

Abstract
Targeted, translational LacZ fusions provided the initial support for the signal sequence hypothesis in prokaryotes and allowed for selection of the mutations that identified the Sec translocon. Many of these selections relied on the fact that expression of targeted, translational lacZ fusions like malE-lacZ and lamB-lacZ42-1 causes lethal toxicity as folded LacZ jams the translocation pore. However, there is another class of targeted LacZ fusions that do not jam the translocon. These targeted, nonjamming fusions also show toxic phenotypes that may be useful for selecting mutations in genes involved in posttranslocational protein folding and targeting; however, they have not been investigated to the same extent as their jamming counterparts. In fact, it is still unclear whether LacZ can be fully translocated in these fusions. It may be that they simply partition into the inner membrane where they can no longer participate in folding or assembly. In the present study, we systematically characterize the nonjamming fusions and determine their ultimate localization. We report that LacZ can be fully translocated into the periplasm, where it is toxic. We show that this toxicity is likely due to LacZ misfolding and that, in the absence of the periplasmic disulfide bond catalyst DsbA, LacZ folds in the periplasm. Using the novel phenotype of periplasmic β-galactosidase activity, we show that the periplasmic chaperone FkpA contributes to LacZ folding in this nonnative compartment. We propose that targeted, nonjamming LacZ fusions may be used to further study folding and targeting in the periplasm of Escherichia coli.
INTRODUCTION
Translational LacZ fusions have been used to study protein targeting in Escherichia coli for over 35 years. The hybrid proteins produced by such fusions comprise an N-terminal targeting sequence fused to a C-terminal, often functional fragment of LacZ. The targeting sequence may consist of an outer membrane protein (OMP) like LamB, a soluble periplasmic protein, or an integral inner membrane (IM) protein like MalF. Each of these hybrid proteins has in common an N-terminal signal sequence, which targets it to the Sec translocon at the inner membrane (IM).
The first lamB-lacZ fusions were described in 1977, and a subset of these were found to confer sensitivity to the inducer maltose (1). Induction of targeted hybrid proteins like LamB-LacZ42-1 causes massive accumulation of the precursor forms of envelope proteins in the cytoplasm (2). It seems that the hybrid proteins jam the secretion machinery, causing a global reduction in translocation efficiency. Jamming in turn leads to proteolytic degradation of the essential components of the Sec translocase and eventually to cell death (3). Screens for mutations that relieve lethal jamming without preventing synthesis of the LamB-LacZ42-1 hybrid protein (Malr Lac+) return mutations in lamB that prevent fusion targeting by altering the signal sequence (4). The Δ60lamB-lacZ fusion, which contains a lamB signal sequence deletion, was identified in this way (5). Such mutants provided important support for the signal sequence hypothesis in bacteria. Together, findings with the lamB- and malE-lacZ (2) fusions made possible the selections and screens that identified the sec and prl alleles, which defined the components of the Sec translocon during the 1980s (6–9).
The role of LacZ in hybrid jamming was determined in studies with two derivatives of lamB-lacZ42-1, which show that envelope protein precursors do not accumulate if LacZ folding is prevented prior to translocation. The first fusion, lamB-lacZX90, contains an ochre mutation that prevents folding by truncating the last 10 amino acids of LacZ (10, 11). The second fusion, H*lamB-lacZ, prevents folding by altering the targeting pathway of the hybrid protein. H* denotes a mutant lamB signal sequence with increased hydrophobicity, which reroutes the fusion from the usual posttranslational, SecB-dependent targeting pathway to the cotranslational, signal recognition particle (SRP) pathway (12). Because translation and translocation are coupled in the SRP pathway, LacZ cannot fold prior to translocation (12). Both the lacZX90 and H*lamB mutations relieve the lethal jamming caused by LamB-LacZ42-1, and because neither fusion jams the secretion machinery, they must allow LacZ sequences to exit the Sec translocon (11, 12).
Despite the fact that the X90 and H* mutations relieve jamming, induction of either fusion is lethal (11, 12). The toxicity conferred by these fusions is manifest in the periplasm as evidenced by the fact that overproduction of the periplasmic protease DegP relieves this toxicity (11, 12). In marked contrast, the lethal jamming caused by the LamB-LacZ42-1 hybrid protein is not relieved by overproduction of DegP (11, 12). Periplasmic toxicity is likely due to the oxidizing nature of the periplasm, which causes LacZ to misfold and aggregate (11, 13, 14). The LamB-LacZX90 hybrid protein forms periplasmic disulfide-bonded aggregates that can be disrupted by removing the disulfide bond catalyst DsbA, although this does not relieve maltose sensitivity (see below). LacZ is also known to misfold in strains expressing the H*lamB-lacZ fusion, because it does not confer β-galactosidase activity (12).
The same is true of another cotranslationally targeted lacZ fusion, malF-lacZ102, which encodes a hybrid protein comprising a folding-competent LacZ moiety fused to the third transmembrane segment of the integral IM protein MalF (14). MalF topology suggests that LacZ is exposed to the periplasm when this hybrid protein partitions into the membrane. Interestingly, selection for β-galactosidase activity in strains expressing the malF-lacZ102 fusion returns extragenic (unlinked) mutations that inactivate the dsbA gene (15). Because LacZ is known to fold only in the cytoplasm in vivo, restoration of β-galactosidase activity in strains expressing malF-lacZ102 fusion has been attributed to the relocalization of LacZ to the cytoplasm, although it has also been suggested that LacZ may be folding in the periplasm (16). According to the cytoplasmic model, the LacZ moiety in MalF-LacZ102 partitions into the membrane along with MalF, adopting an inactive, transmembrane structure that is stabilized by intra-LacZ disulfide bonds at the periplasmic face of the IM. In the absence of DsbA, the exergonic folding reaction pulls LacZ through the membrane into the cytoplasm where it can become active (15).
Perhaps, LacZ contains cryptic stop-transfer sequences that prevent complete translocation of any hybrid protein containing LacZ. Indeed, it is not clear whether the LamB-LacZX90 or H*LamB-LacZ hybrid proteins are fully translocated into the periplasm either. Fractionation of these hybrid proteins has proven difficult, because their misfolding promotes aggregation. The best evidence for full translocation is the fact that DegP overexpression relieves inducer sensitivity in strains expressing these targeted, nonjamming fusions (11, 12); however, this result does not preclude a model whereby hybrid protein partitions into the IM and confers toxicity through exposed periplasmic domains. Neither the toxicity conferred by high-level production of the MalF-LacZ102 hybrid protein nor the effects of DegP overproduction in these fusion strains have been systematically examined.
If the β-galactosidase activity exhibited by the MalF-LacZ102 hybrid protein in the absence of DsbA reflects periplasmic localization of LacZ activity, then this strain should grow on lactose in the absence of the lactose permease, LacY. However, fusion toxicity complicates this simple genetic test because low levels of cell lysis caused by fusion toxicity can support growth by releasing soluble β-galactosidase from the cytoplasm (1). On the other hand, biochemical experiments demonstrating full translocation of the β-galactosidase portion of the MalF-LacZ102 hybrid protein are complicated by the fact that the hybrid protein is membrane bound and thus insoluble.
In the present study, we seek to determine whether LacZ can be fully translocated into the periplasm, and, if so, whether it can fold and what periplasmic factors might be involved. We suggest that the H*lamB-lacZ and malF-lacZ102 fusions provide a useful tool to study protein quality control pathways in the periplasm that help maximize the production of heterologous proteins that require secretion and oxidation in E. coli.
MATERIALS AND METHODS
Strains and plasmids.
Strains and plasmids used in this study are listed in Table 1. All strains are derivatives of NR754, an Ara+ revertant of Escherichia coli MC4100. Marked alleles ΔdsbA::Kanr, ΔfkpA::Cmr, and lacY::Tn9 were moved by transduction with bacteriophage P1 (17). CWB299 is merodiploid, containing both the H*lamB-lacZ fusion and a copy of the lamB gene with the H* signal sequence (Fig. 1A). We found that the extra copy of the Pmal promoter (a divergent promoter) led to a significant decrease in levels of hybrid protein. To facilitate comparison with the other fusion strains, we created a haploid version of H*lamB-lacZ (JCM944) by recombineering (18) (Fig. 1F). To generate the H*lamB-lacZ deletion series, a fragment of the fusion was cloned into pBR322 (pRSD02), and regions of mature lamB sequence were removed using a plasmid-based PCR method (19). Fusions were then amplified from the plasmid by PCR and recombineered into JCM932 as described for JCM944.
TABLE 1.
Strains, plasmids, and phage
| Strain, plasmid, or phage | Genotype or relevant description | Reference(s) or source(s) |
|---|---|---|
| NR754 | MC4100 F− araD+ Δ(argF-lac)U169 rpsL150 relA1 flbB5301 deoC1 ptsF25 thi | 43, 44 |
| CWB299 | MC4100 Φ(H*lamB′-′lacZ) Hyb42-1 [λp1(209)] H*lamB | 12 |
| JCM911 | JCM913 pKD46 | This study |
| JCM912 | NR754 Φ(lamB′-′lacZ) Hyb42-1 [λp1(209)] | This study; 1 |
| JCM913 | NR754 Φ(lamBΔ60′-′lacZ) Hyb42-1 [λp1(209)] | This study; 5 |
| JCM914 | NR754 Φ(lamB-lacZX90) Hyb42-1 [λp1(209)] | This study; 11 |
| JCM932 | NR754 Φ(PlamB Δ230::Kanr lamB′-′lacZ) Hyb42-1 [λp1(209)] pKD46 | This study |
| JCM944 | NR754 Φ(H*lamB′-′lacZ) Hyb42-1 [λp1(209)] | This study |
| RSD106 | NR754 pNG102 | This study |
| RSD107 | NR754 Φ(H*SS + 90lamB′-′lacZ) Hyb42-1 [λp1(209)] | This study |
| RSD108 | NR754 Φ(H*SS + 49lamB′-′lacZ) Hyb42-1 [λp1(209)] | This study |
| RSD109 | NR754 Φ(H*SS + 43lamB′-′lacZ) Hyb42-1 [λp1(209)] | This study |
| RSD110 | NR754 Φ(H*SS + 27lamB′-′lacZ) Hyb42-1 [λp1(209)] | This study |
| RSD111 | NR754 Φ(H*SS + 5lamB′-′lacZ) Hyb42-1 [λp1(209)] | This study |
| RSD112 | JCM913 pDegP | This study |
| RSD113 | JCM912 pDegP | This study |
| RSD114 | JCM914 pDegP | This study |
| RSD115 | JCM944 pDegP | This study |
| RSD116 | RSD106 pDegP | This study |
| RSD117 | JCM913 dsbA::Kanr | This study |
| RSD118 | JCM912 dsbA::Kanr | This study |
| RSD119 | JCM914 dsbA::Kanr | This study |
| RSD120 | JCM944 dsbA::Kanr | This study |
| RSD121 | RSD106 dsbA::Kanr | This study |
| RSD122 | JCM913 lacY::Tn9 | This study |
| RSD123 | JCM944 lacY::Tn9 | This study |
| RSD124 | RSD117 lacY::Tn9 | This study |
| RSD125 | RSD120 lacY::Tn9 | This study |
| RSD126 | RSD121 Δskp yafC::Tn10 | This study |
| RSD127 | RSD121 ΔfkpA::Cmr | This study |
| pNG102 | Ampr LacZ− PmalmalE′ Φ(malF-lacZ) Hyb102 | 14 |
| pDegP | Cmr pACYC184::degP | 45 |
| pKD4 | Ampr Kanr | 18 |
| pKD46 | Ampr ParaB λ-Red recombinase γ β exo | 18 |
| pCWB36 | H*lamB | 12 |
| pRSD02 | pBR322::′H*lamB-lacZ′ Tetr | This study |
| λp1(209) | ::(+Mu′)trp′BA′–W209 lac ′O Z U118Y A′ | 43 |
FIG 1.

Construction of JCM944. (A, B, D, and F) Note that the λ insertion is λp1 (209). (A) CWB299. CWB299 is an H*lamB merodiploid, containing a copy of H*lamB in addition to the H*lamB-lacZ fusion. (B) JCM911. Construction of a haploid H*lamB-lacZ strain began with a strain containing the Δ60lamB-lacZ fusion and pKD46. (C to E) JCM911 was recombineered with the linear DNA fragment depicted in panel C. The fragment was obtained by PCR amplification of the Kanr cassette present on pKD4 with primers having 5′ homology to malK and lamB. The primers were designed to generate a Kanr insertion-deletion removing 27 nucleotides upstream of the lamB open reading frame and 200 nucleotides downstream (Δ230::Kanr). The result of the recombineering reaction is JCM932, depicted in panel D. The Δ230::Kanr insertion-deletion is polar on the lac operon, preventing expression of lacY. As a result, JCM932 cannot grow on minimal melibiose at 42°C. JCM932 was then recombineered with the linear DNA fragment depicted in panel E. The fragment was obtained by PCR amplification of malK and H*lamB from pCWB36. Recombinants that replaced the Δ230::Kanr insertion-deletion and restored lacY expression were selected on minimal melibiose at 42°C and subsequently screened for Kans. (F) JCM944 was identified as a Mel+ Kans recombinant.
Medium and growth conditions.
All fusion strains were cultured overnight at 37°C with aeration in minimal M63 medium supplemented with 1% (wt/vol) glucose to repress expression of the fusion and 0.4% (vol/vol) LB unless otherwise noted (17). All subsequent experiments were carried out at 37°C with aeration unless noted otherwise. Chloramphenicol (20 μg/ml), kanamycin (25 μg/ml), and ampicillin (125 μg/ml) (Sigma) were included in overnight cultures and plates as necessary to select for plasmid retention or recombination.
Maltose disc diffusion assays.
Overnight cultures were resuspended in M63 minimal medium. One-hundred-microliter aliquots of resuspended cells were then mixed with 3 milliliters molten M63 top agar and spread evenly on a plate containing M63 bottom agar. Bottom and top agar were supplemented with 0.2% glycerol, 0.4% LB, and antibiotics as needed. Filter discs infused with 15 μl of 20% maltose (Sigma) were placed in the center of the plates once the top agar solidified. Plates were incubated overnight, and the diameters of zones of inhibition were measured. Measurements exclude the diameter of the filter disc. Representative experiments are shown.
β-Galactosidase assays.
Overnight cultures were resuspended and subcultured 1:50 into LB. Cultures were grown for 2 to 3 h to mid-log phase. One-milliliter aliquots of culture were pelleted and permeabilized using chloroform and SDS. β-Galactosidase assays were performed in triplicate according to the method of Miller (20). Spectrophotometric readings were taken at 1-min intervals over the course of a 15-min o-nitrophenyl-β-d-galactopyranoside (ONPG) hydrolysis reaction, and arbitrary units were calculated from measurements of Vmax after normalization to cell culture density as determined by readings of optical density at 600 nm (OD600).
Growth rate determination.
Overnight cultures were washed twice in minimal M63 medium and subcultured 1:50 in 5 ml M63 minimal medium containing either 1% glycerol or 1% lactose. Medium was not supplemented with LB. Growth was measured by an OD600 reading every hour over an 8-h period. Growth rates were determined during exponential growth using three data points.
Spheroplasting.
Overnight cultures were resuspended and subcultured 1:50 into 50-ml liquid LB cultures. Cultures were grown for 2 to 3 h until reaching mid-log phase. Cultures were then split and subjected to either spheroplasting or lysis in SDS-PAGE sample buffer (21). After spheroplasts were pelleted, supernatants containing soluble periplasmic components were filtered and subjected to β-galactosidase assay, SDS-PAGE, and Western blotting alongside whole-cell lysates.
Western blotting.
Samples resuspended in SDS-PAGE sample buffer were boiled for 10 min and subjected to electrophoresis on a 12% polyacrylamide gel. Immunoblotting was performed with maltose-binding protein (MBP) (1:30,000 dilution) and CpxR (1:10,000 dilution) antisera, and detection was performed with the ECL antibody detection kit (Amersham) and Hyblot CL film (Denville Scientific).
RESULTS
Targeted, nonjamming fusions confer periplasmic toxicity.
To determine whether LacZ can be fully translocated into the periplasm, we first set out to see if the nonjamming fusions confer similar phenotypes. We reasoned that if the malF-lacZ102 fusion also confers maltose sensitivity, some significant portion of LacZ might be exposed to the periplasm in this strain. Using a disc diffusion assay to measure maltose sensitivity, we found that the malF-lacZ102 fusion does indeed confer maltose sensitivity (Table 2). Because malF-lacZ102 is plasmid borne, the size of the zone of clearing cannot be compared directly to those of the other fusion strains, except to say that strains harboring this fusion are certainly Mals.
TABLE 2.
Inducer sensitivity profiles of fusion strains
| Fusion | Zone of inhibition (mm) |
||
|---|---|---|---|
| Wild typea | pDegPb | dsbA::Kanr | |
| Δ60lamB-lacZ | 0 | 0 | 0 |
| lamB-lacZ42-1 | 25 | 19 | 23 |
| lamB-lacZX90 | 23 | 0 | 24 |
| H*lamB-lacZ | 11 | 0 | 4 |
| malF-lacZ102 | 28 | 0 | 2 |
“Wild type” indicates strains with fusions present in the NR754 background without plasmids or markers. See fusion strains JCM913, JCM912, JCM914, JCM944, and RSD06.
“pDegP” indicates fusion strains containing a high-copy-number degP plasmid. See fusion strains RSD112 to RSD116.
“dsbA::Kanr” refers to strains RSD117 to RSD121.
Previously, it was shown that overexpression of the periplasmic protease DegP is sufficient to relieve the Mals phenotype conferred by the H*lamB-lacZ and lamB-lacZX90 fusions. To ensure that the maltose sensitivity conferred by the malF-lacZ102 fusion results from periplasmic toxicity rather than from some deleterious effect in the IM, we determined the effect of DegP overexpression on the fusion strain. Using maltose disc diffusion assays, we found that DegP overexpression caused strains expressing the malF-lacZ102 fusion to become Malr (Table 2). Note that DegP does not relieve the Mals phenotype of the jamming lamB-lacZ42-1 fusion, which does not reach the periplasm. The fact that all three nonjamming fusions confer a Mals phenotype that can be relieved by DegP overexpression suggests that despite differences in targeting sequences and translocation pathways, the hybrid protein is exposed to the periplasm in all three fusions.
Toxicity of targeted, nonjamming fusions is caused by misfolded LacZ in the periplasm.
The fact that the malF-lacZ102 fusion confers periplasmic toxicity suggests that the toxicity in all strains may be unrelated to targeting steps downstream of translocation. MalF should not cause the MalF-LacZ102 hybrid protein to titrate periplasmic chaperones or to interfere with the essential OMP assembly machinery because (i) as an IM protein it should not contain any signal for downstream interactions and (ii) it should tether the hybrid protein to the IM, where it cannot interact with OMPs. To test whether the toxicity conferred by the H*lamB-lacZ fusion is also a result of localizing LacZ to the periplasm, a series of deletion constructs were made, removing lamB sequence from the fusion while leaving the signal sequence intact. These fusions should still be targeted to the translocon, but any downstream targeting signals contained in the lamB fragment should be removed by successive deletions. We found no discernible change in the Mals phenotype of any of the deletion constructs, even when only 5 amino acids of mature lamB were included in the fusion (Table 3). This result supports the conclusion that periplasmic LacZ is the sole source of toxicity in these targeted, nonjamming fusions.
TABLE 3.
Inducer sensitivity of the H*lamB-lacZ deletion series
| Strain | Mature LamB sequencea | Zone of inhibition (mm) |
|---|---|---|
| JCM944 | 173 | 7 |
| RSD107 | 90 | 7 |
| RSD108 | 49 | 6 |
| RSD109 | 43 | 7 |
| RSD110 | 27 | 8 |
| RSD111 | 5 | 7 |
Number of mature LamB residues included in addition to the H*LamB signal sequence.
Since the LamB-LacZX90 hybrid protein is known to form disulfide-bonded aggregates in the periplasm, we wondered if disulfide bond formation may contribute to the periplasmic toxicity of LacZ. Indeed, LacZ contains a total of 16 cysteine residues, any of which could participate in aberrant intra- or intermolecular disulfide bonds. To test this hypothesis, we deleted the gene encoding the periplasmic disulfide bond catalyst DsbA. We found by disc diffusion assay that the dsbA::Kanr insertion-deletion partially relieves the Mals phenotype conferred by the H*lamB-lacZ and malF-lacZ102 fusions; however, it does not relieve the Mals phenotype of cells expressing the lamB-lacZX90 fusion (Table 2). The lamB-lacZX90 result is consistent with a previous report showing that dsbA deletion does not affect maltose sensitivity in this strain (11). Together, these findings suggested that LacZ must be folding competent in order for dsbA disruption to relieve maltose sensitivity. We hypothesized that in the absence of DsbA, LacZ can fold in the periplasm, which reduces its toxicity in turn.
In the absence of DsbA, LacZ folds in the periplasm.
To determine whether LacZ is folding in the absence of DsbA, β-galactosidase assays were performed on whole-cell lysates derived from the various fusion strains. As expected, the Δ60lamB-lacZ and lamB-lacZ42-1 fusions, which lead to retention of LacZ in the cytoplasm, exhibited β-galactosidase activity whether or not DsbA was present (Fig. 2). In the presence of DsbA, none of the targeted, nonjamming fusions conferred β-galactosidase activity; however, strains expressing the H*lamB-lacZ and malF-lacZ102 fusions showed significant β-galactosidase activity upon dsbA disruption (Fig. 2). Coupled with the reduction in maltose sensitivity, this increase in β-galactosidase activity in targeted, nonjamming fusions supports the hypothesis that misfolded LacZ is toxic in the periplasm.
FIG 2.
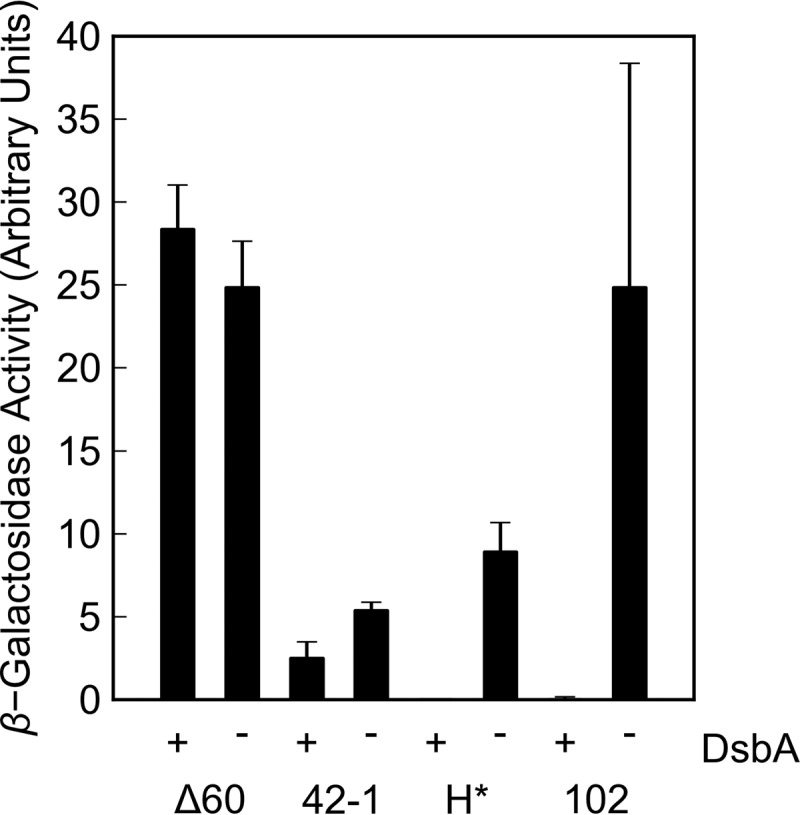
Effect of DsbA on whole-cell β-galactosidase activity. Overnight cultures were washed and subcultured 1:50 into liquid LB cultures. Cultures were incubated with aeration at 37°C for 2 to 3 h until reaching mid-log phase. β-Galactosidase activity was determined from Vmax of whole-cell lysates in a kinetic Miller assay. Δ60 indicates strains JCM913 (dsbA+) and RSD117 (dsbA::Kanr). 42-1 indicates strains JCM912 (dsbA+) and RSD118 (dsbA::Kanr). H* indicates strains JCM944 (dsbA+) and RSD120 (dsbA::Kanr), and 102 indicates strains RSD06 (dsbA+) and RSD121 (dsbA::Kanr). Error bars show standard deviations calculated from three independent biological replicates.
We took two complementary approaches to determine whether the β-galactosidase activity exhibited by strains expressing the H*lamB-lacZ or malF-lacZ102 fusion is localized to the periplasm. In the first approach, we took advantage of the fact that otherwise wild-type strains require the inner membrane lactose permease LacY to grow in minimal lactose medium. In the absence of LacY, lactose cannot reach the cytoplasm where LacZ activity normally resides. We reasoned that if LacZ were instead localized and active in the periplasm, it could cleave periplasmic lactose into galactose and glucose, which could be imported by other means, thereby conferring growth in minimal lactose even in the absence of LacY. To this end, we tested the ability of strains expressing the H*lamB-lacZ and malF-lacZ102 fusions to grow in minimal lactose in three genetic backgrounds: lacY+ dsbA+, lacY::Tn9 dsbA+, and lacY::Tn9 dsbA::Kanr. The Δ60lamB-lacZ fusion was used as a control exhibiting cytoplasmic β-galactosidase activity. As expected, strains expressing the Δ60lamB-lacZ fusion could grow only in the presence of LacY (Fig. 3A). Like Δ60lamB-lacZ, neither the H*lamB-lacZ nor the malF-lacZ102 fusion conferred a Lac+ phenotype in a lacY::Tn9 dsbA+ background; however, these strains became Lac+ in the absence of DsbA (Fig. 3A)—the same condition under which they exhibit β-galactosidase activity. This evidence supports the hypothesis that LacZ can fold in the periplasm in the absence of DsbA. We note that growth in minimal lactose due to cell lysis is not consistent with these findings. If lysis were occurring, we would expect it to be higher in the dsbA+ background, since the dsbA::Kanr allele relieves maltose sensitivity. Yet, significant growth is observed only under the low-lysis condition. We conclude that the hydrolysis of lactose in the medium by extracellular LacZ cannot explain the observed growth.
FIG 3.

Effect of DsbA on periplasmic β-galactosidase activity. (A and B) Error bars show standard deviations calculated from three independent biological replicates. (A) Bypassing lacY. Overnight cultures were washed and subcultured in minimal medium containing glycerol or lactose as the sole carbon source. Δ60 indicates strains containing the Δ60lamB-lacZ fusion in a wild-type background (JCM913, white), a lacY::Tn9 background (RSD122, gray), or a lacY::Tn9 dsbA::Kanr background (RSD124, black). H* indicates strains containing the H*lamB-lacZ fusion in a wild-type background (JCM944, white), a lacY::Tn9 background (RSD123, gray), or a lacY::Tn9 dsbA::Kanr background (RSD125, black). 102 indicates strains containing the malF-lacZ102 fusion in a wild-type background without lacY (RSD06, gray) or a dsbA::Kanr background (RSD121, black). (B and C) Overnight cultures were washed and subcultured 1:50 into liquid LB cultures. Cultures were incubated with aeration at 37°C for 2 to 3 h until reaching mid-log phase. Δ60 refers to RSD117 (dsbA::Kanr), and H* refers to RSD120 (dsbA::Kanr). (B) Releasing soluble periplasmic β-galactosidase activity. β-Galactosidase activity was determined from Vmax of whole-cell lysates or filtered spheroplast supernatants in a kinetic Miller assay. Periplasmic β-galactosidase activity is reported as a fraction of whole-cell β-galactosidase activity. (C) Spheroplasting lysis control. Whole-cell lysates or filtered spheroplast supernatants were subjected to SDS-PAGE and Western blotting. MBP is used as a marker for soluble periplasmic proteins. CpxR is used as a marker of soluble cytoplasmic proteins.
In the second approach, we determined whether β-galactosidase activity could be released along with other soluble periplasmic proteins during spheroplast formation. Because the MalF moiety tethers the MalF-LacZ102 hybrid protein to the IM, only the H*lamB-lacZ fusion was used in this experiment. As expected, we found that cells expressing the Δ60lamB-lacZ fusion released less than 1% of whole-cell β-galactosidase activity upon spheroplast formation (Fig. 3B). Conversely, cells expressing the H*lamB-lacZ fusion released 52% of whole-cell β-galactosidase activity upon spheroplast formation (Fig. 3B). To ensure that release of β-galactosidase activity into the soluble fraction was not due to cell lysis, we attempted to detect cytoplasmic protein in spheroplast supernatants by Western blotting. While we identified the cytoplasmic protein CpxR in whole-cell samples, it was absent from spheroplast supernatants, indicating that the IM was still intact and that cytoplasmic proteins were not released by spheroplasting (Fig. 3C). Why roughly half of whole-cell β-galactosidase activity is retained in spheroplasts is unclear, although other studies have reported partial release of periplasmic proteins upon spheroplast formation (22). It is possible that a fraction of the hybrid protein is tightly associated with the IM or that translocation efficiency is reduced in the absence of DsbA. In any case, it is clear that in the absence of DsbA strains expressing the H*lamB-lacZ fusion harbor folded, active LacZ in the periplasm.
FkpA contributes to LacZ folding.
We thought it likely that some periplasmic factor must be assisting LacZ folding. Because previous studies have implicated the periplasmic chaperones skp and fkpA in the folding of heterologous proteins (23–29), we chose to see what effect disruption of these genes might have on the β-galactosidase activity of a strain expressing the malF-lacZ102 fusion in the absence of DsbA. While disruption of skp did not decrease β-galactosidase activity, we found that disruption of fkpA reduced it by a factor of 2.6 (Fig. 4). This result firmly supports the conclusion that the LacZ moiety in nonjamming targeted fusions is fully translocated into the periplasm, where it can fold in the absence of DsbA.
FIG 4.
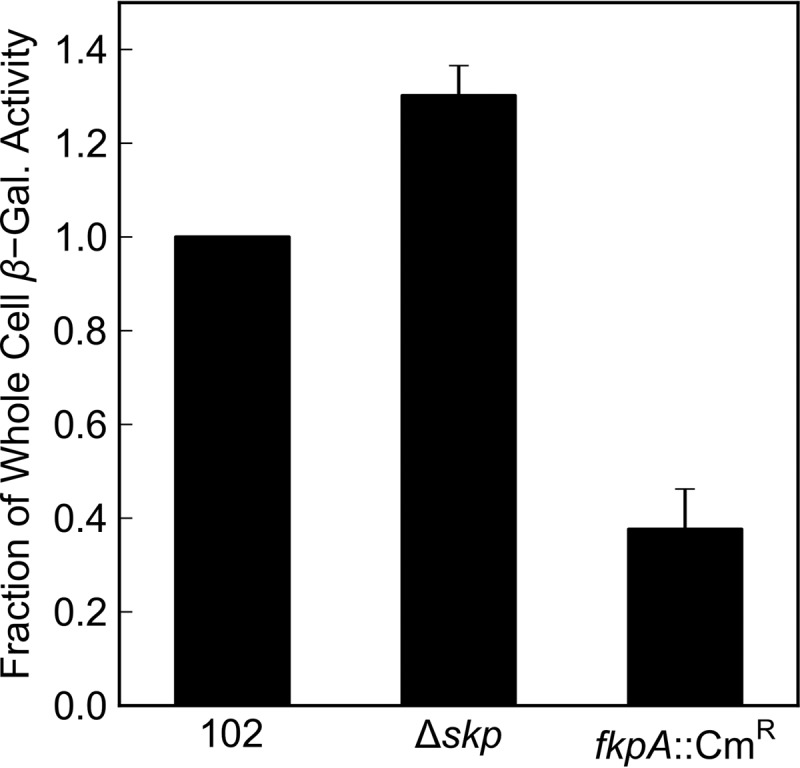
Effect of periplasmic chaperones on periplasmic β-galactosidase activity. Overnight cultures were washed and subcultured 1:50 into liquid LB cultures. Cultures were incubated with aeration at 30°C for 2 to 3 h until reaching mid-log phase. β-Galactosidase activity was determined from Vmax of whole-cell lysates in a kinetic Miller assay. β-Galactosidase activity is normalized to RSD121. Error bars show standard deviations calculated from three independent biological replicates. 102 indicates RSD121. Δskp indicates RSD126. fkpA::Cmr indicates RSD127.
DISCUSSION
Three lines of evidence support the conclusion that LacZ is fully translocated into the periplasm in targeted, nonjamming fusions. These three lines of evidence correspond to three periplasmic proteins, each of which alters the phenotypes conferred by the targeted, nonjamming fusions. First, we showed that malF-lacZ102 confers maltose sensitivity like the H*lamB-lacZ and lamB-lacZX90 fusions and that the periplasmic chaperone/protease DegP can relieve this sensitivity. As DegP is itself localized to the periplasm and is a key component of the Cpx and σE stress responses in this compartment (30–32), we conclude that in all cases the toxicity conferred by targeted, nonjamming fusions is periplasmic and that LacZ is most likely fully translocated by extension.
The second line of evidence supporting full translocation concerns the periplasmic disulfide bond catalyst DsbA, which we found promotes the misfolding and toxicity of the H*LamB-LacZ and MalF-LacZ102 hybrid proteins. In the absence of DsbA, the LacZ moiety in these proteins can fold and become active in the periplasm, reducing its toxicity. Of course, disruption of dsbA has no phenotypic effect on the lamB-lacZX90 fusion, because the X90 mutation prevents LacZ from folding in any compartment. This led us to conclude that it is misfolded LacZ in particular that is toxic in the periplasm. This hypothesis was confirmed when we found that a derivative of the H*lamB-lacZ fusion encoding only the signal sequence and first 5 amino acids of LamB fused to LacZ was just as toxic as the full-length fusion. Interestingly, it has been reported that the signal sequence alone may not be sufficient for targeting and that more than 27 amino acids of mature LamB are needed in addition to the signal sequence to promote SecB-dependent export of the LamB-LacZ hybrid protein (33, 34). It has also been suggested that the signal sequence may not play a role in targeting proteins to the Sec translocon per se (35). Our findings suggest that a signal sequence is both necessary and sufficient for targeting via the SRP pathway.
We believe that the H* signal sequence may present an ideal method for targeting exogenous proteins for folding in the oxidizing environment of the E. coli periplasm. While SecB-dependent targeting sequences like those derived from MBP are sufficient to target hybrid protein to the translocon, they risk lethal jamming. The H* signal sequence avoids jamming by directing hybrid protein to the cotranslational SRP pathway, which should allow for greater induction and higher yield of exogenous proteins. Also, the fact that the signal sequence alone seems to be sufficient for targeting means that little if any targeting sequence need remain after signal sequence cleavage.
The third line of evidence supporting full translocation concerns the effect of FkpA on LacZ folding. FkpA is one of four known peptidyl-prolyl cis-trans isomerases in the periplasm (36). It functions as a chaperone in vivo, promoting assembly of OMPs. FkpA function overlaps Skp function at least in part, and both seem to be involved in assembly of the essential OMP LptD (37), which is in turn responsible for inserting lipopolysaccharide (LPS) into the OM of Gram-negative bacteria like E. coli (38, 39). In the present study, we found that disruption of fkpA leads to a 2- to 3-fold reduction in whole-cell β-galactosidase activity in cells expressing the malF-lacZ102 fusion in the absence of DsbA. This finding is consistent with previous studies, which have implicated FkpA in the folding of exogenous proteins in the periplasm, and like our findings with DegP and DsbA, it confirms that LacZ is fully translocated into the periplasm. Whether the peptidyl-prolyl isomerase (PPIase) activity of FkpA contributes to LacZ folding remains unclear, although LacZ does contain proline residues. We note that FkpA has previously been shown to prevent protein aggregation in the periplasm independent of its PPIase function (40).
The fact that a periplasmic factor contributes to LacZ folding is not surprising given the obstacles that the periplasm presents to folding. LacZ is much larger than most periplasmic proteins, and it must tetramerize to form active enzyme (41). In the cytoplasm, folding is driven by energy-dependent folding factors like GroEL and DnaJ/K; however, there are no homologs of these generalized folding factors in the periplasm, nor are there nucleotide triphosphates in the periplasm to drive energy-dependent folding (42). As previously discussed, the periplasm is also an oxidizing environment, and this is true even in the absence of DsbA as evidenced by the fact that dsbA is not essential under aerobic growth conditions. Given that dsbA disruption only partially relieves the Mals phenotype of the H*lamB-lacZ and malF-lacZ102 fusions, we think it likely that some significant fraction of translocated LacZ is still misfolding even in the absence of DsbA. LacZ must have at least two fates in the periplasm then, folded or misfolded, with FkpA and DsbA, respectively, contributing to these fates.
We believe that this newly discovered periplasmic β-galactosidase activity will be extremely useful in designing screens for periplasmic folding factors. It is likely that additional factors contribute to LacZ folding, since the malF-lacZ102 fusion still confers β-galactosidase activity in the absence of FkpA. Screening for folding factors in an fkpA null background will help overcome the functional redundancy of periplasmic chaperones. Identifying such periplasmic folding factors and efficiently targeting recombinant proteins to the periplasm are keys to the effective expression of exogenous proteins requiring disulfide bonds in E. coli.
ACKNOWLEDGMENTS
This work was supported by National Institute of General Medical Sciences grant GM34821 awarded to T.J.S. J.B. and D.B. were supported by National Institute of General Medical Sciences grant GMO41883.
Footnotes
Published ahead of print 7 July 2014
REFERENCES
- 1.Silhavy TJ, Shuman H, Beckwith J, Schwartz M. 1977. Use of gene fusions to study outer membrane protein localization in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 74:5411–5415. 10.1073/pnas.74.12.5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bassford PJ, Jr, Silhavy TJ, Beckwith JR. 1979. Use of gene fusion to study secretion of maltose-binding protein into Escherichia coli periplasm. J. Bacteriol. 139:19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Stelten J, Silva F, Belin D, Silhavy TJ. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753–756. 10.1126/science.1172221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emr SD, Schwartz M, Silhavy TJ. 1978. Mutations altering the cellular localization of the phage lambda receptor, an Escherichia coli outer membrane protein. Proc. Natl. Acad. Sci. U. S. A. 75:5802–5806. 10.1073/pnas.75.12.5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emr SD, Silhavy TJ. 1980. Mutations affecting localization of an Escherichia coli outer membrane protein, the bacteriophage lambda receptor. J. Mol. Biol. 141:63–90. 10.1016/S0022-2836(80)80029-5 [DOI] [PubMed] [Google Scholar]
- 6.Kumamoto CA, Beckwith J. 1983. Mutations in a new gene, secB, cause defective protein localization in Escherichia coli. J. Bacteriol. 154:253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emr SD, Hanley-Way S, Silhavy TJ. 1981. Suppressor mutations that restore export of a protein with a defective signal sequence. Cell 23:79–88. 10.1016/0092-8674(81)90272-5 [DOI] [PubMed] [Google Scholar]
- 8.Oliver DB, Beckwith J. 1981. E. coli mutant pleiotropically defective in the export of secreted proteins. Cell 25:765–772. 10.1016/0092-8674(81)90184-7 [DOI] [PubMed] [Google Scholar]
- 9.Danese PN, Silhavy TJ. 1998. Targeting and assembly of periplasmic and outer-membrane proteins in Escherichia coli. Annu. Rev. Genet. 32:59–94. 10.1146/annurev.genet.32.1.59 [DOI] [PubMed] [Google Scholar]
- 10.Newton WA, Beckwith JR, Zipser D, Brenner S. 1965. Nonsense mutants and polarity in the Lac operon of Escherichia coli. J. Mol. Biol. 14:290–296. 10.1016/S0022-2836(65)80250-9 [DOI] [PubMed] [Google Scholar]
- 11.Snyder WB, Silhavy TJ. 1995. Beta-galactosidase is inactivated by intermolecular disulfide bonds and is toxic when secreted to the periplasm of Escherichia coli. Microbiology 177:953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowers CW, Lau F, Silhavy TJ. 2003. Secretion of LamB-LacZ by the signal recognition particle pathway of Escherichia coli. J. Bacteriol. 185:5697–5705. 10.1128/JB.185.19.5697-5705.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall MN, Schwartz M, Silhavy TJ. 1982. Sequence information within the IamB gene is required for proper routing of the bacteriophage lambda receptor protein to the outer membrane of Escherichia coli K-12. J. Mol. Biol. 156:93–112. 10.1016/0022-2836(82)90461-2 [DOI] [PubMed] [Google Scholar]
- 14.Froshauer S, Green GN, Boyd D, McGovern K, Beckwith J. 1988. Genetic analysis of the membrane insertion and topology of MalF, a cytoplasmic membrane protein of Escherichia coli. J. Mol. Biol. 200:501–511. 10.1016/0022-2836(88)90539-6 [DOI] [PubMed] [Google Scholar]
- 15.Bardwell JC, McGovern K, Beckwith J. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589. 10.1016/0092-8674(91)90532-4 [DOI] [PubMed] [Google Scholar]
- 16.Beckwith J. 2013. Fifty years fused to lac. Annu. Rev. Microbiol. 67:1–19. 10.1146/annurev-micro-092412-155732 [DOI] [PubMed] [Google Scholar]
- 17.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 18.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansson MD, Rzeznicka K, Rosenbäck M, Hansson M, Sirijovski N. 2008. PCR-mediated deletion of plasmid DNA. Anal. Biochem. 375:373–375. 10.1016/j.ab.2007.12.005 [DOI] [PubMed] [Google Scholar]
- 20.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 21.Schäfer U, Beck K, Müller M. 1999. Skp, a molecular chaperone of gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J. Biol. Chem. 274:24567–24574. 10.1074/jbc.274.35.24567 [DOI] [PubMed] [Google Scholar]
- 22.Heppel LA. 1967. Selective release of enzymes from bacteria. Science 156:1451–1455. 10.1126/science.156.3781.1451 [DOI] [PubMed] [Google Scholar]
- 23.Wang R, Xiang S, Feng Y, Srinivas S, Zhang Y, Lin M, Wang S. 2013. Engineering production of functional scFv antibody in E. coli by co-expressing the molecule chaperone Skp. Front. Cell. Infect. Microbiol. 3:72. 10.3389/fcimb.2013.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonoda H, Kumada Y, Katsuda T, Yamaji H. 2011. Effects of cytoplasmic and periplasmic chaperones on secretory production of single-chain Fv antibody in Escherichia coli. J. Biosci. Bioeng. 111:465–470. 10.1016/j.jbiosc.2010.12.015 [DOI] [PubMed] [Google Scholar]
- 25.Gunnarsen KS, Kristinsson SG, Justesen S, Frigstad T, Buus S, Bogen B, Sandlie I, Løset GÅ 2013. Chaperone-assisted thermostability engineering of a soluble T cell receptor using phage display. Sci. Rep. 3:1162. 10.1038/srep01162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levy R, Ahluwalia K, Bohmann DJ, Giang HM, Schwimmer LJ, Issafras H, Reddy NB, Chan C, Horwitz AH, Takeuchi T. 2013. Enhancement of antibody fragment secretion into the Escherichia coli periplasm by co-expression with the peptidyl prolyl isomerase, FkpA, in the cytoplasm. J. Immunol. Methods 394:10–21. 10.1016/j.jim.2013.04.010 [DOI] [PubMed] [Google Scholar]
- 27.O'Reilly AO, Cole AR, Lopes JLS, Lampert A, Wallace BA. 2014. Chaperone-mediated native folding of a β-scorpion toxin in the periplasm of Escherichia coli. Biochim. Biophys. Acta 1840:10–15. 10.1016/j.bbagen.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bothmann H, Pluckthun A. 2000. The periplasmic Escherichia coli peptidylprolyl cis,trans-isomerase FkpA. I. Increased functional expression of antibody fragments with and without cis-prolines. J. Biol. Chem. 275:17100–17105. 10.1074/jbc.M910233199 [DOI] [PubMed] [Google Scholar]
- 29.Missiakas D, Betton J-M, Raina S. 1996. New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol. Microbiol. 21:871–884. 10.1046/j.1365-2958.1996.561412.x [DOI] [PubMed] [Google Scholar]
- 30.Danese PN, Silhavy TJ. 1997. The sigma(E) and the Cpx signal transduction systems control the synthesis of periplasmic protein-folding enzymes in Escherichia coli. Genes Dev. 11:1183–1193. 10.1101/gad.11.9.1183 [DOI] [PubMed] [Google Scholar]
- 31.Snyder WB, Davis LJ, Danese PN, Cosma CL, Silhavy TJ. 1995. Overproduction of NlpE, a new outer membrane lipoprotein, suppresses the toxicity of periplasmic LacZ by activation of the Cpx signal transduction pathway. J. Bacteriol. 177:4216–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danese PN, Snyder WB, Cosma CL, Davis LJ, Silhavy TJ. 1995. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev. 9:387–398. 10.1101/gad.9.4.387 [DOI] [PubMed] [Google Scholar]
- 33.Moreno F, Fowler AV, Hall M, Silhavy TJ, Zabin I, Schwartz M. 1980. A signal sequence is not sufficient to lead beta-galactosidase out of the cytoplasm. Nature 286:356–359. 10.1038/286356a0 [DOI] [PubMed] [Google Scholar]
- 34.Benson SA, Silhavy TJ. 1983. Information within the mature LamB protein necessary for localization to the outer membrane of E. coli K12. Cell 32:1325–1335. 10.1016/0092-8674(83)90313-6 [DOI] [PubMed] [Google Scholar]
- 35.Gouridis G, Karamanou S, Gelis I, Kalodimos CG, Economou A. 2009. Signal peptides are allosteric activators of the protein translocase. Nature 462:363–367. 10.1038/nature08559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Justice SS, Hunstad DA, Harper JR, Duguay AR, Pinkner JS, Bann J, Frieden C, Silhavy TJ, Hultgren SJ. 2005. Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J. Bacteriol. 187:7680–7686. 10.1128/JB.187.22.7680-7686.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwalm J, Mahoney TF, Soltes GR, Silhavy TJ. 2013. Role for Skp in LptD assembly in Escherichia coli. J. Bacteriol. 195:3734–3742. 10.1128/JB.00431-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu T, McCandlish AC, Gronenberg LS, Chng S-S, Silhavy TJ, Kahne D. 2006. Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 103:11754–11759. 10.1073/pnas.0604744103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chng S-S, Ruiz N, Chimalakonda G, Silhavy TJ, Kahne D. 2010. Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc. Natl. Acad. Sci. U. S. A. 107:5363–5368. 10.1073/pnas.0912872107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arié JP, Sassoon N, Betton JM. 2001. Chaperone function of FkpA, a heat shock prolyl isomerase, in the periplasm of Escherichia coli. Mol. Microbiol. 39:199–210. 10.1046/j.1365-2958.2001.02250.x [DOI] [PubMed] [Google Scholar]
- 41.Jacobson RH, Zhang XJ, DuBose RF, Matthews BW. 1994. Three-dimensional structure of beta-galactosidase from E. coli. Nature 369:761–766. 10.1038/369761a0 [DOI] [PubMed] [Google Scholar]
- 42.Gething MJ, Sambrook J. 1992. Protein folding in the cell. Nature 355:33–45. 10.1038/355033a0 [DOI] [PubMed] [Google Scholar]
- 43.Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J. Mol. Biol. 104:541–555. 10.1016/0022-2836(76)90119-4 [DOI] [PubMed] [Google Scholar]
- 44.Ruiz N, Gronenberg LS, Kahne D, Silhavy TJ. 2008. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 105:5537–5542. 10.1073/pnas.0801196105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cosma CL, Crotwell MD, Burrows SY, Silhavy TJ. 1998. Folding-based suppression of extracytoplasmic toxicity conferred by processing-defective LamB. J. Bacteriol. 180:3120–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]