

Abstract
Lately, researchers have been actively investigating Escherichia coli lptD mutants, which exhibit reduced transport of lipopolysaccharide to the cell surface. In this issue of the Journal of Bacteriology, Sutterlin et al. (H. A. Sutterlin, S. Zhang, and T. J. Silhavy, J. Bacteriol. 196:3214–3220, 2014) now reveal an important functional role for phosphatidic acid in fortifying the outer membrane permeability barrier in certain lptD mutant backgrounds. These findings come on the heels of the first reports of two LptD crystal structures, which now provide a structural framework for interpreting lptD genetics.
TEXT
The outer membranes of enteric Gram-negative bacteria like Escherichia coli provide a permeability barrier to lipophilic antibiotics and detergents. A large part of this barrier to permeability is a consequence of the outer membrane's asymmetric lipid organization, where phospholipids line the inner leaflet and lipopolysaccharide (LPS) molecules line the external leaflet (1). The LPS layer often repels lipophilic molecules that can normally penetrate phospholipid bilayers because ionic bonds bridge neighboring molecules together in a phalanx formation where Mg2+ ions neutralize negatively charged groups located between the lipid A and inner core sugar regions of LPS (2). However, partial loss-of-function mutations in the essential gene lptD cause cells to deliver insufficient LPS to the cell surface, resulting in the anomalous ectopic exposure of phospholipid domains that drift among the LPS and lead to the phenotype of increased membrane permeability (3, 4). By selecting for second-site suppressors in lptD mutants of E. coli, Sutterlin et al., in this issue of the Journal of Bacteriology (5), demonstrate that phosphatidic acid (PA) can fortify the outer membrane permeability barrier.
The genetic selection devised by Sutterlin et al. (5) started with a subset of lptD alleles specified as lptDI54E, lptDV52E-I54E, and lptDΔD57ΔK60, which all affect residues located in the periplasmic β-jellyroll domain near the N-terminal end of LptD (6–8). The LptD β-jellyroll domain is the terminal subunit of a transperiplasmic filament that interconnects with the homologous β-jellyroll domains of the periplasmic LptA and the inner membrane LptC subunits (9). The LptD β-jellyroll mutations all map to the contact interface with LptA (10) and likely interfere with the filament docking interaction (Fig. 1). The LptD β-jellyroll completes a continuous lipophilic groove for lipid A that spirals across the periplasm and provides a track to deliver LPS into the lumen of the much larger outer membrane LptD β-barrel domain (8). Composed of 26 membrane-spanning β-strands, the LptD β-barrel is the largest yet described and folds into two lobes, one plugged by the lipoprotein LptE, which probably functions to reorient the LPS sugar chain into a vertical position. The other lobe, located adjacent to the β-jellyroll, remains empty on the periplasmic side so that it can transiently accommodate LPS on its passage into the external leaflet of the outer membrane. The empty lobe of the LptD β-barrel is closed at the cell surface by the L4 loop, which likely repositions itself in order to create a transitory opening where the LPS sugar chain can emerge. Two combined point mutations in LptE were recently reported to decrease its ability to disaggregate LPS (lptER91D/K136D), and both residues in the complex map to the cell surface near the L4 loop of LptD (11). The LPS lipid A region then diffuses laterally into the external leaflet of the outer membrane through a weakly hydrogen-bonded gate located between β-strands 1 and 26 (7). As such, the lptD β-jellyroll mutants employed by Sutterlin et al. (5) are distinctly different from the better-known alleles lptD4213 (lptDΔ330–352) and lptD208 (lptDΔ335–359), which are 23- and 25-residue deletions, respectively (12, 13), which map to the L4 loop and could potentially expose the lumen of the LptD β-barrel to extracellular solutes (Fig. 1).
FIG 1.

Structure of the LptDE complex for delivering and inserting LPS into the bacterial outer membrane. (A) The LptDE complex from Shigella flexneri reveals the route for LPS delivery and insertion into the outer membrane (blue arrow). The LPS structure shown is devoid of an attached O antigen, but the sugar chain of LPS is positioned by the β-jellyroll to engage first with LptE, which plugs the distal cavity of the LptD β-barrel where it probably reorients the LPS into a vertical position. Delivery of the LPS into the open proximal cavity is followed by insertion into the outer membrane external leaflet by lateral migration through an opening between the first and final β-strands, 1 and 26, in the β-barrel. A belt of 11 ordered detergent molecules (shown in cyan) surrounds the lipid-exposed exterior of the LptD β-barrel, and two more are buried inside the β-jellyroll. LptE is modified by a distinct lipid moiety (Lipo Cys), which is anchored in the inner leaflet of the outer membrane. Residues V52, I54, D57, and K60 associated with the lptD β-jellyroll alleles are shown in green, while the lptD β-barrel L4 loop deletion alleles include residues 330 to 352 (lptD4213) and 335 to 359 (lptD208), which are shown in magenta. (B) View rotated 180° from the orientation shown in panel A. (C) View from the periplasm. (D) View from the cell surface.
The distinction between lptD alleles is important to understand because at least 3 different mechanisms can possibly explain the phenotype of increased membrane permeability in lptD mutants (2). First, as mentioned above, the presence in lptD mutants of localized phospholipid bilayer domains that drift among the insufficient LPS provides a point of access for the lipophilic antibiotics and detergents that would normally be repelled by a continuous Mg2+-bridged sheet of LPS (1). However, certain large polar antibiotics like vancomycin are simultaneously too large to enter any of the common β-barrel porins and insufficiently lipophilic to penetrate phospholipid bilayers, but they still appear to permeate into lptD mutants. To explain this observation, Hiroshi Nikaido has suggested a second mechanism, which he attributes to “cracks” or, perhaps more specifically, “phase boundary defects” that likely occur at sites where the LPS and phospholipid phases encounter one another (14). The third mechanism, the conversion of LptD into a large mutant “porin,” might possibly be occurring with the lptD L4 loop deletion alleles. The “phase boundary defect” mechanism and the LptD “porin” mechanism can be distinguished genetically, but the two are expected to display similar permeabilities to large polar solutes.
Sutterlin et al. (5) screened for second-site suppressors that restore vancomycin resistance, so the judicious choice of the lptD β-jellyroll alleles helps to exclude the LptD “porin” mechanism and leaves the “phase boundary defect” mechanism the preferred choice for focusing any explanations of a fortified permeability barrier. The use of vancomycin rather than, say, erythromycin, which is similarly large and water soluble, but with ribosomes as its cytoplasmic target, is also a judicious choice because the antibiotic target of vancomycin is the d-alanyl-d-alanine terminus of lipid II-linked peptidoglycan pentapeptides, which are encountered instead in the periplasm where they lie extrinsic to the inner membrane (3). Therefore, any fortification of the permeability barrier resulting from the selection of vancomycin resistance is most likely exerted in the outer membrane. So, precisely what is it that Sutterlin et al. (5) found? In every case, the second-site suppressors of the lptD β-jellyroll alleles that were recovered mapped to another essential gene, cdsA, which encodes CTP:PA cytidylyltransferase. CdsA converts CTP and PA into the phospholipid precursor CDP-diacylglycerol, but the “leaky” partial loss-of-function cdsA alleles that were recovered all result in the accumulation of PA in the membranes. The question of how the accumulation of PA in membranes functions to suppress the phase boundary defect responsible for outer membrane vancomycin uptake in lptD mutants thus remains.
Perhaps, the question should be phrased more generally, because Sutterlin et al. (5) go on to demonstrate that any means of increasing PA levels in E. coli, even in wild-type cells, serves to buttress the bacteria against vancomycin. Indeed, when Ganong and Raetz first isolated cdsA mutants of E. coli and observed the membrane accumulation of PA in the early 1980s, they similarly reported an increase in resistance, but, in this case, to erythromycin, so it was not possible for them to ascertain whether the resistance was exerted at the level of the inner or the outer membrane, or both (15, 16). The question then should be how does PA accumulation generally reinforce the membranes against vancomycin and erythromycin. Before attempting any explanation, it is important to consider that PA is not the only phospholipid that accumulates in outer membranes under conditions that shore up the permeability barrier against antibiotics. Dalebroux et al. have recently reported that cardiolipin and palmitoylphosphatidylglycerol (PPG) accumulate in the outer membranes of Salmonella enterica serovar Typhimurium under conditions of PhoP/PhoQ activation known to render the cells resistant to cationic antimicrobial peptides (CAMPs) (17).
In the contexts of both E. coli and S. Typhimurium, the membrane phospholipid composition is approximately 70 to 80% phosphatidylethanolamine (PE), 20 to 25% phosphatidylglycerol (PG), and 5% cardiolipin (Fig. 2). PA is a trace phospholipid precursor that is normally found only at 0.2%, but it can accumulate in cdsA mutants to 5% or more, mostly at the expense of PG and cardiolipin (15). Among these phospholipids, only PE is zwitterionic (the rest are anionic), but PG has the greatest tendency to spontaneously form lamellar bilayer structures because PG has the requisite cylindrical shape (18). PE has a conical shape that causes it to spontaneously form hexagonal-phase structures, but when placed in the context of a bilayer environment, PE has a strong tendency to induce negative membrane curvature because lateral pressure becomes greater in the hydrophobic core region than in the membrane interface. Cardiolipin and PA are unusual because on their own they both are cylindrical like PG and form bilayers, but under physiological conditions (in the presence of Ca2+ and Mg2+), the effective size of their polar head group is reduced by charge neutralization and dehydration, which causes both to become conical so they also function with PE in inducing negative membrane curvature (19). As such, the membrane lipid composition of E. coli is spring-loaded with a tendency to adopt intrinsic negative membrane curvature. One possible explanation for this loading of negative membrane curvature into E. coli phospholipid bilayers is that it adapts them to absorb the positive membrane curvature induced by molecules such as lysophospholipids, which possess an inverted cone shape and function like detergents, CAMPs, and certain other “facial amphiphiles” in disrupting bacterial membranes (20). Therefore, regulated increases in non-bilayer-forming phospholipid species like PA and cardiolipin could, conceivably, help provide resistance to certain membrane-active antibiotic molecules.
FIG 2.
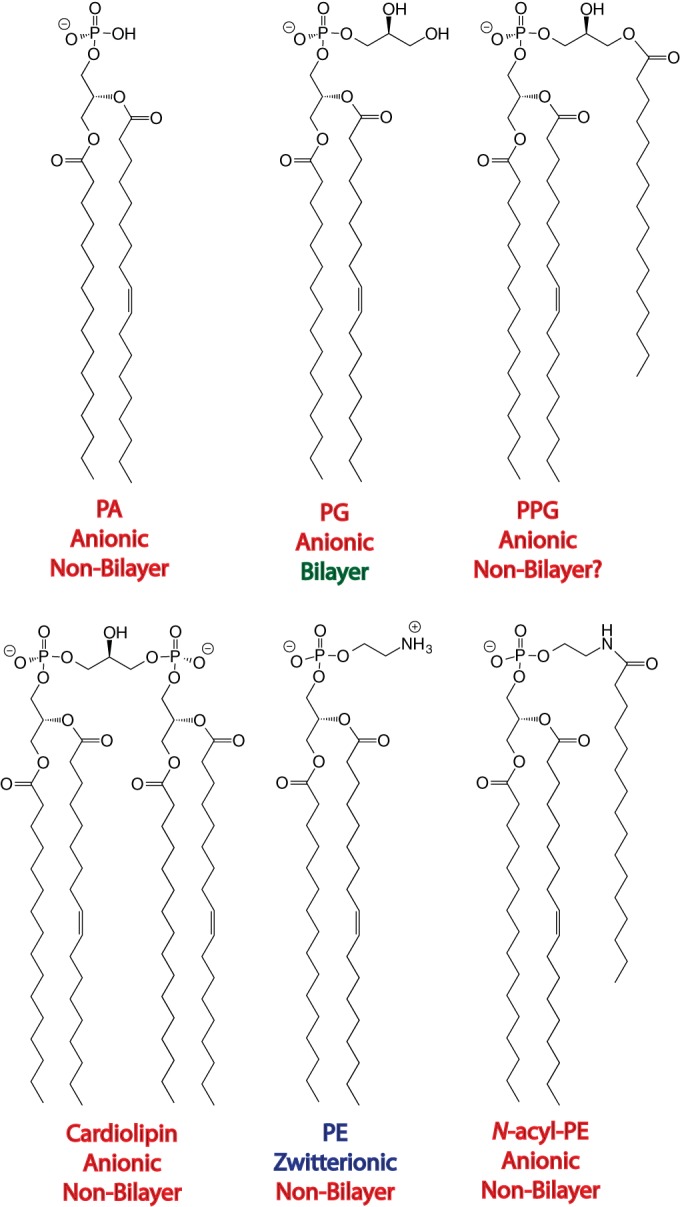
Structures of PA, cardiolipin, PG, PPG, PE, and N-acyl-PE in E. coli. Although PA and cardiolipin have a tendency to form bilayers in the absence of divalent cations, these ions tend to be replete in the outer membrane where PA and cardiolipin are thus expected to display non-bilayer properties.
By virtue of their negative charge, anionic lipids can also interact electrostatically with membrane proteins and cationic antibiotics in ways that zwitterionic lipids cannot. Based on this principle, another mechanism for providing resistance to polycationic facial amphiphiles like CAMPs is known as “anionic lipid clustering” (21). In the presence of an excess of a zwitterionic phospholipid species like PE, as is the case in E. coli and S. Typhimurium, the remaining pool of anionic phospholipids will be evenly dispersed within the bilayer. However, a membrane-active polycationic antibiotic will induce the anionic lipids to cluster around it, thereby causing them to separate from the zwitterionic species into two distinct lipid phases where a phase boundary defect will then emerge and compromise the permeability barrier. This mechanism does an excellent job of explaining why some bacteria that possess large amounts of anionic phospholipids and little PE are insensitive to certain CAMPs that effectively halt the growth of E. coli (22). One can easily imagine how increasing the relative abundance of an anionic phospholipid species like PA or cardiolipin might demand a corresponding increase in CAMP concentration to sufficiently induce the lipids to separate into the distinct phases from which the permeability-compromising phase boundary defect then emerges. While anionic lipid clustering can explain sensitivity to CAMPs, it does not likely explain sensitivity to vancomycin and erythromycin, neither of which is polycationic.
Perhaps, there is something special about PA and cardiolipin, because increasing PE should increase negative membrane curvature and increasing PG should increase negative charge, but it is only an increase in the anionic non-bilayer lipids that appears to be conferring antibiotic resistance. Recent evidence suggests that cardiolipin tends to accumulate at the poles and septal regions where membrane curvature is most pronounced (23, 24). PA has been suggested to function similarly to cardiolipin (23), so perhaps it can also help to cure the phase boundary defects that introduce “cracks” in the outer membranes of lptD mutants. A similar role for PPG can be speculated. Discovered by Olsen and Ballou in 1971 as a minor phospholipid in S. Typhimurium (25), the membrane biophysical properties of head group-acylated PGs have remained largely unexplored. We can say for certain that acyl-PGs are synthesized in E. coli by two very different acyltransferases. The inner membrane PldB is largely unspecific for the acyl chain that is transferred from its lysophospholipid donor to the PG polar head group (26, 27), but the outer membrane PhoP/PhoQ-dependent PagP uses a phospholipid donor and is very specific for palmitate (17). PagP is the same enzyme responsible for the palmitoylation of lipid A, and the recent report by Dalebroux et al. (17) reveals that PagP is, in fact, a bifunctional enzyme, which is also responsible for producing PPG in the outer membrane. The lysophospholipid by-product of the PagP reaction is rapidly transported to the inner membrane and reacylated (28), but PPG is formed at the expense of PG and thus depletes the outer membrane of its key bilayer-forming lipid species. On the structural spectrum, PPG appears to lie somewhere between PG and cardiolipin (Fig. 2), so it could, in principle, prove to increase negative membrane curvature, cure phase boundary defects, or localize to polar/septal domains in a manner similar to that described for cardiolipin and PA (23). In fact, PPG is structurally most like N-acyl-PE, which is a minor phospholipid that accumulates in E. coli mutants deficient in PG and cardiolipin and, along with accumulated PA, compensates for the absence of the major anionic phospholipids (23). Although N-acyl-PE has not been implicated in antibiotic resistance, its characterization as an anionic non-bilayer phospholipid like PA and cardiolipin strongly suggests that PPG belongs in the same category (18). Cardiolipin and PPG accumulation in the outer membrane is clearly regulated by PhoP/PhoQ in S. Typhimurium (17), but it remains to be determined whether any mechanism of regulation can actually modulate PA or N-acyl-PE levels in wild-type enterobacteria.
In summary, increased production of what appear to be anionic non-bilayer lipids (PA, cardiolipin, and PPG), can fortify the outer membranes of enteric Gram-negative bacteria against certain antibiotics ranging in structure from CAMPs to vancomycin. The anionic bilayer-forming PG and the zwitterionic non-bilayer PE have not been so implicated. These recent developments in our understanding of bacterial membrane phospholipid composition have hinged on clever genetic screens combined with advances in mass spectrometry applied to bacterial lipidomics. Since Jones and Osborn first established in 1977 that phospholipids are rapidly exchanging between the inner and outer membranes (29), we still await a structural explanation for how the phospholipids are so effectively shuttled back and forth. The Silhavy lab has broken ground in this area by implicating an ABC transport system in energizing a retrograde phospholipid transport process (30), but so far the specific nature of the anterograde phospholipid transport machinery remains to be revealed. Perhaps, the emerging awareness of how phospholipid transport can control antibiotic resistance will spur on the discovery of these key components. Based on the unidirectional nature of LPS transport to the outer membrane (31), we can anticipate that the phospholipid transport machinery will turn out to be of a distinctly different nature than the Lpt machinery, which has finally yielded some of its structural secrets (6–8).
ACKNOWLEDGMENTS
I am grateful to Richard Epand for his thoughtful suggestions on the initial version of the manuscript. I apologize to the many authors whose work could not be cited due to the brevity of this commentary.
Work in my laboratory is funded by the Canadian Institutes of Health Research operating grant MOP-125979.
Footnotes
Published ahead of print 14 July 2014
The views expressed in this Commentary do not necessarily reflect the views of the journal or of ASM.
REFERENCES
- 1.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593–656. 10.1128/MMBR.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruiz N, Kahne D, Silhavy TJ. 2006. Advances in understanding bacterial outer-membrane biogenesis. Nat. Rev. Microbiol. 4:57–66. 10.1038/nrmicro1322 [DOI] [PubMed] [Google Scholar]
- 3.Ruiz N, Falcone B, Kahne D, Silhavy TJ. 2005. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121:307–317. 10.1016/j.cell.2005.02.014 [DOI] [PubMed] [Google Scholar]
- 4.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245. 10.1016/j.cell.2005.02.015 [DOI] [PubMed] [Google Scholar]
- 5.Sutterlin HA, Zhang S, Silhavy TJ. 2014. Accumulation of phosphatidic acid increases vancomycin resistance in Escherichia coli. J. Bacteriol. 196:3214–3220. 10.1128/JB.01876-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop RE. 2014. Lipopolysaccharide rolls out the barrel. Nature 511:37–38. 10.1038/nature13508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong H, Xiang Q, Gu Y, Wang Z, Paterson NG, Stansfeld PJ, He C, Zhang Y, Wang W, Dong C. 2014. Structural basis for outer membrane lipopolysaccharide insertion. Nature 511:52–56. 10.1038/nature13464 [DOI] [PubMed] [Google Scholar]
- 8.Qiao S, Luo Q, Zhao Y, Zhang XC, Huang Y. 2014. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 511:108–111. 10.1038/nature13484 [DOI] [PubMed] [Google Scholar]
- 9.Whitfield C, Trent MS. 2014. Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem. 83:99–128. 10.1146/annurev-biochem-060713-035600 [DOI] [PubMed] [Google Scholar]
- 10.Suits MD, Sperandeo P, Deho G, Polissi A, Jia Z. 2008. Novel structure of the conserved gram-negative lipopolysaccharide transport protein A and mutagenesis analysis. J. Mol. Biol. 380:476–488. 10.1016/j.jmb.2008.04.045 [DOI] [PubMed] [Google Scholar]
- 11.Malojcic G, Andres D, Grabowicz M, George AH, Ruiz N, Silhavy TJ, Kahne D. 2014. LptE binds to and alters the physical state of LPS to catalyze its assembly at the cell surface. Proc. Natl. Acad. Sci. U. S. A. 111:9467–9472. 10.1073/pnas.1402746111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sampson BA, Misra R, Benson SA. 1989. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122:491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braun M, Silhavy TJ. 2002. Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol. Microbiol. 45:1289–1302. 10.1046/j.1365-2958.2002.03091.x [DOI] [PubMed] [Google Scholar]
- 14.Nikaido H. 2005. Restoring permeability barrier function to outer membrane. Chem. Biol. 12:507–509. 10.1016/j.chembiol.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 15.Ganong BR, Leonard JM, Raetz CR. 1980. Phosphatidic acid accumulation in the membranes of Escherichia coli mutants defective in CDP-diglyceride synthetase. J. Biol. Chem. 255:1623–1629 [PubMed] [Google Scholar]
- 16.Ganong BR, Raetz CR. 1982. Massive accumulation of phosphatidic acid in conditionally lethal CDP-diglyceride synthetase mutants and cytidine auxotrophs of Escherichia coli. J. Biol. Chem. 257:389–394 [PubMed] [Google Scholar]
- 17.Dalebroux ZD, Matamouros S, Whittington D, Bishop RE, Miller SI. 2014. PhoPQ regulates acidic glycerophospholipid content of the Salmonella Typhimurium outer membrane. Proc. Natl. Acad. Sci. U. S. A. 111:1963–1968. 10.1073/pnas.1316901111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dowhan W. 2013. A retrospective: use of Escherichia coli as a vehicle to study phospholipid synthesis and function. Biochim. Biophys. Acta 1831:471–494. 10.1016/j.bbalip.2012.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dowhan W. 1997. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 66:199–232. 10.1146/annurev.biochem.66.1.199 [DOI] [PubMed] [Google Scholar]
- 20.Koller D, Lohner K. 2014. The role of spontaneous lipid curvature in the interaction of interfacially active peptides with membranes. Biochim. Biophys. Acta 10.1016/j.bbamem.2014.05.013 [DOI] [PubMed] [Google Scholar]
- 21.Epand RM, Rotem S, Mor A, Berno B, Epand RF. 2008. Bacterial membranes as predictors of antimicrobial potency. J. Am. Chem. Soc. 130:14346–14352. 10.1021/ja8062327 [DOI] [PubMed] [Google Scholar]
- 22.Epand RM, Epand RF. 2011. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 17:298–305. 10.1002/psc.1319 [DOI] [PubMed] [Google Scholar]
- 23.Mileykovskaya E, Ryan AC, Mo X, Lin CC, Khalaf KI, Dowhan W, Garrett TA. 2009. Phosphatidic acid and N-acylphosphatidylethanolamine form membrane domains in Escherichia coli mutant lacking cardiolipin and phosphatidylglycerol. J. Biol. Chem. 284:2990–3000. 10.1074/jbc.M805189200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Renner LD, Weibel DB. 2011. Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc. Natl. Acad. Sci. U. S. A. 108:6264–6269. 10.1073/pnas.1015757108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olsen RW, Ballou CE. 1971. Acyl phosphatidylglycerol. A new phospholipid from Salmonella typhimurium. J. Biol. Chem. 246:3305–3313 [PubMed] [Google Scholar]
- 26.Karasawa K, Kudo I, Kobayashi T, Sa-Eki T, Inoue K, Nojima S. 1985. Purification and characterization of lysophospholipase L2 of Escherichia coli K-12. J. Biochem. 98:1117–1125 [DOI] [PubMed] [Google Scholar]
- 27.Hsu L, Jackowski S, Rock CO. 1991. Isolation and characterization of Escherichia coli K-12 mutants lacking both 2-acyl-glycerophosphoethanolamine acyltransferase and acyl-acyl carrier protein synthetase activity. J. Biol. Chem. 266:13783–13788 [PubMed] [Google Scholar]
- 28.Hsu L, Jackowski S, Rock CO. 1989. Uptake and acylation of 2-acyl-lysophospholipids by Escherichia coli. J. Bacteriol. 171:1203–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones NC, Osborn MJ. 1977. Translocation of phospholipids between the outer and inner membranes of Salmonella typhimurium. J. Biol. Chem. 252:7405–7412 [PubMed] [Google Scholar]
- 30.Malinverni JC, Silhavy TJ. 2009. An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc. Natl. Acad. Sci. U. S. A. 106:8009–8014. 10.1073/pnas.0903229106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osborn MJ, Gander JE, Parisi E. 1972. Mechanism of assembly of the outer membrane of Salmonella typhimurium. Site of synthesis of lipopolysaccharide. J. Biol. Chem. 247:3973–3986 [PubMed] [Google Scholar]