

Abstract
The localization of the LapA protein to the cell surface is a key step required by Pseudomonas fluorescens Pf0-1 to irreversibly attach to a surface and form a biofilm. LapA is a member of a diverse family of predicted bacterial adhesins, and although lacking a high degree of sequence similarity, family members do share common predicted domains. Here, using mutational analysis, we determine the significance of each domain feature of LapA in relation to its export and localization to the cell surface and function in biofilm formation. Our previous work showed that the N terminus of LapA is required for cleavage by the periplasmic cysteine protease LapG and release of the adhesin from the cell surface under conditions unfavorable for biofilm formation. We define an additional critical region of the N terminus of LapA required for LapG proteolysis. Furthermore, our results suggest that the domains within the C terminus of LapA are not absolutely required for biofilm formation, export, or localization to the cell surface, with the exception of the type I secretion signal, which is required for LapA export and cell surface localization. In contrast, deletion of the central repetitive region of LapA, consisting of 37 repeats of 100 amino acids, results in an inability to form a biofilm. We also used single-molecule atomic force microscopy to further characterize the role of these domains in biofilm formation on hydrophobic and hydrophilic surfaces. These studies represent the first detailed analysis of the domains of the LapA family of biofilm adhesin proteins.
INTRODUCTION
Bacteria in environmental, clinical, and industrial settings predominantly associate with surfaces and persist in biofilms (1, 2). In order to form a biofilm, cells must transition from a planktonic to a sessile mode of life and commit to stable surface attachment. This transition, known as irreversible attachment, is the first committed step in biofilm formation. The mechanisms that foster the transition to a sessile mode of life are quite diverse, as are the environmental signals and cues to which bacteria respond (3–12). A similar trend across many bacterial species, however, is that biofilm formation coincides with the synthesis of the signaling molecule bis-(3′-5′)-cyclic dimeric GMP (c-di-GMP).
Pseudomonas fluorescens Pf0-1, a model organism for biofilm research, functions as a biological control agent that promotes plant growth by forming biofilms on roots (13). The LapA protein, identified by our group, is a bona fide adhesin, as analyzed by atomic force microscopy (AFM) (14–16), and is conserved in many environmental pseudomonads, such as P. fluorescens and Pseudomonas putida (17). In P. putida, LapA is required for biofilm formation on plant surfaces (18), while the P. fluorescens Pf0-1 LapA adhesin is required for initial attachment and biofilm formation on every abiotic surface tested to date, including hydrophobic and hydrophilic surfaces such as plastic and glass, respectively (6, 17).
LapA is a very large protein, with an estimated molecular mass of ∼520 kDa. P. fluorescens LapA belongs to a newly identified subfamily of proteins including other widely studied large putative adhesins with characteristic amino acid repeat regions, repeats-in-toxins (RTX) sequences, and a type I secretion system (T1SS) signal (19). The large repetitive RTX adhesins include LapA, a key determinant of bacterial adhesion to corn seeds (20) and of root colonization by P. putida (18); LapF, a protein required for late-stage biofilm formation in P. putida (21); FrhA, which is associated with hemagglutination, adherence to epithelial cells, biofilm formation, and chitin binding in Vibrio cholerae (22); SiiE in Salmonella enterica, which functions in epithelial cell adhesion (23); RtxA in Legionella pneumophila, which is involved in adhesion and entry into macrophages and amoebae (24); and the biofilm-promoting factor BpfA in Shewanella oneidensis (25). Little is known regarding how these domains contribute to biofilm formation and localization and the association of LapA with the bacterial cell surface.
Studies from our group have identified regulation through posttranslational modification of LapA that occurs in response to conditions unfavorable for biofilm formation, such as low inorganic phosphate (Pi) concentrations (26–28). Briefly, under conditions of low Pi concentrations, the periplasmic cysteine protease LapG cleaves LapA from the cell surface, thus releasing the adhesin into the supernatant and preventing attachment (27). The inner membrane c-di-GMP effector protein LapD regulates LapG's protease activity in a c-di-GMP-dependent manner. Upon binding c-di-GMP, LapD undergoes conformational changes (26, 28), and through an inside-out signaling mechanism, LapD binds LapG, preventing LapG-dependent cleavage of LapA from the cell surface. Thus, under conditions favorable for biofilm formation, LapA remains at the cell surface, thus promoting biofilm formation (27). While we have a detailed picture of the LapD-LapG c-di-GMP control circuit, from the environmental Pi input to the LapA output that regulates biofilm formation by P. fluorescens Pf0-1, determinants by which LapG recognizes LapA to promote cleavage are still poorly understood.
In this study, we address the mechanism of LapA release from the cell surface by determining a critical region required for LapG-dependent cleavage of LapA and, thus, localization of this adhesin to the cell surface. We also present a mutagenesis analysis characterizing the predicted domains of LapA using genetic, biochemical, and AFM analyses to assess how each domain contributes to LapA localization, LapG-dependent release from the cell surface, and biofilm formation. These studies represent the first detailed analysis of this large family of bacterial adhesins.
MATERIALS AND METHODS
Strains and media.
The strains and plasmids used in this study are listed in Table 1 and Table S1 in the supplemental material, respectively. Strain SMC4798, described previously (27, 29, 30), is the wild-type (WT) P. fluorescens Pf0-1 strain used in this study. P. fluorescens strain SMC4798 produces a fully functional chromosomally encoded LapA protein carrying three-hemagglutinin (HA) epitope tags inserted after residue 4093. This HA-tagged, fully functional LapA protein was used in all the studies described here.
TABLE 1.
Strains used in this study
| Organism and strain | Genotype and/or description | Source or reference |
|---|---|---|
| Escherichia coli | ||
| Top-10 | recA1 araD139 Δ(ara-leu)7697 | Life Technologies |
| SMC117 | S17-1(λpir); thi pro hsdR hsdM+ ΔrecA RP4-2::TcMu-Km::Tn7 | 65 |
| SMC31 | DH5α; supE44 lacU169(80lacZM15) hsdR17 thi-1 relA1 recA1 | 66 |
| Pseudomonas fluorescens | ||
| SMC6370 | Pseudomonas fluorescens Pf0-1; wild type | 67 |
| SMC4798 | P. fluorescens Pf0-1 expressing LapA-HA, inserted after residue 4093; designated the wild type for this study | 30 |
| SMC5207 | ΔlapG; SMC4798 with an unmarked deletion of lapG | 27 |
| SMC5152 | ΔlapD; SMC4798 with an unmarked deletion of lapD | 28 |
| SMC6414 | Δhelix; SMC4798 with an unmarked deletion of lapA codons A81–95A | This study |
| SMC6458 | Δhelix lapG::pKO3-Tetr; SMC4798 with an unmarked deletion of LapA amino acids A81–95A and single-crossover knockout mutation disrupting the lapG gene | This study |
| SMC5847 | ΔRpts; SMC4798 with an unmarked deletion of LapA amino acids D273–3982S | This study |
| SMC5849 | ΔCTerm; SMC4798 with an unmarked deletion of LapA amino acids T4018–5151N | This study |
| SMC5853 | ΔvWA; SMC4798 with an unmarked deletion of LapA amino acids N4736–4963S | This study |
| SMC5851 | ΔRTX; SMC4798 with an unmarked deletion of LapA amino acids G5020–5144W | This study |
| SMC5855 | ΔT1SS; SMC4798 with an unmarked deletion of LapA amino acids N5151–5218S | This study |
| SMC5917 | lapA AA108–109RR; SMC4798 with point mutation AA108–109RR | 27 |
| SMC5915 | Pf0-1 lapG::pKO3-Tetr; SMC4798 with a single-crossover knockout mutation disrupting the lapG gene | 27 |
| SMC5927 | lapA A108W; SMC4798 with point mutation A108D | This study |
| SMC5923 | lapA AA108–109DD; SMC4798 with point mutation AA108–109DD | This study |
| SMC5925 | lapA A109D; SMC4798 with point mutation A109D | This study |
| SMC6400 | Pf0-1 lapG::pKO3-Tetr; SMC6370 with a single-crossover knockout mutation disrupting the lapA gene | 30 |
| SMC5869 | lapA ΔA108–120A; SMC4798 with an unmarked deletion of lapD and an unmarked deletion of LapA amino acids A108–120A | This study |
P. fluorescens and Escherichia coli were routinely cultured in liquid LB medium or on solidified LB medium with 1.5% agar and grown at 30°C or 37°C, respectively. Gentamicin (Gm) was used at 30 μg/ml for P. fluorescens and at 10 μg/ml for E. coli. Tetracycline (Tet) was used at 30 μg/ml on plates and at 15 μg/ml in liquid culture for P. fluorescens and E. coli. Chloramphenicol (Cm) was used at 20 μg/ml on plates for P. fluorescens. Arabinose was used to induce expression of the PBAD promoter from pMQ72 at 0.2% (vol/vol) for overexpression experiments. P. fluorescens was grown in K10T-1 medium, previously described as a phosphate-rich medium (31), consisting of 50 mM Tris-HCl (pH 7.4), 0.2% (wt/vol) Bacto tryptone, 0.15% (vol/vol) glycerol, 0.61 mM MgSO4, and 1 mM K2HPO4 for biofilm assays, LapA localization, and protein expression.
The K10T-1 medium used in these studies contains calcium at low concentrations. We measured the calcium concentration by inductively coupled plasma mass spectrometry (ICP-MS), as reported previously (30), and found that K10T-1 medium contains ∼15 μM calcium. K10T-1 medium contains 1.2 mM inorganic phosphate, as reported previously (30). We modeled the amounts of calcium and phosphate that would precipitate from the medium using the publically available software VisualMINTEQ. Using the known concentrations of phosphate and calcium and the known pH of the medium, thermodynamic considerations are consistent with the possibility that hydroxyapatite could precipitate in this solution. However, based on our calculations, only ∼36% of the total Ca2+ and <0.3% of the total phosphate are likely to precipitate as a hydroxyapatite phase, leaving ∼8 μM calcium and ∼1 mM phosphate in solution. Furthermore, we have performed experiments in which we added calcium to K10T-1 medium at concentrations of up to 500 μM, and we saw no changes in biofilm formation or LapA localization (32). Finally, we have also shown that LapG is a calcium-dependent enzyme and requires calcium for LapA cleavage (32). LapG functions in K10T-1 medium without supplementation with additional calcium, also suggesting that this medium is not calcium free, a finding consistent with our ICP-MS analysis.
Saccharomyces cerevisiae strain InvSc1 (Life Technologies; formerly Invitrogen, Carlsbad, CA) was routinely cultured on yeast extract-peptone-dextrose (YPD) medium. When selecting for plasmids carrying the URA3 gene, this strain was grown on yeast nitrogen base (YNB) with the complete supplemental mixture minus uracil.
Biofilm formation assays. (i) Hydrophobic plastic surfaces.
Biofilm formation assays were performed as previously described (29). Briefly, a polyvinyl chloride 96-well round-bottom microtiter plate (catalog number 2797; Corning) containing 100 μl of K10T-1 medium per well was inoculated with 1.5 μl of a liquid culture of P. fluorescens grown overnight in LB medium. Biofilms were grown statically for 6 h at 30°C. At the completion of the assay, medium and unattached bacterial cells were removed, the wells were stained with a 0.1% (wt/vol) solution of crystal violet in water, and the plate was rinsed with water. Staining, imaging, and quantification of attached biofilm biomass were also performed as previously described (30). Briefly, 150 μl of a solution containing 30% methanol and 10% acetic acid was added to each biofilm well to solubilize the crystal violet and determine biofilm biomass. One hundred twenty-five microliters was transferred into a flat-bottom microtiter plate, and a Spectra Max M2 microplate reader (Molecular Devices, Sunnyvale, CA) was used to read the absorbance at 550 nm (30). Quantification of 8 to 10 wells for at least three biological replicates was performed for each P. fluorescens strain tested.
(ii) Hydrophilic glass surfaces.
Borosilicate glass tubes (Fisherbrand culture tubes, catalog number 14-961-27) were used as a model hydrophilic surface. The glass tubes containing 9.850 ml of K10T-1 medium were each inoculated with 150 μl of a liquid culture of P. fluorescens grown overnight in LB medium. Biofilms were grown statically for 6 h at 30°C. At the completion of the assay, medium and unattached bacterial cells were removed, and then adherent cells were stained with a 0.1% (wt/vol) solution of crystal violet in water and the tubes were rinsed with water. Two milliliters of a solution containing 30% methanol and 10% acetic acid was added to each well to solubilize the crystal violet. One milliliter was transferred into a cuvette, and the absorbance was read as described above. Quantification of 6 to 8 glass tubes, in triplicate experiments, was performed for each P. fluorescens strain tested.
Constructs for clean deletion of LapA domains and LapA site-directed mutagenesis.
Saccharomyces cerevisiae strain InvSci (Life Technologies) was used to construct plasmids for clean deletion and site-directed mutagenesis through in vivo homologous recombination, as previously described, using the pMQ30 allelic replacement vector, which carries the PBAD promoter (33). All primers used in this study are listed in Table S2 in the supplemental material. All chromosomal deletion mutants and chromosomal site-directed mutants were constructed by the same basic strategy as that described previously (27, 29). Briefly, two adjacent fragments of homologous DNA, located directly upstream and downstream of the intended deleted region or the region to be replaced with missense mutations, were amplified by PCR (500 to 2,000 bp each). Primers also contained 20 to 40 additional base pairs to facilitate recombination of upstream and downstream fragments with the pMQ30 vector. Primers specific for site-directed mutagenesis contained a sequence to encode missense mutations.
Allelic replacement was performed as previously described (33). Conjugation between P. fluorescens SMC4798, carrying an HA-tagged variant of LapA, and E. coli followed by selection on LB plates containing Gm and Cm was necessary for the selection of P. fluorescens transconjugants. Single-crossover events were confirmed by PCR, and strains were cultured overnight in the absence of antibiotics and then plated onto LB medium with 5% (wt/vol) sucrose to select for the second crossover event. All mutations were verified by PCR and sequencing of purified genomic DNA to confirm the deletion or missense mutation. Phenotypes of mutant strains were examined in three separate clones.
Constructs for overexpression of NTerm-LapA variants.
Overexpression studies were performed by using plasmid pMQ72, which carries the arabinose-inducible PBAD promoter, as previously described (27, 29). N-terminal LapA (NTerm-LapA) variants were made as previously described, using site-directed mutagenesis (27). Briefly, two fragments were amplified from the pMQ72-NTerm-LapA plasmid template containing a C-terminal six-histidine (6×His) tag, using a primer homologous to the vector backbone and a primer adjacent to the region of the missense mutation. Adjacent primers contained additional bases encoding the missense mutations and a homologous sequence to aid in yeast recombination. Yeast cloning resulted in plasmids identical to pMQ72-NTerm-LapA, except for the incorporation of the desired missense mutation. Plasmid DNA was sequenced to ensure successful construction of mutant variants of NTerm-LapA. Expression of the WT and mutant variants of NTerm-LapA was under the control of the arabinose-inducible PBAD promoter.
Construct for single-crossover knockout of the lapG gene.
A single-crossover knockout of the lapG gene in SMC6414, a strain with a clean deletion of the predicted α-helical region spanning residues A81 to 95A, ensured that cleavage of this LapA variant was due to LapG protease function. Cloning to obtain a single-crossover knockout of the lapG gene was performed as previously described for SMC5917 (27). Briefly, an E. coli strain expressing pKO3-lapG(Tetr) (27, 30) was conjugated with strain SMC6414, and the single-crossover event (e.g., Campbell recombination) was selected for by plating onto LB plates containing Tet, which selected for the single-crossover insertion event in the lapG gene, and Cm, which counterselected against the E. coli donor strain.
LapA localization.
For Western blot and dot blot assays to detect the LapA protein, we used strains with an internal HA tag engineered into the chromosomal lapA gene. For Western blot assays for NTerm-LapA, we used strains that expressed this plasmid-encoded NTerm-LapA construct with a C-terminal His tag. Culturing conditions, preparation of clarified whole-cell extracts and cellular supernatants, detection of cell surface LapA by dot blotting, and analysis of samples for LapA localization were performed as previously described (27, 28, 30). Briefly, 1 ml of a bacterial culture grown overnight was subcultured in 50 ml of K10T-1 medium (1:50 dilution) and grown with shaking at 220 rpm at 30°C for 6 h. For preparation of clarified whole-cell extracts and supernatant fractions, cells were harvested by centrifugation (5,000 × g for 12 min at 4°C). For preparation of the supernatant fraction, 15 ml of the supernatant was passed through a 0.22-μm filter (catalog number UFC910096; Millipore) to remove residual cells, and the supernatant was then concentrated by using a 100-kDa Amicon centrifugal filter unit (catalog number UFC910096; Millipore). Cell pellets were resuspended in 750 μl of LapA buffer (20 mM Tris-HCl [pH 8], 10 mM MgCl2) and sonicated on ice to lyse cells (30% power applied 4 times for a duration of 10 s, with a cooling period of 10 s after each treatment). Debris was pelleted by centrifugation (13,000 × g for 10 min at 4°C). The supernatant was retained as the whole-cell fraction. Total protein concentrations of the cell and supernatant fractions were determined by using the Pierce bicinchoninic acid (BCA) protein assay according to the manufacturer's instructions (catalog number 23225; Thermo Scientific).
For preparation of cells for dot blot assays, we utilized aliquots from the same cultures as those used for preparation of the cell and supernatant fractions. Cultures were normalized to the same optical density. Cells were pelleted (13,000 × g for 1 min at 4°C), washed twice in K10T-1 medium, and resuspended in K10T-1 medium in a final volume of 1,000 μl. Five-microliter aliquots were spotted onto a nitrocellulose membrane and dried prior to probing for the HA epitope tag on LapA by Western blotting.
Outer membrane fractions for dot blot analysis were prepared as reported previously (34) and as described below. Bacterial cultures grown overnight were subcultured and harvested identically as described above for LapA localization. The cell pellets were resuspended in 750 μl of buffer C (50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 2 mM MgCl2) and sonicated on ice to lyse cells (30% power applied 4 times for a duration of 10 s, with a cooling period of 10 s after each treatment). Debris was pelleted by centrifugation (13,000 × g for 10 min at 4°C), and the supernatants were separated into soluble cytosolic and insoluble total membrane fractions by ultracentrifugation (100,000 × g for 1 h at 4°C). Cytoplasmic contaminants were removed by washing the total membrane pellets with 750 μl of buffer B (20 mM Tris-HCl, pH 7.6). The pellets were resuspended in 200 μl buffer B, and an equal volume of 4% Sarkosyl in buffer B was added for a 2% final Sarkosyl concentration. Samples were incubated at room temperature for 20 min with gentle agitation, and the inner and outer membrane fractions were separated by ultracentrifugation (100,000 × g for 1 h at 4°C). The outer membrane-containing pellets were resuspended in 50 μl buffer B. Protein concentrations were determined by using the Pierce BCA protein assay according to the manufacturer's instructions. A total of 0.001 mg of the outer membrane fraction was brought to a 5 μl total volume in buffer B and spotted onto a nitrocellulose membrane. The dried nitrocellulose membrane was probed for HA epitope-tagged LapA by Western blotting.
SDS-PAGE and Western blotting.
SDS loading buffer was mixed with samples, followed by heat denaturation for 5 min at 80°C, and samples were resolved by SDS-PAGE. Totals of 0.005 mg and 0.01 mg of the supernatant and cell fractions, respectively, were loaded onto a 4-to-15% gradient Mini-Protean TGX gel (catalog number 456-1084; Bio-Rad Laboratories). Proteins were transferred onto a nitrocellulose membrane by using a Mini Trans-Blot cell (Bio-Rad Laboratories, Hercules, CA) and probed with either an anti-HA antibody to detect full-length chromosomal LapA (catalog number MMS-101R-500; Covance) or an anti-His antibody to detect NTerm-LapA (catalog number 34660; Qiagen). A goat anti-mouse horseradish peroxidase-conjugated secondary antibody was used in all studies (catalog number 170-6516; Bio-Rad Laboratories). ECL detection reagents were used to develop Western blots according to the manufacturer's instructions (catalog number NEL104001EA; PerkinElmer).
Quantification of LapA dot blot assays.
Image J software (http://rsb.info.nih.gov/ij/) was used to detect pixel density. Arbitrary units were assigned, and the WT value was set to 100 arbitrary units.
Cleavage of NTerm-LapA variants.
Bacterial cultures were grown, and clarified whole-cell extracts were prepared as described above for LapA localization studies. To assess the site specificity of LapG cleavage, cell fractions derived from the WT or a ΔlapG mutant strain carrying the pNTerm-LapA variant expression plasmid were analyzed by SDS-PAGE and Western blotting to detect the C-terminally tagged His-NTerm-LapA variant, as described above. A total of 0.04 mg of protein was loaded onto a 15% Ready Gel Tris-HCl gel (catalog number 161-1157; Bio-Rad Laboratories) to resolve the full-length and cleaved NTerm-LapA proteins.
Statistical analysis.
Data were analyzed by one-way analysis of variance followed by Tukey's posttest comparison. Significance was set at the traditional 95% confidence interval.
Biofilm assays and optical microscopy on atomic force microscopy substrates.
Hydrophobic surfaces were immersed in 24-well plates containing 2 ml K10T-1 medium. For cell adhesion and biofilm formation, 25 μl of bacteria from an culture grown overnight in LB medium was added to each well, and the plate was incubated for 8 h at 30°C with shaking (200 rpm). To promote cell detachment, substrates were further incubated for 8 h at 30°C at 200 rpm in plates containing 2 ml K10Tπ (low-Pi) medium (K10T-1 medium without the addition of 1 mM K2HPO4) (31). For optical images, surfaces were rinsed by three consecutive baths of fresh medium before or after incubation in low-Pi medium and directly imaged by using an inverted optical microscope (Axio Observer Z1; Zeiss) equipped with a Hamamatsu C10600 camera.
Atomic force microscopy.
For preparation of hydrophobic substrates, glass coverslips coated with a thin layer of gold were immersed overnight in a solution of 1 mM 1-dodecanethiol (Sigma) and then rinsed with ethanol and dried under N2, as described previously (15, 16). The properties of the hydrophobic substrate used for the AFM studies are similar to those of the plastic used in the biofilm assays; that is, both surfaces show similar water contact angles of ∼90° (35).
AFM experiments were performed at room temperature (20°C) in low-Pi medium by using a Nanoscope VIII Multimode atomic force microscope (Bruker Corporation, Santa Barbara, CA) and oxide-sharpened microfabricated Si3Ni4 cantilevers (Microlevers; Bruker Corporation) with a nominal spring constant of ∼0.01 N m−1 (measured by using the thermal noise method from Picoforce [Bruker]). Force measurements were carried out as previously described (15, 16). Briefly, substrates were rinsed by three consecutive baths of fresh low-Pi medium after cell detachment and mounted onto sample pucks while avoiding dewetting. AFM tips were functionalized with monoclonal anti-HA antibodies (200 μg/ml) (HA.11 clone 16B12; Covance) by using polyethylene glycol (PEG)-benzaldehyde linkers (36) and a previously described protocol (15, 16). Adhesion maps were obtained by recording 32-by-32 force-distance curves on areas of 5 by 5 μm, calculating the adhesion force for each force curve and displaying the adhesive events as gray pixels. All force curves were recorded with a maximum applied force of 250 pN, using a constant approach and retraction speeds of 1 μm s−1.
RESULTS
Conserved residues flanking the LapG cleavage site of LapA are not required for function.
Previously, we demonstrated that LapG cleaves LapA from the cell surface under conditions unfavorable for biofilm formation (27). This previous report supported a model in which cleavage of the first 108 amino acids from the N terminus of LapA by LapG is the mechanism required to release LapA from the cell surface in vivo. We noted that the lapA AA108–109RR (AA-RR) mutation completely eliminates cleavage of the NTerm-LapA polypeptide in vitro, suggesting that LapG recognizes a specific cleavage site in vitro. Surprisingly, this mutation only partially blocks LapG cleavage of LapA in vivo by ∼80% (27).
Because the AA-RR mutation only partially blocks LapG cleavage of LapA in vivo, we predicted that LapG cleavage was occurring at additional sites or that the AA-RR mutation was not sufficient to completely block cleavage at residues 108 and 109. A BLASTP search performed previously with the LapG sequence revealed several LapA-like proteins encoded near genes encoding LapG homologs in other bacterial species (27). Alignment of the N termini of these LapA-like proteins revealed not only that alanines 108 and 109 are conserved residues but also that their position relative to the N terminus is also conserved (27). In addition to the pair of alanine residues at positions 108 and 109 (Fig. 1A, double asterisks), the aligned LapA-like N termini contain many other conserved features relative to the pair of alanine residues (AA) at positions 108 and 109, including additional pairs of AA residues (Fig. 1A, arrows), conserved residues flanking the double alanine residues at positions 108 and 109 (Fig. 1A, highlighted in black), and a conserved predicted α-helical secondary structure found in 8 of 9 proteins with the closest homologs (Fig. 1A, underlined). We predicted that these conserved elements might function in LapG-dependent cleavage of LapA.
FIG 1.

Mutational analysis of the LapG cleavage site. (A) Alignment of the region flanking the LapG cleavage site of putative adhesins identified by their close proximity to genes encoding LapG homologs. The LapG cleavage site of LapA is indicated (**), as are putative alternative cleavage sites (arrows). Conserved residues are highlighted in gray or black, and the helical domain predicted by Phyre (37) is underlined. LapA, P. fluorescens Pf0-1; PFL_0133, P. fluorescens Pf-5; PP_0168, P. putida KT2440; PSEEN0141, Pseudomonas entomophila L-48; ECA3266, Pectobacterium atrosepticum SCRI1043; DP0516, Desulfotalea psychrophila LSv54; Rfer_3766, Rhodoferax ferrireducens DSM 15236; Patl_2528, Pseudoalteromonas atlantica T6c; Sden_0384, Shewanella denitrificans OS217; Bpro_0306, Polaromonas sp. strain JS666. An extended alignment is shown in Fig. S2 in the supplemental material. (B) Representative Western blot developed with antibodies directed against 6×His-tagged NTerm-LapA with a mutation in a conserved residue or deletion of the α-helical motif and expressed in the WT or the ΔlapG mutant. Full-length NTerm-LapA mutants migrate at ∼25 kDa, while the product of LapG processing migrates at ∼15 kDa. A full list of mutants analyzed is shown in Table S3 in the supplemental material.
To assess the role of the conserved elements in LapG-dependent cleavage of LapA, His-tagged NTerm-LapA mutants were constructed. Conserved residues T107 and G110 were each replaced with alanine, residues A108 and A109 were either singly or doubly mutated to other charged or bulky amino acids, and the region predicted to encode the α-helical secondary structure was deleted (this region spans residues A81 to 95A in LapA and the mutant designated Δhelix). Cellular extracts were prepared from both WT and ΔlapG mutant strains expressing mutant NTerm-LapA variants, and NTerm-LapA cleavage was assessed in vitro by Western blotting. A shift in the molecular mass of the His-tagged NTerm-LapA variants from ∼25 kDa to ∼15 kDa in the presence of LapG (e.g., LapG is present in the WT strain but not in the ΔlapG mutant strain) indicates LapG-dependent cleavage. The results from this analysis are presented in Table S3 in the supplemental material, and representative data are shown in Fig. 1B.
For the WT NTerm-LapA polypeptide, LapG cleaved at a unique site (Fig. 1B), yielding a single product, as previously reported (27). The NTerm-LapA Δhelix, A108R, A109R, A108W, and AA108–109DD variants, in addition to AA-RR, as previously described and thus serving as a control (27), were not cleaved by LapG in vitro. These data indicated that the unique cleavage event mediated by LapG in vitro was blocked in these mutant variants. The T107A and G110A variants were still cleaved in vitro (Fig. 1B; see also Table S3 in the supplemental material) and were not further studied.
To assess whether the NTerm-LapA mutations blocked for cleavage in vitro also affected LapG-dependent cleavage in vivo, we introduced the A108W, AA108–109DD, and A109D point mutations into LapA by mutating the chromosomal copy of the lapA gene by allelic replacement. Strains carrying these point mutations did not display the hyperbiofilm phenotype, accumulate LapA at the cell surface, or prohibit LapG-dependent cleavage of LapA to the degree observed for the ΔlapG mutant or the AA-RR variant (see Fig. S1A to S1C in the supplemental material), and thus, these mutants were not further studied.
Taken together, these data suggest that the conserved residues flanking positions 108 and 109 are not required for LapG-mediated cleavage of LapA in vivo and that both AA residues at positions 108 and 109 must be mutated to block LapG-mediated cleavage.
Multiple AA motifs are likely recognized by LapG for cleavage in vivo.
Our previously reported mutagenesis of alanines 108 and 109 (27) and flanking amino acids (described above) indicate that while these residues are conserved in proteins with N termini similar to that of LapA, no mutation produced a hyperadherent biofilm phenotype, accumulated LapA at the cell surface, or completely blocked LapA release into the supernatant at levels identical to that of the ΔlapG mutant (Fig. 1B; see also Fig. S1A to S1C in the supplemental material). These data indicated that we might not have identified all the determinants required for LapA cleavage or, alternatively, that LapG might have other targets in the cell.
Upon further examination of amino acids in the vicinity of the alanine residues at positions 108 and 109, we noted the occurrence of six additional double alanine residues at positions 83 and 84, 95 and 96, 113 and 114, 119 and 120, 149 and 150, and 153 and 154 (Fig. 1A; see also Fig. S2 in the supplemental material). Furthermore, the AA residue pairs at positions 113 and 114 and at positions 119 and 120 are flanked by small hydrophobic residues, similar to the AA residue pair at positions 108 and 109.
Given that mutation of amino acids 108 and 109 completely blocked cleavage of NTerm-LapA in vitro, but not in vivo, we predicted that in the absence of the preferred cleavage site at alanine residues 108 and 109, LapG might be capable of recognizing and cleaving at additional AA motifs in the context of the full-length protein in vivo. To test the importance of the other double alanine residues as possible LapG cleavage sites, a deletion of the chromosomal lapA gene, removing the segment encoding residues A108 to 120A, was constructed via allelic replacement. This mutation removes the AA motif at positions 108 and 109 as well as two additional flanking AA motifs at positions 113 and 114 and at positions 119 and 120.
Deletion of residues A108 to 120A promoted a hyperbiofilm phenotype similar to that of the ΔlapG mutant (Fig. 2A). The LapA ΔA108–120A mutant was detected at WT levels in the cell fraction and accumulated LapA on the cell surface, and no detectable LapA protein was released into the supernatant, again similar to the phenotypes observed for the ΔlapG mutant (Fig. 2B and C). Taken together, these results suggest that LapG is unable to cleave the LapA ΔA108–120A mutant protein. Thus, we suggest that LapG is a somewhat promiscuous enzyme, and while preferring the AA motif at positions 108 and 109, in the absence of these residues, LapG is apparently capable of cleaving at nearby AA motifs. Importantly, these data confirm that the unique target of LapG is LapA, and it is unlikely that LapG has other targets impacting early biofilm formation.
FIG 2.

Deletion of residues 108 to 120 eliminates LapG-dependent processing of the N terminus of LapA in vivo. (A) Images of microtiter dish biofilm assays using K10T-1 medium (top) and quantification of biofilm (middle). The x axis shows the P. fluorescens strains, and the y axis shows the optical density at 550 nm (OD550) of the solubilized crystal violet used to determine the bacterial biofilm biomass. At the bottom is a Western blot showing the levels of LapA in the whole-cell fraction of the WT; the ΔlapG mutant strain; the ΔlapD mutant strain, included as a biofilm-negative control; and the ΔA108–120A mutant strain. LapA was detected via an engineered HA tag. * indicates a significant difference from the WT (P < 0.05). (B) Cell surface levels of LapA. Shown are representative dot blots developed with antibodies directed against HA-tagged LapA (top) and quantification of the pixel density of four replicates (bottom) to assess the level of cell surface LapA of the indicated strains. * indicates a significant difference from the WT (P < 0.05). (C) LapA in supernatant fractions. Shown is a Western blot of the levels of LapA in the supernatant fraction of the indicated strains. LapA was detected via an engineered HA tag.
The α-helical region of LapA is required for LapG processing.
Inspection of the sequences surrounding the LapG cleavage site at positions 108 and 109 revealed a conserved, predicted α-helical secondary structure located ∼12 amino acids upstream of this site in 8/9 LapA-like N termini in the database. The ninth sequence may also have an α-helical structure, but the confidence in this prediction is only ∼50% by using Phyre (37). This α-helix is required for LapG-dependent cleavage of NTerm-LapA of P. fluorescens in vitro (Fig. 1B). Therefore, we deleted this helix (residues A81 to 95A) and assessed its impact on biofilm formation and LapA localization.
Deletion of the α-helical region (designated Δhelix) phenocopied the hyperbiofilm phenotype of the AA-RR mutant without disrupting the expression of the LapA protein (Fig. 3A). Deletion of the α-helical region promoted accumulation of LapA on the cell surface, as detected by dot blotting (Fig. 3B), but the deletion did not completely prevent the release of the LapA protein into the supernatant (Fig. 3C). However, levels of LapA released into the supernatant in the Δhelix mutant were lower than those observed for the WT strain, suggesting that LapG-dependent release is reduced in the absence of the α-helix. Deletion of lapG in the Δhelix mutant background completely prevented LapA release into the supernatant (data not shown), suggesting that the observed low level of LapA released into the supernatant in the Δhelix mutant is still dependent on LapG-mediated cleavage.
FIG 3.

The α-helical region of LapA is required for LapG processing. (A) Images of microtiter dish biofilm assays in K10T-1 medium (top) and quantification of biofilm (middle). The x axis shows the P. fluorescens strains analyzed, and the y axis shows the optical density at 550 nm (OD550) of the solubilized crystal violet used to determine the bacterial biofilm biomass. At the bottom is a Western blot showing the levels of LapA in the whole-cell fraction of the WT, the ΔlapG mutant, and strains carrying the indicated mutations in the chromosomal copy of the lapA gene. LapA was detected via an engineered HA tag. * indicates a significant difference from the WT (P < 0.05). (B) Cell surface levels of LapA. Shown are representative dot blots developed with antibodies directed against HA-tagged LapA (top) and quantification of the pixel density of four replicates (bottom) to assess the level of cell surface LapA of the indicated strains. * indicates a significant difference from the WT (P < 0.05). (C) Localization of LapA in supernatant fractions. Shown is a Western blot of the levels of LapA in the supernatant (Supe) fraction of the indicated strains. LapA was detected via an engineered HA tag.
Taken together, our data suggest that the AA residues at positions 108 and 109 and the predicted helical region encompassed by residues A81 to 95A are critical for LapG-dependent cleavage of LapA; however, residues immediately flanking the LapG cleavage site are dispensable for LapA cleavage. Furthermore, at least for the phenotypes described here, our findings are consistent with the conclusion that the only target for LapG is the LapA adhesin.
Structural features of P. fluorescens LapA deduced by sequence analysis and bioinformatic prediction.
The LapA protein is a cell surface adhesin required for biofilm formation in P. fluorescens (14, 17, 27). This protein is among the best-studied proteins of a large family of adhesins also identified in a range of other microbes, including pathogens. These adhesins share the N-terminal domain required for LapG processing but also share other domains, particularly a large central set of amino acid repeats and conserved domains across the C terminus (CTerm) of the protein. However, the function of these domains in adhesion function and cell surface localization is largely unexplored.
LapA has an estimated molecular mass of ∼520 kDa and contains an extensive repetitive region consisting of 37 repeats of ∼100 amino acids spanning residues D273 to 3982S (Fig. 4A, top). These repeat sequences are rich in small hydrophobic residues and are predicted to form β-sandwich folds comprised of two antiparallel β-sheets. Unsupervised hierarchical clustering using ClustalW (38) revealed that the 37 repeats could be divided into four groups, which we designated repeat A (rA), repeat B (rB), repeat C (rC), and repeat D (rD), based on sequence similarity (Fig. 4). Comparison of the amino acid sequences of all four repeat types showed a low overall identity of 27%, but the 18 repeat B sequences were 68% identical to each other, while the 16 repeat C sequences were 71% identical to each other, indicating that the repeat B and repeat C sequences comprised discrete subgroups of repeats. Furthermore, the repeat B sequences have similarity to the so-called Bid2 domain, which was identified in the E. coli virulence protein intimin (39). The order of the repeats, each consisting of 100 amino acids from the N terminus to the C terminus of LapA, is rA-rA-(rB-rC)8-rB-rD-(rB-rC)8-rB, that is, two repeat A motifs followed by 8 pairs of repeats B and C, a single rB-rD pair, another 8 pairs of repeats B and C, and a final rB motif.
FIG 4.

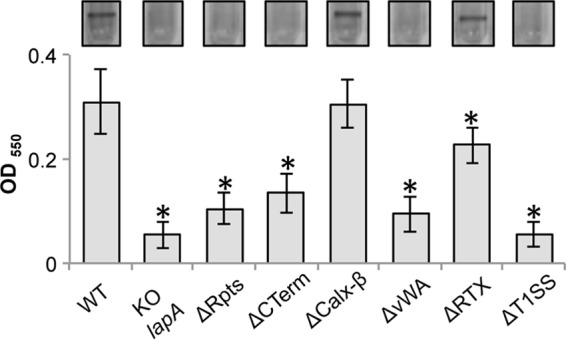
Mutational analysis of LapA domains. (A) The LapA protein (top) is diagrammed to scale, with putative domains and amino acid designations indicated. The three-hemagglutinin (HA) epitope tags inserted after residue 4093 (indicated by the yellow block) are shown, as are the other domains, and their location in the LapA protein sequence is indicated in parentheses. A WebLogo summary of the 37 repeat regions of ∼100 amino acids is shown in the middle, and a WebLogo summary of the 6 RTX repeat sequences of 9 amino acids is shown at the bottom. Gray, rA; blue, rB; red, rC; pink, rD. See the text for a detailed description of the LapA domain designations and abbreviations. (B) Images of microtiter dish biofilm assays in K10T-1 medium (top) and quantification of biofilm (middle). The x axis shows the P. fluorescens strains, and the y axis shows the optical density at 550 nm (OD550) of the solubilized crystal violet used to determine the bacterial biofilm biomass. At the bottom is a Western blot showing the levels of LapA in the whole-cell fraction of the WT strain and LapA domain deletion mutants. LapA was detected via an engineered HA tag. KO, knockout. (C) Representative dot blots developed with antibodies directed against HA-tagged LapA (top) and quantification of the pixel density of four replicates (bottom) to assess the level of cell surface LapA of the indicated strains. (D) Localization of LapA in supernatant fractions. Shown is a Western blot of the levels of LapA in the supernatant fraction of the indicated strains. LapA was detected via an engineered HA tag. (E) Outer membrane fractions of the indicated strains spotted onto a nitrocellulose membrane and detected via an engineered HA tag (top) and quantification of the pixel density of four replicates (bottom) to assess the level of LapA in the indicated strains. The ΔRpts variant of LapA was detected in this fraction, in comparison to the standard dot blot using whole cells shown in panel C, which showed no detection of this variant. * indicates a significant difference from the WT (P < 0.05).
The sequence conservation (overall stack height) and relative frequency of each residue (height of each symbol) are shown as a WebLogo design for all 37 repeats (Fig. 4A, middle) (40). Despite the poor overall sequence similarity among these repeats, there are several conserved residues that may be identified in the WebLogo plot.
Bioinformatics tools predict several conserved domains present at the C-terminal end of the LapA protein, from left to right: a Calx-β domain, a von Willebrand factor type A (vWA) domain, six RTX sequences, and a type I secretion system (T1SS) signal (Fig. 4A, top). The proposed function of each of these domains and their role in LapA function are outlined in detail below.
The Calx-β domain in other proteins has been demonstrated to be involved in calcium binding and regulation (41–43). More specifically, the Calx-β domain was originally described in eukaryotic Na+-Ca2+ exchangers, whereby it is important in expelling calcium from the cytoplasm (41–43). Previously, we showed that deletion of the Calx-β domain spanning amino acids T4049 to 4094D (Fig. 4A, top, black block) did not disrupt LapA localization or biofilm formation on a hydrophobic surface (32).
LapA contains one vWA domain spanning amino acids N4736 to 4963S (Fig. 4A, top, cyan block). The most well-characterized vWA domains are found in extracellular eukaryotic proteins and are involved in cell adhesion and protein-protein interactions (44, 45). Eukaryotic intracellular proteins containing vWA domains are often components of multiprotein complexes. vWA domains may mediate protein-protein interactions or the assembly and function of multiprotein complexes (46). A phyletic distribution study of domains commonly found in eukaryotic signaling proteins revealed that vWA domains are present in prokaryotes, and these domains were hypothesized to function in protein binding (47). The first study to characterize vWA domains in bacteria revealed that the vWA domain in PilA, a pilus-associated adhesin in Streptococcus agalactiae, is essential for mediating adherence to epithelial cells (48). The Pseudomonas aeruginosa vWA-containing protein PilY1 requires this domain for proper localization to the bacterial cell surface (49). We predicted that the vWA domain is required for LapA localization to the cell surface and that it may mediate interactions between the protein and the cell surface.
LapA contains six RTX sequences, spanning amino acids G5020 to 5144W, which consist of glycine-aspartate-rich nonapeptide repeats with the consensus sequence G-G-X-G-(N/D)-D-X-(L/I/F)-X (Fig. 4A, top, orange block). The sequence conservation (overall stack height) and relative frequency of each residue (height of each symbol) are shown as a WebLogo design for all six RTX sequences (Fig. 4A, bottom) (40). RTX sequences are found within proteins belonging to a family of large repetitive RTX adhesins and biofilm-associated proteins (19) and the more classically defined RTX pore-forming cytolysins and hemolysins, such as adenylate cyclase from Bordetella pertussis and HlyA from E. coli (50–52). RTX proteins are transported out of the bacterial cell by an ABC transporter in an unfolded state, where lipopolysaccharide (LPS) molecules packed with Ca2+ and Mg2+ ions are encountered. The RTX repeats bind Ca2+ with high affinity to form a stable parallel β-roll structure, which in turn facilitates protein folding (53, 54). Levels of intracellular Ca2+ are lower than levels of extracellular Ca2+, and therefore, protein folding is prevented prior to secretion (55). RTX repeats have been studied extensively in RTX pore-forming toxins (50, 56–58), but their role in the subfamily of RTX adhesins and biofilm-associated proteins has not been characterized. We hypothesized that the RTX sequences mediate LapA secretion and stability.
Genetic and biochemical studies revealed that LapA is transported to the cell surface by an ABC transporter, encoded by the lapEBC genes (17, 27). LapA contains a T1SS signal that is predicted to be required for transport through the ABC transporter, spanning residues N5151 to 5218S (Fig. 4A, top, purple block). There is an absence of overall sequence conservation among T1SS signals; however, the signal for type I secretion is a noncleaved stretch of amino acid residues found in the last 60 to 80 amino acids of the protein. It includes ∼22 amphipathic amino acid residues, ∼13 uncharged amino acid residues, and ∼8 C-terminal hydrophobic residues (59). We hypothesized that the T1SS signal in the C terminus of LapA is required for transport to the cell surface.
Some domain deletions of LapA perturb function and localization.
With the exception of the Calx-β domain (32), the biological function pertaining to LapA localization and contribution to biofilm formation of the repeat region, vWA domain, RTX sequences, and T1SS sequence has not been experimentally investigated. To assess the requirement of the predicted domains for biofilm formation and LapA localization, chromosomal deletions of the regions encoding the 37 repeats of ∼100 amino acids (this domain spans amino acids D273 to 3982S; designated ΔRpts), the vWA domain (this domain spans amino acids N4736 to 4963S; designated ΔvWA), RTX sequences (this domain spans amino acids G5020 to 5144W; designated ΔRTX), the C terminus (deletion encompassing the Calx-β, vWA, and RTX domains, but not the T1SS, and spanning amino acids T4018 to 5151N; designated ΔCTerm), and the T1SS sequence (this domain spans amino acids N5151 to 5218S; designated ΔT1SS) were constructed in the chromosomal copy of the lapA gene via allelic replacement.
Deletion of the repeat region significantly reduced biofilm biomass compared to that of the WT strain (P < 0.05) (Fig. 4B). The absence of the repeat region did not affect LapA cellular expression, as assessed by Western blotting of the cell fraction of this strain (Fig. 4B, bottom), while levels of LapA on the cell surface were significantly reduced (Fig. 4C); however, LapA release into the supernatant remained apparently unchanged compared to the WT (Fig. 4D).
We reasoned that the decrease in the apparent abundance of cell surface LapA upon deletion of the repeat region was likely caused by an inaccessibility of the internal HA tag in LapA to the HA antibody; that is, the ΔRpts variant of LapA was truncated, and outer membrane components were likely sterically interfering with the detection of the truncated LapA. Indeed, the LapA ΔRpts protein was detected at a level higher than that of WT LapA when the outer membrane fraction of this mutant strain was isolated by detergent fractionation (Fig. 4E). It is not clear why the signal for the ΔRpts variant is higher than that for the WT, but this may be due to the increased exposure of the epitope tag in the shorter LapA variant lacking the repeat region.
The deletion spanning the entire C terminus (the T1SS signal remains intact to maintain LapBCE-dependent transport), deletion of the vWA domain, and deletion of RTX sequences resulted in statistically significant reductions of biofilm biomass in the static microtiter dish biofilm assay compared to the WT strain (P < 0.05) (Fig. 4B). While these reductions in biofilm biomass were ∼50% and statistically significant, the ΔCTerm, ΔvWA, and ΔRTX mutants were still able to form a biofilm, as indicated by the visible ring in the microtiter dish (Fig. 4B, top). LapA levels found in the cell fractions of these mutant proteins were similar to or in some cases higher than that of the WT protein, as detected by Western blotting (Fig. 4B, bottom).
Consistent with a reduction in biofilm biomass, we observed a decrease in the abundance of LapA on the surface of intact cells for the ΔCTerm, ΔvWA, and ΔRTX mutants, as assessed by dot blotting (Fig. 4C), although not all of the reductions were statistically significant. In the supernatant fraction, deletion of the C terminus and the vWA domain resulted in a substantial decrease in the level of LapA; however, deletion of the RTX sequences promoted an increase in the levels of LapA detected in the supernatant fraction.
Finally, deletion of the T1SS significantly reduced biofilm biomass compared to that of the WT (P < 0.05) but did not impact LapA protein stability in the cell fraction (Fig. 4B). Consistent with the prediction that the T1SS sequence is required for transport to the cell surface, deletion of this sequence prevented LapA from localizing to the cell surface (Fig. 4C). Consequently, no LapA was detected in the supernatant fraction (Fig. 4D). These data are consistent with the prediction that the T1SS signal is required for localization to the cell surface by the type I secretion system and biofilm function.
Together, these results demonstrated that the large repeat region of LapA is required for biofilm formation. In contrast, deletion of the entire C terminus or its subdomains, with the exception of the T1SS, did not completely eliminate biofilm formation, suggesting that this region of the protein is not absolutely required for adhesion to this surface and biofilm formation. These results show that deletion of the predicted domains in LapA differentially impact the ability of P. fluorescens to form a biofilm on plastic, the hydrophobic surface tested here.
Identification of LapA domains required for biofilm formation on hydrophilic surfaces.
The ability of P. fluorescens to adhere to a variety of surfaces, including polyvinylchloride, polypropylene, polystyrene, borosilicate glass, and quartz sand, requires the presence of LapA (6, 17), thus establishing that LapA is the main adhesin employed by P. fluorescens to initiate biofilm formation on a wide array of surfaces. In its natural soil environment, P. fluorescens likely encounters many surfaces comprised of different chemical compositions.
As described above, LapA domains differentially contribute to attachment to plastic, a hydrophobic surface (Fig. 4B). For example, the loss of the repeat region (ΔRpts) resulted in a much more drastic loss of biofilm biomass than did the loss of the C-terminal domains. Given the variety and diversity of substrate compositions in the soil environment, we hypothesized that the LapA domains might differentially function in adhesion to different substrates, thus imparting P. fluorescens with the ability to attach to an assortment of surfaces.
To address this hypothesis, the WT and LapA domain deletion mutants were tested for their ability to form biofilms on a model hydrophilic surface: borosilicate glass. Loss of the repeat sequences and the CTerm, vWA, RTX, and T1SS sequences significantly reduced biofilm biomass compared to that of the WT strain (P < 0.05) (Fig. 5). As was observed for the model hydrophobic surface, loss of the repeats or the T1SS resulted in a significant loss of the biofilm. Interestingly, while the loss of the RTX domain still supported biofilm formation on both the hydrophilic and hydrophobic substrata, the C-terminal domain, and specifically the vWA domain, is absolutely required for biofilm formation on the hydrophilic but not on the hydrophobic surface (compare Fig. 5 to 4B). Loss of the Calx-β domain did not affect biofilm formation on this model hydrophilic surface (Fig. 5), as reported previously for a hydrophobic surface (32).
FIG 5.
Biofilm formation on a hydrophilic surface. Shown are images of biofilms on borosilicate glass (a model hydrophilic surface) with the indicated LapA domain deletion strains. * indicates a significant difference from the WT (P < 0.05).
Atomic force microscopy demonstrates the role of the repeat region and the CTerm region in mediating LapA adhesion to hydrophobic substrates.
Single-molecule AFM (35, 60–62) was used to functionally analyze the biophysical properties (distribution, adhesion, and extension) of LapA bacterial “footprints,” i.e., adhesive macromolecules left on substrate surfaces after removal of the attached cells. To generate LapA footprints using physiologically relevant means, P. fluorescens cells were incubated with a model hydrophobic substrate in high-Pi medium for 8 h. Detachment was then induced by reducing the Pi concentration (15, 16). Incubation of the AFM substrates with the WT strain under high-Pi conditions resulted in substantial cell adhesion (surface coverage of ∼25%) (Fig. 6A), while further incubation under low-Pi conditions led to the detachment of most cells (surface coverage of ∼1%) (Fig. 6A, inset). In contrast, incubation of the ΔRpts and ΔCTerm strains under high- and low-Pi conditions led to very poor levels of cell adhesion (surface coverage of ∼1%) (Fig. 6B and C), consistent with microtiter dish biofilm assays, in which the absence of the repeat region and the C terminus decreased biofilm biomass compared to that formed by the WT strain (Fig. 4B). Furthermore, incubation under low-Pi conditions therefore had essentially no effect on how well these mutants attached to this substrate (Fig. 6B and C, insets).
FIG 6.

Single-molecule AFM analysis of the biophysical properties of LapA bacterial footprints on hydrophobic substrates. (A to C) Representative optical microscopy (DIC) images of substrates following 8 h of incubation in high-Pi medium of P. fluorescens WT (A), ΔRpts (B), and ΔCTerm (C) cells. The insets show the images obtained after cell detachment via 8 h of incubation in low-Pi medium. (D to L). Adhesion force maps (5 μm by 5 μm; z range = 300 pN) (D, G, and J), corresponding adhesion force histograms together with representative force curves (E, H, and K), and rupture length histograms (F, I, and L) recorded between an anti-HA tip and hydrophobic substrates following adhesion and detachment of P. fluorescens WT (D to F), ΔRpts (G to I), and ΔCTerm (J to L) cells. The maps in the insets (D, G, and J) are from independent experiments. The cartoons in panels F, I, and L emphasize differences in LapA protein-substrate interactions for the three strains (gray lines, LapA N terminus, red lines, LapA repeats; blue pills, HA tag; purple pills, LapA C terminus).
In order to assess LapA-dependent adhesion to hydrophobic surfaces, AFM tips functionalized with monoclonal anti-HA antibodies were used to map and analyze HA-tagged LapA proteins left on the substrate surface after induced detachment via incubation in low-Pi medium. The adhesion force map, the histogram of adhesion forces, the histogram of rupture lengths, and representative force curves were recorded and are shown in Fig. 6D to F for the WT. The adhesion map revealed a substantial fraction (9%) of detection events, suggesting that LapA molecules were left on the substrate after cell detachment (Fig. 6D). WT force curves showed multiple force peaks with magnitudes of 50 to 500 pN (Fig. 6E) and rupture lengths of 50 to 800 nm (Fig. 6F). Based on previously reported controls employing a strain lacking LapA at the cell surface, these WT signatures were shown to specifically reflect the detection and stretching of the LapA protein (15, 16).
The adhesion of the ΔRpts strain to the hydrophobic surface led to the same detection frequency as that for the WT strain (10%) (Fig. 6G to I), indicating that the number of LapA molecules interacting with the substrate was identical for the WT and the ΔRpts mutant, a somewhat surprising finding given the biofilm formation phenotype of the ΔRpts strain (Fig. 4 to 6). Given that there were many fewer bacteria of the ΔRpts strain attached, these data suggest that the ΔRpts LapA protein, while able to localize to the bacterial cell surface and bind to the substrate (Fig. 4E), is not competent to promote tight bacterial attachment. Interestingly, force profiles of the ΔRpts LapA mutant protein were markedly different from those of the WT LapA protein, in that much lower adhesion forces (∼50 to 200 pN) and much shorter rupture lengths (∼50 to 200 nm) were observed (Fig. 6H and I). Few unfolding events were noted for the ΔRpts mutant LapA protein (Fig. 6I), a finding consistent with previous work (15, 16) and consistent with the lack of the large repeat region. It is important to note that the ΔRpts LapA mutant protein showed a reduction in the number of unfolding events and rupture length, but not elimination of these events, consistent with the facts that the repeat region has been deleted and that the protein is much shorter. However, the remnant parts of the ΔRpts LapA mutant protein could still be detected and stretched. These data provide direct evidence that the multiple force peaks observed for the WT strain reflect the unfolding of the multiple repeats of the LapA protein.
It is important to note that the lack of a regular unfolding pattern for the WT strain suggests that LapA proteins adsorbed on the substrate may be partially denatured, although clear differences in the behaviors of WT and mutant proteins could still be distinguished.
The incubation of the strain expressing the ΔCTerm variant of LapA on the hydrophobic substrates led to very low detection frequencies and adhesion forces (50 to 150 pN) and short extensions (50 to 150 nm) (Fig. 6J to L). These results indicate that the C-terminal region may facilitate the initial attachment of the LapA protein to the substrate. Presumably, without this initial contact, the LapA repeat region is no longer able to interact with the hydrophobic surface as effectively, thus suggesting that LapA-mediated adhesion follows two sequential steps: (i) initial attachment of P. fluorescens is facilitated by the C-terminal domain of LapA, and (ii) irreversible attachment to the substrate is mediated by LapA via the repeat region of this adhesin.
DISCUSSION
Our studies reported here have identified additional critical regions of the N terminus of LapA required for LapG proteolysis and release of LapA from the cell surface, namely, the predicted α-helix and the amino acids between residues 108 and 120. Our data are consistent with the model that LapG preferentially targets the AA motif at positions 108 and 109 in the WT LapA protein; in the absence of this primary cleavage site, LapG can apparently target alternative cleavage sites in vivo. Thus, amino acids in these regions of LapA are required for LapG cleavage and/or LapG recognition of LapA and thus likely participate in the LapG-dependent release of LapA from the cell surface under conditions unfavorable for biofilm formation.
Importantly, our mutational analysis has confirmed that the only target of LapG, at least under our assay conditions, is LapA; that is, removing residues 108 to 120 from LapA completely recapitulated the phenotype of mutating the LapG protease, consistent with LapA being the unique target of LapG during early biofilm formation. Future studies will focus on analyzing, in vitro and in vivo, whether the conserved residues in proteins with N termini similar to those of LapA indicate conserved function, that is, whether these proteins are also targets of LapG.
We also present a mutagenesis analysis to define roles for the domains of LapA and to determine their requirement pertaining to LapA secretion, localization, and adhesin function. This is the first analysis aimed at characterizing domains that are conserved in members of the large subfamily of cell surface RTX adhesins.
RTX adhesins and members of the BAP adhesin family contain extensive repetitive regions (19). Prior to this study, the biological function of these repeats was unknown. Here, we show that extensive repetitive repeats are required for biofilm formation by P. fluorescens on both hydrophobic and hydrophilic surfaces. Interestingly, among the family of RTX adhesins, the sequence similarity of repeat regions can vary in different bacterial species and even in strains of the same species (19), suggesting that the repeats function in biofilm formation but that the repeat region composition may have evolved to facilitate adhesion to a wide variety of surfaces in diverse environments.
Interestingly, while deletion of the vWA domain was sufficient to eliminate biofilm formation on glass, loss of this domain or the entire C-terminal domain of LapA only moderately reduced biofilm formation on the hydrophobic plastic surface. Thus, the vWA domain appears to play a specific role in attachment to hydrophilic surfaces. These data suggest that perhaps different subdomains of the protein play differential roles in the ability of LapA to mediate attachment across a wide range of surfaces, a finding consistent with previous studies from our group (15, 16).
Deletion of the C-terminal RTX sequences reduced biofilm formation on both hydrophobic and hydrophilic surfaces, reduced the localization of LapA to the bacterial cell surface, and promoted greater release into the supernatant. RTX sequences are essential for the catalytic activity of RTX toxins such as HlyA and CyaA and have been implicated in playing an auxiliary role in the secretion of these proteins. However, the precise role of these sequences in RTX toxin secretion remains elusive (56, 57, 63). It has been proposed that RTX repeats may promote folding into a functional conformation in the presence of calcium (53), and it has been shown that calcium binding by the RTX repeats induces a stable protein conformation (58, 64). However, in the absence of the RTX sequences, LapA presumably is still able to fold and function to some degree, given that a strain carrying a LapA variant lacking the RTX sequences is still capable of biofilm formation on both hydrophobic and hydrophilic substrates. Thus, while the RTX motif contributes to LapA function, it is not absolutely required for bacterial adhesion.
Type I secretion systems are predicted to directly transport adhesins to the cell surface. Previously, we showed that mutating lapB, which encodes the inner membrane ATPase and maps near the lapA gene on the chromosome, produced cytoplasmic LapA, but the mutant is unable to transport LapA to the cell surface (17). Here, we show that by specifically deleting the T1SS signal at the C terminus of LapA, not surprisingly, transport of LapA to the cell surface is disrupted, and consequently, a biofilm is not formed on either hydrophobic or hydrophilic surfaces. Thus, of the C-terminal domains of LapA, only the T1SS is absolutely required for biofilm formation on both substrates tested.
We used single-molecule AFM analysis to gain additional insight into how the repeat region and C-terminal domain contribute to biofilm formation on a model hydrophobic surface. The hydrophobic substrate used in the AFM studies differs from the hydrophobic substrate used in the 96-well dish assays; however, our analysis of attachment by differential interference contrast (DIC) imaging shows that the ability of the ΔRpts mutant to attach to this substrate is severely compromised compared to the WT (compare Fig. 6A and B), a finding identical to that for the 96-well dish assays.
AFM analysis indicated that deletion of the C terminus of LapA resulted in much-reduced LapA binding to the hydrophobic substrate (compare Fig. 6D and J), and adhesion to the substrate never matched the degree of adhesive forces observed for the WT. In contrast, the LapA ΔRpts mutant protein bound the substrate to a degree very similar to that of the WT protein (compare Fig. 6D and G), but this mutant protein showed much-reduced adhesion force and could not support bacterial attachment. We take these data to mean that on this hydrophobic surface, perhaps the C terminus of LapA facilitates initial contact with the surface but is not absolutely required to initiate surface adhesion in the context of the whole organism, as strains lacking the C terminus of LapA can still make a biofilm. This finding is consistent with our previous studies of WT LapA showing that the hydrophobic repeat domain is likely the main driver of attachment to hydrophobic surfaces (15, 16). Thus, we propose that the repeat region of LapA drives irreversible surface attachment on hydrophobic surfaces, after the C-terminal region facilitates initial, reversible attachment. In contrast, our biofilm assays presented here (Fig. 5) are consistent with our previous single-cell AFM studies indicating that the C-terminal domain of LapA is critical for the initial attachment of the adhesin to hydrophilic surfaces (15, 16). Finally, a strain carrying the LapA mutant protein lacking the repeats region but retaining the C-terminal domain is also defective for attachment on this hydrophilic surface (Fig. 5), indicating that while the C-terminal domain of LapA may play a key role in initiating hydrophilic surface contact, this domain is not sufficient to establish irreversible surface attachment on such a surface.
Thus, we propose that while the C-terminal domain of LapA likely differentially contributes to the ability of the adhesin to initiate surface contacts on hydrophobic versus hydrophilic surfaces, the large repeat domain is required for irreversible attachment on both of these substrates. Finally, as highlighted in Fig. 4, the LapA protein repeat region is comprised largely of two repeating ∼100-amino-acid repeat motifs (i.e., the rB-rC motif). Our previous AFM studies indicate that it is unlikely that individual repeats of LapA unfold, but instead, the domains unfold two-by-two (15). These data indicate that the rB-rC motif may be quite stable and may serve as the minimal functional unit of LapA required for irreversible attachment under conditions favorable for biofilm formation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Science Foundation grants MCB-9984521 and R01 AI097307-01 to G.A.O. Work at the Université Catholique de Louvain was supported by the National Fund for Scientific Research (FNRS); the Université Catholique de Louvain (Fonds Spéciaux de Recherche); the Federal Office for Scientific, Technical and Cultural Affairs (Interuniversity Poles of Attraction Programme); and the Research Department of the Communauté Française de Belgique (Concerted Research Action).
Y.F.D. is Research Director of the FNRS.
We thank Brian Jackson, Director of the Trace Metal Analysis Core at Dartmouth, for his assistance in measuring the concentration of calcium in our growth medium and Devon Renock, Department of Earth Sciences at Dartmouth College, for his assistance in modeling and calculating the concentrations of calcium and phosphate in our growth medium.
Footnotes
Published ahead of print 16 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01629-14.
REFERENCES
- 1.Costerton JW, Cheng K-J, Geesey GG, Ladd TI, Nickel JG, Dasgupta M, Marrie TJ. 1987. Bacterial biofilms in nature and disease. Annu. Rev. Microbiol. 41:435–464. 10.1146/annurev.mi.41.100187.002251 [DOI] [PubMed] [Google Scholar]
- 2.Davey ME, O'Toole GA. 2000. Microbial biofilms: from ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 64:847–867. 10.1128/MMBR.64.4.847-867.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson DW, Suzuki K, Oakford L, Simecka JW, Hart ME, Romeo T. 2002. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J. Bacteriol. 184:290–301. 10.1128/JB.184.1.290-301.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson DW, Simecka JW, Romeo T. 2002. Catabolite repression of Escherichia coli biofilm formation. J. Bacteriol. 184:3406–3410. 10.1128/JB.184.12.3406-3410.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lugtenberg BJJ, Kravchenko LV, Simons M. 1999. Tomato seed and root exudate sugars: composition, utilization by Pseudomonas biocontrol strains and role in rhizosphere colonization. Environ. Microbiol. 1:439–446. 10.1046/j.1462-2920.1999.00054.x [DOI] [PubMed] [Google Scholar]
- 6.O'Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449–461. 10.1046/j.1365-2958.1998.00797.x [DOI] [PubMed] [Google Scholar]
- 7.O'Toole GA, Gibbs KA, Hager PW, Phibbs PV, Jr, Kolter R. 2000. The global carbon metabolism regulator Crc is a component of a signal transduction pathway required for biofilm development by Pseudomonas aeruginosa. J. Bacteriol. 182:425–431. 10.1128/JB.182.2.425-431.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prigent-Combaret C, Brombacher E, Vidal O, Ambert A, Lejeune P, Landini P, Dorel C. 2001. Complex regulatory network controls initial adhesion and biofilm formation in Escherichia coli via regulation of the csgD gene. J. Bacteriol. 183:7213–7223. 10.1128/JB.183.24.7213-7223.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prigent-Combaret C, Vidal O, Dorel C, Lejeune P. 1999. Abiotic surface sensing and biofilm-dependent regulation of gene expression in Escherichia coli. J. Bacteriol. 181:5993–6002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sauer K, Cullen MC, Rickard AH, Zeef LA, Davies DG, Gilbert P. 2004. Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J. Bacteriol. 186:7312–7326. 10.1128/JB.186.21.7312-7326.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh PK, Parsek MR, Greenberg EP, Welsh MJ. 2002. A component of innate immunity prevents bacterial biofilm development. Nature 417:552–555. 10.1038/417552a [DOI] [PubMed] [Google Scholar]
- 12.Thormann KM, Duttler S, Saville RM, Hyodo M, Shukla S, Hayakawa Y, Spormann AM. 2006. Control of formation and cellular detachment from Shewanella oneidensis MR-1 biofilms by cyclic di-GMP. J. Bacteriol. 188:2681–2691. 10.1128/JB.188.7.2681-2691.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haas D, Defago G. 2005. Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 3:307–319. 10.1038/nrmicro1129 [DOI] [PubMed] [Google Scholar]
- 14.Ivanov IE, Boyd CD, Newell PD, Schwartz ME, Turnbull L, Johnson MS, Whitchurch CB, O'Toole GA, Camesano TA. 2012. Atomic force and super-resolution microscopy support a role for LapA as a cell-surface biofilm adhesin of Pseudomonas fluorescens. Res. Microbiol. 163:685–691. 10.1016/j.resmic.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Kirat-Chatel S, Beaussart A, Boyd CD, O'Toole GA, Dufrene YF. 2014. Single-cell and single-molecule analysis deciphers the localization, adhesion, and mechanics of the biofilm adhesin LapA. ACS Chem. Biol. 9:485–494. 10.1021/cb400794e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Kirat-Chatel S, Boyd CD, O'Toole GA, Dufrene YF. 2014. Single-molecule analysis of Pseudomonas fluorescens footprints. ACS Nano 8:1690–1698. 10.1021/nn4060489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinsa SM, Espinosa-Urgel M, Ramos JL, O'Toole GA. 2003. Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol. Microbiol. 49:905–918. 10.1046/j.1365-2958.2003.03615.x [DOI] [PubMed] [Google Scholar]
- 18.Yousef-Coronado F, Travieso ML, Espinosa-Urgel M. 2008. Different, overlapping mechanisms for colonization of abiotic and plant surfaces by Pseudomonas putida. FEMS Microbiol. Lett. 288:118–124. 10.1111/j.1574-6968.2008.01339.x [DOI] [PubMed] [Google Scholar]
- 19.Satchell KJ. 2011. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu. Rev. Microbiol. 65:71–90. 10.1146/annurev-micro-090110-102943 [DOI] [PubMed] [Google Scholar]
- 20.Espinosa-Urgel M, Salido A, Ramos JL. 2000. Genetic analysis of functions involved in adhesion of Pseudomonas putida to seeds. J. Bacteriol. 182:2363–2369. 10.1128/JB.182.9.2363-2369.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Gil M, Yousef-Coronado F, Espinosa-Urgel M. 2010. LapF, the second largest Pseudomonas putida protein, contributes to plant root colonization and determines biofilm architecture. Mol. Microbiol. 77:549–561. 10.1111/j.1365-2958.2010.07249.x [DOI] [PubMed] [Google Scholar]
- 22.Syed KA, Beyhan S, Correa N, Queen J, Liu J, Peng F, Satchell KJ, Yildiz F, Klose KE. 2009. The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J. Bacteriol. 191:6555–6570. 10.1128/JB.00949-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerlach RG, Jackel D, Stecher B, Wagner C, Lupas A, Hardt WD, Hensel M. 2007. Salmonella pathogenicity island 4 encodes a giant non-fimbrial adhesin and the cognate type 1 secretion system. Cell. Microbiol. 9:1834–1850. 10.1111/j.1462-5822.2007.00919.x [DOI] [PubMed] [Google Scholar]
- 24.Cirillo SL, Yan L, Littman M, Samrakandi MM, Cirillo JD. 2002. Role of the Legionella pneumophila rtxA gene in amoebae. Microbiology 148:1667–1677 http://mic.sgmjournals.org/content/148/6/1667.long [DOI] [PubMed] [Google Scholar]
- 25.Theunissen S, De Smet L, Dansercoer A, Motte B, Coenye T, Van Beeumen JJ, Devreese B, Savvides SN, Vergauwen B. 2010. The 285 kDa Bap/RTX hybrid cell surface protein (SO4317) of MR-1 is a key mediator of biofilm formation. Res. Microbiol. 161:144–152. 10.1016/j.resmic.2009.12.002 [DOI] [PubMed] [Google Scholar]
- 26.Navarro MV, Newell PD, Krasteva PV, Chatterjee D, Madden DR, O'Toole GA, Sondermann H. 2011. Structural basis for c-di-GMP-mediated inside-out signaling controlling periplasmic proteolysis. PLoS Biol. 9:e1000588. 10.1371/journal.pbio.1000588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newell PD, Boyd CD, Sondermann H, O'Toole GA. 2011. A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol. 9:e1000587. 10.1371/journal.pbio.1000587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newell PD, Monds RD, O'Toole GA. 2009. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc. Natl. Acad. Sci. U. S. A. 106:3461–3466. 10.1073/pnas.0808933106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newell PD, Yoshioka S, Hvorecny KL, Monds RD, O'Toole GA. 2011. Systematic analysis of diguanylate cyclases that promote biofilm formation by Pseudomonas fluorescens Pf0-1. J. Bacteriol. 193:4685–4698. 10.1128/JB.05483-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monds RD, Newell PD, Gross RH, O'Toole GA. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol. Microbiol. 63:656–679. 10.1111/j.1365-2958.2006.05539.x [DOI] [PubMed] [Google Scholar]
- 31.Monds RD, Newell PD, Schwartzman JA, O'Toole GA. 2006. Conservation of the Pho regulon in Pseudomonas fluorescens Pf0-1. Appl. Environ. Microbiol. 72:1910–1924. 10.1128/AEM.72.3.1910-1924.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyd CD, Chatterjee D, Sondermann H, O'Toole GA. 2012. LapG, required for modulating biofilm formation by Pseudomonas fluorescens Pf0-1, is a calcium-dependent protease. J. Bacteriol. 194:4406–4414. 10.1128/JB.00642-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from Gram-negative bacteria. Appl. Environ. Microbiol. 72:5027–5036. 10.1128/AEM.00682-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuchma SL, Brothers KM, Merritt JH, Liberati NT, Ausubel FM, O'Toole GA. 2007. BifA, a cyclic-Di-GMP phosphodiesterase, inversely regulates biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J. Bacteriol. 189:8165–8178. 10.1128/JB.00586-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alsteens D, Garcia MC, Lipke PN, Dufrene YF. 2010. Force-induced formation and propagation of adhesion nanodomains in living fungal cells. Proc. Natl. Acad. Sci. U. S. A. 107:20744–20749. 10.1073/pnas.1013893107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebner A, Wildling L, Kamruzzahan AS, Rankl C, Wruss J, Hahn CD, Holzl M, Zhu R, Kienberger F, Blaas D, Hinterdorfer P, Gruber HJ. 2007. A new, simple method for linking of antibodies to atomic force microscopy tips. Bioconjug. Chem. 18:1176–1184. 10.1021/bc070030s [DOI] [PubMed] [Google Scholar]
- 37.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371. 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 38.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelly G, Prasannan S, Daniell S, Fleming K, Frankel G, Dougan G, Connerton I, Matthews S. 1999. Structure of the cell-adhesion fragment of intimin from enteropathogenic Escherichia coli. Nat. Struct. Biol. 6:313–318. 10.1038/7545 [DOI] [PubMed] [Google Scholar]
- 40.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alonso-Garcia N, Ingles-Prieto A, Sonnenberg A, de Pereda JM. 2009. Structure of the Calx-beta domain of the integrin beta4 subunit: insights into function and cation-independent stability. Acta Crystallogr. D Biol. Crystallogr. 65:858–871. 10.1107/S0907444909018745 [DOI] [PubMed] [Google Scholar]
- 42.Matsuoka S, Nicoll DA, Hryshko LV, Levitsky DO, Weiss JN, Philipson KD. 1995. Regulation of the cardiac Na(+)-Ca2+ exchanger by Ca2+. Mutational analysis of the Ca(2+)-binding domain. J. Gen. Physiol. 105:403–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwarz EM, Benzer S. 1997. Calx, a Na-Ca exchanger gene of Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 94:10249–10254. 10.1073/pnas.94.19.10249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colombatti A, Bonaldo P. 1991. The superfamily of proteins with von Willebrand factor type A-like domains: one theme common to components of extracellular matrix, hemostasis, cellular adhesion, and defense mechanisms. Blood 77:2305–2315 [PubMed] [Google Scholar]
- 45.Colombatti A, Bonaldo P, Doliana R. 1993. Type A modules: interacting domains found in several non-fibrillar collagens and in other extracellular matrix proteins. Matrix 13:297–306. 10.1016/S0934-8832(11)80025-9 [DOI] [PubMed] [Google Scholar]
- 46.Whittaker CA, Hynes RO. 2002. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell 13:3369–3387. 10.1091/mbc.E02-05-0259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ponting CP, Aravind L, Schultz J, Bork P, Koonin EV. 1999. Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J. Mol. Biol. 289:729–745 [DOI] [PubMed] [Google Scholar]
- 48.Konto-Ghiorghi Y, Mairey E, Mallet A, Dumenil G, Caliot E, Trieu-Cuot P, Dramsi S. 2009. Dual role for pilus in adherence to epithelial cells and biofilm formation in Streptococcus agalactiae. PLoS Pathog. 5:e1000422. 10.1371/journal.ppat.1000422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuchma SL, Ballok AE, Merritt JH, Hammond JH, Lu W, Rabinowitz JD, O'Toole GA. 2010. Cyclic-di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa: the pilY1 gene and its impact on surface-associated behaviors. J. Bacteriol. 192:2950–2964. 10.1128/JB.01642-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linhartova I, Bumba L, Masin J, Basler M, Osicka R, Kamanova J, Prochazkova K, Adkins I, Hejnova-Holubova J, Sadilkova L, Morova J, Sebo P. 2010. RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 34:1076–1112. 10.1111/j.1574-6976.2010.00231.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanski E, Farfel Z. 1985. Bordetella pertussis invasive adenylate cyclase. Partial resolution and properties of its cellular penetration. J. Biol. Chem. 260:5526–5532 [PubMed] [Google Scholar]
- 52.Short EC, Kurtz HJ. 1971. Properties of the hemolytic activities of Escherichia coli. Infect. Immun. 3:678–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holland IB, Schmitt L, Young J. 2005. Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway (review). Mol. Membr. Biol. 22:29–39. 10.1080/09687860500042013 [DOI] [PubMed] [Google Scholar]
- 54.Lilie H, Haehnel W, Rudolph R, Baumann U. 2000. Folding of a synthetic parallel beta-roll protein. FEBS Lett. 470:173–177. 10.1016/S0014-5793(00)01308-9 [DOI] [PubMed] [Google Scholar]
- 55.Jones HE, Holland IB, Baker HL, Campbell AK. 1999. Slow changes in cytosolic free Ca2+ in Escherichia coli highlight two putative influx mechanisms in response to changes in extracellular calcium. Cell Calcium 25:265–274. 10.1054/ceca.1999.0028 [DOI] [PubMed] [Google Scholar]
- 56.Bauche C, Chenal A, Knapp O, Bodenreider C, Benz R, Chaffotte A, Ladant D. 2006. Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 281:16914–16926. 10.1074/jbc.M601594200 [DOI] [PubMed] [Google Scholar]
- 57.Chenal A, Guijarro JI, Raynal B, Delepierre M, Ladant D. 2009. RTX calcium binding motifs are intrinsically disordered in the absence of calcium: implication for protein secretion. J. Biol. Chem. 284:1781–1789. 10.1074/jbc.M807312200 [DOI] [PubMed] [Google Scholar]
- 58.Sotomayor Perez AC, Karst JC, Davi M, Guijarro JI, Ladant D, Chenal A. 2010. Characterization of the regions involved in the calcium-induced folding of the intrinsically disordered RTX motifs from the Bordetella pertussis adenylate cyclase toxin. J. Mol. Biol. 397:534–549. 10.1016/j.jmb.2010.01.031 [DOI] [PubMed] [Google Scholar]
- 59.Holland IB, Benadbelhak H, Young J, de Lima Pimenta A, Schmitt L, Blight MA. 2003. Bacterial ABC transporters involved in protein translocation, p 209–241 In Cole SP, Kuchler K, Higgens C. (ed), ABC proteins: from bacteria to man. Academic Press, London, United Kingdom [Google Scholar]
- 60.Dupres V, Menozzi FD, Locht C, Clare BH, Abbott NL, Cuenot S, Bompard C, Raze D, Dufrene YF. 2005. Nanoscale mapping and functional analysis of individual adhesins on living bacteria. Nat. Methods 2:515–520. 10.1038/nmeth769 [DOI] [PubMed] [Google Scholar]
- 61.Andre G, Kulakauskas S, Chapot-Chartier MP, Navet B, Deghorain M, Bernard E, Hols P, Dufrene YF. 2010. Imaging the nanoscale organization of peptidoglycan in living Lactococcus lactis cells. Nat. Commun. 1:27. 10.1038/ncomms1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alsteens D, Trabelsi H, Soumillion P, Dufrene YF. 2013. Multiparametric atomic force microscopy imaging of single bacteriophages extruding from living bacteria. Nat. Commun. 4:2926. 10.1038/ncomms3926 [DOI] [PubMed] [Google Scholar]
- 63.Felmlee T, Welch RA. 1988. Alterations of amino acid repeats in the Escherichia coli hemolysin affect cytolytic activity and secretion. Proc. Natl. Acad. Sci. U. S. A. 85:5269–5273. 10.1073/pnas.85.14.5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanchez-Magraner L, Viguera AR, Garcia-Pacios M, Garcillan MP, Arrondo JL, de la Cruz F, Goni FM, Ostolaza H. 2007. The calcium-binding C-terminal domain of Escherichia coli α-hemolysin is a major determinant in the surface-active properties of the protein. J. Biol. Chem. 282:11827–11835. 10.1074/jbc.M700547200 [DOI] [PubMed] [Google Scholar]
- 65.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1:784–791. 10.1038/nbt1183-784 [DOI] [Google Scholar]
- 66.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580. 10.1016/S0022-2836(83)80284-8 [DOI] [PubMed] [Google Scholar]
- 67.Compeau G, Al-Achi BJ, Platsouka E, Levy SB. 1988. Survival of rifampin-resistant mutants of Pseudomonas fluorescens and Pseudomonas putida in soil systems. Appl. Environ. Microbiol. 54:2432–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.