

Abstract
Iron is an essential nutrient for survival and establishment of infection by Neisseria gonorrhoeae. The neisserial transferrin binding proteins (Tbps) comprise a bipartite system for iron acquisition from human transferrin. TbpA is the TonB-dependent transporter that accomplishes iron internalization. TbpB is a surface-exposed lipoprotein that makes the iron uptake process more efficient. Previous studies have shown that the genes encoding these proteins are arranged in a bicistronic operon, with the tbpB gene located upstream of tbpA and separated from it by an inverted repeat. The operon is under the control of the ferric uptake regulator (Fur); however, promoter elements necessary for regulated expression of the genes have not been experimentally defined. In this study, putative regulatory motifs were identified and confirmed by mutagenesis. Further examination of the sequence upstream of these promoter/operator motifs led to the identification of several novel repeats. We hypothesized that these repeats are involved in additional regulation of the operon. Insertional mutagenesis of regions upstream of the characterized promoter region resulted in decreased tbpB and tbpA transcript levels but increased protein levels for both TbpA and TbpB. Using RNA sequencing (RNA-Seq) technology, we determined that a long RNA was produced from the region upstream of tbpB. We localized the 5′ endpoint of this transcript to between the two upstream insertions by qualitative RT-PCR. We propose that expression of this upstream RNA leads to optimized expression of the gene products from within the tbpBA operon.
INTRODUCTION
Neisseria gonorrhoeae is the etiological agent of the sexually transmitted infection (STI) gonorrhea. This disease is the second most commonly reported infectious disease in the United States, with 334,826 cases reported in 2012 (http://www.cdc.gov/nchhstp/newsroom/docs/STD-Trends-508.pdf); however, the CDC estimates that the actual number of gonorrhea cases is over 800,000 per year (http://www.cdc.gov/std/stats/sti-estimates-fact-sheet-feb-2013.pdf). Worldwide, the WHO estimated that there were 106.1 million new cases of gonorrhea in adults in 2008 (1). Infection manifests primarily as urethritis in men and as cervicitis in women; however, asymptomatic infections are common, especially in women. Due to a rapid rise in resistance to previously effective antibiotics (2, http://www.cdc.gov/nchhstp/newsroom/docs/STD-Trends-508.pdf, and http://www.cdc.gov/std/stats/sti-estimates-fact-sheet-feb-2013.pdf), the CDC currently recommends combination therapy with the extended-spectrum cephalosporin ceftriaxone, plus doxycycline or azithromycin, for treatment (3). Thus, the need for new treatments or ideally preventative methods is evident.
Thus far, vaccine development efforts against gonorrhea have been unsuccessful. Previous attempts focused on surface antigens, such as porin (4) or pilin (5, 6), and have been ineffective, in part because of high-frequency antigenic and/or phase variation of most surface-exposed antigens. The transferrin binding proteins (Tbps) are attractive vaccine targets because they are expressed by all gonococcal isolates tested to date (7), are surface accessible, have conserved sequences, and have been shown to be necessary for the establishment of infection in an experimental human male infection model (8). We demonstrated that recombinant Tbp proteins conjugated to the cholera toxin B subunit are capable of inducing antibody responses in the serum and the genital tract of female mice (9, 10), suggesting that these antigens can be components of an efficacious vaccine.
Most pathogenic bacteria require iron in order to sustain essential metabolic processes and to establish successful infections (11). In a mammalian host, bacteria are presented with the challenge of having to compete for iron with the host; thus, they need to be armed with mechanisms that will allow them to appropriate iron or take advantage of the iron transport and storage mechanisms of the host. Many bacteria, like Escherichia coli, are capable of secreting siderophores as their primary mechanism for iron acquisition (12–14). Gonococci do not produce siderophores (15, 16) but are able to obtain iron from human transferrin by expressing transferrin-binding proteins (17, 18).
The gonococcal transferrin iron acquisition system is comprised of two transferrin-binding proteins, TbpA and TbpB. TbpA is an integral outer membrane protein which shares sequence similarity with TonB-dependent outer membrane transporters (18). TbpA is capable of transporting iron across the outer membrane of gonococci, making it essential in the process of iron uptake from transferrin (19, 20). TbpB is a surface-exposed lipoprotein (19, 20) that has the ability to discriminate between holo- and apotransferrin (21, 22). Expression of TbpA is essential for uptake of iron (18), and although expression of TbpB is not essential, its presence makes the iron uptake process more efficient (17).
The tbpA and tbpB genes are arranged in a bicistronic operon, with the tbpB gene located upstream of tbpA (17, 23, 24). The genes are separated by an 86-bp region that contains an inverted repeat. The genes are cotranscribed (24) and the operon is under the control of the Fur protein, resulting in preferential expression of Tbps under iron-limited conditions. However, the genes are differentially expressed. Using a variety of reverse transcription-PCR (RT-PCR) and fusion techniques, we previously demonstrated that tbpB-specific transcripts are approximately 2-fold more prevalent than tbpA-specific transcripts under iron-stressed conditions (24). Given that TbpB binds specifically to holotransferrin, it may be advantageous to the gonococcus to produce more TbpB and present it on the cell surface as an iron-sequestering mechanism.
In this study, we sought to characterize the mechanisms that coordinately control expression of the two genes within the tbpBA operon. We identified the promoter elements required for tbpB and tbpA expression and further identified a novel repeat-rich region that influences expression of these genes. A long RNA species apparently is located upstream of tbpB in a polarity opposite that of the genes encoding the Tbps. A mutant in which this RNA is insertionally interrupted showed dysregulation of the tbp genes. Therefore, we propose that this long RNA is critical to the optimized expression of the components of the transferrin receptor complex. A complete understanding of the conditions under which this system is expressed is imperative in order to exploit their potential as vaccine candidates.
MATERIALS AND METHODS
Bacterial strains.
Bacterial strains used in this study are listed in Table 1. Neisseria gonorrhoeae strains were routinely maintained on GC medium base (Difco) agar with Kellogg's supplement I (25) and 12 μM Fe (NO3)3 at 37°C in a 5% CO2 atmosphere. When required, iron stress was imposed by overnight growth of strains on GC agar plates containing Kellogg's supplement I and 10 μM deferoxamine mesylate (DFO) (Desferal; Sigma) with no additional iron.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Description | Reference |
|---|---|---|
| N. gonorrhoeae | ||
| FA19 | Wild type (TbpB+/TbpA+) | 7 |
| FA1090 | Wild type (TbpB+/TbpA+) | 52 |
| F62 | Wild type (TbpB+/TbpA+) | 53 |
| MS11 | Wild type (TbpB+/TbpA+) | 54 |
| FA6815 | tbpB::Ω (TbpB−/TbpA−) | 17 |
| MCV108 | tbpA-lacZ fusion (TbpB+/TbpA−) | 24 |
| MCV109 | tbpB-lacZ fusion (TbpB−/TbpA+) | 24 |
| MCV113 | FA19 tbpBΩ−447 | This study |
| MCV114 | FA19 tbpBΩ−82 | This study |
| MCV117 | −10 Promoter element replaced by SmaI site | This study |
| MCV118 | −35 Promoter element replaced by BamHI site | This study |
| MCV119 | Fur box sequence replaced by SmaI site | This study |
| MCV120 | tbpA-lacZ fusion in MCV113 | This study |
| MCV121 | tbpB-lacZ fusion in MCV113 | This study |
| MCV122 | tbpA-lacZ fusion in MCV114 | This study |
| MCV123 | tbpB-lacZ fusion in MCV114 | This study |
| Plasmids | ||
| pCRII-TOPO | Ampr Kanr | Invitrogen |
| pCR2.1-TOPO | Ampr Kanr | Invitrogen |
| pHSS6-GCU (Kanr) | Vector containing gonococcal uptake sequence | 27 |
| pVCU108 | lacZ-ermC′ insertion in the MluI site of tbpA | 24 |
| pVCU109 | lacZ-ermC′ insertion in the PmlI site of tbpB | 24 |
| pVCU122 | pCRII-TOPO containing a 609-bp segment of tbpBA upstream region with novel SmaI site at position −447 relative to TbpB start codon | This study |
| pVCU123 | pCRII-TOPO containing a 609-bp segment of tbpBA upstream region with novel SmaI site at position −82 relative to TbpB start codon | This study |
| pVCU124 | pCRII-TOPO containing a 609-bp segment of tbpBA upstream region with an Ω insertion at position −447 relative to TbpB start codon | This study |
| pVCU125 | pCRII-TOPO containing a 609-bp segment of tbpBA upstream region with an Ω insertion at position −82 relative to TbpB start codon | This study |
| pVCU126 | pHSS6-GCU containing EcoRI fragment of VCU124 | This study |
| pVCU127 | pHSS6-GCU containing EcoRI fragment of VCU125 | This study |
PCR amplification, sequencing, and DNA sequence alignment.
Wild-type gonococcal strains FA19, MS11, and F62 were propagated on GC agar plates as described above. Single colonies were resuspended in 100 μl of distilled water. Samples were boiled at 100°C for 5 min. PCR amplification was conducted using primers oVCU151 and oVCU153 (see Table S1 in the supplemental material) and Platinum Taq DNA polymerase (Invitrogen). The size of the PCR amplicon was confirmed by gel electrophoresis visualization. The PCR product was cloned into pCR2.1-TOPO (Invitrogen) and sequenced by the Nucleic Acids Research Facility at Virginia Commonwealth University. DNA sequence alignments were produced and visualized with ClustalW.
Construction of promoter region mutants.
Promoter region mutants were generated by gene splicing using overlap extension PCR (26). The putative −10 region was replaced by an SmaI site using primers oVCU258 and oVCU259. The putative −35 region was replaced by a BamHI site using primers oVCU256 and oVCU257. Six residues in the putative Fur box were replaced by an SmaI site using primers oVCU260 and oVCU261. Mutagenized PCR products were subcloned into pHSS6-GCU (27) to provide the gonococcal uptake sequence, which is necessary for transformation. The resulting plasmids were then used to transform wild-type gonococcal strain FA19. Successful recombination into the chromosome was confirmed by PCR amplification followed by digestion with either SmaI or BamHI, as necessary. PCR products derived from amplification of chromosomal DNA from recombinant strains were sequenced by the Nucleic Acids Research Facility at Virginia Commonwealth University. The resulting mutant strains were named MCV117 (−10 mutant), MCV118 (−35 mutant), and MCV119 (Fur box mutant), as described in Table 1.
Construction of Ω cassette insertion mutants.
The Ω cassette insertion mutations upstream of tbpB were generated by gene splicing using overlap extension PCR (26). Novel SmaI sites were generated at positions −447 and −82 relative to the tbpB transcriptional start site. The mutated upstream regions were cloned into pCRII-TOPO (Invitrogen) and designated pVCU122 and pVCU123 (corresponding to positions −447 and −82, respectively). The resulting plasmids were digested with SmaI and then ligated to the Ω fragment, which encodes streptomycin resistance (28). The ligation mixture was transformed into E. coli, generating pVCU124 and pVCU125, and the sequences were confirmed by PCR and DNA sequencing. The tbpBA upstream regions containing the Ω fragments from pVCU124 and pVCU125 were subcloned into pHSS6-GCU, generating plasmids pVCU126 and pVCU127, respectively. The plasmids were linearized and then transformed into gonococcal strain FA19. Transformants were selected on GC agar containing streptomycin, confirmed by PCR, and designated MCV113 and MCV114, respectively.
Generation of iron stress conditions for whole-cell lysate preparation.
N. gonorrhoeae strains were cultivated on plates as described above. For iron-depleted cultures, colonies were picked from plates containing GC medium base with supplement I and DFO and used to inoculate GC broth containing Kellogg's supplement I only. Cultures were grown at 37°C at 5% CO2 with vigorous shaking. Culture densities were monitored, and after one doubling, the cultures were supplemented with 250 μM DFO. Cultures were then allowed to grow for 4 h, after which the final cell densities were measured and samples standardized to the same density. Samples were centrifuged for 10 min at 13,000 rpm. Pellets were resuspended in Laemmli solubilizing buffer (29), and lysates were stored at −20°C.
SDS-PAGE and Western blot analysis.
Whole-cell lysates, prepared as described above, were treated with 5% β-mercaptoethanol and boiled for 2 min. Solubilized samples were subjected to SDS-PAGE, and proteins were transferred to nitrocellulose membranes (GE Healthcare Life Sciences) in 20 mM Tris base, 150 mM glycine, and 20% methanol in a submerged transfer apparatus (Bio-Rad). For TbpA detection, membranes were blocked with 5% nonfat dry milk in high-salt Tris-buffered saline (TBS) and 0.05% Tween 20 (Sigma). Membranes were probed with primary anti-TbpA polyclonal antibodies (16), washed with high-salt TBS and 0.05% Tween 20, and incubated with a goat-anti-rabbit alkaline phosphate-conjugated (Bio-Rad) secondary antibody. For TbpB detection, membranes were blocked with 5% nonfat dry milk in low-salt Tris-buffered saline. Membranes were probed with primary anti-TbpB polyclonal antibodies (30) and washed with low-salt TBS and 0.05% Tween 20, followed by goat-anti-rabbit alkaline phosphatase-conjugated secondary antibody. All blots were developed using the nitroblue tetrazolium–5-bromo-4-chloro-3-indolylphosphate colorimetric system (Sigma). Blots were scanned with Adobe Photoshop, and band intensity was quantified using NIH Image J software (31). Each TbpA and TbpB signal was normalized to the loading control from the same sample. The normalized values were then averaged.
Transferrin-iron utilization assays.
The promoter mutants were tested for transferrin utilization by inoculating strains onto chelexed defined media (CDM) (32) supplemented with 30% iron-saturated human transferrin (Sigma) as the sole iron source. Plates were incubated at 37°C and 5% CO2, and bacterial growth was monitored after 24 to 48 h.
Construction of transcriptional lacZ fusion strains.
Upstream insertion mutants MCV113 and MCV114 were transformed by plasmids pVCU109 and pVCU108 (24), which contain promoterless lacZ genes fused to either tbpB or tbpA, respectively. Allelic replacement mutants in which the lacZ gene was located downstream of the polar Ω mutations were selected for by growth on erythromycin, resistance to which is encoded by the promoterless lacZ cassette. The resulting strains were named as described in Table 1.
β-Galactosidase assays.
At each time point, aliquots were removed and mixed with Z-buffer (33). Cells were lysed with SDS and chloroform, and the β-galactosidase assay was performed according to the method of Miller (33). All strains were analyzed in triplicate at every time point. Values shown represent the means from at least four independent experiments conducted on different days.
RNA isolation.
Total RNA was isolated from gonococcal cultures grown for 2 h under iron-depleted or iron-replete conditions using an RNeasy maxi kit as directed by the manufacturer (Qiagen). For qualitative reverse transcription-PCR (RT-PCR) experiments, gonococcal cultures were treated with RNAprotect bacterial reagent (Qiagen) immediately prior to RNA isolation. Purified RNA was treated twice with RQ1 RNase-free DNase (Promega) at 37°C for 1 h prior to storage at −80°C.
Primer extension analysis.
Primer oVCU151 (see Table S1 in the supplemental material), located 37 bases downstream of the TbpB start codon, was end labeled with [γ-32P]ATP using T4 polynucleotide kinase. The radioactive primer was hybridized with 100 μg total RNA, isolated as described above, and treated with avian myeloblastosis virus (AMV) reverse transcriptase (GE Healthcare). The resulting primer extension products were analyzed on a sequencing gel containing 8% polyacrylamide and 8 M urea. Sequencing reactions using the same primer were loaded alongside the primer extension products to serve as a marker.
Relative RT-PCR.
RNA was isolated as described above and treated with DNase. The Thermoscript RT-PCR system (Invitrogen) was utilized in combination with a specific primer designed to prime reverse transcription of an upstream RNA species (oVCU735C; see Table S1 in the supplemental material) or with random hexamers. Following reverse transcription, cDNAs were amplified with gene-specific primers (see Table S1 in the supplemental material) using Platinum Taq polymerase (Invitrogen). PCR products were separated on 2.5% agarose gels and visualized by ethidium bromide staining.
Quantitative, real-time RT-PCR.
Total RNA was isolated from iron-depleted gonococcal cultures as described above. cDNA was generated by reverse transcription of 100 ng of total RNA using the primers listed in Table S1 in the supplemental material and the Accuscript high-fidelity 1st strand cDNA synthesis kit (Agilent Technologies). cDNA was amplified with the CFX96 real-time system (Bio-Rad) utilizing the SensiMix SYBR No-ROX kit (Bioline). The expression level of the target genes were normalized to porB1A expression as an internal control. All oligonucleotides used are listed in Table S1 in the supplemental material. Control wells without template were conducted for every reaction in every experiment. Each assay was performed at least in triplicate, and cDNA from 3 independent RNA preparations was analyzed. Relative expression values for each gene were calculated using the 2−ΔΔCT method (34, 35).
RNA-Seq analysis.
Recently, we (J. L. Kandler and W. M. Shafer, unpublished) performed a transcriptional profiling comparison by RNA sequencing (RNA-Seq) using enriched mRNA prepared from wild-type strain FA19 and an isogenic transformant bearing an insertionally inactivated copy of the misR gene (annotated NGO0177 in strain FA1090 [www.genome.ou.edu/gono.html]), which is part of a two-component regulatory system in pathogenic Neisseria species (36–38). The details of this comparison will be published elsewhere. However, from analysis of the transcripts produced by these strains, we detected equal levels of a 1.8-kb RNA species that would be located upstream of the tbpBA operon (see Results). The procedure for RNA-Seq relevant to the work presented here is described below. Bioinformatic analysis used the transcriptional profiling data from the RNA-Seq work performed on strain FA19 misR::kan. This strain was grown in liquid culture in triplicate to late-log phase in GC broth containing Kellogg's supplement I and iron, 0.043% (wt/vol) sodium bicarbonate, and 10 mM MgCl2 in a 37°C water bath with shaking at 150 rpm. At late-log phase, 8 ml was harvested from each of the three cultures and processed in RNAprotect bacterial reagent (Qiagen) according to the manufacturer's instructions. Total RNA was harvested from culture pellets using an RNeasy minikit (Qiagen) according to the manufacturer's instructions. Removal of any contaminating DNA was accomplished by treatment with TURBO DNA-free (Ambion) according to the manufacturer's instructions. rRNA was depleted using two rounds of treatment with the MICROBExpress bacterial mRNA enrichment kit (Ambion) according to the manufacturer's instructions. RNA purity was confirmed by analysis on an Agilent 2100 Bioanalyzer (Agilent Technologies) after each step. cDNA libraries were prepared from enriched FA19 mRNA transcriptomes using the SuperScript double-stranded cDNA synthesis kit (Invitrogen) and random hexameric primers (Invitrogen) according to the manufacturer's instructions. Because the RNase H provided in the SuperScript kit digests RNA only in DNA-RNA hybrids, samples were treated with 10 μg of RNase A (Novagen) for 10 min at 37°C to ensure complete removal of any remaining, unhybridized RNA. cDNA was purified using phenol extraction and ethanol precipitation. cDNA libraries were amplified by 15 cycles of PCR for the addition of adaptors with TruSeq indexes using the Illumina TruSeq sample preparation kit according to the manufacturer's instructions. After quantitation and dilution, the cDNA libraries representing the FA19 and FA19 misR::kan mutant late-log transcriptomes were clustered in a single lane and sequenced on an Illumina Genome Analyzer IIx instrument with 50-bp paired-end (PE) reads. The raw reads were trimmed by removing adapter sequences and ambiguous nucleotides. Reads with quality scores of less than 20 and a length below 30 bp all were trimmed. The resulting high-quality reads from each of the 3 replicate samples were mapped onto the N. gonorrhoeae FA1090 reference genome (GenBank accession no. AE004969.1) using CLC Genomics Workbench software. For the reference mapping, at least 95% of the bases were required to align to the reference genome, and a maximum of 2 mismatches were allowed. The total number of reads mapped for each transcript was determined and then normalized to detect the reads-per-kilobase-per-million-reads (RPKM) measure. Transcribed regions of the FA19 misR::kan genome were visualized using SeqMan software (DNASTAR).
Statistical analysis.
Statistical significance was evaluated using a two-tailed, unpaired Student's t test. Statistical significance is noted when P ≤ 0.05.
Accession numbers.
The new sequence information presented in this report has been submitted to GenBank under the following accession numbers: F62 sequence upstream of tbpB, KJ579423; MS11 sequence upstream of tbpB, KJ579424; and FA19 sequence upstream of tbpB, KJ579425. The complete data set of the sequence reads can be accessed through GEO accession number GSE50184 and SRA accession number SRP029218.
RESULTS
tbpBA promoter mapping.
While the genetic arrangement and cotranscription of the tbpBA operon has been described (17, 24), little is known about cis- or trans-acting factors that control expression of the operon. We first sought to identify the transcriptional start site of tbpB using primer extension analysis. A primer extension product was observed using RNA isolated from gonococcal strain FA19 grown under iron-restricted growth conditions (Fig. 1A). This primer extension product was not detectable from gonococcal cultures grown under iron-replete conditions. The primer extension product detected under iron-depleted growth conditions corresponded to the C residue located 31 bp upstream of the TbpB start codon (Fig. 1A and B).
FIG 1.

Identification of promoter elements controlling expression of the tbpBA operon. (A) Primer extension analysis demonstrating the transcriptional start site for tbpB in the wild-type and upstream insertion mutants. Total RNA was isolated from gonococcal cultures grown under iron-depleted (−) and iron-replete (+) conditions as indicated at the top. A sequencing ladder generated with the same primer is shown on the left. The arrow marks the location of the primer extension product. This analysis compared the transcriptional start sites of the wild-type strain (FA19) to two Ω insertion mutants (MCV113 and MCV114; see the text). (B) Mutagenesis of promoter elements. The tbpB transcriptional start site (+1) is shown. The TbpB initiation codon is underlined. The mutagenized promoter elements (−10, −35, and Fur box) are labeled; the new nucleotides in the mutants are shown above or below the wild-type sequence. (C) Expression of Tbps in the promoter region mutants. Gonococci were grown under iron-replete (+) or iron-depleted (−) conditions and then lysed. The whole-cell lysates were subjected to SDS-PAGE, and proteins were transferred to nitrocellulose. Blots were probed with α-TbpA or α-TbpB antibodies as labeled on the left. MCV117 is the −10 mutant, MCV118 is the −35 mutant, and MCV119 is the Fur box mutant. (D) Gonococcal mutants were tested for growth in human transferrin by plating on CDM containing 30% saturated transferrin as the sole iron source. FA19 is the positive control and FA6815 (tbpBΩ) is the negative control. MCV117 is the −10 mutant, MCV118 is the −35 mutant, and MCV119 is the Fur box mutant.
To identify putative promoter elements, the upstream region of N. gonorrhoeae wild-type strain FA19 (Fig. 1B) was compared to consensus E. coli sequences (39). A putative −10 region was identified 7 bp upstream of the identified transcriptional start site and matched the E. coli consensus sequence (5′-TATAAT-3′) at five of six positions. A putative −35 region matched the E. coli consensus sequence (5′-TTGACA-3′) at four of six positions. These two motifs were separated by 16 nucleotides. We also identified a predicted Fur binding site (Fig. 1B), which overlaps the −10 sequence and matched the E. coli consensus sequence (5′-GATAATGATAATCATTATC-3′) at 13 of 19 positions. The identified site is a perfect match with the consensus Fur binding site identified for Neisseria species (40).
To confirm that the putative −10, −35, and Fur box elements were indeed responsible for iron-regulated promoter activity, mutational analysis was conducted. Using PCR and the mutagenic primers listed in Table S1 in the supplemental material, the putative promoter elements were changed as shown in Fig. 1B. We evaluated TbpA and TbpB protein expression in the mutants by Western blotting. As expected, TbpA and TbpB were detected in the wild-type strain under iron-depleted conditions. Mutagenesis of the −10 region (strain MCV117) abolished expression of the Tbps even under low-iron conditions (Fig. 1C). Mutagenesis of the −35 region (strain MCV118) almost completely abrogated expression of both Tbps, although TbpA was weakly detectable in the blot probed with the α-TbpA antibody. Mutation of the putative Fur binding site (strain MCV119) resulted in expression of both Tbps regardless of iron availability during growth. This phenotype is consistent with that observed using a fur point mutant (41).
The promoter region mutants were also tested for growth on human transferrin as a sole iron source (Fig. 1D). The wild-type strain FA19 was able to grow on the plates and served as the positive control. Strain FA6815 (Table 1) was the negative control, as this strain is TbpA and TbpB deficient (17). The −10 region mutant (MCV117) was incapable of growth on transferrin even after 48 h, and the Fur box mutant (MCV119) grew much like the wild-type strain on transferrin. The −35 region mutant (MCV118) also grew on transferrin (Fig. 1D), demonstrating that even a very low level of TbpA expression apparently was sufficient to support transferrin-dependent growth. Taken together, these data confirm that the predicted tbpBA promoter elements and Fur box do indeed control the production of TbpA and TbpB.
Characterization of an extended intergenic region upstream of tbpB.
The validated promoter extends to 66 bp upstream of the TbpB start codon (Fig. 1B). However, in the genome sequence of gonococcal strain FA1090 (accession number NC_002946), there is an extremely long (approximately 1.9 kb) intergenic region that separates the tbp operon from the next proximal upstream gene (NGO1499) (Fig. 2). This region contains two short, annotated hypothetical open reading frames (ORFs) (NGO1497 and NGO1498), but it is unclear whether either is actually translated into protein. A third, unannotated ORF could be identified further upstream of NGO1498, as shown in Fig. 2. Because the ORFs are within repeated regions, they share sequence identity and are paralogous. A similar distance separates the tbp locus from the next upstream open reading frame in Neisseria meningitidis (42, 43). Several dRS3 repeats (43), which have the consensus sequence of ATTCCC(N8)GGGAAT, are located within this intergenic region (Fig. 2). These repeats, which are also located upstream of the meningococcal tbpB gene, have been proposed to promote recombination with exogenously acquired DNA (42). Within 1 kb of the tbpB promoter region, we identified three direct repeats in the FA1090 DNA sequence, each of which could encode a hypothetical, short open reading frame (Fig. 2). Repeat 1 (R1) is 366 nt in length, R2 is 365 nt in length, and R3 is 237 nt in length. These tandem repeats share extensive sequence identity; R1 is 97.8% identical to R2, and R1 is 84.2% identical to R3 (Fig. 3). Each of the repeats is also flanked by at least one inverted repeat sequence of CTCTCTCCC(N3)GGGAGAGAG (Fig. 2). Figure 4 shows the intergenic DNA sequence in the FA1090 genome between tbpB and the NGO1499 open reading frame with relevant repeats and primer sequences highlighted. The particular arrangement of tandem, direct, and short inverted repeats downstream of the dRS3 units (Fig. 2 and 4) is unique to the tbpB upstream region in the gonococcal chromosome, as we were unable to find similar arrangements within the genomes of the other Neisseria species. Moreover, the arrangement of direct repeat units (R1, R2, and R3) located upstream of tbpB is unique to this locus and not found elsewhere in the gonococcal genome. However, partial matches for R1 can be found near pilC1 (NGO0055) and carA (NGO0053). We next explored the possibility that these sequences form part of an extended regulatory region for the tbpBA operon.
FIG 2.
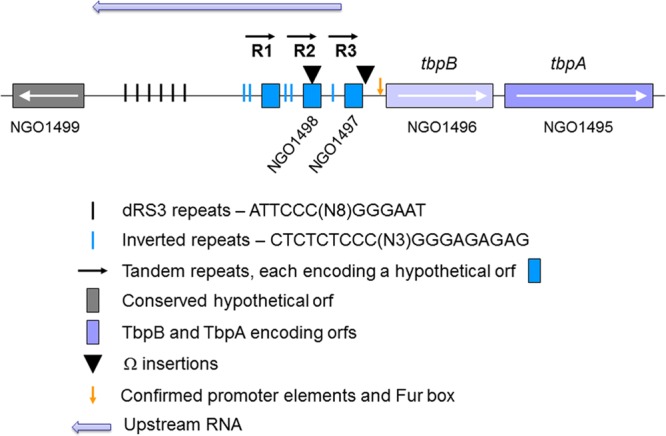
Representation of the region (approximately 1.9 kb) between tbpB and NGO1499 from the FA1090 genome database (www.genome.ou.edu/gono.html). Six dRS3 repeats are located 5′ of tbpB (short, black lines). Three tandem, direct repeats (horizontal arrows) are located between the dRS3 repeats and the tbpB start codon. Each repeat unit contains a short hypothetical ORF (blue boxes) and is flanked at the 5′ end by at least one inverted repeat (short blue lines). ▼, Locations of Ω cassette insertions. Genes and intergenic regions are not drawn to scale.
FIG 3.

Comparison of repeat sequences upstream of the tbpBA operon. Repeats are between 237 and 366 nucleotides in length. Repeat 1 (R1) shares 97.8% identity with repeat 2 (R2). R1 shares 84.2% identity with repeat 3 (R3). Repeat 2 shares 84.5% identity with repeat 3. Multiple-sequence alignment of R1, R2, and R3 demonstrates that the greatest level of sequence identity is centrally located. Asterisks mark positions of identity between the aligned sequences.
FIG 4.

Sequence upstream of tbpB gene in FA1090 genome with repeats and positions of Ω insertions highlighted. Yellow highlighting shows predicted G4 sequences (see the text). Dotted lines below sequences indicate C-rich domains that are complementary to G4 sequences. Dark green shows the position of oligonucleotide oVCU153, used for cloning. Light green highlighting indicates the nucleotide immediately 3′ of Ω insertion positions. Cyan highlighting, repeat 1 sequence; open box, repeat 2 sequence; gray highlighting, repeat 3 sequence; inverted arrows, inverted repeats; blue boldfaced text, predicted start codons for hypothetical ORFs; pink highlighting, dRS3 repeats. The boxed, red G residue indicates the start of long RNA predicted from the RNA-Seq result. Upstream RNA extends 1.8 kb in the orientation opposite that of tbpB. Red text shows the −35, −10, and transcriptional start sites for the tbpBA operon. Brackets with blue arrows show positions of primers used for relative RT-PCR.
We rationalized that if the repeats were involved in regulation of the operon, they would be highly conserved among gonococcal strains. Accordingly, based upon the DNA sequence from strain FA1090, we designed primers to amplify the upstream intergenic region from other gonococcal strains, including our type strain, FA19. We were only able to amplify and clone a product of approximately 600 bp from gonococcal strain FA19. Repeated attempts to amplify and clone regions longer than 600 bp failed. The amplicon that we were able to clone and sequence (Fig. 5) was flanked by primers oVCU151 and oVCU153 (see Table S1 in the supplemental material) and included only two of the repeat units identified in the genome sequence of FA1090 (Fig. 2 and 4). We subsequently utilized these same oligonucleotides to amplify the homologous regions from gonococcal strains MS11 and F62. Alignment of the resulting sequences revealed that one repeated region was well conserved in all strains examined, while the other repeat was not as well conserved and was largely absent from strain F62 (Fig. 5). The sequences shown in Fig. 5 are not present in the partially completed genome sequences of strains FA19, MS11, and F62 (Broad Institute). These areas represent gaps in the partial genome sequences, which is perhaps consistent with our difficulties in cloning DNA from this repeat-rich intergenic region.
FIG 5.

Alignment of sequence upstream of the tbpBA operon from four wild-type N. gonorrhoeae strains. Regions of approximately 600 bp upstream of tbpB were cloned and sequenced from wild-type strains FA19, MS11, and F62. The resulting sequences were aligned with the homologous region from strain FA1090. Dashes represent spaces introduced into the alignment. The tbpB transcriptional start site is highlighted in yellow. The conserved promoter elements are highlighted in blue. The Fur box is shown in red text. Part of R2, highlighted by the single horizontal line, is included in the aligned sequence. All of R3, which is highlighted by the double horizontal line, is included in the aligned sequence. The blue text (TTG) designates the predicted start codons for two hypothetical ORFs upstream of tbpB. The green highlighting shows the nucleotides in FA19 that are immediately 3′ of the Ω insertions created in this study. The gray shading shows the predicted G4 sequences (see the text).
Disruption of the region upstream of the tbpB promoter affects transcript levels.
To elucidate whether the repeat units impacted the expression of the tbp operon, the upstream region was disrupted by insertion of Ω cassettes. An Ω cassette was inserted 447 bp upstream of the tbpB transcriptional start site (strain MCV113) (Table 1). A second Ω cassette was inserted 82 bp upstream of the tbpB transcriptional start site (strain MCV114) (Table 1). The Ω cassette in MCV113 disrupts R2, and the cassette in MCV114 is just 3′ of the end of R3 (Fig. 4). Insertional mutagenesis did not alter the transcriptional start site for the operon (Fig. 1A).
Transcriptional lacZ fusions were created for each tbp gene in the Ω−447 insertion background (MCV120 and MCV121) as well as for the Ω−82 background (MCV122 and MCV123) (Table 1) in order to assay transcript levels. Strains MCV108 and MCV109 were previously generated (24) and contain lacZ insertions into the tbpA and tbpB genes, respectively. These fusions have wild-type upstream regions; thus, they were used as positive controls for tbp transcript levels. All strains were grown in GC base media supplemented with Kellogg's supplement I. DFO was added to all cultures in order to generate iron-depleted conditions. β-Galactosidase assays showed that tbpB transcript levels decreased in the Ω−447 insertion mutant compared to the positive-control strain (Fig. 6A). tbpA transcript levels were also decreased relative to wild-type tbpA levels. Similarly, tbpB transcript levels appeared decreased in the Ω−82 insertion mutant compared to the positive control (Fig. 6B). However, the modest decrease in tbpA transcript levels in the Ω−82 insertion mutant did not reach the level of statistical significance in this assay. Overall, these results indicated that the Ω cassette inserted 447 bp upstream of the tbpB transcriptional start site had a dramatic, negative effect on tbpB and tbpA transcript levels. In contrast, the Ω insertion located closer to the start of tbpB (−82) had less of an impact on tbp transcript levels.
FIG 6.
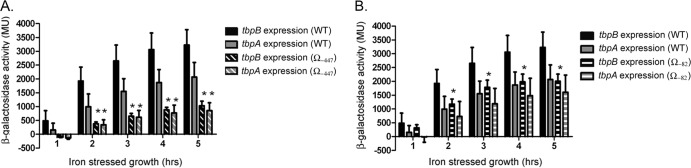
Disruption of the upstream region decreases transcription of tbpB and tbpA. In strain MCV113, repeat 2 of wild-type strain FA19 was disrupted by insertion of an Ω cassette 447 bp upstream of the tbpB transcriptional start site. In strain MCV114, an Ω cassette was inserted 82 bp upstream of the tbpB transcriptional start site. tbpB and tbpA transcript levels in upstream insertion mutants were analyzed by β-galactosidase assay after insertion of a transcriptional lacZ fusion into tbpB or tbpA in the MCV113 and MCV114 backgrounds. (A) tbpB and tbpA transcription levels when the upstream sequence is wild type (solid bars) compared to when the Ω cassette is inserted 447 bp upstream of the tbpB transcriptional start site (striped). (B) tbpB and tbpA transcription levels when the upstream sequence is wild type (solid bars) compared to when the Ω cassette is inserted 82 bp upstream of the tbpB transcriptional start site (striped). For both panels, each bar represents the means from at least four assays conducted on different days; error bars represent standard deviation. For both panels, MU represents Miller units. Asterisks denote P ≤ 0.01 compared to the wild-type transcript levels for the corresponding gene.
We confirmed that these results were not an artifact due to the orientation of the polar Ω cassette and its internal termination signals by evaluating the cassette in the opposite direction, which resulted in the same outcomes (data not shown). Additionally, we confirmed our findings by means of qRT-PCR. RNA extracted from iron-stressed cultures was utilized to detect changes in tbp gene expression. Our results confirmed that the Ω cassette insertion mutants did indeed demonstrate decreased tbp gene expression compared to the wild-type strain (Table 2). Similar to the results of the lacZ fusion analysis, transcript levels of tbpB and tbpA in MCV113 were more profoundly affected than in MCV114. Indeed, tbpB transcript levels were 6.3-fold lower in the MCV113 mutant than in the wild-type strain. tbpB transcript levels were only 2-fold lower in the MCV114 strain than in the wild-type strain. tbpA transcript levels were 6-fold lower in MCV113 and 2-fold lower in MCV114 than in the wild type.
TABLE 2.
Fold changes in tbp expression in the Ω cassette insertion mutants compared to the wild-type strain determined by qRT-PCR
| Strain |
porBIA normalization ofa: |
|
|---|---|---|
| tbpB | tbpA | |
| WT/MCV113 | 6.30 (4.20–9.58) | 6.07 (3.94–9.32) |
| WT/MCV114 | 1.98 (1.07–3.20) | 1.86 (1.04–2.48) |
Numbers in boldface are the means from three experimental replicates from each of three biological replicates conducted on different days (total of nine samples). Numbers in parentheses indicate the range of values obtained by analysis with the 2−ΔΔCT method (34, 35). All fold differences comparing the wild-type strain to the mutants for both tbpB and tbpA were statistically significant (P < 0.05).
Upstream Ω insertions result in increased TbpB and TbpA protein levels.
We analyzed TbpB and TbpA protein levels in the upstream Ω insertion mutants (MCV113 and MCV114) by Western blotting. As shown in Fig. 7, TbpB and TbpA protein levels were increased in the Ω−447 mutant (MCV113) relative to the parent, FA19. Protein levels in MCV114 appeared to be between wild-type and MCV113 levels. When Western blots were scanned and imaged, we determined that TbpB protein levels increased 240% in MCV113 and 220% in MCV114. Similarly, TbpA levels increased 290% in MCV113 and 150% in MCV114. All of the increases in protein levels in the mutants reached statistical significance (P < 0.01) relative to the wild type, except for the TbpA levels in MCV114 (P > 0.05). This result is similar to that with the lacZ transcriptional fusion activity (Fig. 6), in which the change in tbpA levels did not reach the level of statistical significance when comparing the wild-type strain to MCV114. Cumulatively, the results from the insertional mutagenesis studies indicated that an Ω insertion 447 bp upstream of the tbpB start site resulted in decreased transcript levels for both tbp genes but increased levels of both proteins. The impact of the insertion closer to the tbpB start site (−82) was similar in effect but of diminished magnitude.
FIG 7.
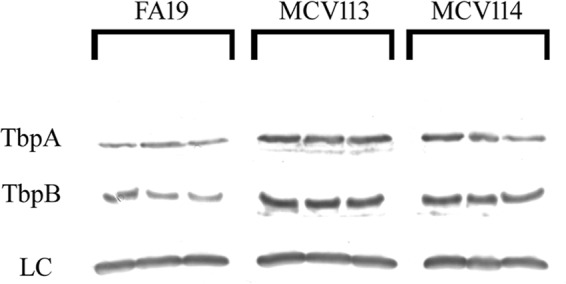
Western blot demonstrating that TbpA and TbpB are produced at higher levels in the Ω insertion mutants. Expression of TbpA and TbpB in wild-type (FA19) and upstream Ω insertion mutants, MCV113 (Ω−447) and MCV114 (Ω−82). The three lanes per strain represent biological replicates grown under iron-stressed conditions on different days. Lanes were loaded equivalently as assessed by the standardized culture density of the whole-cell lysates and Ponceau S staining of the nitrocellulose membrane, and the loading control (LC) is shown. The loading control is a cross-reactive band that is detected when probing whole-cell lysates with the polyclonal anti-TbpA antibody used in this assay. The loading control recapitulates the equivalent staining in each lane detected by Ponceau S.
A long RNA is synthesized from within the region impacted by the Ω insertions upstream of tbpB.
The region upstream of tbpB potentially carried three short, hypothetical open reading frames, all oriented in the same direction as tbpB transcription (Fig. 2 and 4). However, visual analysis of RNA-Seq data obtained from FA19 misR::kan demonstrated that the nucleotides comprising NGO1497 (Fig. 2) were not transcribed under our in vitro conditions (iron-replete GC medium). In contrast, we did observe transcription of a 1.8-kb RNA (mapping to FA1090 nucleotides 1465413 to 1467214; see File S1 in the supplemental material) that encompasses NGO1498 and the undesignated ORF region (Fig. 8). Coverage was quite high near the tbpB gene but waned as the distance increased. This suggested that the observed long RNA was transcribed in the orientation opposite that required for translation of the hypothetical proteins and the tbpBA operon, as areas of high coverage in promoter regions are usually followed by declining coverage moving toward the 3′ end of the RNA. This RNA species is not anticipated to encode any proteins, as no conserved protein domains were detectable in either orientation upon analysis with the Conserved Domain Database search engine (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Although a few short, hypothetical proteins are predicted to be encoded by sequence in the FA1090 genome database between tbpB and NGO1499, the vast majority of the 1.8-kb transcript is not anticipated to be translated.
FIG 8.
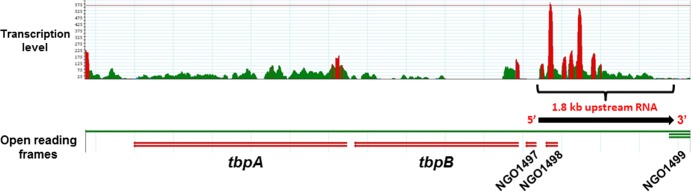
An RNA species (1.8 kb) is transcribed upstream and in the opposite orientation to that of the tbpBA operon. RNA-Seq analysis, which maps and quantifies transcribed regions of a genome, revealed that a long RNA is transcribed from a region mapping to nucleotides 1465413 to 1467214 of strain FA1090. The transcription level (top) represents the number of sequencing reads at each base pair (green, transcribed areas that do not exceed 100 sequencing reads; red, transcribed areas that exceed 100 sequencing reads at the corresponding sequence). The genomic position (bottom) of the annotated open reading frames for tbpB, tbpA, NGO1497, NGO1498, and NGO1499 can be seen in relation to the 1.8-kb RNA (black arrow). Context of the 1.8-kb RNA was visualized using SeqMan Pro software (DNASTAR).
The 5′ end of the upstream 1.8-kb RNA species is located between the positions into which the Ω cassettes were inserted.
We next used qualitative RT-PCR to discern whether the upstream Ω insertions impacted expression of the 1.8-kb RNA species. We predicted that Ω−82 (in strain MCV114) would not interrupt expression of the regulatory RNA species, whereas Ω−447 (in strain MCV113) would, based upon the predicted 5′ end of the RNA. When RT-PCR was conducted on RNA isolated from strains FA19, MCV113, and MCV114 using primers oVCU199C and oVCU735C (Fig. 4; also see Table S1 in the supplemental material), we detected an amplification product only in FA19 and MCV114 (Fig. 9). The predicted-sized product (238 bp; arrow in Fig. 9) was not detected when RNA from strain MCV113 was subjected to RT-PCR. The RT-PCR product detected in FA19 and MCV114 was not sensitive to iron concentrations, unlike the control tbpA gene (Fig. 9). Other controls shown in Fig. 9 include no reverse transcriptase (negative control) and 16S rRNA (positive control). The results shown in Fig. 9 confirm that an RNA species is located upstream and in the orientation opposite that of tbpB in gonococcal strain FA19. Furthermore, this analysis maps the 5′ endpoint of the RNA to between −82 and −447 upstream of the tbpB start site.
FIG 9.
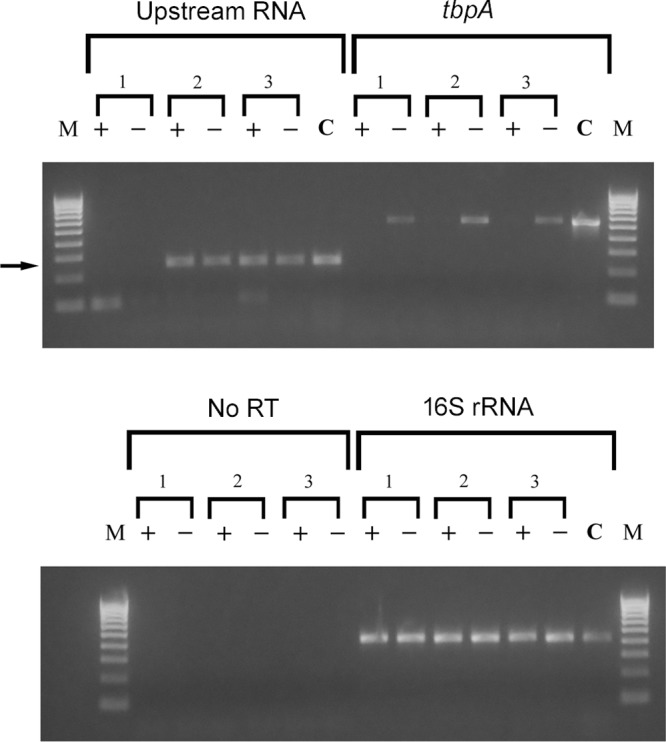
RT-PCR experiment demonstrating that Ω insertion into R2 (−447 relative to the transcriptional start site) prevents expression of the upstream RNA species. Cultures of MCV113 (1), MCV114 (2), and wild-type FA19 (3) were grown under iron-replete (+) and iron-depleted (−) conditions. RNA was isolated from each culture and subjected to reverse transcription. oVCU735C was used to reverse transcribe the upstream RNA, and random hexamers were used to reverse transcribe tbpA and 16S rRNA. A negative control in which no reverse transcriptase was added, along with random hexamers, was also conducted (no RT). For PCRs, the following oligonucleotides were utilized: oVCU735C and oVCU199C to amplify the upstream RNA, oVCU186 and oVCU187 to amplify tbpA, and oVCU110 and oVCU111 to amplify 16S rRNA and no-RT reactions. Chromosomal DNA from FA19 (C) also was amplified with the same primer sets to demonstrate the wild-type size of each amplicon. Molecular mass markers are shown on the left and right of each ethidium bromide-stained gel. Markers (M) range from 100 bp to 1,000 bp and increase in increments of 100 bp. The arrow indicates the position of the amplicon that was amplified with primers oVCU735C and oVCU199C, corresponding to the upstream RNA.
DISCUSSION
Studies on the gonococcal transferrin receptor have focused mainly on deciphering the structure-function relationships of its components (19, 20), and little is known about the precise mechanisms that coordinately control expression of these proteins. A putative promoter for the operon had previously been proposed (17) but never experimentally defined. The purpose of this study was to characterize the extent of the functional promoter for the operon. Using primer extension analysis, we mapped the start site of tbp transcription at a position 31 bp upstream of the TbpB start codon. The C residue, representing the transcriptional start site for the tbpBA operon, was preceded by a C residue at position −1 and followed by a T residue at position +2. These nucleotides are most commonly found at positions −1 and +2 in E. coli promoters (39, 44). Tbp protein expression was abolished or severely decreased when promoter elements were mutagenized, confirming that the sequences of these regions are necessary for transcription of the operon. Although mutagenesis of the −35 promoter element did not completely abrogate expression of the Tbps, it did lead to a severe decrease in the detectable levels of the proteins. In transcription, the −35 motif serves as a point of initial contact between the RNA polymerase and the DNA, but it is the −10 region where the DNA starts to unwind to initiate transcription. It is possible that the mutagenesis of the −35 element was enough to disturb gene expression but not eliminate it entirely, since the −10 region was still intact. Additionally, the Fur binding site motif was similarly identified by homology and mutagenized. The resulting mutant demonstrated Tbp expression regardless of the iron availability during growth. This is the first report to experimentally define the promoter for the gonococcal tbpBA operon. Thus, our results confirm and extend our findings that the genes are cotranscribed from a common upstream promoter and that the operon is under the control of the Fur protein (24).
Analysis of the region upstream of the tbpBA operon revealed approximately 1.9 kb of sequence with several repeat regions and three short, hypothetical open reading frames. In the 600 bp directly upstream of the tbpB transcriptional start site, we identified two direct repeats. A third repeat is located immediately upstream of this region in the complete FA1090 genome database (www.genome.ou.edu/gono.html). The other gonococcal genomes that have been sequenced by the Broad Institute have not been closed or completely annotated, and each is lacking sequence data in the region immediately upstream of the tbp genes. Thus, the sequences presented here for strains FA19, MS11, and F62 are new and not currently in any other database. The repeats within this upstream region resulted in our cloning difficulties and also likely contributed to the failure of genome sequencing in this area of the chromosome.
The repeat-rich region immediately upstream of tbpB is unique to this locus and is not found in the analogous position in N. meningitidis or the commensal Neisseria genomes. This suggests that the regulatory region described here is specific to the gonococcal tbp locus and important for appropriate, coordinated expression of the gonococcal Tbp proteins. We have previously reported that the ratio of tbp transcripts is 2 tbpB transcripts to 1 tbpA transcript in wild-type FA19, even though the genes are cotranscribed as a bicistronic operon. The mechanism by which this ratio is achieved is not completely clear, but we proposed that the intergenic region between tbpB and tbpA, which includes a putative secondary structure, contributes to increased tbpB transcripts relative to tbpA transcripts. In the current study, we also found that insertionally interrupting expression of an upstream RNA resulted in a ratio of nearly 1 tbpB transcript to 1 tbpA transcript (Fig. 6A), suggesting that preventing expression of this long RNA alters the relative amounts of tbpB and tbpA transcripts that are detectable.
Regulatory RNAs have been detected widely in bacteria, utilizing RNA-Seq and other transcriptomics approaches (for reviews, see references 45–47). While the best-characterized examples of regulatory RNAs in bacteria are relatively short (between 50 and 300 nucleotides [48]), it has become clear that bacteria also produce relatively long RNAs with the capability to alter gene expression by affecting both transcript and protein levels. cis-encoded regulatory RNAs generally overlap the genes they regulate and share a great deal of sequence complementarity (45, 48). On the other hand, trans-encoded regulatory RNAs are transcribed from loci far removed from those genes they regulate and share less sequence complementarity (46). The RNA species transcribed upstream of tbpB, in the opposite orientation, appears to be distinct from either of the two well-characterized paradigms of how regulatory RNAs impact gene expression. The RNA transcribed upstream of tbpB is predicted to be large, at 1.8 kb, and is produced from a locus adjacent to the genes it regulates. However, this cis-encoded RNA species does not share extended regions of complementarity with tbpB or tbpA and does not overlap the 5′ end of the tbpB transcript, which is typical of classical cis-encoded regulatory RNAs. Thus, the RNA upstream of the tbpBA operon, first described in this study, has features that distinguish it from regulatory RNAs previously characterized in other bacteria.
G4 sequences, or G-quadruplexes, are guanine-rich sequences that have been identified in many genomes and have wide-ranging impacts on DNA recombination and gene expression, among others (for a review, see references 49 and 50). Cahoon and Seifert (51) have recently shown that pilin antigenic variation in N. gonorrhoeae is affected by a proximal G4 sequence, which directs the expression of a noncoding RNA upstream of pilE. Several predicted G4 sequences are located upstream of the tbpB gene (Fig. 4) and could have effects on transcription of the long RNA detected in the current study. In addition to the guanine-rich repeats, we identified complementary cytosine-rich sequences (Fig. 4) that could base pair with the G4 sequences, potentially altering the folding of the planar G-quadruplexes and their effects on the surrounding genes. While we did not directly demonstrate folding of these G4 sequences or their impacts on expression of the Tbps, their presence in this complicated, repeat-rich regulatory region is intriguing and worthy of further study.
We found that the repeats upstream of tbpB were well conserved in several N. gonorrhoeae strains. In strain F62, the region between two putative G4 sequences was deleted, consistent with these repeats contributing to recombination and impacting the number and length of repeats upstream of tbpB. Deletion of this region in F62 would be expected to impact the expression and potentially the length of the upstream regulatory RNA. The impact of this alteration in strain F62 is unclear but would be expected to result in changes in expression of the transferrin-binding proteins. While sequence upstream of tbpB in FA19, FA1090, and MS11 all have the potential to encode the short, atypical ORFs (Fig. 5; TTG start codons are highlighted in blue), it is anticipated that these short peptides, which would start with leucine rather than methionine, are not actually produced, since the long RNA detected by RNA-Seq appeared to be in the orientation opposite that in which tbpB is expressed. The orientation of the only RNA detectable upstream of tbpB is opposite that of the upstream ORFs, and the 5′ endpoint of the RNA does not encompass NGO1497, precluding its production under the in vitro conditions used in this study.
Insertions in and around repeats 2 and 3 upstream of tbpB decreased both tbpB and tbpA transcript levels but, conversely, increased Tbp protein levels relative to the wild-type strain. However, the insertion at −447 clearly had the greatest impact, both on transcript and protein levels. The Ω insertion at position −447 also clearly interrupts the expression of the upstream RNA, whereas the Ω insertion at −82 was not anticipated to insertionally inactivate the upstream RNA. The RT-PCR results from this study map the 5′ end of the upstream RNA species to between −447 and −82 nucleotides upstream of the tbpB transcriptional start site, consistent with the RNA-Seq data. The Ω insertion at −447 results in decreased tbpBA transcript levels, as assessed by qRT-PCR and lacZ fusion analysis. This result suggests that expression of the RNA upstream of tbpB results in enhanced expression or stability of the tbpBA cotranscript. Furthermore, the Ω insertion at −447 resulted in increased production of the Tbp proteins. Thus, expression of the upstream RNA appears to negatively influence translation of the Tbps. These results are both potentially achievable if the regulatory RNA located upstream of tbpB enhances transcription or mRNA stability but negatively affects the translatability of this message into protein. Using both Target RNA (http://snowwhite.wellesley.edu/targetRNA/index_2.html) and RNA Predator (http://rna.tbi.univie.ac.at/RNApredator2/target_search.cgi), modest regions of alignment between the sequence of the upstream RNA (see File S1 in the supplemental material) and tbpB and tbpA genes can be detected (data not shown). Our working hypothesis is that hybridization between the long RNA and the tbp locus influences both transcript and protein levels. Clearly, further studies are necessary to extend and validate this proposed model.
Overall, the results of this study provide insight into the coordinated regulation of the tbpBA operon. We identified the promoter elements required for iron-regulated expression of both TbpA and TbpB. Our data suggest that both the quantity and stoichiometry of the tbp gene products are affected by interruption of an extended, repeat-filled regulatory region, which is located immediately upstream of tbpB. While this region generally is well conserved within the gonococcal genome sequences analyzed, some heterogeneity was identified, particularly in proximity to predicted G4 sequences. This repeat-filled region is unique to this location in gonococcal genomes. We detected the expression of a regulatory RNA upstream of tbpB in the orientation opposite that of tbpB. This RNA species possesses several unique features, including its relatively long length and the fact that it is cis encoded but does not share significant complementarity with the genes it apparently regulates, the tbpBA locus. Wild-type gonococcal strain FA19 expresses this RNA regardless of iron status. Wild-type expression of this regulatory RNA species appears to enhance transcription or transcript levels of tbpB and tbpA; however, in contrast, wild-type expression of the regulatory RNA also results in decreased translatability of the tbpBA mRNA. Although the precise mechanisms by which these phenomena are achieved as a consequence of expression of this regulatory RNA are currently unclear, our results indicate that wild-type expression of this novel RNA species is important for coordinated, optimized expression of the gonococcal transferrin binding proteins. Further study of this system and the conditions that influence their expression is warranted, as the transferrin binding proteins are potential vaccine candidates for the prevention of gonococcal infections.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by Public Health Service grants R01 AI047141, R01 AI84400, R37 AI21150, and U19 AI31496, all from the National Institute of Allergy and Infectious Diseases, National Institutes of Health. W.M.S. received funding from a VA Merit Award from the Medical Research Service of the Department of Veterans Affairs. In addition, W.M.S. was supported by a Senior Research Career Scientist Award from the Medical Research Service of the Department of Veterans affairs. R. Vélez Acevedo was supported by a diversity supplement to R01 AI047141.
We acknowledge outstanding technical support from Abena Watson-Siroboe. We also thank David Dyer of the University of Oklahoma for his helpful contributions in the analysis of the upstream repeats in strain FA1090. We thank Timothy D. Read, Karla D. Passalacqua, and M. Ryan Weil of the Emory Genomics Core for their assistance in the preparation of transcriptome libraries and bioinformatic analysis of the RNA-Seq data. We thank Kim M. Gernert (Emory University BimCore) for expert help with SeqMan and visualization of the tbp locus. We also thank Gregory H. Doho (Emory Integrated Genomics Core) for his assistance in the publication of the RNA-Seq data sets.
Footnotes
Published ahead of print 16 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01693-14.
REFERENCES
- 1.WHO. 2012. The global incidence and prevalence of selected curable sexually transmitted infections–2008. WHO, Geneva, Switzerland [Google Scholar]
- 2.Unemo M, Shafer WM. 2011. Antibiotic resistance in Neisseria gonorrhoeae: origin, evolution, and lessons learned for the future. Ann. N. Y. Acad. Sci. 1230:E19–E28. 10.1111/j.1749-6632.2011.06215.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.CDC. 2012. Update to CDC's Sexually Transmitted Diseases Treatment Guidelines, 2010: oral cephalosporins no longer a recommended treatment for gonococcal infections. MMWR Morb. Mortal. Wkly. Rep. 61:590–594 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6131a3.htm [PubMed] [Google Scholar]
- 4.Wetzler LM, Blake MS, Barry K, Gotschlich EC. 1992. Gonococcal porin vaccine evaluation: comparison of Por proteosomes, liposomes, and blebs isolated from rmp deletion mutants. J. Infect. Dis. 166:551–555. 10.1093/infdis/166.3.551 [DOI] [PubMed] [Google Scholar]
- 5.Boslego JW, Tramont EC, Chung RC, McChesney DG, Ciak J, Sadoff JC, Piziak MV, Brown JD, Brinton CC, Jr, Wood SW, Bryan JR. 1991. Efficacy trial of a parenteral gonococcal pilus vaccine in men. Vaccine 9:154–162. 10.1016/0264-410X(91)90147-X [DOI] [PubMed] [Google Scholar]
- 6.Johnson SC, Chung RC, Deal CD, Boslego JW, Sadoff JC, Wood SW, Brinton CC, Jr, Tramont EC. 1991. Human immunization with Pgh 3-2 gonococcal pilus results in cross-reactive antibody to the cyanogen bromide fragment-2 of pilin. J. Infect. Dis. 163:128–134. 10.1093/infdis/163.1.128 [DOI] [PubMed] [Google Scholar]
- 7.Mickelsen PA, Sparling PF. 1981. Ability of Neisseria gonorrhoeae, Neisseria meningitidis, and commensal Neisseria species to obtain iron from transferrin and iron compounds. Infect. Immun. 33:555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornelissen CN, Kelley M, Hobbs MM, Anderson JE, Cannon JG, Cohen MS, Sparling PF. 1998. The transferrin receptor expressed by gonococcal strain FA1090 is required for the experimental infection of human male volunteers. Mol. Microbiol. 27:611–616. 10.1046/j.1365-2958.1998.00710.x [DOI] [PubMed] [Google Scholar]
- 9.Price GA, Masri HP, Hollander AM, Russell MW, Cornelissen CN. 2007. Gonococcal transferrin binding protein chimeras induce bactericidal and growth inhibitory antibodies in mice. Vaccine 25:7247–7260. 10.1016/j.vaccine.2007.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price GA, Russell MW, Cornelissen CN. 2005. Intranasal administration of recombinant Neisseria gonorrhoeae transferrin binding proteins A and B conjugated to the cholera toxin B subunit induces systemic and vaginal antibodies in mice. Infect. Immun. 73:3945–3953. 10.1128/IAI.73.7.3945-3953.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briat J-F. 1992. Iron assimilation and storage in prokaryotes. J. Gen. Microbiol. 138:2475–2483. 10.1099/00221287-138-12-2475 [DOI] [PubMed] [Google Scholar]
- 12.Neilands JB. 1981. Microbial iron compounds. Annu. Rev. Biochem. 50:715–731. 10.1146/annurev.bi.50.070181.003435 [DOI] [PubMed] [Google Scholar]
- 13.Perry RD, San Clemente CL. 1979. Siderophore synthesis in Klebsiella pneumoniae and Shigella sonnei during iron deficiency. J. Bacteriol. 140:1129–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty R, Storey E, van der Helm D. 2007. Molecular mechanism of ferric siderophore passage through the outer membrane receptor proteins of Escherichia coli. Biometals 20:263–274. 10.1007/s10534-006-9060-9 [DOI] [PubMed] [Google Scholar]
- 15.McKenna WR, Mickelsen PA, Sparling PF, Dyer DW. 1988. Iron uptake from lactoferrin and transferrin by Neisseria gonorrhoeae. Infect. Immun. 56:785–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.West SEH, Sparling PF. 1985. Response of Neisseria gonorrhoeae to iron limitation: alterations in expression of membrane proteins without apparent siderophore production. Infect. Immun. 47:388–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson JE, Sparling PF, Cornelissen CN. 1994. Gonococcal transferrin-binding protein 2 facilitates but is not essential for transferrin utilization. J. Bacteriol. 176:3162–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornelissen CN, Biswas GD, Tsai J, Paruchuri DK, Thompson SA, Sparling PF. 1992. Gonococcal transferrin-binding protein 1 is required for transferrin utilization and is homologous to TonB-dependent outer membrane receptors. J. Bacteriol. 174:5788–5797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noinaj N, Buchanan SK, Cornelissen CN. 2012. The transferrin-iron import system from pathogenic Neisseria species. Mol. Microbiol. 86:246–257. 10.1111/mmi.12002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noinaj N, Easley NC, Oke M, Mizuno N, Gumbart J, Boura E, Steere AN, Zak O, Aisen P, Tajkhorshid E, Evans RW, Gorringe AR, Mason AB, Steven AC, Buchanan SK. 2012. Structural basis for iron piracy by pathogenic Neisseria. Nature 483:53–58. 10.1038/nature10823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boulton IC, Gorringe AR, Allison N, Robinson A, Gorinsky B, Joannou CL, Evans RW. 1998. Transferrin-binding protein B isolated from Neisseria meningitidis discriminates between apo and diferric human transferrin. Biochem. J. 334:269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cornelissen CN, Sparling PF. 1996. Binding and surface exposure characteristics of the gonococcal transferrin receptor are dependent on both transferrin-binding proteins. J. Bacteriol. 178:1437–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Legrain M, Mazarin V, Irwin SW, Bouchon B, Quentin-Millet M-J, Jacobs E, Schryvers AB. 1993. Cloning and characterization of Neisseria meningitidis genes encoding the transferrin-binding proteins Tbp1 and Tbp2. Gene 130:73–80. 10.1016/0378-1119(93)90348-7 [DOI] [PubMed] [Google Scholar]
- 24.Ronpirin C, Jerse AE, Cornelissen CN. 2001. The gonococcal genes encoding transferrin binding proteins (Tbp) A and B are arranged in a bicistronic operon but are subject to differential expression. Infect. Immun. 69:6336–6347. 10.1128/IAI.69.10.6336-6347.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kellogg DS, Jr, Peacock WL, Jr, Deacon WE, Brown L, Pirkle CI. 1963. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J. Bacteriol. 85:1274–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 27.Elkins C, Thomas CE, Seifert HS, Sparling PF. 1991. Species-specific uptake of DNA by gonococci is mediated by a 10-base-pair sequence. J. Bacteriol. 173:3911–3913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prentki P, Krisch HM. 1984. In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29:303–313. 10.1016/0378-1119(84)90059-3 [DOI] [PubMed] [Google Scholar]
- 29.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 30.Price GA, Hobbs MM, Cornelissen CN. 2004. Immunogenicity of gonococcal transferrin binding proteins during natural infections. Infect. Immun. 72:277–283. 10.1128/IAI.72.1.277-283.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.West SEH, Sparling PF. 1987. Aerobactin utilization by Neisseria gonorrhoeae and cloning of a genomic DNA fragment that complements Escherichia coli fhuB mutations. J. Bacteriol. 169:3414–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller JH. 1992. β-Galactosidase activity, a short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 34.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 35.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- 36.Tzeng YL, Kahler CM, Zhang X, Stephens DS. 2008. MisR/MisS two-component regulon in Neisseria meningitidis. Infect. Immun. 76:704–716. 10.1128/IAI.01007-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tzeng YL, Zhou X, Bao S, Zhao S, Noble C, Stephens DS. 2006. Autoregulation of the MisR/MisS two-component signal transduction system in Neisseria meningitidis. J. Bacteriol. 188:5055–5065. 10.1128/JB.00264-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao S, Montanez GE, Kumar P, Sannigrahi S, Tzeng YL. 2010. Regulatory role of the MisR/S two-component system in hemoglobin utilization in Neisseria meningitidis. Infect. Immun. 78:1109–1122. 10.1128/IAI.00363-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harley CB, Reynolds RP. 1987. Analysis of E. coli promoter sequences. Nucleic Acids Res. 15:2343–2361. 10.1093/nar/15.5.2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grifantini R, Sebastian S, Frigimelica E, Draghi M, Bartolini E, Muzzi A, Rappuoli R, Grandi G, Genco C. 2003. Identification of iron-activated and -repressed Fur-dependent genes by transcriptome analysis of Neisseria meningitidis group B. Proc. Natl. Acad. Sci. USA 100:9542–9547. 10.1073/pnas.1033001100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas CE, Sparling PF. 1996. Isolation and analysis of a fur mutant of Neisseria gonorrhoeae. J. Bacteriol. 178:4224–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bentley SD, Vernikos GS, Snyder LA, Churcher C, Arrowsmith C, Chillingworth T, Cronin A, Davis PH, Holroyd NE, Jagels K, Maddison M, Moule S, Rabbinowitsch E, Sharp S, Unwin L, Whitehead S, Quail MA, Achtman M, Barrell B, Saunders NJ, Parkhill J. 2007. Meningococcal genetic variation mechanisms viewed through comparative analysis of serogroup C strain FAM18. PLoS Genet. 3:e23. 10.1371/journal.pgen.0030023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marri PR, Paniscus M, Weyand NJ, Rendon MA, Calton CM, Hernandez DR, Higashi DL, Sodergren E, Weinstock GM, Rounsley SD, So M. 2010. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PLoS One 5:e11835. 10.1371/journal.pone.0011835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hawley DK, McClure WR. 1983. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res. 11:2237–2255. 10.1093/nar/11.8.2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628. 10.1016/j.cell.2009.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caldelari I, Chao Y, Romby P, Vogel J. 2013. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect. Med. 3:a010298. 10.1101/cshperspect.a010298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Georg J, Hess WR. 2011. cis-Antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 75:286–300. 10.1128/MMBR.00032-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bobrovskyy M, Vanderpool CK. 2013. Regulation of bacterial metabolism by small RNAs using diverse mechanisms. Annu. Rev. Genet. 47:209–232. 10.1146/annurev-genet-111212-133445 [DOI] [PubMed] [Google Scholar]
- 49.Bochman ML, Paeschke K, Zakian VA. 2012. DNA structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 13:770–780. 10.1038/nrg3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ehrat EA, Johnson BR, Williams JD, Borchert GM, Larson ED. 2012. G-quadruplex recognition activities of E. coli MutS. BMC Mol. Biol. 13:23. 10.1186/1471-2199-13-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cahoon LA, Seifert HS. 2013. Transcription of cis-acting, noncoding, small RNA is required for pilin antigenic variation in Neisseria gonorrhoeae. PLoS Pathog. 9:e1003074. 10.1371/journal.ppat.1003074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen MS, Cannon JG, Jerse AE, Charniga LM, Isbey SF, Whicker LG. 1994. Human experimentation with Neisseria gonorrhoeae: rationale, methods, and implications for the biology of infection and vaccine development. J. Infect. Dis. 169:532–537. 10.1093/infdis/169.3.532 [DOI] [PubMed] [Google Scholar]
- 53.Maness MJ, Sparling PF. 1973. Multiple antibiotic resistance due to a single mutation in Neisseria gonorrhoeae. J. Infect. Dis. 128:321–330. 10.1093/infdis/128.3.321 [DOI] [PubMed] [Google Scholar]
- 54.Swanson J, Kraus SJ, Gotschlich EC. 1971. Studies on gonococcus infection I. Pili and zones of adhesion: their relation to gonococcal growth patterns. J. Exp. Med. 134:886–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.