

Abstract
Expression of the surface protein Cnm has been directly implicated in the ability of certain strains of Streptococcus mutans to bind to collagen and to invade human coronary artery endothelial cells (HCAEC) and in the killing of Galleria mellonella. Sequencing analysis of Cnm+ strains revealed that cnm is located between the core genes SMU.2067 and SMU.2069. Reverse transcription-PCR (RT-PCR) analysis showed that cnm is cotranscribed with SMU.2067, encoding a putative glycosyltransferase referred to here as PgfS (protein glycosyltransferase of streptococci). Notably, Cnm contains a threonine-rich domain predicted to undergo O-linked glycosylation. The previously shown abnormal migration pattern of Cnm, the presence of the threonine-rich domain, and the molecular linkage of cnm with pgfS lead us to hypothesize that PgfS modifies Cnm. A ΔpgfS strain showed defects in several traits associated with Cnm expression, including collagen binding, HCAEC invasion, and killing of G. mellonella. Western blot analysis revealed that Cnm from the ΔpgfS mutant migrated at a lower molecular weight than that from the parent strain. In addition, Cnm produced by ΔpgfS was highly susceptible to proteinase K degradation, in contrast to the high-molecular-weight Cnm version found in the parent strain. Lectin-binding analyses confirmed the glycosylated nature of Cnm and strongly suggested the presence of N-acetylglucosamine residues attached to Cnm. Based on these findings, the phenotypes observed in ΔpgfS are most likely associated with defects in Cnm glycosylation that affects protein function, stability, or both. In conclusion, this study demonstrates that Cnm is a glycoprotein and that posttranslational modification mediated by PgfS contributes to the virulence-associated phenotypes linked to Cnm.
INTRODUCTION
Streptococcus mutans is a natural inhabitant of the oral cavity and a major etiological agent of dental caries (1, 2). Strains of S. mutans are classified in four serotypes (c, e, f, and k), with approximately 70% of strains isolated from dental plaque belonging to serotype c, over 20% belonging to serotype e, and less than 5% belonging to serotypes f and k (3, 4). In addition to dental caries, S. mutans is frequently associated with nonoral diseases, such as infective endocarditis (IE) and, possibly, atherosclerotic coronary heart diseases (ACHD) (5, 6). Combined, staphylococci, enterococci, and viridans group streptococci are responsible for more than 80% of all cases of IE, and it is estimated that S. mutans is responsible for ∼20% of cases caused by viridans group streptococci (7, 8). Meanwhile, an association between S. mutans and ACHD, the leading cause of death in the United States (9), has been suggested based on the frequent detection of S. mutans DNA in atherosclerotic plaque (10, 11). In proatherogenic ApoEnull mice, systemic infection with the highly invasive S. mutans strain OMZ175 accelerated atherosclerotic plaque formation and led to increased levels of inflammation (12). Thus, the pathogenic potential of S. mutans is not restricted to the oral cavity, and the identification and characterization of virulence factors involved in extraoral diseases are of great relevance.
The ability of oral streptococci to interact with host components of the extracellular matrix (ECM) is regarded as a key virulence trait involved in adherence and colonization of host tissues (e.g., heart valves) (7, 13). In S. mutans, Cnm is a proven virulence factor that mediates binding to both collagen and laminin (14, 15). The cnm gene is found in approximately 10% of S. mutans clinical isolates but is more frequently detected (approximately ∼40%) among strains isolated from blood, atheromas, and extirpated heart valves (5, 16, 17). Notably, cnm is unevenly distributed among the four different serotypes, being found in the highest proportion in the less prevalent serotypes in the oral cavity (e, f, and k) but rarely present in serotype c strains (16, 18). Not surprisingly, a correlation between Cnm-expressing strains belonging to serotypes e, f, and k and systemic infections has been observed (5, 16, 19). Studies from our group revealed that cnm is essential for S. mutans ability to bind to collagen and laminin and to invade human coronary artery endothelial cells (HCAEC) (15, 18). Strains expressing Cnm were also found to have increased virulence in Galleria mellonella, an invertebrate model of systemic infection (15, 18). Finally, expression of cnm in the serotype c strain UA159 significantly increased its ability to bind to collagen and laminin, invade HCAEC, and kill G. mellonella, which further confirms the importance of Cnm in S. mutans-host interactions (15).
Microbial surface components recognizing adhesive matrix molecules (MSCRAMMs), such as Cnm, are of growing interest, as they are viewed as desirable targets for therapeutic interventions (20). In some cases, bacterial MSCRAMMs, such as the fimbria-associated protein Fap1 from Streptococcus parasanguinis and AIDA-1 from Escherichia coli, undergo posttranslational modifications, specifically, glycosylation (21, 22). Similar to its role in eukaryotes, protein glycosylation contributes to protein folding and secondary structure formation, cell adhesion, thermodynamic stability, modulation of immune recognition, and protection against degradation by proteases in bacteria (23). In S. parasanguinis, glycosylation of Fap1 at its serine-rich domain is important for proper fimbrial assembly, adhesion, and biofilm formation (24, 25). On the other hand, glycosylation of E. coli AIDA-1 is required for proper conformation and increased resistance to degradation by proteases but does not appear to affect binding to epithelial cells (26, 27).
In this study, we demonstrate that Cnm is modified by its immediate downstream gene product, a putative glycosyltransferase that we named PgfS (for protein glycosyltransferase of streptococci). We specifically show that PgfS is responsible for Cnm glycosylation and that the attached glycoconjugate is wheat germ agglutinin reactive. A strain lacking pgfS showed defects in phenotypes associated with Cnm expression. Absence of PgfS-dependent modification of Cnm led to decreased collagen binding and decreased resistance to protease degradation, suggesting that glycosylation of Cnm is required for both function and protein stability.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Strains used in this study are listed in Table 1. E. coli strains were routinely grown in Luria-Bertani (LB) broth medium at 37°C. When required, 100 μg ml−1 ampicillin or 100 μg ml−1 kanamycin was added to LB broth or plates. S. mutans strains were routinely cultured in brain heart infusion (BHI) medium at 37°C in a humidified 5% CO2 atmosphere. When required, 1 mg ml−1 kanamycin or 10 μg ml−1 erythromycin was added to BHI broth or plates.
TABLE 1.
Streptococcus mutans strains used in this study
| Strain | Serotype | Relevant phenotype | Reference |
|---|---|---|---|
| OMZ175 | f | Cnm+ | 19 |
| B14 | e | Cnm+ | 19 |
| 11060 | e | Cnm+ | 19 |
| LM7 | e | Cnm+ | 19 |
| OM50E | f | Cnm+ | 19 |
| Δcnm mutant | f | Cnm− Kanr | 19 |
| UA159+cnm | c | Cnm+ Kanr | 15 |
| UA159+cnmΔpgfS | c | Cnm+ PgfS− Kanr Ermr | This study |
| OMZ175ΔpgfS | f | Cnm+ PgfS− Kanr | This study |
| UA159ΔpgfS | c | Cnm+ PgfS− Kanr | This study |
| 11060ΔpgfS | e | Cnm+ PgfS− Kanr | This study |
| B14ΔpgfS | e | Cnm+ PgfS− Kanr | This study |
| LM7ΔpgfS | e | Cnm+ PgfS− Kanr | This study |
| OM50EΔpgfS | f | Cnm+ PgfS− Kanr | This study |
| OMZ175CpgfS | f | Cnm+ PgfS+ Ermr | This study |
Genetic manipulation of S. mutans.
The SMU.2067 gene (referred to here as pgfS) from S. mutans OMZ175 was mutated by allelic replacement with a nonpolar kanamycin (NPKan) marker (28) using a PCR ligation mutagenesis strategy (29). The NPKan cassette contains a promoterless aphA3 Kmr gene without transcription termination sequences to allow transcription readthrough into downstream sequences. The primers used for gene inactivation are listed in Table 2. Briefly, HindIII sites were introduced into DNA fragments containing the 5′ and the 3′ regions of pgfS using primers pgfS F1/R1 and pgfS F2/R2, respectively. PCR products were digested with HindIII and then ligated to a HindIII-digested nonpolar kanamycin cassette. S. mutans OMZ175 was transformed with the resulting ligation product, and transformants were selected on plates containing kanamycin. In addition to OMZ175, pgfS inactivation in other Cnm+ invasive S. mutans strains was achieved by amplifying a DNA fragment containing the pgfS mutation from OMZ175 by PCR using the pgfS F1/R2 primer pair. The desired mutations were confirmed by PCR and sequencing of the insertion site.
TABLE 2.
Primers used in this study
| Primer | Sequencea | Analysis |
|---|---|---|
| pgfS F1 | Forward: 5′-GTCACTTCTCGGTCTTG-3′ | Inactivation of pgfS |
| pgfS R1 | Reverse: 5′-CTGAAGCTGGGATCCCTGACCC-3′ | |
| pgfS F2 | Forward: 5′-GCGACCGCGGATCCTTGTAGATG-3′ | |
| pgfS R2 | Reverse: 5′-GAGCATAGGCCGTTGC-3′ | |
| cnm F1 | Forward: 5′- TGGAACCTTGCCATCA-3′ | RT-PCR |
| cnm R1 | Reverse: 5′- CGACCATTGAACCTTCGAC −3′ | |
| pgfS F3 | Forward: 5′-TGGATGGCGATTTGCAGCAC-3′ | RT-PCR |
| pgfS R3 | Reverse: 5′-TCTCCTGCTTCACGAACTTG-3′ |
Restriction sites introduced for cloning purposes are underlined.
Complementation of pgfS was achieved by restoring the original sequence at the same location. Briefly, a PCR product containing the intact pgfS and flanking region was amplified from OMZ175 and used along with pCJK96 (30), which confers erythromycin resistance, to transform the ΔpgfS strain. The pCJK96 plasmid was used to monitor competent cells that were able to pick up exogenous DNA and to counterselect for erythromycin-positive/kanamycin-negative clones. Upon transformation, selection was done on plates containing erythromycin. Resulting colonies were patched on plates containing erythromycin or kanamycin. Colonies that grew in erythromycin but not in kanamycin were screened by PCR. Reintroduction and restoration of pgfS expression were confirmed by sequencing of the pgfS gene and flanking region and by RT-PCR analysis, respectively.
In silico analysis.
The Prokaryotic Promoter Predictor software (http://bioinformatics.biol.rug.nl/websoftware/ppp/) was used to determine putative promoters driving transcription of cnm and pgfS, and ARNold software (http://rna.igmors.u-psud.fr/toolbox/arnold/) was used to determine putative transcriptional terminators. NetOGlyc 3.1 software was used to predict protein glycosylation (http://www.cbs.dtu.dk/services/NetOGlyc-3.1/).
RT-PCR analysis.
RNA was extracted from S. mutans cultures grown to mid-exponential phase (optical density at 600 nm [OD600] ∼ 0.5) as previously described (31). cDNA from 0.5 μg of RNA was synthesized using a high-capacity cDNA reverse transcriptase kit containing random primers (Applied Biosystems, Foster, CA). Primers (Table 2) specific for cnm and pgfS coding regions were used to determine the expression and transcriptional organization of these genes.
Western blot analysis.
Whole-cell protein lysates were obtained by homogenization in the presence of 0.1-mm glass beads using a bead beater (Biospec, Bartlesville, OK). Protein lysates were separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA). Cnm detection was performed using rabbit anti-rCnmA (recombinant A domain of Cnm) polyclonal antibody (15) diluted 1:2,000 in 1× phosphate-buffered saline (PBS) plus 0.1% Tween 20 and anti-rabbit horseradish peroxidase (HRP)-coupled antibody (Sigma-Aldrich, St. Louis, MO).
ECM binding and HCAEC invasion.
In vitro assays for collagen and laminin binding and HCAEC invasion were performed as previously described (15). For ECM binding assays, 100 μl of PBS-washed bacterial suspensions containing approximately 1 × 109 CFU ml−1 was added to each well of a microtiter plate containing immobilized type I collagen from rat tail (Sigma-Aldrich) or mouse laminin (Becton, Dickinson, Franklin Lakes, NJ). Adherent cells were stained with 0.05% crystal violet (CV) solution, and the OD575 was measured. For HCAEC invasion, 1 ml of 2% fetal bovine serum (FBS)-endothelial basal medium (EBM-2) containing 1 × 107 CFU ml−1 of S. mutans was used to infect HCAEC-containing wells at a multiplicity of infection (MOI) of 100:1. The percentage of invasion for each strain was calculated based on the initial inoculum and the intracellular bacteria recovered from HCAEC lysates. All experiments were performed at least in triplicate. A one-way analysis of variance (ANOVA) was performed to verify the significance of binding and invasion. P values of ≤0.05 were considered significant.
Galleria mellonella infection.
For the G. mellonella infection model, 5-μl aliquots containing 1 × 108 CFU ml−1 of overnight-grown cultures of S. mutans in sterile saline were injected into the hemocoel of each larva via the last left proleg, as detailed elsewhere (19). Larvae injected with heat-inactivated S. mutans strains (30 min at 80°C) were used as controls. After injection, larvae were kept in the dark at 37°C, and survival was recorded at selected intervals. Experiments were performed in triplicate. Kaplan-Meier killing curves were plotted, and estimations of differences in survival were compared using the log rank test. P values of ≤0.05 were considered significant.
Microscopic analysis.
Cultures of S. mutans were grown overnight in BHI, washed once in PBS, and incubated with anti-rCnmA antibodies diluted 1:250 in PBS at room temperature for 1 h. For immunofluorescence labeling, after incubation with primary antibody, cells were washed in PBS and incubated with 10 μg ml−1 anti-rabbit IgG coupled with Alexa Fluor-488 dye (Life Technologies, Grand Island, NY) in PBS at room temperature for 1 h. After three washes with PBS, cells were examined using an Olympus BX41 fluorescence microscope, and images were captured with the QCapture Software.
For immunoelectron microscopy analysis, after incubation with anti-rCnmA, cells were washed in PBS and incubated with anti-rabbit IgG coupled with 12-nm colloidal gold particles (Jackson ImmunoResearch Laboratories, West Grove, PA) diluted 1:25 in PBS at room temperature for 1 h. Next, cells were washed with PBS and fixed with 2% glutaraldehyde for 30 min, followed by a rinse with 0.5× PBS (pH 7.2). Upon fixation, samples were embedded for sectioning before loading them onto grids. The grids were examined using a Hitachi 7650 transmission electron microscope, and the images were captured using the attached Gatan Erlangshen 11 megapixel digital camera at the University of Rochester Medical Center Electron Microscopy core facility.
Proteinase K susceptibility assay.
Susceptibility of Cnm to protease degradation was determined as described elsewhere (27). Briefly, overnight cultures of S. mutans strains were pelleted and resuspended in 1× PBS pH 7.2 containing increasing amounts of proteinase K (Sigma-Aldrich). After 30 min incubation on ice, protease activity was neutralized by the addition of 1× proteases inhibitor cocktail (Thermo Scientific). Bacterial cells were washed once with PBS after proteinase K neutralization, and Cnm stability was analyzed by Western blotting.
Cnm purification from S. mutans OMZ175 and the ΔpgfS strain.
Native Cnm was purified from S. mutans strains using a custom immuno-affinity column. Briefly, an agarose resin with N-hydroxysuccinimide (NHS)-reacting groups (Thermo Scientific, Rockford, IL) was used to immobilize purified rCnmA (recombinant A domain of Cnm) (15) and used as bait for the purification of anti-rCnmA-specific IgG antibodies from rabbit sera. Purified anti-rCnmA polyclonal antibodies were then chemically coupled to a new NHS-activated resin, and non-cross-linked antibodies were washed off the resin using stringent elutions (0.1 M glycine, pH 2.5). Whole-cell lysates of S. mutans strains grown in BHI to stationary phase were obtained as described above in the presence of 1× proteases inhibitor cocktail (Thermo Scientific). The soluble protein fraction was then bound to the NHS–anti-rCnmA column overnight at 4°C. Then, the column was washed with 15 ml of 1× PBS (pH 7.2), and Cnm was eluted using 0.1 M glycine buffer (pH 2.5) for 5 min. Elutions were immediately neutralized with 1/10 volume 1 M Tris (pH 8.0). Purified Cnm from OMZ175 and the ΔpgfS mutant was concentrated, dialyzed against 100 mM ammonium bicarbonate, and quantified for subsequent analysis.
Enzymatic deglycosylation.
One microgram of purified native Cnm from S. mutans OMZ175 and the ΔpgfS mutant were treated with a mixture of glycosidases (Prozyme, San Leandro, CA) as recommended by the manufacturer. Briefly, 1 μg of Cnm was incubated for 20 h at 37°C under denaturing conditions with a cocktail of enzymes containing: 0.005 U of N-glycosidase F (PNGase F), 0.005 U of sialidase A, 0.00125 U of endo-α-N-acetylgalactosaminidase (O-glycanase), 0.002 U of β(1-4)-galactosidase, and 0.04 U of β-N-acetylglucosaminidase. Samples were then analyzed by SDS-PAGE and Western blotting using anti-rCnmA antibodies.
Lectin binding analysis.
Whole-cell lysates of S. mutans strains were separated by SDS-PAGE and transferred to a PVDF membrane as described above. After blocking with 5% bovine serum albumin (BSA) for 1 h at room temperature, the membranes were incubated with 20 μg ml−1 of biotinylated wheat germ agglutinin (WGA), succinylated wheat germ agglutinin (sWGA), concanavalin A (ConA), or peanut agglutinin (PNA) (Vector Laboratories, Burlingame, CA) in PBS containing 0.5% BSA for 1 h at room temperature. Membranes were washed three times with PBS containing 0.1% Tween 20, followed by incubation with HRP-conjugated streptavidin (Cell Signaling Technology, Danvers, MA). Bound lectins were visualized using an enhanced chemiluminescent detection kit (GE Life Sciences, Pittsburgh, PA).
RESULTS
cnm is cotranscribed with SMU.2067, a putative glycosyltransferase.
In all strains investigated thus far, cnm is part of a conserved 5-kbp region located between the core genes SMU.2067 and SMU.2069 (Fig. 1A). Recently, we showed that this 5-kbp region contains two additional genes encoding putative surface proteins, CnaB and CbpA, but neither of these proteins was associated with the expression of Cnm-related phenotypes (15). Despite the presence of a putative Rho-independent transcriptional terminator after cnm and a putative σA type promoter upstream of SMU.2067 (data not shown), RT-PCR analyses revealed that cnm is cotranscribed with SMU.2067 (Fig. 1A). While Northern blots show cnm expressed mostly as a monocistronic message (data not shown), we also observed higher-molecular-weight bands hybridizing to the cnm probe. Considering the presence of a transcriptional terminator after cnm, the most reasonable interpretation of these results is that transcription termination is not 100% efficient.
FIG 1.
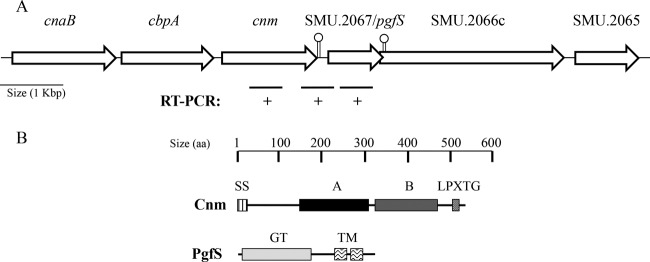
Genetic linkage of cnm and pgfS in S. mutans. (A) In all Cnm+ strains, cnm is located within a unique 5-kbp region between the core genes SMU.2067 and SMU.2069. Two adjacent genes (cnaB and cbpA) present in all cnm+ strains were described previously (15). SMU.2067 (pgfS) encodes a putative glycosyltransferase, whereas SMU.2066c encodes an integral membrane protein with homology to yfhO of B. subtilis. A summary of semiquantitative RT-PCR analysis revealed the cotranscription of cnm and pgfS. Putative Rho-independent transcriptional terminators are indicated with lollypops. (B) Conserved predicted domains of Cnm and PgfS. SS, secretion signal; A, collagen-binding domain; B, threonine-rich repeat domain; LPXTG, cell wall anchor motif; GT, DPM1-like glycosyltransferase domain; TM, transmembrane domain.
Bioinformatic analysis revealed that the SMU.2067 gene encodes a 36-kDa protein that belongs to the superfamily of glycosyltransferase A (GT-A type). The majority of proteins in this superfamily catalyze the transfer of carbohydrate moieties from an activated nucleotide-sugar donor to an acceptor molecule, such as a lipid or protein (32). In both eukaryotic and prokaryotic organisms, protein glycosylation generally occurs at asparagine residues (N-linked glycosylation) or serine/threonine residues (O-linked glycosylation) (23). Notably, the C-terminal B-domain of Cnm is rich in threonine and contains multiple TTTEAP repeat sequences. This threonine-rich domain was predicted to undergo O-linked glycosylation based on analysis using the NetOGlyc algorithm (see Fig. S1 in the supplemental material). This pattern of glycosylation was predicted for all Cnm variants available in the sequence repository, although the number of TTTEAP repeats may vary between strains (data not shown). In addition, we and others showed that Cnm migrates at ∼120 kDa on SDS-PAGE despite a predicted molecular mass of 51 kDa (15, 33). Collectively, these observations suggest that Cnm undergoes O-linked glycosylation and that sugar side chains attached to Cnm by SM.2067 may be responsible, at least in part, for the unexpected migration pattern.
Characterization of the ΔpgfS strain.
To begin to assess the role of SMU.2067, referred to here as PgfS (for protein glycosyltransferase of streptococci), in the expression of Cnm-related traits (e.g., collagen binding, HCAEC invasion, and killing of G. mellonella), the pgfS gene was replaced by a nonpolar kanamycin resistance cassette. When the ΔpgfS strain was tested for its ability to bind to ECM proteins, a modest (∼20%) yet significant decrease in collagen binding was observed (Fig. 2A). However, no obvious change in binding to laminin was noted (Fig. 2B). In agreement with the impaired ability to bind to collagen, invasion of HCAEC was significantly reduced in the ΔpgfS strain compared to the parent strain (Fig. 2C). Finally, the mortality rates of G. mellonella were significantly lower in larvae infected with the ΔpgfS strain than in those infected with the parental OMZ175 strain (Fig. 2D). Collectively, with the exception of laminin binding, the ΔpgfS strain showed intermediate phenotypes compared to the Δcnm strain and OMZ175 (Fig. 2).
FIG 2.

Phenotypic characterization of the pgfS deletion strain. (A and B) Binding of S. mutans OMZ175 and its Δcnm and ΔpgfS derivatives to collagen type I (A) and laminin (B). (C) Antibiotic protection assay showing the percentage of HCAEC invasion for each strain after 5 h of infection. (D) Killing of G. mellonella larvae infected with S. mutans strains. *, P < 0.05.
PgfS modifies Cnm in S. mutans.
As indicated above, Cnm migrates at ∼120 kDa on SDS-PAGE, although in silico analysis, based solely on amino acid sequence, predicts Cnm to migrate at 51 kDa. In agreement with a role of PgfS in Cnm modification, a reduced mobility indicating a size of nearly 30 kDa was observed for Cnm in the ΔpgfS strain (Fig. 3A). Similarly, when the pgfS gene was inactivated in other Cnm+ strains from our collection, the same pattern of Cnm migration was observed (see Fig. S2 in the supplemental material). Reintroduction of pgfS at its native locus (CpgfS) restored expression of the 120-kDa Cnm version (Fig. 3A). Immunoelectron microscopy (IEM) analysis of the ΔpgfS mutant showed no obvious differences in surface-exposed Cnm compared to OMZ175, indicating that glycosylation by PgfS is not required for extracellular transport and anchoring to the cell wall (Fig. 3B). We conclude that the aberrant electrophoretic mobility of Cnm is, at least in part, due to PgfS-mediated modification.
FIG 3.

PgfS modifies Cnm. Detection of Cnm in S. mutans OMZ175 and its ΔpgfS and CpgfS derivatives. (A) Western blot analysis using anti-rCnmA shows a band at 120 kDa corresponding to native Cnm in OMZ175. A shift in size of Cnm in the ΔpgfS strain compared to parental OMZ175 indicates PgfS-dependent modification that is restored in the CpgfS strain. (B) IEM analysis and surface localization of Cnm in OMZ175 and the ΔpgfS strain using anti-rCnmA antiserum (1:250).
Cnm from ΔpgfS mutant is highly susceptible to proteinase K degradation.
Protein modification via the action of glycosyltransferases functions as a fine-tuning mechanism that modulates protein functionality and/or stability. The phenotypes of the ΔpgfS strain in relation to collagen binding, HCAEC invasion, and killing of G. mellonella indicate that Cnm function is impaired in the absence of PgfS (Fig. 2). To test whether Cnm modification by PgfS confers increased protein stability, whole cells from OMZ175 or the ΔpgfS mutant were treated with increasing concentrations of proteinase K and analyzed by Western blotting. The 120-kDa Cnm version produced from OMZ175 was degraded in a concentration-dependent manner, although significant amounts of intact protein were still detected at the highest concentration of protease (Fig. 4). However, the 90-kDa Cnm protein produced by the ΔpgfS mutant was hypersensitive to the proteinase K treatment. The 90-kDa Cnm produced by the ΔpgfS mutant was almost completely degraded even at the lowest concentration of proteinase K, with no intact Cnm detected at higher concentrations (Fig. 4). Similar results were obtained with the other ΔpgfS mutants from our collection of Cnm+ strains, indicating that the increased susceptibility of Cnm in the absence of PgfS is conserved (see Fig. S2 in the supplemental material). Proteinase K resistance of Cnm was partially restored in the CpgfS strain (Fig. 4).
FIG 4.
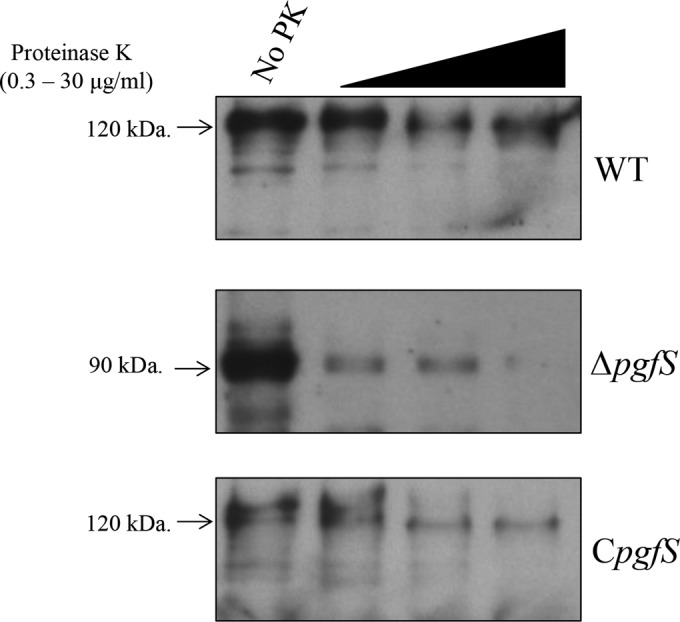
Increased susceptibility of Cnm from the ΔpgfS strain to proteinase K degradation. Whole cells from S. mutans OMZ175 and its ΔpgfS and CpgfS derivatives were treated with increasing concentrations of proteinase K (0, 0.3, 3, or 30 μg ml−1) for 30 min. Upon treatment, whole-cell lysates were prepared, and Cnm degradation was monitored by Western blotting.
To demonstrate the biological relevance of PgfS-dependent modification of Cnm, proteinase K-treated cells from S. mutans OMZ175 and the ΔpgfS and CpgfS strains were used in a collagen-binding assay. Treatment of OMZ175 or the CpgfS strain with proteinase K (3 μg ml−1) caused a small decrease in their ability to bind to collagen (Fig. 5A). However, when the ΔpgfS strain was treated with the same concentration of proteinase K, collagen binding was nearly abolished (Fig. 5A). A similar trend was observed for the ability of Cnm to mediate invasion of HCAEC in vitro, whereby the ΔpgfS strain was virtually unable to invade HCAEC upon treatment with proteinase K (Fig. 5B). In contrast, at the concentration tested, proteinase K had no significant effect on the ability of OMZ175 or the CpgfS strain to invade HCAEC (Fig. 5B). As expected, the reduced levels of binding in the ΔpgfS strain correlated well with increased degradation of Cnm, as assessed by immunofluorescent labeling (see Fig. S3 in the supplemental material). These results further support the idea that PgfS plays an essential role in Cnm modification and that such modification confers increased proteolytic stability to Cnm.
FIG 5.
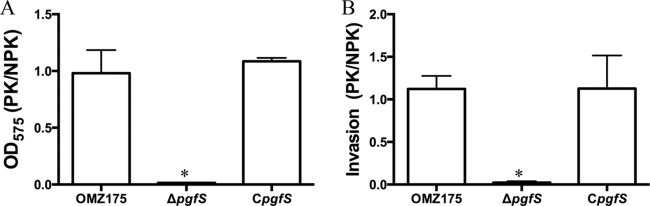
Pretreatment with proteinase K nearly abolishes the ability of the ΔpgfS strain to bind to collagen and invade HCAEC. OMZ175 and its ΔpgfS and CpgfS derivatives were treated with 3 μg ml−1 of proteinase K for 30 min, and their abilities to bind to collagen (A) and invade HCAEC (B) were assessed. Results are expressed as the ratio of binding by proteinase K-treated cells to that by untreated cells. *, P < 0.05.
Lectin binding analysis confirms the glycosylated nature of Cnm.
Electrophoretic and functional characterization of Cnm in OMZ175 and the ΔpgfS strain strongly suggests that PgfS modifies Cnm, and based on the presence of multiple threonine residues at the Cnm B domain, it is likely that Cnm undergoes O-linked glycosylation. To identify the carbohydrates attached to Cnm, we evaluated the ability of three different lectins (WGA, PNA, and ConA) to interact with whole-cell lysates of S. mutans OMZ175 and its derivatives. A band of the predicted size of Cnm was detected with WGA (Fig. 6A) but not with PNA or ConA (data not shown). Most importantly, Cnm detection by WGA was not observed in the Δcnm and ΔpgfS strains (Fig. 6A). These results are consistent with the interpretation that Cnm is a glycoprotein and that PgfS is essential for the glycosylation of Cnm.
FIG 6.

WGA and sWGA lectin analysis of Cnm. Detection and identification of carbohydrates attached to Cnm in S. mutans strains using biotinylated lectins is shown. Whole-cell lysates were separated by SDS-PAGE, transferred onto PVDF membranes, and probed with 20 μg ml−1 WGA (A) or 20 μg ml−1 sWGA (C). (B) Purified Cnm was treated with sialidase for 20 h and probed with WGA. (D) WGA detection of purified Cnm was blocked using increasing concentrations of N-acetylglucosamine.
The interaction of Cnm with WGA suggests that there could be at least two types of sugars attached to Cnm: sialic acid and N-acetylglucosamine (34). To distinguish between these two sugar moieties, native Cnm was purified from OMZ175 and the ΔpgfS strain using a custom-made affinity column (see Fig. S4 in the supplemental material). To test for the presence of sialic acid residues, the purified proteins were exposed to sialidase prior to WGA detection. However, no decrease in binding to WGA was observed after sialidase treatment (Fig. 6B), indicating that the sugar residues attached to Cnm are not sialic acid. The absence of sialic acid residues was further confirmed by using succinylated WGA, which recognizes only N-acetylglucosamine residues with no reactivity to sialic acid (Fig. 6C). Detection of Cnm by WGA was not affected by lysozyme and mutanolysin treatment during cell lysis, demonstrating that WGA reactivity was not due to residual N-acetylglucosamine from cell wall material (data not shown). Finally, when supplied in excess, N-acetylglucosamine inhibited Cnm binding to WGA in a concentration-dependent manner (Fig. 6D). Collectively, these data support the idea that PgfS is involved in Cnm modification, most likely through the addition of N-acetylglucosamine residues to the threonine-rich B-domain of Cnm.
DISCUSSION
S. mutans is an important human pathogen capable of causing infections in different sites of the body, such as the oral cavity and the heart. Recently, we showed that the collagen (and laminin)-binding protein Cnm is a major virulence factor of invasive S. mutans strains (15, 19). Cnm is composed of an N-terminal conserved collagen-binding domain (A domain), a unique threonine-rich B-repeat domain (B domain), and a C-terminal sortase signal LPXTG motif for cell wall anchoring (14). Initially, Cnm was identified as the factor responsible for the cold agglutination phenotype with a predicted molecular mass of 54 kDa (33). However, Cnm has been shown to migrate at ∼120 kDa in SDS-PAGE, and such a discrepancy in size was, until now, poorly understood (15, 33).
Our interest in the association of cnm and pgfS was initially sparked by transcriptional analyses showing cotranscription of these two genes, by the presence of threonine residues in the B-domain of Cnm and by the fact that pgfS encodes a putative glycosyltransferase. The linkage between Cnm and PgfS is somewhat unexpected given that pgfS, unlike cnm, is part of the core genome of S. mutans (35). A PgfS homolog, termed CsbB, was first described for Bacillus subtilis (36). In B. subtilis, csbB expression is controlled by the alternative σB factor and is predicted to participate in cell envelope biogenesis (36). Although the genomes of streptococcal species lack the σB factor, the link between PgfS and cell envelope biogenesis in S. mutans remains to be determined.
Aberrant electrophoretic migration patterns and increased tolerance to proteases are common traits of glycoproteins present in all domains of life (37). In this study, we showed that Cnm migrates at a lower molecular mass in the ΔpgfS strain (∼90 kDa) than its parent strain OMZ175 (∼120 kDa). However, Cnm has a predicted molecular mass of ∼54 kDa, indicating that, even in the absence of PgfS, Cnm has an abnormal migration pattern. It is worth mentioning that a recombinant form of Cnm expressed in E. coli migrated at the same mobility as Cnm expressed by the ΔpgfS strain (∼90 kDa) (data not shown). It is highly unlikely that the laboratory strains of E. coli possess the necessary machinery to glycosylate Cnm (38). Thus, such a discrepancy between the electrophoretic mobility and the predicted mass is likely due to the unique biochemical properties of the B domain of Cnm, a highly acidic repeat region with a negative net charge (pI < 3.75). Such a regular distribution of negative charges in the large B domain could repel SDS binding and artificially reduce the charge-to-mass ratio of Cnm in SDS-PAGE analysis. In accordance with this interpretation, a truncated version of Cnm, obtained via recombinant expression and lacking the threonine-rich B domain, migrated at the predicted molecular mass of 33 kDa (data not shown). Thus, it appears that the aberrant electrophoretic migration pattern of Cnm is due to both PgfS-dependent glycosylation and the high negative charge density of the B domain.
Modification of the Cnm B domain appears to modulate the functional properties of the collagen-binding A domain, as lack of PgfS glycosylation significantly affected Cnm-mediated binding to collagen. In addition, absence of PgfS affected Cnm susceptibility to proteinase K degradation, suggesting that Cnm modification by PgfS is necessary for Cnm function and proteolytic stability. These results are on a par with other bacterial protein glycosylation systems that directly or indirectly affect protein folding, modulation of conformation, proteolytic and thermodynamic stability, fimbrial assembly, binding to ECM molecules, adhesion to epithelial cells, and protein-protein interactions (23, 24, 26, 39, 40).
To further characterize the modification of Cnm by PgfS, we performed a series of experiments to confirm the glycosylated nature and type(s) of carbohydrate(s) associated with Cnm. Enzymatic deglycosylation using a cocktail of five of the major glycosidases yielded no noticeable changes in Cnm, suggesting that the attached carbohydrates synthesized by S. mutans may not be typical glycan structures. We also performed liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify carbohydrates that might be associated with Cnm. Despite our using a variety of proteases (trypsin, Glu-C, and pepsin) to digest natively purified Cnm, no peptides corresponding to the B domain were identified (data not shown). Similar observations have been made with other bacterial glycoproteins, for which enzymatic deglycosylation and standard MS analyses were not successful (41, 42). Nevertheless, the glycosylated nature of Cnm and the likely presence of N-acetylglucosamine residues were confirmed using lectin-binding assays.
The glycosylation biogenesis of bacterial surface components with host-binding abilities in closely related organisms has been described (40). For example, glycosylation of the serine-rich protein Fap-1 in S. parasanguinis is initiated by two glycosyltransferases, Gtf1 and Gtf2, which are responsible for the addition of a N-acetylglucosamine residue (43). Upon attachment of N-acetylglucosamine to Fap1, four other glycosyltransferases (Gly, Gtf3, GalT1, and GalT2) mediate the addition of other sugar residues, such as glucose, N-acetylgalactosamine, and rhamnose (44–46). Other accessory proteins, such as Gap1, Gap2, and Gap3, are also involved in the formation of a stable complex, which allows proper Fap1 maturation and secretion (25). Similarly complex mechanisms for glycosylation of the human platelet-binding protein GspB and the fibrinogen-binding protein Srr1 have been observed in Streptococcus gordonii and Streptococcus agalactiae, respectively (41, 47). While homologs of these glycosylation systems are found in other Gram-positive organisms (40), none of the genes encoding these glycosyltransferases are present in S. mutans. Hence, the interaction between Cnm-PgfS represents a potential novel protein glycosylation system in streptococci. At this time, we do not know whether the addition of N-acetylglucosamine moieties to Cnm is mediated solely by PgfS, or if this modification involves multiple proteins as seen in other streptococci. It also remains unclear whether Cnm modification occurs by the addition of a single monosaccharide or an entire oligosaccharide molecule with a more complex carbohydrate composition. Investigations to determine the carbohydrate composition and how these carbohydrates are added to Cnm are under way.
During our initial attempts to complement the ΔpgfS strain, several strategies restored Cnm-dependent phenotypes poorly or not at all (data not shown). For example, inserting a copy of pgfS at the gtfA locus using the integration vector pBGE (48) resulted in partial protection against proteinase K treatment yet no complementation of the defects in collagen binding or HCAEC invasion. Western blot analysis using anti-rCnmA and WGA revealed that Cnm is inefficiently glycosylated in the pBGE::CpgfS strain (data not shown). Although we have used a well-characterized nonpolar kanamycin cassette (28) to replace the pgfS gene in OMZ175, we initially speculated that failure to complement the ΔpgfS strain with the pBGE integration vector could be due to unexpected polar effects on the genes downstream of pgfS. To rule out this possibility, we performed qRT-PCR analysis on the two genes (SMU.2066c and SMU.2065) immediately downstream of pgfS. No differences in the mRNA levels of SMU.2066c and SMU.2065 were observed between parent and ΔpgfS strains (data not shown). However, we also noticed that pgfS and SMU.2066c overlap by four nucleotides and, therefore, are predicted to be cotranslated. Thus, while transcription of these genes was apparently not affected by insertion of the nonpolar kanamycin cassette, it is possible that the pgfS and SMU.2066c mRNAs are not efficiently translated or are less stable in the pBGE::CpgfS strain. This led us to restore the pgfS gene at the original locus by replacing the mutated fragment with an intact copy of pgfS, thereby creating the CpgfS strain. In this strain, most phenotypes affected by the pgfS mutation (HCAEC invasion, proteinase K degradation, and WGA binding) were complemented, suggesting that the genetic arrangement cnm-pgfS-SMU.2066c-SMU.2065 must remain intact for optimal expression. Incidentally, a recent study in B. subtilis reported that the interaction between a homolog of SMU.2066c, yfhO, and csbB interactively affects SigM activity (48). Generally, protein glycosylation machineries tend to be located at the bacterial membrane, where glycosylation is thought to occur (23). Since both PgfS and SMU.2066c are predicted to be membrane associated, it would be interesting to determine the functional relationship between these proteins.
Our findings suggest that pgfS has a conserved function across cnm+ strains, since all isolates analyzed thus far showed a similar pattern of PgfS-dependent glycosylation of Cnm. Furthermore, the same pattern was observed when pgfS was deleted in the UA159+cnm background strain, demonstrating that PgfS function is conserved in S. mutans, since cnm is absent in this laboratory strain. Interestingly, genes encoding putative collagen-binding proteins with threonine-rich domains followed by pgfS and SMU.2066c orthologs are present in several other streptococcal species (i.e., Streptococcus ratti, S. suis, S. infantis, and S. parasanguinis), raising the possibility of a novel and conserved mechanism for protein glycosylation in this bacterial group (see Fig. S5 in the supplemental material).
In conclusion, we showed that Cnm is a glycoprotein and that the downstream gene pgfS is directly involved in its modification. The inability of the ΔpgfS strain to glycosylate Cnm significantly impaired Cnm proteolytic stability, thereby affecting the expression of several Cnm-dependent phenotypes, such as collagen binding, HCAEC invasion, and killing of G. mellonella. To our knowledge, this is the first time that protein glycosylation has been linked to virulence expression in S. mutans.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the American Heart Association (10GRNT4210049) and NIH-NIDCR (R01 DE022559). A.A.-R. was supported by NIH-NIDCR training grant in oral sciences T90 DE021985.
We thank Christine Szymanski and Harald Nothaft from University of Alberta for helpful discussions.
Footnotes
Published ahead of print 16 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01783-14.
REFERENCES
- 1.Loesche WJ. 1986. Role of Streptococcus mutans in human dental decay. Microbiol. Rev. 50:353–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowen WH, Koo H. 2011. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res. 45:69–86. 10.1159/000324598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakano K, Nomura R, Matsumoto M, Ooshima T. 2010. Roles of oral bacteria in cardiovascular diseases—from molecular mechanisms to clinical cases: cell-surface structures of novel serotype k Streptococcus mutans strains and their correlation to virulence. J. Pharmacol. Sci. 113:120–125. 10.1254/jphs.09R24FM [DOI] [PubMed] [Google Scholar]
- 4.Nakano K, Lapirattanakul J, Nomura R, Nemoto H, Alaluusua S, Gronroos L, Vaara M, Hamada S, Ooshima T, Nakagawa I. 2007. Streptococcus mutans clonal variation revealed by multilocus sequence typing. J. Clin. Microbiol. 45:2616–2625. 10.1128/JCM.02343-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakano K, Nemoto H, Nomura R, Homma H, Yoshioka H, Shudo Y, Hata H, Toda K, Taniguchi K, Amano A, Ooshima T. 2007. Serotype distribution of Streptococcus mutans a pathogen of dental caries in cardiovascular specimens from Japanese patients. J. Med. Microbiol. 56:551–556. 10.1099/jmm.0.47051-0 [DOI] [PubMed] [Google Scholar]
- 6.Nakano K, Nomura R, Ooshima T. 2008. Streptococcus mutans and cardiovascular diseases. Jpn. Dent. Sci. Rev. 44:29–37. 10.1016/j.jdsr.2007.09.001 [DOI] [Google Scholar]
- 7.Que YA, Moreillon P. 2011. Infective endocarditis. Nat. Rev. Cardiol. 8:322–336. 10.1038/nrcardio.2011.43 [DOI] [PubMed] [Google Scholar]
- 8.Banas JA. 2004. Virulence properties of Streptococcus mutans. Front. Biosci. 9:1267–1277. 10.2741/1305 [DOI] [PubMed] [Google Scholar]
- 9.Minino AM, Murphy SL, Xu J, Kochanek KD. 2011. Deaths: final data for 2008. Natl. Vital Stat Rep. 59:1–126 http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_10.pdf [PubMed] [Google Scholar]
- 10.Nakano K, Inaba H, Nomura R, Nemoto H, Takeda M, Yoshioka H, Matsue H, Takahashi T, Taniguchi K, Amano A, Ooshima T. 2006. Detection of cariogenic Streptococcus mutans in extirpated heart valve and atheromatous plaque specimens. J. Clin. Microbiol. 44:3313–3317. 10.1128/JCM.00377-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakano K, Nemoto H, Nomura R, Inaba H, Yoshioka H, Taniguchi K, Amano A, Ooshima T. 2009. Detection of oral bacteria in cardiovascular specimens. Oral Microbiol. Immunol. 24:64–68. 10.1111/j.1399-302X.2008.00479.x [DOI] [PubMed] [Google Scholar]
- 12.Kesavalu L, Lucas AR, Verma RK, Liu L, Dai E, Sampson E, Progulske-Fox A. 2012. Increased atherogenesis during Streptococcus mutans infection in ApoE-null mice. J. Dent. Res. 91:255–260. 10.1177/0022034511435101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nobbs AH, Lamont RJ, Jenkinson HF. 2009. Streptococcus adherence and colonization. Microbiol. Mol. Biol. Rev. 73:407–450. 10.1128/MMBR.00014-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato Y, Okamoto K, Kagami A, Yamamoto Y, Igarashi T, Kizaki H. 2004. Streptococcus mutans strains harboring collagen-binding adhesin. J. Dent. Res. 83:534–539. 10.1177/154405910408300705 [DOI] [PubMed] [Google Scholar]
- 15.Aviles-Reyes A, Miller JH, Simpson-Haidaris PJ, Lemos JA, Abranches J. 2014. Cnm is a major virulence factor of invasive Streptococcus mutans and part of a conserved three-gene locus. Mol. Oral Microbiol. 29:11–23. 10.1111/mom.12041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakano K, Nomura R, Taniguchi N, Lapirattanakul J, Kojima A, Naka S, Senawongse P, Srisatjaluk R, Gronroos L, Alaluusua S, Matsumoto M, Ooshima T. 2010. Molecular characterization of Streptococcus mutans strains containing the cnm gene encoding a collagen-binding adhesin. Arch. Oral Biol. 55:34–39. 10.1016/j.archoralbio.2009.11.008 [DOI] [PubMed] [Google Scholar]
- 17.Nomura R, Naka S, Nemoto H, Inagaki S, Taniguchi K, Ooshima T, Nakano K. 2013. Potential involvement of collagen-binding proteins of Streptococcus mutans in infective endocarditis. Oral Dis. 19:387–393. 10.1111/odi.12016 [DOI] [PubMed] [Google Scholar]
- 18.Abranches J, Miller JH, Martinez AR, Simpson-Haidaris PJ, Burne RA, Lemos JA. 2011. The collagen-binding protein Cnm is required for Streptococcus mutans adherence to and intracellular invasion of human coronary artery endothelial cells. Infect. Immun. 79:2277–2284. 10.1128/IAI.00767-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakano K, Hokamura K, Taniguchi N, Wada K, Kudo C, Nomura R, Kojima A, Naka S, Muranaka Y, Thura M, Nakajima A, Masuda K, Nakagawa I, Speziale P, Shimada N, Amano A, Kamisaki Y, Tanaka T, Umemura K, Ooshima T. 2011. The collagen-binding protein of Streptococcus mutans is involved in haemorrhagic stroke. Nat. Commun. 2:485–494. 10.1038/ncomms1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kline KA, Falker S, Dahlberg S, Normark S, Henriques-Normark B. 2009. Bacterial adhesins in host-microbe interactions. Cell Host Microbe 5:580–592. 10.1016/j.chom.2009.05.011 [DOI] [PubMed] [Google Scholar]
- 21.Wu H, Mintz KP, Ladha M, Fives-Taylor PM. 1998. Isolation and characterization of Fap1, a fimbriae-associated adhesin of Streptococcus parasanguis FW213. Mol. Microbiol. 28:487–500. 10.1046/j.1365-2958.1998.00805.x [DOI] [PubMed] [Google Scholar]
- 22.Benz I, Schmidt MA. 2001. Glycosylation with heptose residues mediated by the aah gene product is essential for adherence of the AIDA-I adhesin. Mol. Microbiol. 40:1403–1413. 10.1046/j.1365-2958.2001.02487.x [DOI] [PubMed] [Google Scholar]
- 23.Szymanski CM, Wren BW. 2005. Protein glycosylation in bacterial mucosal pathogens. Nat. Rev. Microbiol. 3:225–237. 10.1038/nrmicro1100 [DOI] [PubMed] [Google Scholar]
- 24.Wu H, Zeng M, Fives-Taylor P. 2007. The glycan moieties and the N-terminal polypeptide backbone of a fimbria-associated adhesin, Fap1, play distinct roles in the biofilm development of Streptococcus parasanguinis. Infect. Immun. 75:2181–2188. 10.1128/IAI.01544-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou M, Zhu F, Li Y, Zhang H, Wu H. 2012. Gap1 functions as a molecular chaperone to stabilize its interactive partner Gap3 during biogenesis of serine-rich repeat bacterial adhesin. Mol. Microbiol. 83:866–878. 10.1111/j.1365-2958.2012.07970.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cote JP, Charbonneau ME, Mourez M. 2013. Glycosylation of the Escherichia coli TibA self-associating autotransporter influences the conformation and the functionality of the protein. PLoS One 8:e80739. 10.1371/journal.pone.0080739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charbonneau ME, Girard V, Nikolakakis A, Campos M, Berthiaume F, Dumas F, Lepine F, Mourez M. 2007. O-linked glycosylation ensures the normal conformation of the autotransporter adhesin involved in diffuse adherence. J. Bacteriol. 189:8880–8889. 10.1128/JB.00969-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kremer BH, van der Kraan M, Crowley PJ, Hamilton IR, Brady LJ, Bleiweis AS. 2001. Characterization of the sat operon in Streptococcus mutans: evidence for a role of Ffh in acid tolerance. J. Bacteriol. 183:2543–2552. 10.1128/JB.183.8.2543-2552.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau PC, Sung CK, Lee JH, Morrison DA, Cvitkovitch DG. 2002. PCR ligation mutagenesis in transformable streptococci: application and efficiency. J. Microbiol. Methods 49:193–205. 10.1016/S0167-7012(01)00369-4 [DOI] [PubMed] [Google Scholar]
- 30.Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc. Natl. Acad. Sci. U. S. A. 104:3508–3513. 10.1073/pnas.0608742104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kajfasz JK, Rivera-Ramos I, Abranches J, Martinez AR, Rosalen PL, Derr AM, Quivey RG, Lemos JA. 2010. Two Spx proteins modulate stress tolerance, survival, and virulence in Streptococcus mutans. J. Bacteriol. 192:2546–2556. 10.1128/JB.00028-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coutinho PM, Deleury E, Davies GJ, Henrissat B. 2003. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328:307–317. 10.1016/S0022-2836(03)00307-3 [DOI] [PubMed] [Google Scholar]
- 33.Sato Y, Okamoto K, Kagami A, Yamamoto Y, Ohta K, Igarashi T, Kizaki H. 2004. Application of in vitro mutagenesis to identify the gene responsible for cold agglutination phenotype of Streptococcus mutans. Microbiol. Immunol. 48:449–456. 10.1111/j.1348-0421.2004.tb03535.x [DOI] [PubMed] [Google Scholar]
- 34.Monsigny M, Roche AC, Sene C, Maget-Dana R, Delmotte F. 1980. Sugar-lectin interactions: how does wheat-germ agglutinin bind sialoglycoconjugates? Eur. J. Biochem. 104:147–153. 10.1111/j.1432-1033.1980.tb04410.x [DOI] [PubMed] [Google Scholar]
- 35.Ajdic D, McShan WM, McLaughlin RE, Savic G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. U. S. A. 99:14434–14439. 10.1073/pnas.172501299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akbar S, Price CW. 1996. Isolation and characterization of csbB, a gene controlled by Bacillus subtilis general stress transcription factor sigma B. Gene 177:123–128. 10.1016/0378-1119(96)00287-9 [DOI] [PubMed] [Google Scholar]
- 37.Geyer H, Geyer R. 2006. Strategies for analysis of glycoprotein glycosylation. Biochim. Biophys. Acta 1764:1853–1869. 10.1016/j.bbapap.2006.10.007 [DOI] [PubMed] [Google Scholar]
- 38.Guarino C, DeLisa MP. 2012. A prokaryote-based cell-free translation system that efficiently synthesizes glycoproteins. Glycobiology. 22:596–601. 10.1093/glycob/cwr151 [DOI] [PubMed] [Google Scholar]
- 39.Tang G, Ruiz T, Mintz KP. 2012. O-Polysaccharide glycosylation is required for stability and function of the collagen adhesin EmaA of Aggregatibacter actinomycetemcomitans. Infect. Immun. 80:2868–2877. 10.1128/IAI.00372-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou M, Wu H. 2009. Glycosylation and biogenesis of a family of serine-rich bacterial adhesins. Microbiology 155:317–327. 10.1099/mic.0.025221-0 [DOI] [PubMed] [Google Scholar]
- 41.Bensing BA, Gibson BW, Sullam PM. 2004. The Streptococcus gordonii platelet binding protein GspB undergoes glycosylation independently of export. J. Bacteriol. 186:638–645. 10.1128/JB.186.3.638-645.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Woude AD, Mahendran KR, Ummels R, Piersma SR, Pham TV, Jimenez CR, de Punder K, van der Wel NN, Winterhalter M, Luirink J, Bitter W, Houben EN. 2013. Differential detergent extraction of Mycobacterium marinum cell envelope proteins identifies an extensively modified threonine-rich outer membrane protein with channel activity. J. Bacteriol. 195:2050–2059. 10.1128/JB.02236-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bu S, Li Y, Zhou M, Azadin P, Zeng M, Fives-Taylor P, Wu H. 2008. Interaction between two putative glycosyltransferases is required for glycosylation of a serine-rich streptococcal adhesin. J. Bacteriol. 190:1256–1266. 10.1128/JB.01078-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, Bu S, Newell P, Chen Q, Fives-Taylor P. 2007. Two gene determinants are differentially involved in the biogenesis of Fap1 precursors in Streptococcus parasanguis. J. Bacteriol. 189:1390–1398. 10.1128/JB.00836-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou M, Zhu F, Dong S, Pritchard DG, Wu H. 2010. A novel glucosyltransferase is required for glycosylation of a serine-rich adhesin and biofilm formation by Streptococcus parasanguinis. J. Biol. Chem. 285:12140–12148. 10.1074/jbc.M109.066928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mistou MY, Dramsi S, Brega S, Poyart C, Trieu-Cuot P. 2009. Molecular dissection of the secA2 locus of group B Streptococcus reveals that glycosylation of the Srr1 LPXTG protein is required for full virulence. J. Bacteriol. 191:4195–4206. 10.1128/JB.01673-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng L, Burne RA. 2009. Transcriptional regulation of the cellobiose operon of Streptococcus mutans. J. Bacteriol. 191:2153–2162. 10.1128/JB.01641-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inoue H, Suzuki D, Asai K. 2013. A putative bactoprenol glycosyltransferase, CsbB, in Bacillus subtilis activates SigM in the absence of transcribed YfhO. Biochem. Biophys. Res. Commun. 436:6–11. 10.1016/j.bbrc.2013.04.064 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.