

Abstract
In Gram-negative bacteria, lipopolysaccharide (LPS) contributes to the robust permeability barrier of the outer membrane, preventing entry of toxic molecules such as antibiotics. Mutations in lptD, the beta-barrel component of the LPS transport and assembly machinery, compromise LPS assembly and result in increased antibiotic sensitivity. Here, we report rare vancomycin-resistant suppressors that improve barrier function of a subset of lptD mutations. We find that all seven suppressors analyzed mapped to the essential gene cdsA, which is responsible for the conversion of phosphatidic acid to CDP-diacylglycerol in phospholipid biosynthesis. These cdsA mutations cause a partial loss of function and, as expected, accumulate phosphatidic acid. We show that this suppression is not confined to mutations that cause defects in outer membrane biogenesis but rather that these cdsA mutations confer a general increase in vancomycin resistance, even in a wild-type cell. We use genetics and quadrupole time of flight (Q-TOF) liquid chromatography-mass spectrometry (LC-MS) to show that accumulation of phosphatidic acid by means other than cdsA mutations also increases resistance to vancomycin. We suggest that increased levels of phosphatidic acid change the physical properties of the outer membrane to impede entry of vancomycin into the periplasm, hindering access to its target, an intermediate required for the synthesis of the peptidoglycan cell wall.
INTRODUCTION
The outer membrane (OM) of Gram-negative bacteria serves as a robust permeability barrier (1). Composed of lipopolysaccharide (LPS) molecules in the outer leaflet and glycerophospholipids (GPLs) in the inner leaflet, the OM protects the cell from harmful agents, such as detergents and antibiotics (1–3). LPS is synthesized at the inner membrane (IM), transported across the aqueous periplasm, and finally assembled into the outer leaflet of the OM (4). The Lpt proteins are responsible for this transport and assembly (5). Glycerophospholipids (GPLs) are synthesized at the IM, but little is known about their transport across the periplasm and assembly into the OM (6). The coordination of LPS and GPL assembly is important to maintain an effective OM permeability barrier (1).
Because of the high density of LPS in the outer leaflet, the OM carries a net negative charge (7). The human innate immune system takes advantage of this property of the OM by producing cationic antimicrobial peptides (CAMPs) (8). Positively charged CAMPs associate with the negatively charged cell surface and insert into the OM, killing the bacterial cell (8). Bacteria have systems in place, such as the PhoPQ virulence system in Salmonella enterica serovar Typhimurium, that modify lipid A and the PL content to decrease the negative charge of the OM, thereby decreasing the affinity of CAMPs for the OM (9–11). This increases bacterial resistance to CAMPs and enables evasion of the immune system.
Like LPS, GPLs can contribute to the electric charge of the membrane. Escherichia coli membranes are composed of three major GPLs: phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and cardiolipin (CL). The relative abundances of each of these GPL species are similar in the IM and OM (PE, 75%; PG, 20%; CL, 5%) (12). PE is a zwitterionic GPL with a neutral charge, while PG and CL are acidic GPLs that carry negative charges (6). Although these GPLs have different chemical properties due to their head groups, they are synthesized from the same precursor molecule, phosphatidic acid (PA) (6, 12). The conversion of PA to CDP-diacylglycerol is the last step before commitment to either the zwitterionic GPL pathway or the acidic GPL pathway (6, 12).
In this work, we focus on E. coli mutants defective in LPS assembly that exhibit increased sensitivity to antibiotics. Mutations that compromise the function of LptD, the beta-barrel component of the Lpt machinery, are often sensitive to vancomycin. In order to act on its cell wall target, vancomycin must traverse the OM. Once in the periplasm, vancomycin binds to the two terminal d-alanine residues on the cell wall precursor lipid II, thus preventing polymerization of the peptidoglycan cell wall (13, 14). lptD mutants exhibit increased sensitivity to vancomycin because they allow for more efficient entry of the drug into the periplasm as a result of a compromised OM barrier.
We report here the isolation and characterization of vancomycin-resistant suppressors of a subset of lptD mutations. These suppressors map to the essential gene cdsA. Vancomycin-resistant suppression is not specific to the lptD mutants; the cdsA mutations actually increase vancomycin resistance even in an otherwise wild-type cell. These cdsA mutations cause a recessive, partial loss of function, and they accumulate PA like other cdsA mutants previously described (15, 16). We suggest that increased PA alters the OM and causes increased resistance to vancomycin.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and chemicals.
All strains used in the work are isogenic derivatives of NR754, an araD+ revertant of MC4100 (17, 18). Strains were constructed by generalized P1 transduction or transformation (19). Suppressor mutations were mapped using pools of random Tn10 insertions as described previously (20). If necessary, kan cassettes were excised using the Flp recombinase (21). Strains and plasmids used in this work are listed in Table 1. All cultures were grown in Luria-Bertani (LB) broth or agar at 37°C. For mass spectrometry (MS), high-pressure liquid chromatography (HPLC)-grade solvents (water, methanol, and isopropanol), acetic acid, and ammonium acetate were obtained from Fisher Scientific (Pittsburgh, PA).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant features | Reference and/or source |
|---|---|---|
| Strains | ||
| MC4100 | F− araD139 Δ(argF-lac)U169 rpsL150 relA1 flbB5301 deoC1 ptsF25 rbsR thi | 17 |
| NR754 | MC4100 Ara+ | 18 |
| HC334 | NR754 ΔlptD pET2342::lptDI54E | 21, 48, 49; this study |
| HC338 | NR754 ΔlptD pET2342::lptDV52E-I54E | 21, 48, 49; this study |
| HC340 | NR754 ΔlptD pET2342::lptD | 21, 48, 49; this study |
| HC342 | NR754 cdsA7 dgt::Tn10 | This study |
| HC351 | HC334 ΔclsA::kan | 34; this study |
| HC386 | HC334 cdsA7 dgt::Tn10 | This study |
| HC509 | NR754 ΔlptD pET2342::lptDΔD57ΔK60 | 21, 48, 49; this study |
| HC510 | HC334 pgsA444 zed-3069::Tn10 | 39, 50, 51; this study |
| HC530 | HC509 cdsA7 dgt::Tn10 | This study |
| HC571 | NR754 ΔclsA::kan | 34; this study |
| HC575 | NR754 pgsA444 zed-3069::Tn10 | 39, 50, 51; this study |
| HC630 | HC509 pgsA444 zed-3069::Tn10 | 39, 50, 51; this study |
| HC960 | HC334 cdsA9 dgt::Tn10 | This study |
| HC961 | HC509 cdsA9 dgt::Tn10 | This study |
| HC962 | NR754 cdsA9 dgt::Tn10 | This study |
| HC1045 | NR754 pASKA::plsC | 35; this study |
| HC1198 | JAS193 cdsA7 dgt::Tn10 | This study |
| HC1199 | JAS193 cdsA9 dgt::Tn10 | This study |
| HC1240 | HC342 pASKA::cdsA | 35; this study |
| HC1241 | HC962 pASKA::cdsA | 35; this study |
| JAS193 | MC4100 ΔbamE::kan | J. Schwalm, unpublished data |
| Plasmids | ||
| pET2342::lptD | lptD cloned into pET2342 | 48 |
| pASKA::cdsA | IPTG inducible, Camr | 35 |
| pASKA::plsC | IPTG inducible, Camr | 35 |
Antibiotic sensitivity assays.
Efficiency of plating assays (EOPs) or disk diffusion assays were used to determine antibiotic sensitivity. For EOPs, strains were grown overnight, serially diluted, and then replica plated on LB agar or LB agar supplemented with vancomycin (70 mg/liter, 120 mg/liter, 140 mg/liter, 150 mg/liter, or 200 mg/liter), 625 mg/liter bacitracin, 1 mg/liter gentamicin, or 10 mg/liter rifampin. For disk diffusion assays, the disks contained the following amounts of antibiotics: 1.25 mg ampicillin, 320 μg cetyltrimethylammonium bromide, 125 μg erythromycin, 30 μg moxalactam, 0.1 μg norfloxacin, 260 μg paraquat, 300 units polymyxin B, or 1 mg vancomycin. Isopropyl-β-d-1-thiogalactopyranoside (IPTG; 0.1 mM) was present in the LB agar if indicated.
Growth rate determination.
Overnight cultures were diluted 1:100 in 100 μl of LB medium in 96-well plates. One hundred microliters of mineral oil was overlaid to prevent evaporation. Optical density at 600 nm (OD600) was measured every 20 min with intermittent orbital shaking at 37°C. These 4-hour OD600 growth curves were used to determine the growth rate. OD600 was plotted against time on a logarithmic scale, and linear regression analysis was used to determine the growth rate of each culture. For each strain, three technical replicates of three biological replicates were averaged and the mean of the averages was calculated and reported with standard error of the mean.
Cell size measurements.
Overnight cultures were diluted 1:100 and grown to an OD600 of approximately 0.4. Cells were suspended on a 1% agarose pad and imaged using a 100× 1.4-numerical-aperture (NA) objective on a Nikon 90i microscope equipped with a QImaging Rolera XR camera and NIS Elements software. Cell dimensions were quantified with MicrobeTracker version 0.936 (22), using MATLAB version R2013a. Approximately 100 cells were examined for each of three independent experiments. P values were calculated using Student's t test.
Bligh Dyer lipid extraction.
Lipids were extracted using an acidic Bligh Dyer method as previously described (23, 24). One-hundred-milliliter cultures were grown to an OD600 of 1.5, and then the cells were harvested by centrifugation. Cell pellets were washed once with phosphate-buffered saline (PBS) and then resuspended in 1 ml of 0.1 N HCl. Methanol (2.5 ml) and chloroform (1.25 ml) were added to form a single-phase solution (chloroform-methanol-0.1 N HCl [1:2:0.8 {vol/vol/vol}]) and incubated for 30 min at room temperature with sporadic vortexing. Then, 0.1 N HCl (1.25 ml) and chloroform (1.25 ml) were added to form a two-phase solution (chloroform-methanol-0.1 N HCl [2:2:1.8 {vol/vol/vol}]). These solutions were then centrifuged at 3,000 × g for 25 min at room temperature. The lower phase was recovered and dried under nitrogen stream.
Mass spectrometry.
Samples were reconstituted into 500 μl 1:1:0.3 methanol-chloroform-water. Mass spectrometric analyses were performed on a 6550 Accurate-Mass quadrupole time of flight (Q-TOF) mass spectrometer (Agilent Technologies, Inc., Santa Clara, CA), equipped with an electrospray ionization (ESI) source operated in both negative- and positive-ion mode. Liquid chromatography (LC) was performed using a 1290 Infinity HPLC system (Agilent Technologies, Inc., Santa Clara, CA) coupled to the mass spectrometer, which scans from m/z 200 to 1,000 at 2-GHz extended dynamic range. Nitrogen was used as drying gas, nebulizer gas, and collision gas. Mass spectrometer operation conditions were as follows: capillary voltage, 4,000 V; fragmentor voltage, 225 V; nebulizer gas, 35 lb/in2; drying gas, 13 liter/min; gas temperature, 275°C. The instrument control, data acquisition, and data analysis were performed by the Agilent Mass Hunter software (Agilent Technologies; version B.05.00), which also controlled the chromatography system.
The LC parameters were as follows: column, Agilent Poroshell-120 EC-C18 (2.7 μm, 2.1 by 150 mm); autosampler temperature, 5°C; injection volume, 10 μl; column temperature, 60°C; and solvent flow rate, 150 μl/min. Solvent A is 1 mM ammonium acetate plus 0.2% acetic acid in 90:10 water-methanol, and solvent B is 1 mM ammonium acetate plus 0.2% acetic acid in 2:98 methanol–2-propanol. The gradient was 0 min, 25% B; 2 min, 25% B; 4 min, 65% B; 16 min, 100% B; 20 min, 100% B; 21 min, 25% B; 22 min, 25% B. Postrun time was 6 min.
RESULTS
Vancomycin-resistant suppressors of lptD mutations map to cdsA.
Escherichia coli lpt mutants that exhibit defects in transport and assembly of LPS often exhibit sensitivity to detergents and antibiotics (18, 25–31). lptDI54E, lptDV52E-I54E, and lptDΔD57ΔK60 harbor the denoted mutations that alter the soluble N terminus of the OM beta-barrel protein LptD and confer sensitivity to vancomycin to differing degrees. Spontaneously arising vancomycin-resistant suppressors were isolated on LB medium supplemented with 120 mg/liter vancomycin.
With strains carrying each of these mutations, survivors arose at a frequency of 10−8, and all suppressors analyzed mapped to an essential gene, cdsA. CdsA is an inner membrane protein responsible for the conversion of PA to CDP-diacylglycerol in GPL biosynthesis (6, 15, 32). Figure 1A shows the location of the different cdsA mutations on a predicted CdsA structure (33). Most mutations map to the transmembrane helices, and some may affect protein stability in the membrane. Amino acid changes I203S (cdsA7) and R263L (cdsA9) were chosen as representative examples for this study because they are located in different parts of the protein, and unlike some of the other cdsA suppressors isolated, they do not exhibit any growth defects.
FIG 1.
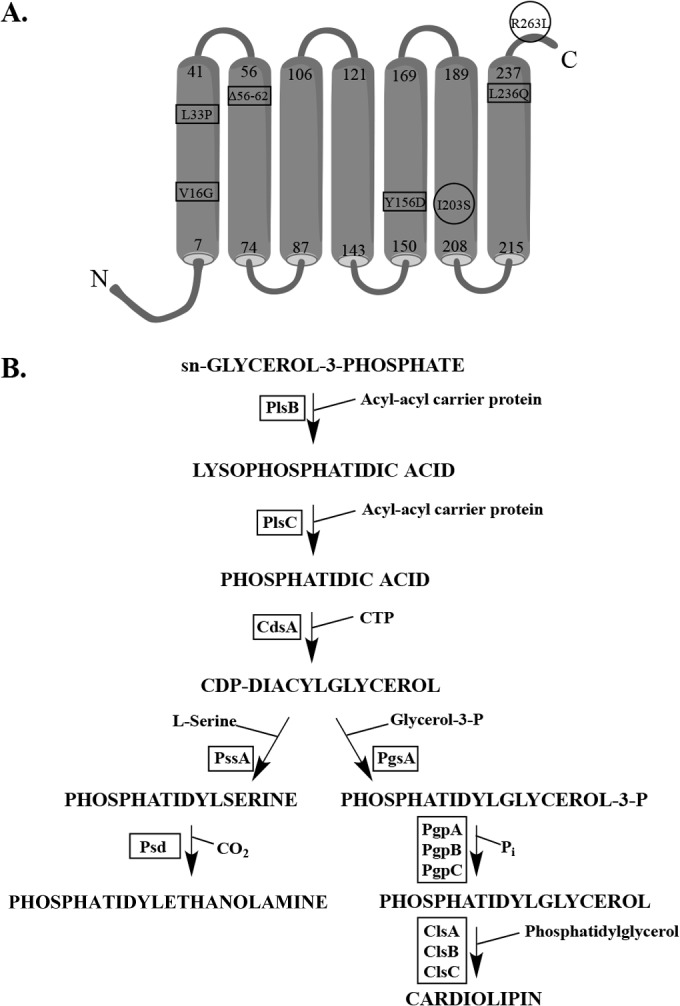
Vancomycin-resistant lptD suppressors map to cdsA, the gene responsible for the conversion of phosphatidic acid to CDP-diacylglycerol. (A) The inner membrane CdsA protein is predicted to have seven transmembrane domains (33). Vancomycin-resistant cdsA suppressors mapped mainly within these domains. Mutations V16G, L33P, Δ56–62, Y156D, and L236Q were isolated in an lptDV52E-I54E background. I203S was isolated in an lptDI54E background, and R263L was isolated in an lptDΔD57ΔK60 background. The mutations enclosed in circles, I203S (cdsA7) and R263L (cdsA9), are characterized further in this study. (B) Biosynthetic pathway for major phospholipids in Escherichia coli. Protein names are in boxes, and they are next to the arrow that signifies the step at which they act.
Because cdsA is an essential gene (16, 34), the suppressors cannot be null mutations. We used complementation analysis to determine whether the cdsA7 and cdsA9 mutations were dominant or recessive. The cdsA clone from the “A Complete Set of E. coli K-12 ORF Archive” (ASKA) collection was introduced into the cdsA7 and cdsA9 backgrounds (35). Basal-level (uninduced) expression of a wild-type copy of cdsA on the ASKA plasmid decreased vancomycin resistance back to wild-type levels (see Fig. S1 in the supplemental material). Because cdsA7 and cdsA9 are recessive, we conclude that both are partial loss-of-function mutations.
The cdsA mutations do not alter cell size.
Previously, it has been shown that mutations that confer LPS transport defects can be suppressed by mutations that alter fatty acid biosynthesis (36), and we wanted to determine if our suppressors acted in a similar manner. fabH and accD mutations can suppress certain lpt mutations by reducing cell size and growth rate, which in turn decrease the rate of growth of the cell envelope (36). The cdsA7 and cdsA9 mutants did not exhibit a decrease in cell volume (see Fig. S2 in the supplemental material), and their growth rates did not differ significantly from that of the wild type (see Table S1). We conclude from these results that these cdsA mutations do not suppress lptD mutations by decreasing the rate of growth of the cell envelope.
Partial loss-of-function cdsA mutants generally increase vancomycin resistance of E. coli strains.
Because these cdsA suppressors were isolated in different lptD mutant backgrounds, we asked whether this mode of suppression was specific to each lptD mutant. For simplicity, we chose to analyze two of the cdsA mutants (cdsA7 and cdsA9) in two of the lptD mutant backgrounds (lptDI54E and lptDΔD57ΔK60). cdsA7 and cdsA9 are both able to cross-suppress lptDI54E and lptDΔD57ΔK60, regardless of which lptD background they were isolated in (Fig. 2). This demonstrates that these cdsA suppressors are not allele specific but rather increase vancomycin resistance in a more generalized manner.
FIG 2.

Partial loss-of-function cdsA mutants and pgsA444 suppress the vancomycin sensitivity of lptD mutants. (A) Efficiency of plating assay (EOP) on LB agar plates supplemented with 70 mg/liter, 140 mg/liter, or 150 mg/liter vancomycin and grown at 37°C overnight. Suppression is shown for lptDI54E. Note that the chromosomal lptD allele is ΔlptD, while the lptD allele listed for each EOP is present in pET2342::lptD, which has a leaky T7 promoter. T7 RNA polymerase is not present in these strains. (B) EOPs as described for panel A. Suppression is shown for lptDΔD57ΔK60.
Mutants in the beta-barrel assembly machinery (Bam) increase sensitivity to vancomycin due to impaired assembly of OM proteins (37, 38). The ΔbamE::kan allele is a null mutation that removes BamE, one of the lipoproteins in the Bam complex. Strains carrying the ΔbamE::kan allele exhibit mild OM protein assembly defects and display increased sensitivity to vancomycin (37). When cdsA7 and cdsA9 were introduced into a ΔbamE::kan strain, vancomycin resistance increased (Fig. 3A). This suggests that cdsA7 and cdsA9 increase vancomycin resistance in a general manner in any strain with a compromised OM.
FIG 3.
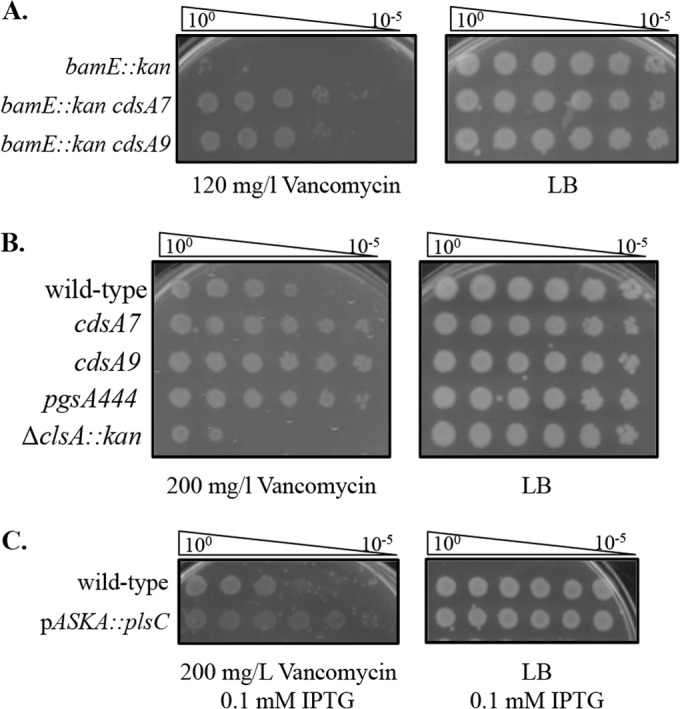
cdsA mutants, pgsA444, and plsC overexpression increase resistance to vancomycin. (A) Partial loss-of-function cdsA mutants suppress the vancomycin sensitivity of other cell envelope mutants. EOPs of strains supplemented with 120 mg/liter vancomycin. (B) Partial loss-of-function cdsA mutants and pgsA444 increase vancomycin resistance in wild-type cells. EOPs of strains supplemented with 200 mg/liter vancomycin. (C) Overproduction of PlsC increases vancomycin resistance in wild-type cells. EOPs of strains supplemented with 200 mg/liter vancomycin and 0.1 mM IPTG.
To test whether cdsA7 and cdsA9 increase resistance to vancomycin in wild-type strains, we evaluated these mutants in an otherwise wild-type background. On LB plates supplemented with 200 mg/liter vancomycin, cdsA7 and cdsA9 mutants were able to survive better than the wild type (Fig. 3B). This suggests that the cdsA7 and cdsA9 strains are altered in a way that protects the cell from vancomycin. Previously isolated cdsA mutants showed increased resistance to erythromycin in the strain background tested, but the effect of the mutations on sensitivity to other antibiotics was not reported (16). Using disk diffusion assays, we have checked the cdsA mutants against a battery of other antibiotics. We see a very slight increase in resistance to erythromycin but not to any other drug tested (norfloxacin, paraquat, cetyltrimethylammonium bromide, bacitracin, and polymyxin B). Strikingly, however, we do see that the cdsA mutations cause a clear increase in sensitivity to ampicillin, moxalactam, gentamicin, and rifampin.
Changes in phospholipid content confer vancomycin resistance.
The enzymes and intermediates involved in GPL biosynthesis were identified in the lab of Eugene Kennedy (Fig. 1B). Strains harboring partial loss-of-function mutations in cdsA have been shown to accumulate PA (15, 16). We would also expect a concomitant decrease in CDP-diacylglycerol levels in cdsA mutants, which would result in a decrease in acidic GPL biosynthesis, since CDP-diacylglycerol has a higher affinity for PssA than for PgsA (15, 16).
We hypothesized that a change in the phospholipid content in the OM could result in this increased resistance to vancomycin, and we wondered whether it was due to the accumulation of PA or the decrease in acidic GPLs. To distinguish between these two possibilities, we introduced pgsA444, a partially inactivating mutation in the phosphatidylglycerophosphate synthetase (39), as well as ΔclsA::kan from the Keio collection (34) into our lptD mutant strains (note that we were unable to construct an lptDΔD57ΔK60 ΔclsA::kan double mutant, so these data are not shown). ClsA is responsible for a significant portion of CL produced in the cell, and cls mutants have been shown to have lower levels of CL (24, 40). We found that pgsA444 partially suppresses the vancomycin sensitivity of the lptD mutants, while ΔclsA::kan does not (Fig. 2). This suggests that the increase in negatively charged PA species in the membrane and not a decrease in acidic GPLs was responsible for the increase in vancomycin resistance. It is likely that pgsA444 only partially suppresses the vancomycin sensitivity of these lptD mutants because it does not accumulate as much PA as the cdsA mutants. We also discovered that pgsA444 confers increased vancomycin resistance in an otherwise wild-type background (Fig. 3B). The ΔclsA::kan allele does not confer this increased resistance and in fact renders cells more sensitive to vancomycin.
cdsA mutants accumulate PA.
To confirm our genetic results, we investigated the GPL content of strains harboring cdsA7, cdsA9, and the pgsA444 mutations. GPLs were extracted from whole cells using an acidic Bligh Dyer method (23, 24). Lipids were then separated and quantified by Agilent quadrupole time of flight (Q-TOF) liquid chromatography-mass spectrometry (LC-MS). Distinct PA species were summed, and each sum was normalized to total PS content for each strain. PS levels did not change significantly in all strains tested. We found that PA accumulates in cdsA7, cdsA9, and pgsA444 strains (Fig. 4A). We hypothesize that the cdsA9 mutation compromises CdsA function more severely than does cdsA7 because PA levels are much higher in the cdsA9 mutant. This correlates with the suppression seen with the lptD mutants (Fig. 2); at higher vancomycin concentrations, cdsA9 suppresses vancomycin sensitivity better than does cdsA7, suggesting that greater PA accumulation results in better suppression. The pgsA444 mutant accumulates less PA than either the cdsA7 or cdsA9 mutant does, and this correlates with the fact that pgsA444 does not suppress vancomycin sensitivity as well (Fig. 2). The cdsA7, cdsA9, and pgsA444 mutants all show decreased CL levels compared to the wild type (Fig. 4B), most likely because PA is accumulating at the expense of CL.
FIG 4.

PA accumulates in cdsA mutants and pgsA444 and when plsC is overexpressed. PA was quantified by Q-TOF LC-MS. The average fold differences of all PA species ± standard errors from three experiments are shown (top). The changes of PA/PS are reported ± standard errors from three experiments (bottom). (B) CL levels decrease in cdsA mutants and pgsA444. CL was quantified by Q-TOF LC-MS. The average fold differences of all CL species ± standard errors from three experiments are shown (top). The changes of CL/PS are reported ± standard errors from three experiments (bottom).
Overexpression of plsC increases PA and increases vancomycin resistance.
We have shown that loss-of-function mutations in the phospholipid biosynthesis pathway that cause accumulation of PA increase vancomycin resistance. We wondered whether overproduction of the protein directly upstream of PA synthesis, PlsC, which should cause the accumulation of PA, would also increase vancomycin resistance. We used the plsC ASKA clone to overexpress plsC in an otherwise wild-type background (35). When plsC expression is induced with 0.1 mM IPTG, an increase in vancomycin resistance is observed (Fig. 3C). Q-TOF LC-MS confirms that this level of induction of plsC expression results in an accumulation of PA (Fig. 4A). These results confirm that accumulation of PA increases vancomycin resistance in E. coli.
DISCUSSION
We show here that OM permeability defects associated with lptD mutants can be suppressed by partial loss-of-function mutations in the essential gene, cdsA. The results of this selection for increased vancomycin resistance were striking: suppression was rare and the seven suppressors analyzed altered seven different codons in a single essential gene. These cdsA mutations increase vancomycin resistance in lptD mutants, bam mutants, and even in an otherwise wild-type strain. However, they do not markedly increase resistance to any other antibiotic tested. Thus, these suppressors are not gene or pathway specific; rather, they are drug specific. Using classic genetic methods and Q-TOF LC-MS, we show that increasing PA in three different ways (using the cdsA suppressors, the pgsA444 mutation, and overexpression of plsC) increases vancomycin resistance in E. coli. We conclude that vancomycin resistance increases as a consequence of PA accumulation.
It is difficult to speculate how the accumulation of PA results in increased vancomycin resistance primarily because we do not know how vancomycin enters the periplasm to bind its target, lipid II (13, 14). When the lpt system is compromised, LPS transport and assembly slow, allowing for PLs to flip into the outer leaflet of the OM (20). These PLs self-associate to form rafts of PL bilayer in the OM that allow for diffusion of hydrophobic molecules into the periplasm (41). It is not obvious that a large, hydrophilic molecule like vancomycin would be able to cross the membrane in this way. Instead, it is possible that vancomycin enters the cell through “cracks” in the bilayer at disordered LPS-PL junctions (41). This predicts that an lpt mutant would be more vancomycin sensitive due to the increase in LPS-PL junctions, i.e., “cracks” in the membrane.
Ganong et al. have shown that cdsA mutants accumulate PA in both the IM and the OM (15). While it is true that there is roughly four times as much PA in the IM as in the OM, PA concentrations in the OM are more than 10-fold higher in their cdsA mutant than in the wild type (15). Because vancomycin needs only to traverse the OM to act on its target in the periplasm, it seems unlikely that PA accumulation in the IM is contributing to vancomycin resistance. Rather, we suggest that PA accumulation in the OM impedes efficient entry of vancomycin into the periplasm by reducing the size or altering the properties of the “cracks.” Since PA accumulation also increases vancomycin resistance in an otherwise wild-type cell as well, we must propose the presence of a small amount of PLs in the outer leaflet that produce “cracks” in the OM of wild-type strains.
It is possible that the accumulation of PA in the OM reduces the size of “cracks” by increasing LPS assembly, by increasing the activity of the Lpt system, or by decreasing the amount of PLs in the outer leaflet by increasing the activity of PldA or the Mla transport system (20). However, since PA is really an intermediate in PL biosynthesis, and since it is present normally at low levels, we think that this is unlikely.
We propose that the accumulation of PA increases vancomycin resistance by altering the biophysical properties of the OM. PA lacks a head group and therefore increases membrane curvature, and this could affect the size or properties of the “cracks” (42, 43). In addition to increased membrane curvature, PA increases the negative charge of the OM, and this may increase vancomycin association with the OM (43). While this may seem counterintuitive, other studies have shown that CL microdomains interact with CAMPs and prevent them from interacting with their targets, thereby increasing resistance (44, 45).
Changes in GPL composition have been previously shown to alter resistance and susceptibility to CAMPs and antibiotics (9, 46). In Staphylococcus aureus, fmtC and lysC mutations, which reduce lysyl-PG, increase resistance to moenomycin and vancomycin (46). The increased net negative charge of these mutant membranes may increase interaction with these drugs and prevent them from interacting with their targets. In Salmonella Typhimurium, the PhoPQ system responds to acidic pH and CAMPs by increasing levels of cardiolipin and palmitoylated acyl-PG (9). This increase in these GPLs correlates with an increase in resistance to CAMPs that helps evade the host immune system (9). There is speculation that this resistance may be due to increased membrane curvature as well as CAMP association with CL microdomains, thereby occluding CAMP interactions with their targets (9, 44, 45).
The mode of resistance shown here is inherently different from that regulated by the PhoPQ system. When this system is turned on, palmitoylation of PG increases, which results in a reduction in polarity and an increase in the hydrophobicity and saturation of the membrane (9, 10). In this work, we show an overall increase in PA, regardless of the chain length or saturation state (see Fig. S3 in the supplemental material). As a result of the accumulation of PA, we also show a reduction in CL levels. This also contrasts with the increase in CL levels observed in Salmonella when the PhoPQ system is activated (9). Accumulation of PA is yet another way in which the OM can be modified to increase resistance to antibiotics.
The emergence of multidrug-resistant bacteria is a serious, life-threatening problem (47). Owing to the barrier properties of the OM, this problem is especially acute for Gram-negative pathogens. Many antibiotics that work well for infections caused by Gram-positive bacteria fail when used against Gram-negative bacteria because of the effectiveness of the OM barrier, and we do not know how to modify these agents so that they can reach their targets. A better understanding of the permeability properties of the OM and how they can be modified should help facilitate the design of new antibacterials.
Supplementary Material
ACKNOWLEDGMENTS
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE1148900 (H.A.S.). This work was also supported by the National Institute of General Medical Sciences grant GM34821 (T.J.S.) and NIH/NIGMS grant P50 GM071508 (S.Z.).
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
We thank the Silhavy lab, present and past, for helpful discussion and comments. We also thank Mark Rose and Zemer Gitai for helpful discussion and critical analysis. We are grateful to Thomas Bartlett for help with imaging and cell size analysis as well as Jaclyn Schwalm for strains.
Footnotes
Published ahead of print 23 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01876-14.
REFERENCES
- 1.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2:a000414. 10.1101/cshperspect.a000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593–656. 10.1128/MMBR.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bos MP, Robert V, Tommassen J. 2007. Biogenesis of the gram-negative bacterial outer membrane. Annu. Rev. Microbiol. 61:191–214. 10.1146/annurev.micro.61.080706.093245 [DOI] [PubMed] [Google Scholar]
- 4.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635–700. 10.1146/annurev.biochem.71.110601.135414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruiz N, Kahne D, Silhavy TJ. 2009. Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat. Rev. Microbiol. 7:677–683. 10.1038/nrmicro2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dowhan W. 2013. A retrospective: use of Escherichia coli as a vehicle to study phospholipid synthesis and function. Biochim. Biophys. Acta 1831:471–494. 10.1016/j.bbalip.2012.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang YM, Rock CO. 2008. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 6:222–233. 10.1038/nrmicro1839 [DOI] [PubMed] [Google Scholar]
- 8.Wiesner J, Vilcinskas A. 2010. Antimicrobial peptides: the ancient arm of the human immune system. Virulence 1:440–464. 10.4161/viru.1.5.12983 [DOI] [PubMed] [Google Scholar]
- 9.Dalebroux ZD, Matamouros S, Whittington D, Bishop RE, Miller SI. 2014. PhoPQ regulates acidic glycerophospholipid content of the Salmonella Typhimurium outer membrane. Proc. Natl. Acad. Sci. U. S. A. 111:1963–1968. 10.1073/pnas.1316901111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Needham BD, Trent MS. 2013. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 11:467–481. 10.1038/nrmicro3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murata T, Tseng W, Guina T, Miller SI, Nikaido H. 2007. PhoPQ-mediated regulation produces a more robust permeability barrier in the outer membrane of Salmonella enterica serovar Typhimurium. J. Bacteriol. 189:7213–7222. 10.1128/JB.00973-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cronan JE. 2003. Bacterial membrane lipids: where do we stand? Annu. Rev. Microbiol. 57:203–224. 10.1146/annurev.micro.57.030502.090851 [DOI] [PubMed] [Google Scholar]
- 13.Pootoolal J, Neu J, Wright GD. 2002. Glycopeptide antibiotic resistance. Annu. Rev. Pharmacol. Toxicol. 42:381–408. 10.1146/annurev.pharmtox.42.091601.142813 [DOI] [PubMed] [Google Scholar]
- 14.Barna JC, Williams DH. 1984. The structure and mode of action of glycopeptide antibiotics of the vancomycin group. Annu. Rev. Microbiol. 38:339–357. 10.1146/annurev.mi.38.100184.002011 [DOI] [PubMed] [Google Scholar]
- 15.Ganong BR, Leonard JM, Raetz CR. 1980. Phosphatidic acid accumulation in the membranes of Escherichia coli mutants defective in CDP-diglyceride synthetase. J. Biol. Chem. 255:1623–1629 [PubMed] [Google Scholar]
- 16.Ganong BR, Raetz CR. 1982. Massive accumulation of phosphatidic acid in conditionally lethal CDP-diglyceride synthetase mutants and cytidine auxotrophs of Escherichia coli. J. Biol. Chem. 257:389–394 [PubMed] [Google Scholar]
- 17.Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J. Mol. Biol. 104:541–555. 10.1016/0022-2836(76)90119-4 [DOI] [PubMed] [Google Scholar]
- 18.Ruiz N, Gronenberg LS, Kahne D, Silhavy TJ. 2008. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 105:5537–5542. 10.1073/pnas.0801196105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 20.Malinverni JC, Silhavy TJ. 2009. An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc. Natl. Acad. Sci. U. S. A. 106:8009–8014. 10.1073/pnas.0903229106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 22.Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. 2011. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 80:612–627. 10.1111/j.1365-2958.2011.07579.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can. J. Physiol. Pharmacol. 37:911–917. 10.1139/y59-099 [DOI] [PubMed] [Google Scholar]
- 24.Tan BK, Bogdanov M, Zhao J, Dowhan W, Raetz CR, Guan Z. 2012. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc. Natl. Acad. Sci. U. S. A. 109:16504–16509. 10.1073/pnas.1212797109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grabowicz M, Yeh J, Silhavy TJ. 2013. Dominant negative lptE mutation that supports a role for LptE as a plug in the LptD barrel. J. Bacteriol. 195:1327–1334. 10.1128/JB.02142-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freinkman E, Chng SS, Kahne D. 2011. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc. Natl. Acad. Sci. U. S. A. 108:2486–2491. 10.1073/pnas.1015617108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freinkman E, Okuda S, Ruiz N, Kahne D. 2012. Regulated assembly of the transenvelope protein complex required for lipopolysaccharide export. Biochemistry 51:4800–4806. 10.1021/bi300592c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villa R, Martorana AM, Okuda S, Gourlay LJ, Nardini M, Sperandeo P, Deho G, Bolognesi M, Kahne D, Polissi A. 2013. The Escherichia coli Lpt transenvelope protein complex for lipopolysaccharide export is assembled via conserved structurally homologous domains. J. Bacteriol. 195:1100–1108. 10.1128/JB.02057-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sampson BA, Misra R, Benson SA. 1989. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122:491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sperandeo P, Pozzi C, Deho G, Polissi A. 2006. Non-essential KDO biosynthesis and new essential cell envelope biogenesis genes in the Escherichia coli yrbG-yhbG locus. Res. Microbiol. 157:547–558. 10.1016/j.resmic.2005.11.014 [DOI] [PubMed] [Google Scholar]
- 31.Ruiz N, Falcone B, Kahne D, Silhavy TJ. 2005. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121:307–317. 10.1016/j.cell.2005.02.014 [DOI] [PubMed] [Google Scholar]
- 32.Icho T, Bulawa CE, Raetz CR. 1985. Molecular cloning and sequencing of the gene for CDP-diglyceride hydrolase of Escherichia coli. J. Biol. Chem. 260:12092–12098 [PubMed] [Google Scholar]
- 33.Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Heijne G. 2005. Global topology analysis of the Escherichia coli inner membrane proteome. Science 308:1321–1323. 10.1126/science.1109730 [DOI] [PubMed] [Google Scholar]
- 34.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12:291–299. 10.1093/dnares/dsi012 [DOI] [PubMed] [Google Scholar]
- 36.Yao Z, Davis RM, Kishony R, Kahne D, Ruiz N. 2012. Regulation of cell size in response to nutrient availability by fatty acid biosynthesis in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 109:E2561–E2568. 10.1073/pnas.1209742109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rigel NW, Schwalm J, Ricci DP, Silhavy TJ. 2012. BamE modulates the Escherichia coli beta-barrel assembly machine component BamA. J. Bacteriol. 194:1002–1008. 10.1128/JB.06426-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ricci DP, Hagan CL, Kahne D, Silhavy TJ. 2012. Activation of the Escherichia coli beta-barrel assembly machine (Bam) is required for essential components to interact properly with substrate. Proc. Natl. Acad. Sci. U. S. A. 109:3487–3491. 10.1073/pnas.1201362109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishijima M, Raetz CR. 1979. Membrane lipid biogenesis in Escherichia coli: identification of genetic loci for phosphatidylglycerophosphate synthetase and construction of mutants lacking phosphatidylglycerol. J. Biol. Chem. 254:7837–7844 [PubMed] [Google Scholar]
- 40.Shibuya I, Miyazaki C, Ohta A. 1985. Alteration of phospholipid composition by combined defects in phosphatidylserine and cardiolipin synthases and physiological consequences in Escherichia coli. J. Bacteriol. 161:1086–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikaido H. 2005. Restoring permeability barrier function to outer membrane. Chem. Biol. 12:507–509. 10.1016/j.chembiol.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 42.McMahon HT, Gallop JL. 2005. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438:590–596. 10.1038/nature04396 [DOI] [PubMed] [Google Scholar]
- 43.Kooijman EE, Burger KN. 2009. Biophysics and function of phosphatidic acid: a molecular perspective. Biochim. Biophys. Acta 1791:881–888. 10.1016/j.bbalip.2009.04.001 [DOI] [PubMed] [Google Scholar]
- 44.Tran TT, Panesso D, Mishra NN, Mileykovskaya E, Guan Z, Munita JM, Reyes J, Diaz L, Weinstock GM, Murray BE, Shamoo Y, Dowhan W, Bayer AS, Arias CA. 2013. Daptomycin-resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane phospholipids. mBio 4(4):e00281–13. 10.1128/mBio.00281-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hartmann M, Berditsch M, Hawecker J, Ardakani MF, Gerthsen D, Ulrich AS. 2010. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob. Agents Chemother. 54:3132–3142. 10.1128/AAC.00124-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishi H, Komatsuzawa H, Fujiwara T, McCallum N, Sugai M. 2004. Reduced content of lysyl-phosphatidylglycerol in the cytoplasmic membrane affects susceptibility to moenomycin, as well as vancomycin, gentamicin, and antimicrobial peptides, in Staphylococcus aureus. Antimicrob. Agents Chemother. 48:4800–4807. 10.1128/AAC.48.12.4800-4807.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dortet L, Poirel L, Nordmann P. 2014. Worldwide dissemination of the NDM-type carbapenemases in gram-negative bacteria. Biomed. Res. Int. 2014:249856. 10.1155/2014/249856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu T, McCandlish AC, Gronenberg LS, Chng SS, Silhavy TJ, Kahne D. 2006. Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 103:11754–11759. 10.1073/pnas.0604744103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chng SS, Ruiz N, Chimalakonda G, Silhavy TJ, Kahne D. 2010. Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc. Natl. Acad. Sci. U. S. A. 107:5363–5368. 10.1073/pnas.0912872107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol. Rev. 53:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nichols BP, Shafiq O, Meiners V. 1998. Sequence analysis of Tn10 insertion sites in a collection of Escherichia coli strains used for genetic mapping and strain construction. J. Bacteriol. 180:6408–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.