsA | 52 ± 5 | 0.033 ± 0.001 | 590 ± 72 |
| NAD+ | 240 ± 15 | 0.052 ± 0.002 | 220 ± 31 |
Data are from previously published literature. The peptide substrate of BsAcuA was the C-terminal 31 residues of the BsAcsA protein (LPKTRSGKIMRRVLKAWELNLPAGDLSTMED) (27). The H3 peptide substrate of Gcn5 was ARTKQTARKSTGGKAPPKQLC, corresponding to the 20 amino-terminal residues of human histone H3 and an additional carboxyl-terminal cysteine (28). Boldface indicates acetylated lysine.
In vitro, SacAcsA is acetylated at four lysine residues.
To determine the acetylation sites of SacAcsA protein, we cloned the SacacsA gene and purified recombinant SacAcsA (668 amino acids). SacAcsA was incubated with SacAcuA and Ac-CoA for 2 h. The in vitro-acetylated SacAcsA protein was subjected to trypsin digestion, and the resulting peptides were analyzed by tandem mass spectrometry. Lysine-acetylated peptides may be identified as they have a mass increment of 42 Da compared with unacetylated peptides. Four peptides were acetylated, the sequences of which are TK(237)TDVEWNDGR, TFMK(380)WGAEIPAR, DHVAHEIGPIAK(611)PR, and SGK(628)IMR (see Fig. S3 in the supplemental material). Lysine 237, lysine 380, lysine 611, and lysine 628 (in boldface) were identified as the acetylated residues in in vitro-acetylated SacAcsA (Fig. 3A). In our study, multiple acetylation sites were observed in S. erythraea SacAcsA, but three of these sites (K237, K380, and K611) were not identified in any of the other studies, and these lysines were not conserved in Acs homologues in other organisms (Fig. 3B). In S. erythraea SacAcsA, K628 is a conserved active-site residue (conserved putative acylation motif PXXXXGK), similar to Lys609 of SeAcs from S. enterica, Lys606 of RpAcs from R. palustris, Lys617 of MtAcs from M. tuberculosis, and Lys549 of BsAcsA from B. subtilis. K237 and K380 lysine residues are located in the CoA-binding domain and acetate-binding domain.
FIG 3.

Acetylation sites of SacAcsA. (A) Predicted domains of SacAcsA are shown in differently colored boxes. Red lines show the positions of acetylated lysine residues. (B) Multiple sequence alignment of the conserved putative acylation motif in acetyl-CoA synthetases from different species. The active-site lysine of all species is boxed in red. (C) The effect of SacAcuA on SacAcsA wild type and four variants (SacAcsK237Q, SacAcsK380Q, SacAcsK611Q, and SacAcsK628Q). Unacetylated SacAcsA wild type and four variants were incubated with SacAcuA and Ac-CoA at 37°C for 2 h. After incubation, samples were collected and analyzed by SDS-PAGE, and the acetylation levels were determined by Western blotting.
To confirm the sites of acetylation and investigate the effects of these residues on the acetylation level of SacAcsA, we created substitution mutations at these positions to generate K237Q, K380Q, K611Q, and K628Q variants of SacAcsA (see Fig. S4 in the supplemental material). Glutamine (Q) abolishes the positive charge and serves as a structural mimic for acetyl-lysine. SacAcsAK237Q, SacAcsAK380Q, SacAcsAK611Q, SacAcsAK628Q, and SacAcsAWT were incubated with SacAcuA enzyme and Ac-CoA. Western blotting was performed to detect the acetylation level of these AcsA variants with anti-AcK antibody. As shown in Fig. 3C, all SacAcsA variants were acetylated by SacAcuA, indicating that multiple SacAcsA lysine residues were modified by SacAcuA, in agreement with the results obtained mass spectrometry. SacAcsAK237Q, SacAcsAK380Q, SacAcsAK611Q, and SacAcsAK628Q mutants revealed similar acetylation levels, which were decreased slightly compared with the level of SacAcsAWT.
For comparison, SacAcsAWT and four variants were also incubated with SePat from S. enterica and BsAcuA from B. subtilis. SePat and BsAcuA acetylated SacAcsAWT, SacAcsAK237Q, SacAcsAK380Q, and SacAcsAK611Q but did not acetylate SacAcsAK628Q, suggesting that SePat and BsAcuA modified only the conserved lysine residue K628 in the active site (Fig. 4A). No acetylation was identified at the three residues K237, K380, and K611 in SacAcsA. As shown in Fig. 4B, SacAcuA enzyme can also acetylate SeAcs from S. enterica and BsAcsA from B. subtilis. However, SeAcs and BsAcsA revealed similar acetylation levels, but these acetylation levels were decreased drastically compared with the SacAcsA level, indicating that SeAcs and BsAcsA were poor substrates for SacAcuA. To further investigate whether SacAcuA modified the multiple lysine residues in other acetyl-CoA synthetases, we determined the acetylation sites of SeAcs and BsAcsA modified by SacAcuA. The in vitro-acetylated acetyl-CoA synthetases were digested with trypsin, and the resulting peptides were analyzed by tandem mass spectrometry. The results showed that lysine residues 29, 44, 56, 585, and 609 (in boldface) were acetylated in peptides YK(29)QSINDPDTFWGEQGK, QSINDPDTFWGEQGK(44)ILDWITPYQK, VK(56)NTSFAPGNVSIK, K(585)EIGPLATPDVLHWTDSLPK, and SGK(609)IMR of SeAcs (see Fig. S5 in the supplemental material); lysine residues 16, 98, 320, 524, and 549 were acetylated in peptides ALPAIEGDHNLK(16)NYEETYR, YGNVEK(98)GDR, MLMGAGDEMAAK(320)YDLTSLR, LFVK(524)QGLAAHAAPR, and SGK(549)IMR of BsAcsA (see Fig. S6 in the supplemental material). It was reported that only the conserved active-site residue of acetyl-CoA synthetase (Lys609 of SeAcs or Lys549 of BsAcsA) was acetylated by cognate acetyltransferases in S. enterica and B. subtilis (5, 12). The acetyltransferase SacAcuA appears to have evolved to acetylate multiple lysine residues, unlike other previously reported bacterial acetyltransferases which acetylate only one lysine residue in the conserved putative acylation motif PXXXXGK in AMP-forming acyl-CoA synthetases.
FIG 4.
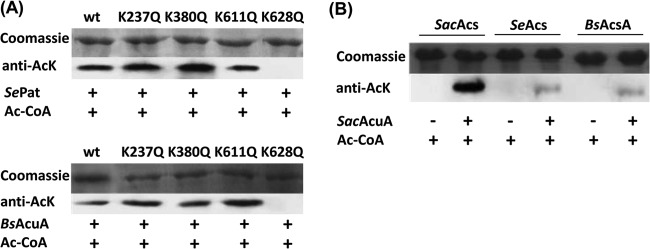
SePat and BsAcuA acetylate only one lysine residue. (A) The effect of SePat and BsAcuA on SacAcsA wild type and four variants (SacAcsK237Q, SacAcsK380Q, SacAcsK611Q, and SacAcsK628Q). Unacetylated SacAcs wild type and four variants were incubated with SePat or BsAcuA in the presence of Ac-CoA at 37°C for 2 h. After incubation, samples were collected and analyzed by SDS-PAGE, and the acetylation levels were determined by Western blotting. (B) SacAcuA acetylated SacAcsA, SeAcs, and BsAcsA. After incubation with SacAcuA and Ac-CoA at 37°C for 2 h, samples were analyzed by SDS-PAGE and Western blotting.
The previously reported bacterial acetyltransferases exhibit the specificity of lysine sites for protein acetylation. There is the proposed acetylation motif (PXXXXGK) found in AMP-forming acyl-CoA synthetases (20). GNAT acetyltransferases recognize this motif and acetylate the last lysine residue of PXXXXGK. It is unclear why these acetyltransferases acetylate a lysine only in a specific position. We speculate that such site specificity is attributed to other functional domains of multidomain acetyltransferases or a substrate-binding cleft composed of motifs A and D. As shown in Fig. S1 in the supplemental material, motif C is involved in orienting the protein substrate, and motif D forms part of the β-sheet core and, together with motif A, makes up half of the catalytic site (29). There are more α-helixes and β-sheets in the core region of BsAcuA, such as α3, β6, α4, β7, α7, and α8, and this region forms a substrate binding pocket for the PXXXXGK motif and therefore likely plays a crucial role in determining substrate specificity. In SacAcuA, the absence of such α-helixes and β-sheets maybe leaves larger or more flexible cleft benefits for the access of the variant lysine sites of substrates to be acetylated. Thus, SacAcuA has broad specificity for lysine acetylation, allowing it to acetylate multiple lysine residues. The substrate recognition and acetylation mechanism of specialized lysine sites by acetyltransferases remain to be determined.
SacAcsA is inactive mainly upon acetylation of K628.
To examine the effects of acetylation lysine residues on SacAcsA activity, the activities of SacAcsAWT and SacAcsA variants were determined. As shown in Fig. 5A, two SacAcsA variants were less active to some extent, while lysine acetylation significantly inhibited SacAcsA activity. The results showed that mutations at the K238 or K380 site produced activity similar to that of the SacAcsAWT, while mutation of K611 resulted in a 30% decrease in acetyl-CoA synthetase activity compared to the wild-type enzyme. In contrast, the K628Q mutant was essentially inactive, with <10% of the wild-type SacAcsA activity. These results indicate that residue K628 is critical for catalytic activity. Substitution of K628 effectively abolished activity of AcsA, in agreement with the results that acetylation of K628 inhibited SacAcsA activity.
FIG 5.
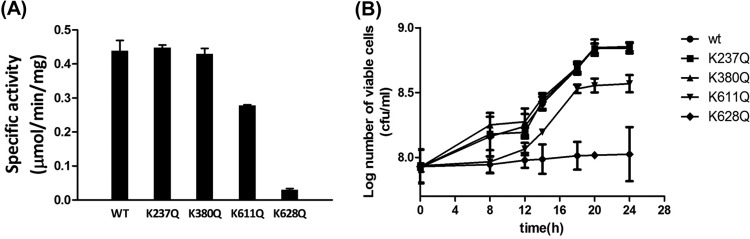
Effect of acetylated sites on enzyme activity. (A) The activity of AcsA (50 pM) incubated with ATP (8 mM), CoA (0.2 mM), and 100 mM acetate at 37°C. (B) Growth curves of the S. enterica Δacs strains complemented with SacAcsA wild-type or mutant genes in acetate minimal medium (10 mM acetate as a sole carbon source). Expression of the acs gene was induced by addition of l-(+)-arabinose (250 μM). Cell density measurements at 600 nm were acquired. Graphed points represent the means of three independent measurements.
To test the effects of substitutions at the four different Lys residues in SacAcsA in vivo, alleles encoding wild-type and variant SacAcsA proteins were introduced into an S. enterica Δacs strain carrying a chromosomal deletion of the acs gene. The resulting strains were grown at 37°C in 5 ml of the minimal acetate medium (10 mM acetate as a sole carbon source), and growth was monitored in triplicate at an OD600 (30). As shown in Fig. 5B, differences in growth behavior were observed. For example, the strain synthesizing SacAcsAK628Q failed to grow on 10 mM acetate as a sole carbon and energy source; growth of the strain that synthesized SacAcsAK611Q was strongly reduced, reaching only 60% of the cell density of the strain synthesizing SacAcsAWT; strains that synthesized SacAcsAK237Q or SacAcsAK380Q grew as well as the strain synthesizing SacAcsAWT. The in vivo experimental results were consistent with the activity of the SacAcsA variants measured in vitro (Fig. 5A).
To further investigate the mechanism by which each acetylation site reduces SacAcsA activity, enzyme kinetic studies of the SacAcsAWT and its variants were performed. The results are shown in Table 2. The mutants at the K237, K380, and K611 sites exhibited kcat values of 35 to 40 s−1, which were very similar to the turnover number for the wild-type enzyme (33.5 s−1). The kcat values of SacAcsAK628Q was 2.7 s−1, about 12.4-fold lower than the value for SacAcsAWT. These results indicated that it was the K628 site, not K237, K380, or K611, that was directly involved in enzyme catalytic activity. The crystal structure of benzoyl-CoA synthetase from Burkholderia xenovorans (RCSB Protein Data Bank [PDB] 2V7B) shows that a conserved lysine residue (such as K628) in its active site forms two hydrogen bonds with the carboxylate group of the substrate and orients the acid substrate in the active site. This process is critical to the first step of the SacAcsA catalysis reaction, which ordinarily consumes ATP to form an acyl-adenylate intermediate, releasing pyrophosphate. The presence of the acetyl moiety would block the interactions between the ε-amino group of lysine and the carboxylate group of the substrate (31). Thus, Lys628 in a putative acylation motif was shown to be important for catalysis of the overall reaction. The K628Q and K611Q mutants increased Km values for acetate (5.5-fold and 2.2-fold increases, respectively), compared with the wild-type protein, whereas the mutants at the K237 and K380 sites revealed Km values similar to the value of the wild-type protein. The above observations indicate a possible role of K628 and K611 in binding the acetate substrate. It is worth noting that the K611 mutants had no obvious effect on the kcat value but increased the Km value, suggesting that K611 may be involved in binding acetate but is not needed for catalysis. Kinetic analysis of the K628Q variants revealed a significantly reduced catalytic efficiency (kcat/Km) value for acetate (67-fold decrease).
TABLE 2.
Kinetic analysis of WT SacAcsA and its variants
| SacAcsA enzyme | Km (acetate [mM]) | kcat (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|
| WT | 3.3 ± 0.3 | 33.5 ± 6.2 | (1.0 ± 0.3) × 104 |
| K237Q | 3.3 ± 0.2 | 37.5 ± 1.2 | (1.0 ± 0.1) × 104 |
| K380Q | 2.8 ± 0.5 | 40.1 ± 1.4 | (1.5 ± 0.3) × 104 |
| K611Q | 7.3 ± 0.1 | 34.8 ± 3.4 | (5.0 ± 0.5) × 103 |
| K628Q | 18.1 ± 0.1 | 2.7 ± 0.1 | (1.5 ± 0.1) × 102 |
SacAcsAAc is deacetylated and reactivated by the sirtuin-type deacetylase SacSrtN.
Acetylation of acetyl-CoA synthetase is reversed by deacetylases. It has been found that there are two types of protein deacetylases responsible for deacetylation of proteins in prokaryotes. Bacterial sirtuin-like deacetylases, reported first in S. enterica (5), use NAD+ as the substrate to deacetylate proteins. In so doing, sirtuins transfer the acetyl group from the protein to ADP-ribose, producing nicotinamide and 2′-O-acetyl-ADP-ribose as products. The other type of AcuC-like deacetylase, reported first in B. subtilis (12), is a zinc-dependent member of the class IIa histone deacetylases that uses water to hydrolyze the acetyl moiety, releasing acetate, as seen in reactions catalyzed by BsAcuC and RpLdaA. Bioinformatics analysis of the S. erythraea genome identified the gene SACE_3798 (predicted to encode a 259-residue protein, here referred to as SacSrtN) as homologous to the S. enterica cobB (35%), which encodes an NAD+-dependent deacetylase sirtuin. We also found gene SACE_1779, which encodes a putative zinc-dependent deacetylase enzyme (predicted to be 391 amino acids long) that is homologous to the B. subtilis acuC gene (40%). A phylogenetic analysis with the full-length sequence of SacSrtN and the eight CobB-like deacetylases previously reported showed that SacSrtN clusters with the Streptomyces coelicolor CobB1 (ScCobB1) and ScCobB2 (Fig. 6A).
FIG 6.

SacSrtN deacetylated acetylated acetyl-CoA synthetase. (A) Phylogenetic analysis with SacSrtN and the eight protein deacetylases: SeCobB (STM1221), RpSrtN (formerly rpa2524), EcCobB (b1120), MsCobB (MSMEG_5175), MtCobB (Rv1151c), ScCobB1 (SCO0452), ScCobB2 (SCO6464), and BsSrtN (yhdZ; BSU09650). (B) Deacetylation of SacAcsA by SacSrtN. Acetylated SacAcsA was incubated with or without SacSrtN and 1 mM NAD+ at 37°C for 3 h. After incubation, samples were collected and analyzed by SDS-PAGE, and the acetylation levels were determined by Western blotting using specific anti-AcK antibody. (C) Effect of deacetylation on the activity of SacAcsA. Data are expressed as means plus standard deviations of three repeated assays. (D) Time-dependent reactivation of acetylated SacAcsA by deacetylation. SacAcsA activity was measured at different time intervals during the incubation with SacSrtN. ■, 0.5 μM SacSrtN and 1 mM NAD+; ●, 0.5 μM SacSrtN; ▲, 5 μM acetylated SacAcsA. Each data point represents the average from three repeated assays.
Previous works demonstrated that the S. enterica SeCobB sirtuin deacetylated efficiently and reactivated acetylated acetyl-CoA synthetase (5). To assess the deacetylase activity of SacSrtN, S. erythraea AcsA (SacAcsA) was acetylated by SacAcuA with acetyl-CoA, and the product of the reaction was used as the substrate for in vitro deacetylation activity assays. The acetylated SacAcsA was incubated with SacSrtN in the absence and presence of NAD+. The acetylation level of SacAcsA was monitored by Western blotting using antibody against acetyl-lysine (Fig. 6B). In the presence of SacSrtN and NAD+, the acetylation level of SacAcsA was found to be significantly decreased (almost undetectable), indicating that SacSrtN removed acetyl groups from acetyl-lysine residues of SacAcsA in an NAD+-dependent manner. These results also showed that SacAcsA acetylation was reversible. After deacetylation, SacAcsA activity was restored to a level comparable to that of the nonacetylated SacAcsA control (Fig. 6C). Time-dependent activation of SacAcsA by SacSrtN deacetylation was also studied. As shown in Fig. 6D, an increase in SacAcsA activity over time was observed during deacetylation by the SacSrtN enzyme. The kinetic parameters of SacSrtN for deacetylation of acetylated SacAcsA were determined. The resulting data are shown in Table 1. The Km and kcat values for SacAcsAAc were 52 μM and 0.03 s−1. Similarly, The Km and kcat values for NAD+ were 240 μM and 0.05 s−1.
SacAcsA is also acetylated in vivo at multiple lysine residues.
We have provided biochemical evidence to show that SacAcsA is a substrate of SacAcuA and SacSrtN in vitro; however, it was important to determine whether SacAcsA could be utilized as a substrate of SacAcuA and SacSrtN in vivo. To investigate whether SacAcsA was posttranslationally modified in vivo, we performed an immunoprecipitation experiment to assess the acetylation state of SacAcsA in S. erythraea. SacAcsA isolated from S. erythraea was subjected to trypsin digestion, and the resulting peptides were analyzed by LC-MS/MS. Sequence determination of the peptides identified K237, K380, K611, and K628 (in boldface) as multiple sites of acetylation of SacAcsA. MS/MS spectra of the peptides TK(237)TDVEWNDGR, TFMK(380)WGAEIPAR, DHVAHEIGPIAK(611)PR, and SGK(628)IMR are shown in Fig. S7 in the supplemental material, in agreement with the results obtained from in vitro acetylation experiments. Based on these data, we inferred that, in vivo, SacAcsA was acetylated at multiple sites by the SacAcuA acetyltransferase.
In vivo, SacAcsA acetylation is influenced by extracellular nutrient availability or lack of enzymes.
To provide more direct evidence for acetylation and deacetylation of endogenous SacAcsA by SacAcuA and SacSrtN in vivo, we also analyzed the acetylation status of endogenous SacAcsA in three strains (the wild-type, ΔSacacuA, and ΔSacsrtN strains). Immunoprecipitation experiments were conducted with 300-μg protein samples from three strains. Anti-SacAcsA antibody was used to monitor the amount of SacAcsA enzyme. Immunoblotting with anti-acetyl-lysine antibody showed that the acetylation level of endogenous SacAcsA in the ΔSacacuA (ΔSACE_5148) strain was much lower than that in the wild-type and ΔSacsrtN (ΔSACE_3798) strains (Fig. 1A and 7A). SacAcsA amounts were comparable in all three strains (Fig. 1A), indicating that endogenous SacAcsA is acetylated in vivo and that it may be a substrate for the SacAcuA acetyltransferase and SacSrtN deacetylase.
FIG 7.
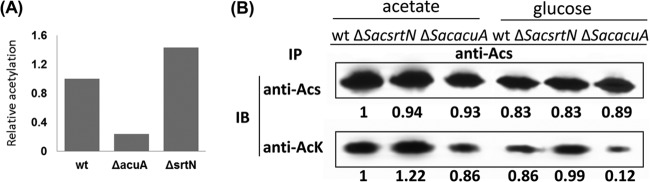
Analysis of SacAcsA acetylation in vivo. (A) Relative acetylation of the results shown in Fig. 1A. (B) WT, ΔSacsrtN, and ΔSacacuA strains of Saccharopolyspora erythraea expressing SacAcsA were grown in minimal medium with acetate or glucose as a carbon source. Culture samples were collected as a function of time, cells were analyzed, and SacAcsA was immunoprecipitated with anti-SacAcsA antibody and subjected to Western analysis. Immunoblotting was performed with anti-SacAcsA and anti-AcK antibodies. Representative blots are shown. The band intensities were quantified by densitometry using ImageJ software.
SacAcsA is involved in carbon metabolism. We therefore investigated the acetylation of SacAcsA under culture conditions with acetate or glucose as carbon and energy sources. Endogenous SacAcsA enzymes were immunoprecipitated from S. erythraea wild-type, ΔSacacuA, and ΔSacsrtN strains grown in minimal medium supplemented with acetate or glucose. The acetylation state of SacAcsA was assessed by immunoblotting with anti-AcK antibody (Fig. 7B). We found that the amount of Acs enzyme in three strains in the presence of acetate increased slightly compared with culture in the presence of glucose. The level of SacAcsA acetylation also revealed a similar pattern. It is worth noting that inactivation of SacAcuA (in the ΔSacacuA strain) led to a significant decrease of acetylation in glucose-grown cells.
In summary, we identified the Gcn5-like protein acetyltransferase SacAcuA responsible for acetylation of acetate-scavenging acetyl-CoA synthetase SacAcsA in S. erythraea. Acetylated AcsA then is deacetylated by a sirtuin-type NAD-dependent deacetylase SacSrtN. In vitro acetylation/deacetylation of SacAcsA enzyme was verified by Western blotting, mass spectrometry, and gene knockout (ΔSacacuA), and acetylated lysine residues including Lys237, Lys380, Lys611, and Lys628 were identified. The acetyltransferase SacAcuA revealed a unique feature that can acetylate multiple lysine sites, unlike other previously reported bacterial acetyltransferases which acetylate only one lysine residue in the conserved putative acylation motif PXXXXGK in AMP-forming acyl-CoA synthetases. The activity of SacAcsA is controlled by lysine acetylation. Site-specific mutagenesis experiments were conducted to investigate the effects of the acetylated lysine residues on the activity of SacAcsA. The immunoprecipitation data showed that in vivo acetylation of SacAcsA is influenced by glucose and acetate availability. These results suggest that reversible acetylation may also be a conserved regulatory PTM strategy in antibiotic-producing actinomycetes.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the China NSF 21276079, SRFDP 20120074110009 of the Chinese Ministry of Education, the National Key Technologies R&D Programs (2007AA02Z331 and 2014AA021502), and the Fundamental Research Funds for the Central Universities. J.C.E.-S. was supported by USPHS grant R01 GM062203.
Footnotes
Published ahead of print 23 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01961-14.
REFERENCES
- 1.Bernal V, Castaño-Cerezo S, Gallego-Jara J, Ecija-Conesa A, de Diego T, Iborra JL, Cánovas M. 15 March 2014. Regulation of bacterial physiology by lysine acetylation of proteins. New Biotechnol. 10.1016/j.nbt.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Ahao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. 2010. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327:1004–1007. 10.1126/science.1179687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thao S, Escalante-Semerena JC. 2011. Control of protein function by reversible Nε-lysine acetylation in bacteria. Curr. Opin. Microbiol. 14:200–2004. 10.1016/j.mib.2010.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiong Y, Guan KL. 2012. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 198:155–164. 10.1083/jcb.201202056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. 2002. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298:2390–2392. 10.1126/science.1077650 [DOI] [PubMed] [Google Scholar]
- 6.Tucker AC, Escalante-Semerena JC. 2013. Acetoacetyl-CoA synthetase activity is controlled by a protein acetyltransferase with unique domain organization in Streptomyces lividans. Mol. Microbiol. 87:152–167. 10.1111/mmi.12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu X, Vellaichamy A, Wang D, Zamdborg L, Kelleher NL, Huber SC, Zhao Y. 2013. Differential lysine acetylation profiles of Erwinia amylovora strains revealed by proteomics. J. Proteomics 79:60–71. 10.1016/j.jprot.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okanishi H, Kim K, Masui R, Karamitsu S. 2013. Acetylome with structural mapping reveals the significance of lysine acetylation in Thermus thermophiles. J. Proteomics Res. 12:3952–3968. 10.1021/pr400245k [DOI] [PubMed] [Google Scholar]
- 9.Lee DW, Kim DI, Lee YJ, Choi JY, Kang S, Pan JG. 2013. Proteomic analysis of acetylation in thermophilic Geobacillus kaustophilus. Proteomics 13:2278–2282. 10.1002/pmic.201200072 [DOI] [PubMed] [Google Scholar]
- 10.Yu BJ, Kim JA, Moon JH, Ryu SE, Pan JG. 2008. The diversity of lysine-acetylated proteins in Escherichia coli. J. Microbiol. Biotechnol. 18:1529–1536 [PubMed] [Google Scholar]
- 11.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. 2009. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8:215–225. 10.1074/mcp.M800187-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardner JG, Grundy FJ, Henkin TM, Escalante-Semerena JC. 2006. Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD+ involvement in Bacillus subtilis. J. Bacteriol. 188:5460–5468. 10.1128/JB.00215-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castaño-Cerezo S, Bernal V, Blanco-Catala J, Iborra JL, Canovas M. 2011. cAMP-CRP co-ordinates the expression of the protein acetylation pathway with central metabolism in Escherichia coli. Mol. Microbiol. 82:1110–1128. 10.1111/j.1365-2958.2011.07873.x [DOI] [PubMed] [Google Scholar]
- 14.Crosby HA, Heiniger EK, Harwood CS, Escalante-Semerena JC. 2010. Reversible N-lysine acetylation regulates the activity of acyl-CoA synthetases involved in anaerobic benzoate catabolism in Rhodopseudomonas palustris. Mol. Microbiol. 76:874–888. 10.1111/j.1365-2958.2010.07127.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu H, Hegde SS, Blanchard JS. 2011. Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry 50:5883–5892. 10.1021/bi200156t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starai VJ, Takahashi H, Boeke JD, Escalante-Semerena JC. 2003. Short-chain fatty acid activation by acyl-coenzyme A synthetases requires SIR2 protein function in Salmonella enterica and Saccharomyces cerevisiae. Genetics 163:545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumari S, Beatty CM, Browning DF, Busby SJ, Simel EJ, Hovel-Miner G, Wolfe AJ. 2000. Regulation of acetyl coenzyme A synthetase in Escherichia coli. J. Bacteriol. 182:4173–4179. 10.1128/JB.182.15.4173-4179.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browning DF, Beatty CM, Sanstad EA, Gunn KE, Busby SJ, Wolfe AJ. 2004. Modulation of CRP-dependent transcription at the Escherichia coli acsP2 promoter by nucleoprotein complexes: anti-activation by nucleoid proteins FIS and IHF. Mol. Microbiol. 51:241–254. 10.1046/j.1365-2958.2003.03824.x [DOI] [PubMed] [Google Scholar]
- 19.Starai VJ, Escalante-Semerena JC. 2004. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J. Mol. Biol. 340:1005–1012. 10.1016/j.jmb.2004.05.010 [DOI] [PubMed] [Google Scholar]
- 20.Crosby HA, Pelletier DA, Hurst GB, Escalante-Semerena JC. 2012. System-wide studies of N-lysine acetylation in Rhodopseudomonas palustris reveal substrate specificity of protein acetyltransferases. J. Biol. Chem. 287:15590–15601. 10.1074/jbc.M112.352104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nambi S, Basu N, Visweswariah SS. 2010. Cyclic AMP-regulated protein lysine acetylases in mycobacteria. J. Biol. Chem. 285:24313–24323. 10.1074/jbc.M110.118398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikulik K, Felsberg J, Kudrnacova E, Bezouskova S, Setinova D, Stodulkova E, Zidkova J, Zidek V. 2012. CobB1 deacetylase activity in Streptomyces coelicolor. Biochem. Cell Biol. 90:179–187. 10.1139/o11-086 [DOI] [PubMed] [Google Scholar]
- 23.Hui L, Huang X-S, Liu D-Q, Ahao W, Fan W, Han S, Zhang B-C. 2009. Rapid chromosomic gene-inactivating technology of Saccharopolyspora erythraea. Bull. Acad. Mil. Med. Sci. 33:365–369 [Google Scholar]
- 24.van den Berg MA, de Jong-Gubbels P, Kortland CJ, va Kijken JP, Pronk JT, Steensma HY. 1996. The two acetyl-coenzyme a synthetases of Saccharomyces cerevisiae differ with respect to kinetic properties and transcriptional regulation. J. Biol. Chem. 271:28953–28959. 10.1074/jbc.271.46.28953 [DOI] [PubMed] [Google Scholar]
- 25.Wisniewski JR, Zougman A, Naqaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat. Methods 6:359–362. 10.1038/nmeth.1322 [DOI] [PubMed] [Google Scholar]
- 26.Takamura Y, Nomura G. 1988. Changes in the intracellular concentration of acetyl-CoA and malonyl-CoA in relation to the carbon and energy metabolism of Escherichia coli K12. J. Gen. Microbiol. 134:2249–2253. 10.1099/00221287-134-8-2249 [DOI] [PubMed] [Google Scholar]
- 27.Gardner JG, Escalante-Semerena JC. 2008. Biochemical and mutational analyses of AcuA, the acetyltransferase enzyme that controls the activity of the acetyl coenzyme a synthetase (AcsA) in Bacillus subtilis. J. Bacteriol. 190:5132–5136. 10.1128/JB.00340-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanner KG, Langer MR, Kim Y, Denu JM. 2000. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J. Biol. Chem. 275:22048–22055. 10.1074/jbc.M002893200 [DOI] [PubMed] [Google Scholar]
- 29.Dyda F, Klein DC, Hickman AB. 2000. GCN5-related N-acetyltransferases: a structural overview. Annu. Rev. Biophys. Biomol. Struct. 29:81–103. 10.1146/annurev.biophys.29.1.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan CH, Garrity J, Crosby HA, Escalante-Semerena JC. 2011. In Salmonella enterica, the sirtuin-dependent protein acylation/deacylation system (SDPADS) maintains energy homeostasis during growth on low concentrations of acetate. Mol. Microbiol. 80:168–183. 10.1111/j.1365-2958.2011.07566.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bains J, Boulanger MJ. 2007. Biochemical and structural characterization of the paralogous benzoate CoA ligases from Burkholderia xenovorans LB400: defining the entry point into the novel benzoate oxidation (box) pathway. J. Mol. Biol. 373:965–977. 10.1016/j.jmb.2007.08.008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.