

Abstract
The gastric pathogen Helicobacter pylori possesses a highly active urease to support acid tolerance. Urea hydrolysis occurs inside the cytoplasm, resulting in the production of NH3 that is immediately protonated to form NH4+. This ammonium must be metabolized or effluxed because its presence within the cell is counterproductive to the goal of raising pH while maintaining a viable proton motive force (PMF). Two compatible hypotheses for mitigating intracellular ammonium toxicity include (i) the exit of protonated ammonium outward via the UreI permease, which was shown to facilitate diffusion of both urea and ammonium, and/or (ii) the assimilation of this ammonium, which is supported by evidence that H. pylori assimilates urea nitrogen into its amino acid pools. We investigated the second hypothesis by constructing strains with altered expression of the ammonium-assimilating enzymes glutamine synthetase (GS) and glutamate dehydrogenase (GDH) and the ammonium-evolving periplasmic enzymes glutaminase (Ggt) and asparaginase (AsnB). H. pylori strains expressing elevated levels of either GS or GDH are more acid tolerant than the wild type, exhibit enhanced ammonium production, and are able to alkalize the medium faster than the wild type. Strains lacking the genes for either Ggt or AsnB are acid sensitive, have 8-fold-lower urea-dependent ammonium production, and are more acid sensitive than the parent. Additionally, we found that purified H. pylori GS produces glutamine in the presence of Mg2+ at a rate similar to that of unadenylated Escherichia coli GS. These data reveal that all four enzymes contribute to whole-cell acid resistance in H. pylori and are likely important for assimilation and/or efflux of urea-derived ammonium.
INTRODUCTION
Helicobacter pylori is a human pathogen that causes chronic infection of the gastric mucosa. Colonization and persistence of this neutralophile in an acidic environment depend upon active intracellular urease, which comprises between 5 and 10% of the total cellular protein (1). Urease catalyzes the hydrolysis of urea into ammonia (NH3), which scavenges protons to mitigate acidity, and carbon dioxide (CO2) (2). A pair of carbonic anhydrases convert the CO2 to HCO3−, buffering the periplasmic space (3, 4).
Urea is required for H. pylori acid survival below pH 4. This molecule is a by-product of human amino acid metabolism and is present in gastric mucus at concentrations ranging from 1 to 8 mM (5, 6). Urea can passively diffuse across the bacterial inner membrane; however, when H. pylori is challenged with high acidity, a urea-specific permease (UreI) opens to allow faster influx of urea (7–9). H. pylori intracellular urease is both catalytically efficient and highly abundant (10, 11), and its activity far exceeds that of other ureases (12–14). As a consequence, H. pylori produces unusually large amounts of urea-derived ammonium.
Most of this urea-derived ammonium is thought to be effluxed outward across the inner membrane via an energy-dependent mechanism rather than by passive diffusion (15–18). Surprisingly, H. pylori lacks homologs to known NH3/NH4+ transporters, and therefore, it is unclear how NH4+, which is charged, can rapidly exit across the inner membrane. One possibility is that UreI may be capable of extruding NH3 and/or NH4+ (19).
A second possible fate for ammonium is its assimilation into organic nitrogen. Previous studies using 15N-labeled urea show that H. pylori incorporates this nitrogen into amino acids, initially and primarily glutamate and glutamine (20). Furthermore, both glutamate dehydrogenase (GDH) and glutamine synthetase (GS) activities have been detected in this organism (2, 21). The glnA gene from H. pylori was successfully used to complement an Escherichia coli glutamine auxotroph (21), substantiating the enzyme's role in glutamine synthesis. GS was further identified as a possible interacting partner with urease, implicating this enzyme as a player in the metabolism of urea-derived nitrogen (22).
In this investigation, we clarified the roles of both GS and GDH as enzymes responsible for ammonium assimilation in H. pylori. We also studied two amino acid deamidases, glutaminase (Ggt) and asparaginase (AsnB). These enzymes have been studied for their roles in host cell cytotoxicity (23, 24) and in suppression of the host immune response (25); however, our interest pertains to their function as periplasmic amino acid deamidases (26). These four enzymes (GS, GDH, Ggt, and AsnB) all appear to be involved in regulating either (i) the hydrolysis of urea inside the cell or (ii) the extent to which this ammonium is extruded versus assimilated. Our study therefore provides a link between urease-mediated acid resistance in H. pylori and the metabolism of this urea-derived ammonium.
MATERIALS AND METHODS
Strains and growth conditions.
H. pylori strains were routinely grown on brucella agar plates (Oxoid Ltd., Hampshire, England) supplemented with 10% defibrinated sheep's blood (QuadFive, Ryegate, MT) (BA plates) or supplemented with 5% newborn calf serum (Atlanta Biologicals, Lawrenceville, GA) (NCS plates). Cells were grown in incubators that maintain temperature and gas concentrations at 37°C and 5% CO2 and 2% O2, with the balance N2. The following antibiotics were supplemented to the plates as needed: chloramphenicol (30 μg/ml), kanamycin (25 μg/ml), and/or erythromycin (15 μg/ml). Escherichia coli strains were grown on Luria-Bertani (LB) plates at 37°C and atmospheric gas concentrations. Plates were supplemented with ampicillin (100 μg/ml) or chloramphenicol (30 μg/ml) as needed.
Construction of EM210 and EM210.
E. coli strain DH5α was used for all plasmid DNA manipulations. The genes glnA (hp0512) and gdhA (hp0380) were amplified from H. pylori 26695 genomic DNA using iProof (Bio-Rad) high-fidelity polymerase, along with the appropriate primers (Table 1). This PCR product was digested using NdeI/XhoI and ligated into a similarly digested pPA plasmid (27) to create pPAgln or pPAgdh. In this construct, the inserted gene is located downstream of the ureA promoter. Digestion of pPAgln or pPAgdh with BglII/XhoI released the fragment, which was subsequently treated with T4 DNA polymerase to create blunt ends. These fragments were ligated into pEU39Cm (28), which had been previously digested with EcoRV and treated with alkaline phosphatase (AP). In this final construct, a sandwich fusion was obtained in which UreAp-hp0512 or UreAp-hp0380 is located between regions of DNA that are homologous to the hp0405 locus. H. pylori 26695 was transformed naturally, and mutant strains were selected for on BA plates containing chloramphenicol. Verification of the desired genotype was performed using PCR and gel electrophoresis, followed by sequencing of the PCR product (Georgia Genomics Facility).
TABLE 1.
Oligonucleotide primers used in this study
| Name | Sequence (5′→3′) |
|---|---|
| Cat-5 | GATATAGATTGAAAAGTGGAT |
| Cat-6 | TTATCAGTGCGACAAACTGGG |
| AphA5 | CAAGACGATAAATGCGTC |
| AphA6 | CTAGGTACTAAAACAATTC |
| ErmCatFus1 | TTACGGAGGATAAATGATGAACAAAAATATAAAATATTC |
| ErmCatFus2 | GAATATTTTATATTTTTGTTCATCATTTATCCTCCGTAA |
| Erm6 | TTATTTCCTCCCGTTAAAT |
| GlnAUP | CCGCGTCATATGATAGTAAGAACTC |
| GlnAREV | CAGAGTCTCGAGTTAGCATGAATAAGTGGTG |
| GdhAUP | CCGCGTCATATGCCGCTCATGTATGTTG |
| GdhAREV | CAGAGTCTCGAGTCAAACCCCTTGCGC |
| HP0511-1 | GCCTTTAATATCGCTGAATC |
| HP0511-2 | ATCCACTTTTCAATCTATATCCATCTTATCATTCCCACC |
| HP0511-3 | CCCAGTTTGTCGCACTGATAAGGGTGGTAGGAATAGGC |
| HP0511-4 | TCTTTCGTGATCGCTCC |
| AsnB1 | TTGGCGCAAGGTAATTG |
| AsnB2 | ATCCACTTTTCAATCTATATCAACCTGCCCCTTCAAAC |
| AsnB3erm | ATTTAACGGGAGGAAATAAGATTCTCTTAAATCACCCA |
| AsnB4 | CTACATGGCTTAATTCTC |
| UreI1 | GAAGAATTAGGGCTTG |
| UreI2 | GTATAACATAGTATCGACATTGCCTTATCCTTCC |
| UreI3 | GAATTGTTTTAGTACCTAGCCTGCTTGGTTACTCTT |
| UreI4 | TTCTAATAACGCTAGCG |
| Ggt1 | AACTCTCTATTTCGCAAG |
| Ggt2 | GTATAACATAGTATCGACCCTTTCAATCAGTTACG |
| Ggt3 | GAATTCTTTTAGTACCTAGAACGGATCCAAGGAAAG |
| Ggt4 | TTGTTCAAGGTGTTCTGC |
Construction of deletion mutants.
All strains except EM210 and EM211 were created using overlapping PCR followed by allelic-exchange mutagenesis of the desired locus. H. pylori 26695 genomic DNA was used as a template to amplify short (400- to 500-bp) fragments located up- and downstream of the target gene. Primers to amplify the upstream region were given the designations 1 and 2, while primers to amplify the dowstream region were designated 3 and 4. Sections of 16 to 20 bp at the 5′ ends of primers 2 and 3 were designed to anneal to the upstream and downstream ends of the antibiotic resistance cassettes, which were amplified using the following primers: Cat-5 and Cat-6 for cat (encoding chloramphenicol resistance) and AphA5 and AphA6 for aphA3 (encoding kanamycin resistance). The cat-ermB cassette (encoding erythromycin resistance) was constructed by placing the ermB gene from Enterococcus faecalis under the control of the cat promoter. To this end, the cat promoter was amplified using primers Cat-5 and ErmCatFus1, while ermB was amplified using primers ErmCatFus2 and Erm6. Overlapping PCR using these two amplicons as templates resulted in the fusion Φcatp-ermB. To create the final DNA fragment for transformation into H. pylori, overlapping PCR was carried out using as templates the regions upstream and downstream of the target locus, along with the desired antibiotic resistance cassette. This fragment was purified following gel electrophoresis and was used to transform the H. pylori parent strain naturally. Mutant strains were selected on BA plates containing the appropriate antibiotic. Mutant genotypes were verified by PCR and gel electrophoresis and by sequencing of the PCR product using either primer 1 or 4 in accordance with the gene locus (Georgia Genomics Facility). Strains and plasmids used are listed in Table 2.
TABLE 2.
Strains and plasmids used in this study
| Strain or plasmid | Characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| H. pylori | ||
| 26695 | Parent strain | ATCC |
| EM210 | hp0405::(ureAp-glnA-cat) Cmr | This study |
| EM211 | hp0405::(ureAp-gdhA-cat) Cmr | This study |
| EM214 | Δggt Kanr | This study |
| EM222 | ΔasnB Ermr | This study |
| EM223 | ΔasnB Δggt Ermr Kanr | This study |
| EM215 | ΔureI Kanr | This study |
| EM216 | EM210 parent; Δggt Cmr Kanr | This study |
| EM224 | EM210 parent; ΔasnB Cmr Ermr | This study |
| EM226 | EM210 parent; ΔasnB Δggt Cmr Ermr Kanr | This study |
| EM217 | EM211 parent; Δggt Cmr Kanr | This study |
| EM225 | EM211 parent; ΔasnB Cmr Ermr | This study |
| EM227 | EM211 parent; ΔasnB Δggt Cmr Ermr Kanr | This study |
| EM230 | ΔureAB Kanr | This study |
| EM231 | EM210 parent; ΔureAB Cmr Kanr | This study |
| EM232 | EM211 parent; ΔureAB Cmr Kanr | This study |
| E. coli DH5α | Cloning strain | This study |
| Plasmids | ||
| pPA | pET21b derivative in which ureAp (ureA promoter region) replaces T7p to promote gene expression in H. pylori; Ampr | 6 |
| pEU39cm | Suicide vector used for homologous recombination into hp0405; Apr Cmr | 31 |
Acid survival assay.
H. pylori strains were grown overnight on BA plates, harvested, and washed once with 20 mM phosphate-buffered saline (PBS; pH 7.0). The acid challenge buffer was made fresh that day and contained 20 mM citrate, 155 mM Na2HPO4, and 20 mM urea (pH 2.8). Cells were suspended in this buffer at a concentration of 5 × 107 CFU/ml and were incubated at 37°C and 2% O2 for a period of 6 h. The viable H. pylori cells were measured at 1.5-h intervals by incubating serial dilutions on NCS plates. Colonies were counted after 4 to 5 days at 37°C and 2% O2, and the results were expressed as CFU/ml. The pH of the medium over time was measured using a pH-sensitive electrode.
Extracellular urea-dependent ammonium production by whole cells.
Ammonium was measured using a protocol modified from that of Weatherburn (29). Following overnight growth on BA plates, H. pylori cells were harvested and washed twice with 40 mM HEPES buffer (pH 7.0), and the optical density at 600 nm (OD600) was adjusted to 1.0. The reaction mixture contained 40 mM HEPES and 5 mM urea and was equilibrated to 37°C. Ammonium production was initiated by adding 3 × 107 bacterial cells (30 μl) to 240 μl of reaction mixture. After 1 or 5 min, cells were removed from the 37°C water bath and centrifuged for 2 min at 6,000 × g. A total of 240 μl of supernatant was removed, and the ammonium concentration was measured as described previously (29). A standard curve containing NH4Cl was used to correlate the absorbance at 625 nm to the concentration of ammonium. The rate of ammonium production over time was calculating based on the difference in ammonium concentrations between the two time points.
Glutamate dehydrogenase assay.
H. pylori glutamate dehydrogenase activity in cell extracts was measured by monitoring α-ketoglutarate-dependent NADPH oxidation over time as previously described (30). H. pylori cells were harvested after overnight growth on BA plates and washed twice in 100 mM Tris-HCl (pH 8.0). Cells were sonicated twice for 10 s, and cell extracts were obtained by collecting the supernatant following centrifugation at 14,000 × g for 10 min. Cell extracts (125 μg) were added directly to a quartz cuvette containing a reaction mixture consisting of 10 mM Tris-HCl (pH 8.5), 10 mM NH4Cl, 2.5 mM α-ketoglutarate, and 0.05 mM NADPH (25°C) and mixed thoroughly with a Pasteur pipette. The rate of disappearance of NADPH was measured by monitoring the decrease in absorbance at 340 nm at 10-s intervals for 5 min. In order to account for endogenous NADPH oxidase activity, the rate of a control reaction lacking α-ketoglutarate was subtracted from the experimental rate in the presence of α-ketoglutarate. The concentration of NADPH was calculated using the millimolar extinction coefficient (ε) of 6.22.
Glutamine synthetase assays.
Glutamine synthetase (GS) activity in cell lysates was measured using the γ-glutamyltransferase forward reaction (31). Cells were grown overnight on BA plates and washed in 100 mM imidazole hydrochloride (pH 7.7), and cell extracts were prepared as described above. The reaction mixture contained the following: 100 mM imidazole hydrochloride (pH 7.7), 50 mM hydroxylamine, 60 mM MgCl2, 100 mM l-glutamate, and 25 μg of cell extract. The reaction mixture was equilibrated to 37°C, and catalysis was initiated by adding ATP at a final concentration of 12 mM. The reaction was quenched using a 2-volume excess of stop solution containing 0.2 M ferric chloride, 0.12 M trichloroacetic acid, and 2% HCl. After 45 min of incubation, samples were centrifuged to remove remaining precipitate, and the absorbance was measured at 540 nm. The concentration of γ-glutamyl hydroxamate (GGH) was calculated by comparing to a standard curve of known GGH concentrations.
Glutamine synthetase activity of purified HP0512 was measured based on modifications of a previous method in which radiolabeled substrate and product are separated from each other using thin-layer chromatography (TLC) (32). A reaction mixture was prepared containing 50 mM imidazole hydrochloride (pH 7.7), 30 mM [14C]glutamate (1 μCi/ml), 30 mM NH4Cl, 40 mM MgCl2 (or 4 mM MnCl2), and 10 ng of purified HP0512. After equilibration to 37°C, the reaction was initiated by adding 10 mM ATP. Aliquots of 100 μl were removed every 5 min and quickly heated to 95°C to quench the reaction. Two microliters of the quenched reaction was spotted onto a 250-μm silica gel TLC plate (Analtech, Newark, DE). TLC plates were developed for 6 h using a previously optimized solvent system (33), which contained tert-butanol-acetic acid-H2O (25:6:10). Amino acid spots were visualized by spraying plates with 2% ninhydrin (wt/vol) in ethanol. Glu and Gln spots were scraped off using a razor blade, and the radioactivity was measured using a scintillation counter.
Glutaminase and asparaginase assays.
Glutamine- or asparagine-dependent ammonium production by whole cells was measured using a combination of two methods (29, 34). H. pylori cells grown overnight on BA plates were harvested and washed in 40 mM HEPES-buffered saline. Cells were resuspended at a final OD600 of 1.0, and 30 μl (3 × 107 CFU/ml) of this suspension was added to 220 μl of a reaction mixture containing 40 mM HEPES and 10 mM glutamine or 10 mM asparagine (pH 7.0). Reactions were quenched by direct addition of stop solution into the 250-μl reaction, and the ammonium concentration was determined as previously described (29). Ammonium was measured at 1 min and 10 min for glutaminase assays and at 1 min and 20 min for asparaginase assays. The rate of ammonium production over time was obtained by calculating the difference in ammonium concentrations between the two time points.
Western blot against urease.
Levels of urease protein were compared between strains using monoclonal anti-UreA rabbit IgG (Santa Cruz Biotechnology, CA). The wild type and mutant strains EM210 and EM211 were grown overnight on BA plates and washed twice in PBS (pH 7.0). Cell extracts were prepared, and 10 μg of protein was separated using SDS-PAGE (10% or 4 to 20% polyacrylamide [NuSep]) along with a prestained molecular weight marker (Bio-Rad). Proteins were transferred onto a 0.45-μm-pore-size nitrocellulose membrane using an electrical transfer apparatus. Transfer was carried out in Towbin buffer (0.025 M Tris, 0.192 M glycine, 15% methanol, 0.1% SDS [pH 8.3]) at 350 mA for 1 h. The membrane was then blocked overnight at 4°C using 3% (wt/vol) gelatin in 0.02 M Tris-HCl (pH 7.6) and 0.1 M NaCl (TBS). The membrane was washed twice using 0.1% Tween 20 in TBS (TTBS), followed by a 1.5-h incubation at room temperature with anti-UreA (1:2,000 dilution) in TTBS containing 1% (wt/vol) gelatin. The membrane was washed three times in TTBS prior to incubation with goat anti-rabbit IgG (H+L)-AP conjugate (Bio-Rad; 1:2,000 dilution). The membrane was briefly rinsed in TBS, and bands containing bound antibody were visualized by incubating the membrane in AP buffer (0.02 M Tris-HCl [pH 9.5], 0.1 M NaCl) in the presence of 125 μg/ml of 5-bromo-4-chloro-30 -indolyl phosphate (BCIP) and 300 μg/ml of Nitro Blue Tetrazolium (NBT). Band intensities were compared using the densitometry analysis program Labworks (Ultra-Violet Products Ltd.).
RESULTS
hp0512 and hp0380 encode the ammonium assimilation enzymes GS and GDH.
GS catalyzes the ATP-dependent synthesis of glutamine from NH4+ and glutamate, while GDH catalyzes the reversible conversion of α-ketoglutarate and NH4+ to glutamate. Homologs to GS and GDH are present in all sequenced H. pylori strains, and their activities have been detected in cell lysates (2, 21). Our attempts to delete the gene encoding GS (hp0512) were unsuccessful, confirming previous reports that hp0512 is essential in H. pylori (21). We did successfully delete the downstream locus (hp0511), implying that the essential nature of glnA is not due to a polar effect on downstream genes.
As an alternative to gene deletion, we pursued glnA overexpression as a strategy for altered strain analysis. We engineered a second chromosomal copy of glnA to be expressed under the control of the strong ureA promoter (EM210) (Table 2). This strain possessed 3-fold-greater GS activity than did the wild type (Fig. 1a). Using a similar strategy, we engineered a second strain (EM211) for elevated expression of GDH (encoded by hp0380). This strain had higher GDH activity than did the wild type (Fig. 1b). These data show that hp0512 and hp0380 encode GS and GDH enzymes in H. pylori.
FIG 1.
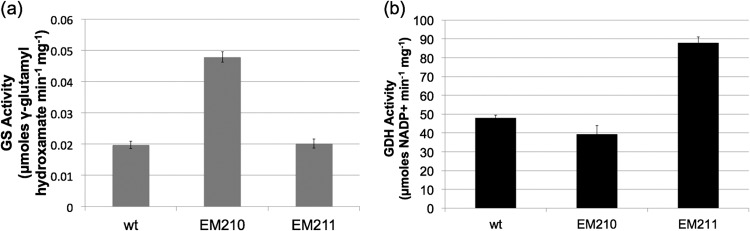
H. pylori strains engineered to possess higher GS or GDH activity. H. pylori strains 26695 (wild type), EM210 (GS elevated), and EM211 (GDH elevated) were grown on BA plates overnight and assayed for GS and GDH activities. (a) GS activity of permeabilized whole cells was measured by monitoring the formation of γ-glutamyl hydroxamate (GGH) from α-ketoglutarate as described by Bender et al. (31). (b) GDH activity of cell lysates was measured spectrophotometrically by monitoring the decrease in NADPH over time at 325 nm. Shown are the means ± SDs of results of at least three independent replicates, and the same trend was seen in a minimum of three independent experiments.
We then sought to confirm that HP0512 does indeed catalyze the assimilation of NH4+ into glutamine, as our initial measurements of GS activity (and those of Garner et al. [21]) relied on a colorimetric assay involving the nonphysiological substrate hydroxylamine (31). We therefore measured GS activity of purified, recombinant HP0512 using a second assay involving the physiologically relevant substrate ammonium (32). In the presence of [14C]glutamate, Mg2+, and NH4+, HP0512 produced [14C]glutamine at a rate of 850 μmol min−1 mg−1, confirming the role of this enzyme as a glutamine synthetase that uses free ammonium as the amide donor (Fig. 2).
FIG 2.
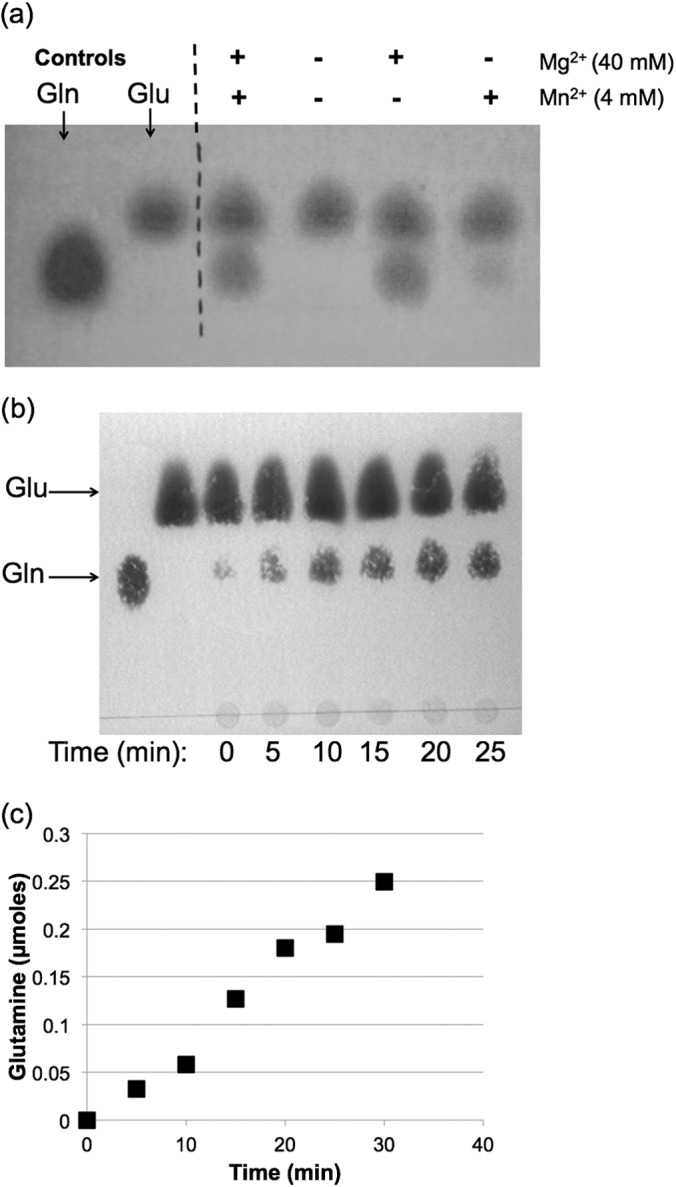
Glutamine synthetase activity of purified HP0512. Purified enzyme was incubated in an imidazole buffer (pH 7.6) containing [14C]glutamine, ATP, NH4+, and Mg2+ (or Mn2+) as indicated. (a) Pure enzyme was incubated for 30 min with substrates, and amino acid spots were visualized using ninhydrin after separation using TLC. Glutamine (lower row) spots were more intense when Mg2+ was used as the divalent cation. (b) TLC plate showing a time course reaction of HP0512 in which Mg2+ was added as the divalent cation. (c) Glutamine production over time by HP0512.
GS-elevated and GDH-elevated strains exhibit enhanced acid survival and elevated rates of urea-dependent ammonium production.
Because GS and GDH both assimilate ammonium, we predicted that the GS-elevated (EM210) and GDH-elevated (EM211) strains would direct a higher proportion of urea-derived ammonium toward assimilation and would accordingly exhibit decreased rates of ammonium production. Concurrent with this logic, we also predicted higher acid sensitivity of strains EM210 and EM211. To our surprise, both strains exhibited enhanced acid survival compared to that of the wild type (Fig. 3a). This phenotype was accompanied by a higher rate of buffer alkalization in the presence of urea (Fig. 3b and c). Rates of extracellular, urea-dependent ammonium production by EM210 and EM211 were 5-fold and 8-fold greater than for the wild type (Table 3).
FIG 3.
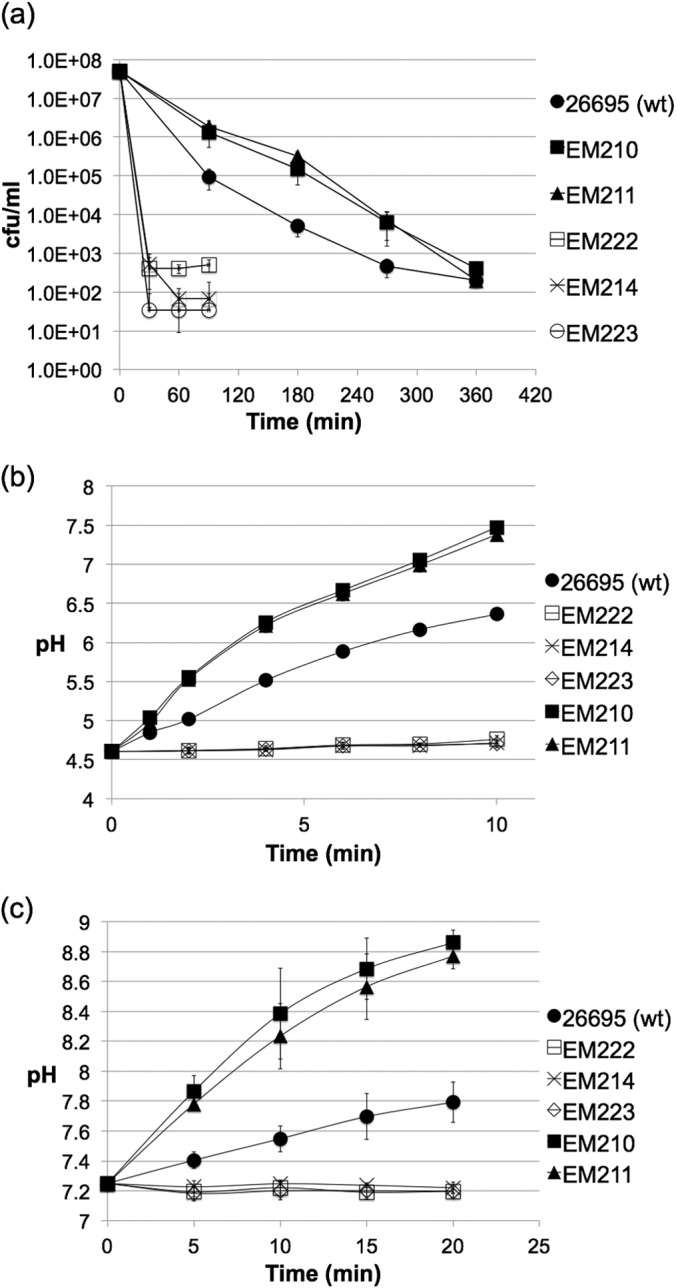
Acid sensitivity and ammonium production of H. pylori strains in the presence of 20 mM urea. (a) H. pylori cells were suspended in citrate-phosphate buffer (pH 3.0) containing 20 mM urea, and viable cell counts were monitored over time. Shown are the means ± SDs of results of three independent replicates, and the same trend was validated in two or more independent experiments. (b and c) Whole cells (OD, 2.0) were incubated in citrate-phosphate buffer (pH 7.2 or 4.5) containing 10 mM urea, and the rise in buffer pH was measured over time.
TABLE 3.
Enzyme activities and rates of urease-dependent extracellular ammonium production by whole cells for all H. pylori strains used in this studya
| Strain | Glutaminase activity (μmol of NH4+ min−1 108 cells−1) | Asparaginase activity (μmol of NH4+ min−1 108 cells−1) | Urea-dependent ammonium production (μmol of NH4+ min−1 108 cells−1) |
|---|---|---|---|
| 26695 (wild type) | 2.6 ± 0.8 | 0.5 ± 0.1 | 2.5 ± 1.3 |
| EM214 (ggt) | BDL | 0.4 ± 0.1 | 0.33 ± 0.2 |
| EM222 (asnB) | 1.9 | BDL | 0.34 ± 0.1 |
| EM223 (asnB ggt) | 0.25 ± 0.1 | BDL | 0.31 ± 0.1 |
| EM210 (GS elevated) | 2.5 ± 0.7 | 0.6 ± 0.1 | 13.4 ± 2.8 |
| EM216 (EM210 ggt) | 0.06 ± 0.2 | 0.2 ± 0.2 | 14.3 ± 2.3 |
| EM224 (EM210 asnB) | 2.3 ± 0.2 | 0.03 ± 0.1 | 13.5 ± 1.5 |
| EM226 (EM210 ggt asnB) | BDL | BDL | 12.1 ± 0.4 |
| EM211 (GDH elevated) | 2.7 ± 0.3 | 0.6 ± 0.1 | 20.1 ± 3.5 |
| EM217 (EM211 ggt) | BDL | 0.4 ± 0.1 | 17.6 ± 5.1 |
| EM225 (EM211 asnB) | 2.0 ± 0.3 | BDL | 11.3 ± 0.7 |
| EM227 (EM211 ggt asnB) | BDL | BDL | 17.3 ± 1.1 |
| EM230 (ureAB) | 2.5 ± 0.2 | 0.4 ± 0.2 | BDL |
| EM231 (EM210 ureAB) | 2.2 ± 0.5 | 0.7 ± 0.1 | BDL |
| EM232 (EM211 ureAB) | 2.0 ± 0.7 | 0.6 ± 0.1 | BDL |
Values are the means ± standard deviations of results from at least three independent experiments performed in triplicate. BDL, below detection limit.
We then asked whether strains EM210 an EM211 produce more ammonium simply because of elevated levels of urease protein compared to that of the wild type. Using cell extracts, we performed a Western blot against the α-subunit of urease. Densitometry analysis of the visualized bands showed that all strains (EM210, EM211, and wild-type H. pylori) possess similar levels of urease protein (Fig. 4a). This result confirms that a larger amount of urease protein cannot explain the enhanced ammonium production phenotype observed in strains EM210 and EM211. We then asked whether this phenotype was perhaps due to other ammonium-evolving reactions independent of urease. To address this, the ureAB locus was deleted from the parent strains EM210 and EM211. The resulting strains (EM231 and EM232) (Table 2) did not produce ammonium in the presence of urea (Table 3), showing that elevated urea-dependent ammonium production seen in strains EM210 and EM211 is also urease dependent.
FIG 4.
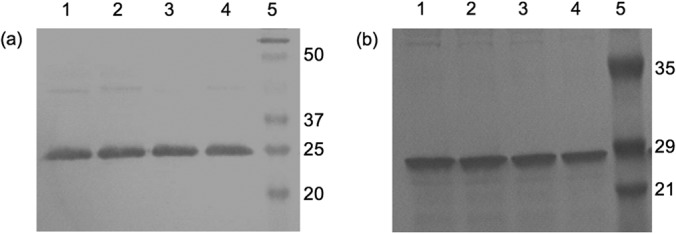
Western blots of H. pylori cell extracts using anti-α-urease antibodies. (a) Lane 1, wild type; lane 2, EM210; lane 3, wild type; lane 4, EM211; lane 5, molecular weight marker. (b) Lane 1, wild type; lane 2, EM214; lane 3, EM222; lane 4, EM223; lane 5, molecular weight marker.
Ggt and AsnB deletion mutants exhibit decreased acid survival and lower rates of urea-dependent ammonium production.
In order to further examine a connection between urea hydrolysis and ammonium metabolism in H. pylori, we considered two of the enzymes in H. pylori that produce rather than assimilate ammonium. Glutaminase (Ggt; encoded by hp1118), and asparaginase (AsnB; encoded by hp0723) are periplasmic enzymes that hydrolyze glutamine and asparagine, respectively, to liberate ammonia (26, 34–36). Gln and Asn are the major products of the assimilation of urea nitrogen by H. pylori (20). While Gln is synthesized by the combined action of GS and GDH, Asn is synthesized by GatCAB amidotransferase, which uses Gln as the amido donor to produce mischarged Asp-tRNAAsn (37). We hypothesized that following their synthesis from the assimilation of urea nitrogen, Gln and Asn could become substrates for periplasmic Ggt and AsnB, respectively. To test this, we constructed an hp1118 deletion mutant (EM214), which we confirmed to lack glutaminase activity (Table 3), and a deletion mutant lacking hp0723 (EM222), which lacked asparaginase activity (Table 3). When incubated at pH 4.5 or pH 7.2 in buffer containing 10 mM urea, strains EM214 and EM222 were unable to alkalize the buffer to a detectable level (Fig. 3b and c). Correspondingly, these three strains were highly sensitive to acidity in the presence of urea to the extent that viable colonies were below the limit of detection after only 30 min (Fig. 3a). A double mutant lacking both Ggt and AsnB activity (EM223) displayed a phenotype identical to that of each single-deletion mutants (Fig. 3).
Extracellular urea-dependent ammonium production in these strains was approximately 8-fold lower than for the wild type (Table 3). Western blotting against the urease α-subunit confirm that EM214, EM222, and EM223 all possess urease protein at levels similar to that of the wild type (Fig. 4b). These results suggest a link between periplasmic amino acid deamidases and urea-mediated ammonium production in H. pylori.
Elevated urease-dependent ammonium production by GS-elevated and GDH-elevated strains does not depend upon Ggt or AsnB activity.
We showed that strains EM210 and EM211 produce an excess of extracellular ammonium in the presence of urea, while strains EM214, EM222, and EM223 have an opposing (decreased ammonium production) phenotype. Additionally, the rate of ammonium production for each strain correlates with its acid tolerance. One explanation for these phenotypes is that urea-derived ammonium is initially assimilated by GDH and GS, elevating levels of glutamine and asparagine, which are then hydrolyzed in the periplasm by Ggt and AsnB. A consequence of these events would be the net efflux of ammonium outward into the periplasm. To test this ammonium efflux hypothesis, we asked whether Ggt and AsnB were essential for urea-dependent ammonium production in a GS-elevated and GDH-elevated strain background. We engineered strains lacking genes for both Ggt and AsnB in either a GS-elevated or GDH-elevated background. To our surprise, this combination of mutations had no effect on the urease-dependent ammonium production seen in the EM210 and EM211 parent strains (Table 3). This shows that production of extracellular urea-derived ammonium in the GS-elevated and GDH-elevated backgrounds does not depend on the activities of periplasmic Ggt and AsnB.
DISCUSSION
Urease-mediated acid resistance in H. pylori is critical for this organism's survival in the host, yet its details have not been fully described. Urea-derived ammonium was shown to be assimilated into amino acids (20); however, studies were not carried out to elaborate on a connection between acid resistance and nitrogen metabolism. Furthermore, although the importance of intracellular ammonium removal by H. pylori during urea exposure has been acknowledged (16–19), an adequate mechanism to account for the efflux of this ammonium is missing.
We showed that H. pylori GDH and GS assimilate ammonium into glutamate and glutamine, respectively. Purified HP0512 produced [14C]glutamine at a rate similar to the values measured for E. coli unadenylated GS (38), supporting previous studies which showed that hp0512 can complement an E. coli GS mutant (21). H. pylori strains engineered to overproduce either GS or GDH possessed elevated levels of extracellular urea-dependent ammonium production, enhanced acid survival, and more rapid buffer alkalization. The expression of hp0512 was previously shown to be under the control of the ArsS acid-responsive regulon (39) and was shown by microarray to be induced at low pH and during in vivo colonization of Mongolian gerbils (40–42). Additionally, a putative interaction was shown between GS and UreA by in vivo tandem affinity purification (22). These previous results, along with our current report, support a link between the assimilation of ammonium and the primary acid resistance mechanism in H. pylori.
We showed that strains lacking the periplasmic amino acid deamidases Ggt and AsnB exhibited decreased extracellular urea-dependent ammonium production, increased acid sensitivity, and an inability to use urea to alkalize the surrounding buffer. These results demonstrate yet another metabolic pathway influencing acid tolerance: the periplasmic amino acid deamidation reactions carried out by Ggt and AsnB. These two periplasmic enzymes have been previously studied for H. pylori (23–26); however, their potential role in acid resistance was not addressed. Here we show that both deamidases (Ggt and AsnB) have roles in supporting urease-mediated acid resistance.
As a whole, our data reveal a connection between urea metabolism and acid resistance. One model to explain these observed strain phenotypes is the existence of an ammonium efflux system that begins with the intracellular assimilation of urea-derived ammonium by GDH and GS and ends with the deamidation of glutamine and asparagine in the periplasm by Ggt and AsnB (Fig. 5). A similar system occurs in certain hepatocytes, wherein Gln is taken up by mitochondria and undergoes deamidation to form Glu, thereby concentrating ammonium within these organelles (43). If such an ammonium efflux system does occur in H. pylori, it would be the first of its kind reported for bacteria. One consequence of this outward NH4 efflux would be the net loss of Glu, Asp, and possibly α-ketoglutarate from the cell. How would these metabolites be replenished? The transporters GltS and DcuA are known in H. pylori to take up Glu and Asp, respectively (26). Furthermore, an E. coli Dcu-type protein has been shown to function as a dicarboxylate antiporter (45). The possibility of Gln/Glu or Asn/Asp antiporters in H. pylori would be an efficient mechanism by which the organism could continually cycle ammonia/ammonium outward while avoiding substrate depletion. Numerous other hypothetical transporters in H. pylori hold potential as metabolite transporters that could contribute to this ammonium efflux model.
FIG 5.
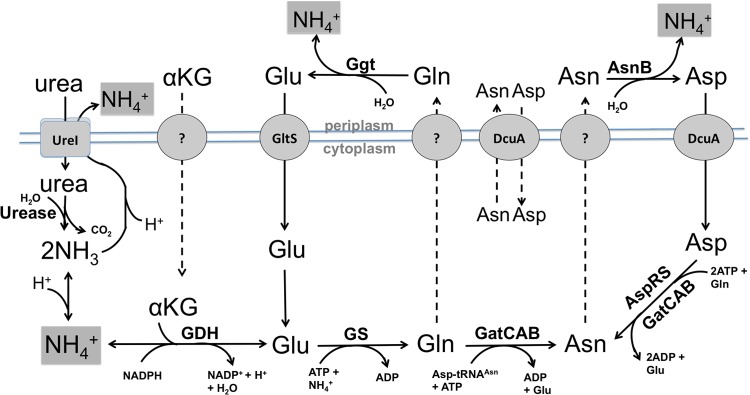
Model for the involvement of nitrogen-containing metabolites in ammonium efflux. Solid arrows indicate verified reactions or processes. Dashed arrows indicate reactions/processes that remain to be verified. Abbreviations: GDH, glutamate dehydrogenase; GS, glutamine synthetase; αKG, α-ketoglutarate; Ggt, glutaminase; AsnB, asparaginase; AspRS, aspartyl-tRNA synthetase; GatCAB, GatCAB aminoacyl-tRNA amidotransferase.
In general, this model will require further investigation, as our data show two pieces of evidence to the contrary. First, a low basal rate of extracellular urea-derived ammonium is produced even in the AsnB/Ggt double mutant (∼0.3 μmol min−1 mg−1). Second, AsnB and Ggt appear to be dispensable in a GS-elevated or GDH-elevated background. Therefore, if our hypothesized ammonium efflux mechanism does exist, it appears to be redundant to another system independent of GS/GDH and Ggt/AsnB. This second system could be the UreI permease, which was shown to transport NH4+ (19). Our attempts to study this possibility using a ureI deletion mutant are confounded by the fact that UreI may serve as an antiporter for urea and ammonium. Thus, the slowed urea-dependent ammonium production seen in a ureI mutant (44) could be due either to slowed urea entry or to slowed ammonium export, these two possibilities being indistinguishable from one another using the methods described herein.
In summary, we have shown a link between ammonium metabolism and the major (urease-dependent) acid resistance mechanism in H. pylori. We showed that both GS and GDH serve as ammonium assimilation enzymes in this organism and that their actions enhance the ability of H. pylori to survive in an acidic environment. The periplasmic amino acid deamidases Ggt and AsnB appear to have a role opposite to that of GS and GDH with regard to acid resistance. Further studies addressing the removal of intracellular ammonium by H. pylori will clarify this fundamental issue about this primary (urease) acid resistance pathway.
ACKNOWLEDGMENTS
We thank Stéphane Benoit and Soumya Vaish for their valuable input and assistance in the experimental portion of the research and for help in editing and preparing the manuscript. We also thank Robert Bender for helpful critique of the manuscript.
Research reported in this publication was supported primarily by the National Institute of Diabetes and Digestive and Kidney Diseases under award number 1F31DK091182-01 to E.F.M. Secondary support came from National Institutes of Health award number RO1-AI077569.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 16 June 2014
REFERENCES
- 1.Hu L, Mobley H. 1990. Purification and N-terminal analysis of urease from Helicobacter pylori. Infect. Immun. 58:992–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrero R, Hazell S, Lee A. 1988. The urease enzymes of Campylobacter pylori and a related bacterium. J. Med. Microbiol. 27:33–40. 10.1099/00222615-27-1-33 [DOI] [PubMed] [Google Scholar]
- 3.Chirica LC, Petersson C, Hurtig M, Jonsson BH, Borén T, Lindskog S. 2002. Expression and localization of [alpha]- and [beta]-carbonic anhydrase in Helicobacter pylori. Biochim. Biophys. Acta 1601:192–199. 10.1016/S1570-9639(02)00467-3 [DOI] [PubMed] [Google Scholar]
- 4.Marcus EA, Moshfegh AP, Sachs G, Scott DR. 2005. The periplasmic α-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J. Bacteriol. 187:729–738. 10.1128/JB.187.2.729-738.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nijevitch AA, Loguinovskaya VV. 2002. Significance of blood urea: gastric ammonia gradient for H. pylori detection in childhood. Dig. Dis. Sci. 47:65–66 (Letter.) 10.1023/A:1013259302767 [DOI] [PubMed] [Google Scholar]
- 6.Neithercut W, Rowe P, El Nujumi A, Dahill S, McColl K. 1993. Effect of Helicobacter pylori infection on intragastric urea and ammonium concentrations in patients with chronic renal failure. J. Clin. Pathol. 46:544–547. 10.1136/jcp.46.6.544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bury-Moné S, Skouloubris S, Labigne A, De Reuse H. 2001. The Helicobacter pylori UreI protein: role in adaptation to acidity and identification of residues essential for its activity and for acid activation. Mol. Microbiol. 42:1021–1034. 10.1046/j.1365-2958.2001.02689.x [DOI] [PubMed] [Google Scholar]
- 8.Weeks DL, Eskandari S, Scott DR, Sachs G. 2000. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287:482–485. 10.1126/science.287.5452.482 [DOI] [PubMed] [Google Scholar]
- 9.Scott DR, Weeks D, Hong C, Postius S, Melchers K, Sachs G. 1998. The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterology 114:58–70. 10.1016/S0016-5085(98)70633-X [DOI] [PubMed] [Google Scholar]
- 10.Dunn BE, Campbell GP, Perez-Perez G, Blaser M. 1990. Purification and characterization of urease from Helicobacter pylori. J. Biol. Chem. 265:9464–9469 [PubMed] [Google Scholar]
- 11.Evans DJ, Evans DG, Kirkpatrick SS, Graham DY. 1991. Characterization of the Helicobacter pylori urease and purification of its subunits. Microb. Pathog. 10:15–26. 10.1016/0882-4010(91)90062-F [DOI] [PubMed] [Google Scholar]
- 12.Todd MJ, Hausinger RP. 1987. Purification and characterization of the nickel-containing multicomponent urease from Klebsiella aerogenes. J. Biol. Chem. 262:5963–5967 [PubMed] [Google Scholar]
- 13.Clemens DL, Lee BY, Horwitz MA. 1995. Purification, characterization, and genetic analysis of Mycobacterium tuberculosis urease, a potentially critical determinant of host-pathogen interaction. J. Bacteriol. 177:5644–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breitenbach JM, Hausinger RP. 1988. Proteus mirabilis urease. Partial purification and inhibition by boric acid and boronic acids. Biochem. J. 250:917–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clyne M, Labigne A, Drumm B. 1995. Helicobacter pylori requires an acidic environment to survive in the presence of urea. Infect. Immun. 63:1669–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stingl K, Uhlemann EM, Schmid R, Altendorf K, Bakker EP. 2002. Energetics of Helicobacter pylori and its implications for the mechanism of urease-dependent acid tolerance at pH 1. J. Bacteriol. 184:3053–3060. 10.1128/JB.184.11.3053-3060.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Athmann C, Zeng N, Kang T, Marcus EA, Scott DR, Rektorschek M, Buhmann A, Melchers K, Sachs G. 2000. Local pH elevation mediated by the intrabacterial urease of Helicobacter pylori cocultured with gastric cells. J. Clin. Invest. 106:339–347. 10.1172/JCI9351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer-Rosberg K, Scott D, Rex D, Melchers K, Sachs G. 1996. The effect of environmental pH on the proton motive force of Helicobacter pylori. Gastroenterology 111:886–900. 10.1016/S0016-5085(96)70056-2 [DOI] [PubMed] [Google Scholar]
- 19.Scott DR, Marcus EA, Wen Y, Singh S, Feng J, Sachs G. 2010. Cytoplasmic histidine kinase (HP0244)-regulated assembly of urease with UreI, a channel for urea and its metabolites, CO2, NH3, and NH4+, is necessary for acid survival of Helicobacter pylori. J. Bacteriol. 192:94–103. 10.1128/JB.00848-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams C, Preston T, Hossack M, Slater C, McColl K. 1996. Helicobacter pylori utilises urea for amino acid synthesis. FEMS Immunol. Med. Microbiol. 13:87–94. 10.1111/j.1574-695X.1996.tb00220.x [DOI] [PubMed] [Google Scholar]
- 21.Garner RM, Fulkerson J, Jr, Mobley HLT. 1998. Helicobacter pylori glutamine synthetase lacks features associated with transcriptional and posttranslational regulation. Infect. Immun. 66:1839–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stingl K, Schauer K, Ecobichon C, Labigne A, Lenormand P, Rousselle JC, Namane A, de Reuse H. 2008. In vivo interactome of Helicobacter pylori urease revealed by tandem affinity purification. Mol. Cell. Proteomics 7:2429–2441. 10.1074/mcp.M800160-MCP200 [DOI] [PubMed] [Google Scholar]
- 23.Kim KM, Lee SG, Park MG, Song JY, Kang HL, Lee WK, Cho MJ, Rhee KH, Youn HS, Baik SC. 2007. [gamma]-Glutamyltranspeptidase of Helicobacter pylori induces mitochondria-mediated apoptosis in AGS cells. Biochem. Biophys. Res. Commun. 355:562–567. 10.1016/j.bbrc.2007.02.021 [DOI] [PubMed] [Google Scholar]
- 24.Scotti C, Sommi P, Pasquetto MV, Cappelletti D, Stivala S, Mignosi P, Savio M, Chiarelli LR, Valentini G, Bolanos-Garcia VM. 2010. Cell-cycle inhibition by Helicobacter pylori L-asparaginase. PLoS One 5:e13892. 10.1371/journal.pone.0013892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, Bauer S, Brenner L, Schmid RM, Gerhard M. 2007. Inhibition of T-cell proliferation by Helicobacter pylori [gamma]-glutamyl transpeptidase. Gastroenterology 132:1820–1833. 10.1053/j.gastro.2007.02.031 [DOI] [PubMed] [Google Scholar]
- 26.Leduc D, Gallaud J, Stingl K, de Reuse H. 2010. Coupled amino acid deamidase-transport systems essential for Helicobacter pylori colonization. Infect. Immun. 78:2782–2792. 10.1128/IAI.00149-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benoit S, Maier RJ. 2003. Dependence of Helicobacter pylori urease activity on the nickel-sequestering ability of the UreE accessory protein. J. Bacteriol. 185:4787–4795. 10.1128/JB.185.16.4787-4795.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olson JW, Mehta NS, Maier RJ. 2001. Requirement of nickel metabolism proteins HypA and HypB for full activity of both hydrogenase and urease in Helicobacter pylori. Mol. Microbiol. 39:176–182. 10.1046/j.1365-2958.2001.02244.x [DOI] [PubMed] [Google Scholar]
- 29.Weatherburn M. 1967. Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 39:971–974. 10.1021/ac60252a045 [DOI] [Google Scholar]
- 30.Schmidt E. 1963. Glutamic dehydrogenase, p 752 In Bergmeyer HU. (ed), Methods of enzymatic analysis. Academic Press, New York, NY [Google Scholar]
- 31.Bender R, Janssen K, Resnick A, Blumenberg M, Foor F, Magasanik B. 1977. Biochemical parameters of glutamine synthetase from Klebsiella aerogenes. J. Bacteriol. 129:1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pahuja SL, Reid TW. 1982. Radioisotope assay for glutamine synthetase using thinlayer chromatography. J. Chromatogr. A 235:249–255. 10.1016/S0021-9673(00)95806-0 [DOI] [Google Scholar]
- 33.Dale T, Court W. 1981. Improved separation of amino acids by thin-layer chromatography. Chromatographia 14:617–620. 10.1007/BF02291097 [DOI] [Google Scholar]
- 34.Stark R, Suleiman MS, Hassan I, Greenman J, Millar M. 1997. Amino acid utilisation and deamidation of glutamine and asparagine by Helicobacter pylori. J. Med. Microbiol. 46:793–800. 10.1099/00222615-46-9-793 [DOI] [PubMed] [Google Scholar]
- 35.Shibayama K, Wachino J, Arakawa Y, Saidijam M, Rutherford NG, Henderson PJF. 2007. Metabolism of glutamine and glutathione via γ-glutamyltranspeptidase and glutamate transport in Helicobacter pylori: possible significance in the pathophysiology of the organism. Mol. Microbiol. 64:396–406. 10.1111/j.1365-2958.2007.05661.x [DOI] [PubMed] [Google Scholar]
- 36.Shibayama K, Takeuchi H, Wachino J, Mori S, Arakawa Y. 2011. Biochemical and pathophysiological characterization of Helicobacter pylori asparaginase. Microbiol. Immunol. 55:408–417. 10.1111/j.1348-0421.2011.00333.x [DOI] [PubMed] [Google Scholar]
- 37.Sheppard K, Akochy PM, Salazar JC, Söll D. 2007. The Helicobacter pylori amidotransferase GatCAB is equally efficient in glutamine-dependent transamidation of Asp-tRNAAsn and Glu-tRNAGln. J. Biol. Chem. 282:11866–11873. 10.1074/jbc.M700398200 [DOI] [PubMed] [Google Scholar]
- 38.Shapiro BM, Kingdon HS, Stadtman E. 1967. Regulation of glutamine synthetase. VII. Adenylyl glutamine synthetase: a new form of the enzyme with altered regulatory and kinetic properties. Proc. Natl. Acad. Sci. U. S. A. 58:642–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pflock M, Finsterer N, Joseph B, Mollenkopf H, Meyer TF, Beier D. 2006. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J. Bacteriol. 188:3449–3462. 10.1128/JB.188.10.3449-3462.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott DR, Marcus EA, Wen Y, Oh J, Sachs G. 2007. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc. Natl. Acad. Sci. U. S. A. 104:7235–7240. 10.1073/pnas.0702300104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wen Y, Marcus EA, Matrubutham U, Gleeson MA, Scott DR, Sachs G. 2003. Acid-adaptive genes of Helicobacter pylori. Infect. Immun. 71:5921–5939. 10.1128/IAI.71.10.5921-5939.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S. 2003. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 71:3529–3539. 10.1128/IAI.71.6.3529-3539.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haüssinger D. 1990. Nitrogen metabolism in liver: structural and functional organization and physiological relevance. Biochem. J. 267:281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skouloubris S, Thiberge J-M, Labigne A, De Reuse H. 1998. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immun. 66:4517–4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleefeld A, Ackermann B, Bauer J, Krämer J, Unden G. 2009. The fumarate/succinate antiporter DcuB of Escherichia coli is a bifunctional protein with sites for regulation of DcuS-dependent gene expression. J. Biol. Chem. 284:265–275 [DOI] [PubMed] [Google Scholar]