

Abstract
Activity-dependent competition of synapses plays a key role in neural organization and is often promoted by GABA; however, its cellular bases are poorly understood. Excitatory synapses of cortical pyramidal neurons are formed on small protrusions known as dendritic spines, which exhibit structural plasticity. We used two-color uncaging of glutamate and GABA in rat hippocampal CA1 pyramidal neurons and found that spine shrinkage and elimination were markedly promoted by the activation of GABAA receptors shortly before action potentials. GABAergic inhibition suppressed bulk increases in cytosolic Ca2+ concentrations, whereas it preserved the Ca2+ nanodomains generated by NMDA-type receptors, both of which were necessary for spine shrinkage. Unlike spine enlargement, spine shrinkage spread to neighboring spines (<15 μm) and competed with their enlargement, and this process involved the actin-depolymerizing factor ADF/cofilin. Thus, GABAergic inhibition directly suppresses local dendritic Ca2+ transients and strongly promotes the competitive selection of dendritic spines.
Introduction
The selection of synapses is key to the reorganization of the CNS during development and in adulthood. Synaptic plasticity is often competitive and dependent on the excitation-inhibition balance of neuronal circuits1, 2. In pyramidal neurons of the cerebral cortices, excitatory synaptic inputs are made on small protrusions of dendrites, called dendritic spines. The sizes of spines are highly variable and correlate with glutamate sensitivity3, 4, and the long-term potentiation (LTP)5, 6 and long-term depression (LTD)7 of glutamatergic synapses are often associated with the enlargement and shrinkage of dendritic spines8, 9. If LTP and LTD are associated with structural changes, they can mediate selection of synapses. In fact, spine generation and elimination have been demonstrated in vivo8, 10.
NMDA receptors are involved in both LTP and LTD, which of these occurs depending on the patterns of neuronal activation because their effective Ca2+ entry is regulated by the timing of glutamate release from the presynaptic terminals and the postsynaptic depolarization that relieves the NMDA receptors’ magnesium block11, 12. For example, LTP is robustly induced when glutamatergic inputs are followed shortly by action potentials13, 14. In contrast, when glutamatergic inputs are delayed behind the spike, they give rise to LTD, possibly owing to moderate increases in cytosolic Ca2+ concentration ([Ca2+]i)15, although the mechanisms of coincidence detection in spike timing–dependent LTD are not well understood.
The competitive selection of synapses often depends on GABA. For example, the opening of the critical period of sensory cortices is governed by the maturation of GABAergic inputs16 and is associated with an enhancement in elimination of spines17. The elimination of climbing fibers of Purkinje cells in the immature cerebellum also depends on GABA2. It has, however, never been clarified how elimination of synapses is promoted by GABAergic inputs.
Two-photon glutamate uncaging allows stimulation of individual spines3 and investigation of spatial spread and interactions underlying spine structural plasticity. Spine enlargement has been localized to the stimulated spines5, 6, 18, 19. Although spine elimination has been demonstrated through the electrical stimulation of presynaptic fibers7, it has never to our knowledge been induced through the stimulation of identified spines, hampering the understanding of the competitive selection of synapses in the dendrite. Here we established conditions whereby prominent shrinkage and elimination of spines were induced using two-color uncaging of glutamate and GABA. Spine shrinkage showed marked spread, leading to heterosynaptic competitive interactions between enlargement and shrinkage of spines in which GABA finely regulated local dendritic Ca2+ signaling without affecting the generation of action potentials.
Results
Spine shrinkage by two-color glutamate and GABA uncaging
We investigated the structural plasticity of individually identified spines on the proximal apical dendrites of pyramidal neurons in the CA1 region of rat hippocampal slice preparations using two-color uncaging of caged glutamate and GABA compounds, which selectively stimulated glutamate and GABA receptors20, respectively (Supplementary Fig. 1 and Supplementary Data Set 1). The laser power for the glutamate uncaging was adjusted to mimic the amplitudes of miniature excitatory postsynaptic currents (EPSCs)3. We investigated spines in secondary to ternary apical dendritic branches, 100–150 μm away from the soma, using a two-photon microscope to image the dendritic structures, which were whole-cell dialyzed with Alexa 594.
Spine enlargement in whole-cell patch-clamped neurons was readily induced by the repetitive pairing of two-photon glutamate uncaging (at 720 nm) and subsequent back-propagating action potentials (bAPs) in a single spine for 80 s at 1 Hz (LTP protocol)6, 18 (Fig. 1a,b; mean ± s.e.m. = 63 ± 21%; ΔVH represents percentage changes in spine volumes relative to those immediately before stimulation).
Figure 1. Spine shrinkage and elimination induced with the spike-timing protocol.
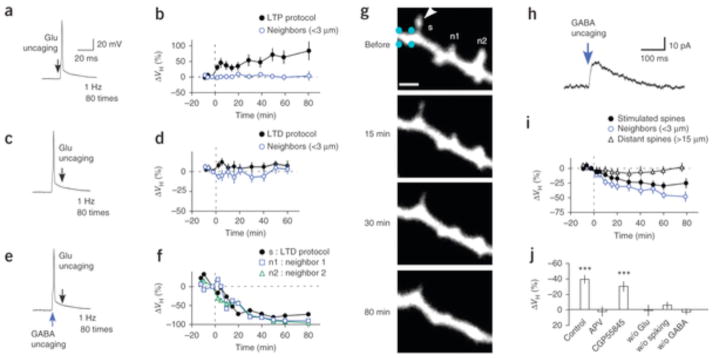
(a,c,e) Pairing of glutamate uncaging and postsynaptic action potentials in the LTP (a) and LTD (c,e) protocols. Scale bars in a also apply to c,e. (b) Spine enlargement induced with the LTP protocol at 1 Hz, 80 times (8 spines, 8 dendrites, 8 slices, 4 rats). (d) The absence of spine shrinkage in the LTD protocol without GABA uncaging (15 spines, 15 dendrites, 8 slices, 5 rats). (f) Time courses of spine shrinkage in the stimulated (s) and neighboring (n1, n2) spines shown in g. (g) Alexa 594 fluorescence images of a dendrite with one spine (arrowhead) to which the LTD protocol was applied with GABA uncaging. GABA uncaging was applied at spike onset as in e, at four points (cyan) around the dendritic shaft for a total duration of 4 ms, each separated by 1 ms. Scale bar, 2 μm. The soma was located to the left. (h) A current trace that was elicited by GABA uncaging at the dendritic shaft shown in g, with it voltage clamped at 0 mV. (i) Spine shrinkage induced with the LTD protocol with GABA uncaging in stimulated (14 spines, 14 dendrites, 14 slices, 10 rats), neighboring (19 spines) and distant spines (9 spines, 5 dendrites, 5 slices, 5 rats). (j) The average spine shrinkage by the LTD protocol in the presence of APV (Supplementary Fig. 3a; 13 spines, 4 dendrites, 4 slices, 2 rats), CGP55845 (22 spines, 5 dendrites, 4 slices, 3 rats), without glutamate uncaging (w/o Glu; 14 spines, 3 dendrites, 3 slices, 2 rats), without the spike (w/o spiking, 33 spines, 6 dendrites, 3 slices, 2 rats) or without GABA uncaging (w/o GABA; 24 spines, 15 dendrites, 8 slices, 5 rats). Data are presented as mean ± s.e.m. ***P < 0.001 versus 0% by Wilcoxon signed-rank test.
Conversely, spine shrinkage was never induced when bAPs preceded glutamate uncaging (LTD protocol, Fig. 1c,d; 2.9 ± 3.7%). Unexpectedly, spine shrinkage was reliably induced when each bAP was paired with GABA uncaging at 458 nm at the dendritic shaft close to the stimulated spine in the LTD protocol (Fig. 1e–i; (38.0 ± 5.2%). We adjusted the power of the blue laser to elicit chloride currents (Fig. 1h) with amplitudes similar to those of miniature inhibitory postsynaptic currents (IPSCs) (Supplementary Fig. 2). Spine shrinkage was eliminated by (2R)-amino-5-phosphonovaleric acid (APV, 50 μM; Fig. 1j and Supplementary Fig. 3a; 2.5 ± 6.5%) and was not induced without spiking or glutamate uncaging (Fig. 1j). These data exclude the possibility that the shrinkage was induced by photodamage and confirm that it required activation of NMDA receptors and spike. Spine shrinkage did not display a strong correlation with initial spine volumes (Supplementary Fig. 3e).
Shrinkage tended to spread to neighboring spines for a distance of up to approximately 15 μm (Figs. 1g and 2a), which did not occur with spine enlargement5, 6, 18, 19. Shrinkage occurred in both distal and proximal spines (Supplementary Fig. 4a) and often resulted in their elimination (Fig. 1g). We also detected a reduction in glutamate sensitivity in stimulated and neighboring spines but not in spines located >15 μm from the uncaged spines (Fig. 2b), indicating that spine shrinkage was associated with homo- and heterosynaptic LTD. These results are consistent with previous observations that LTD depends on GABAA receptors21, 22 and spreads along dendrites23, 24, 25.
Figure 2. Spine shrinkage spread and GABA effects.

14 slices, 10 rats) and neighboring spines at ≤3 μm (15 spines), 3–10 μm (41 spines), 10–15 μm (17 spines) and ≥15 μm (9 spines) from the stimulated spines. (b) The time courses of current amplitudes evoked by glutamate uncaging in stimulated (9 spines, 9 dendrites, 9 slices, 7 rats), neighboring (<3 μm, 10 spines, 9 dendrites, 9 slices, 7 rats) and distant spines (>15 μm, 8 spines, 8 dendrites, 7 slices, 6 rats). The insets show an example of current traces (two-photon stimulation–induced EPSCs (2pEPSCs)) before and after LTD induction. (c) Time points (blue arrows) of GABA uncaging relative to spike onset. (d) Timing dependence of GABA uncaging on spine shrinkage (Supplementary Fig. 3c,d). (e) A drawing of a dendritic shaft where GABA uncaging was applied 25 μm from the stimulated spine. (f) Spine shrinkage was detected when GABA uncaging was within 5 μm of the stimulated spines (33 spines, 14 dendrites, 14 slices, 10 rats) but not when GABA uncaging was more than 25 μm away from the stimulated spines (21 spines, 6 dendrites, 6 slices, 3 rats). Data are presented as mean ± s.e.m. *P < 0.05, **P < 0.01,***P < 0.001 versus 0% by Wilcoxon signed-rank test.
The induction of spine shrinkage required GABA uncaging within a narrow spatiotemporal window. GABA uncaging induced shrinkage only when it occurred within 50 ms of the preceding bAPs (Fig. 2c,d and Supplementary Fig. 3c,d), the time course of which mirrored that of GABA-mediated chloride currents (Fig. 1h). GABA uncaging was also ineffective when it occurred >25 μm away from the stimulated spines (Fig. 2e,f). GABA inhibition must have been mediated by the shunting effects of GABAA receptors because GABA uncaging yielded only small changes (<1 mV) in membrane potentials held at a potential (−65 mV) that was close to the chloride reversal potential. And indeed, spine shrinkage was induced even in the presence of an inhibitor of GABAB receptor, CGP55845 (1 μM; Fig. 1j and Supplementary Fig. 3b; −29.7 ± 4.5%). We were unable to examine the effects of GABAA antagonists on spine shrinkage because the complete blockade of GABAA receptors induced a burst of action potentials during the LTD protocol in our preparations.
Pharmacological properties of spine shrinkage
Shrinkage (−49.3 ± 4.1%) was induced when the GABAA agonist muscimol was applied at 200 nM to the neurons (Fig. 3a–c) using a glass pipette instead of when GABA uncaging was used. Spine shrinkage spread over approximately 15 μm also with muscimol (Fig. 3d and Supplementary Fig. 4b,c). We speculate that spine shrinkage was never induced with the LTD protocol in the absence of the GABAA agonist (Figs. 1d and 3e) partly because the caged-glutamate compounds used for inducing enlargement displayed a mild antagonistic effect on GABAA receptors26, 27 and blocked tonic baseline GABAergic inhibition (Supplementary Fig. 2a)28. In fact, spine shrinkage was induced even with a low muscimol concentration (50 nM; Fig. 3e and Supplementary Fig. 5a; −16 ± 5.3%), which restored tonic GABA-mediated currents (Supplementary Fig. 2b–d). However, the degree of shrinkage with 50 nM muscimol was smaller than that with GABA uncaging or 200 nM muscimol (Fig. 3e), indicating that the synaptic activation of GABAA receptors markedly promoted spine shrinkage. We confirmed that spine shrinkage spread in acute slice preparations with muscimol in the bathing solution (Supplementary Fig. 6), although the extent of shrinkage tended to be smaller, possibly because of the damage from slicing.
Figure 3. Pharmacology of spine shrinkage.

(a) LTD protocol with 200 nM muscimol applied using the same pipette as that used for caged glutamate application. bAPs and glutamate uncaging were repeated 80 times at 5 Hz. (b) Spine shrinkage induced with the LTD protocol with 200 nM muscimol. The spine in which glutamate was uncaged is labeled s; neighboring spines, n1–n3. Scale bar, 2 μm. The soma was located to the left. (c) Average time courses of changes in the volumes of stimulated (21 spines, 21 dendrites, 19 slices, 16 rats) and neighboring spines (27 spines). (d) Average reductions in spine volumes of stimulated (21 spines, 21 dendrites, 19 slices, 16 rats) and neighboring spines at ≤3 μm (27 spines), 3–10 μm (56 spines), 10–15 μm (38 spines) and ≥15 μm (21 spines) from the stimulated spines. (e) Average reductions in spine volumes with the LTD protocol with muscimol (0 nM, 24 spines, 15 dendrites, 8 slices, 5 rats; 50 nM, 28 spines, 11 dendrites, 7 slices, 4 rats; 200 nM, 56 spines, 18 dendrites, 17 slices, 14 rats; Supplementary Fig. 5a) and APV (50 μM, 41 spines, 13 dendrites, 6 slices, 4 rats), MK-801 (1 mM, 14 spines, 5 dendrites, 2 slices, 2 rats), MCPG (1 mM, 28 spines, 10 dendrites, 5 slices, 3 rats), CPA (30 μM, 23 spines, 8 dendrites, 4 slices, 2 rats), Y-27632 Rho-kinase inhibitor (20 μM, 21 spines, 8 dendrites, 3 slices, 3 rats), FK506 (1 μM, 28 spines, 10 dendrites, 5 slices, 4 rats) and P-cofilin peptide (0.5 mM, 18 spines, 5 dendrites, 5 slices, 3 rats) (Supplementary Fig. 5b). Wilcoxon signed-rank test versus 0%: **P < 0.01, ***P < 0.001. Steel’s test versus muscimol 200 nM: 0 nM, 50 nM muscimol, APV, internal MK-801, FK506 and P-cofilin peptide (P < 0.0001), CPA (P = 0.028), MCPG (P = 0.81) and Y-27632 (P = 0.99). (f) Spine volumes during the LTD protocol in controls and with MCPG and FK506 in the bathing solution or MK-801 and P-cofilin peptide in the pipette. The shrinkage amplitudes were averaged among neighboring spines (2–5 spines) in dendrites within 3 μm of the stimulated spines. Data are presented as mean ± s.e.m.
We then studied the pharmacological properties of this spine shrinkage in the presence of 200 nM muscimol (Fig. 3e). Spine shrinkage was abolished when APV (50 μM, 4.8 ± 5.5%) was present in the bathing solution or MK-801 was in the patch pipette (1 mM; 8.4 ± 6.7%), indicating that the shrinkage depended on Ca2+ influx through NMDA receptors (Fig. 3e,f). Spine shrinkage was induced even in the presence of inhibitors of metabotropic glutamate receptors (α-methyl-4-carboxyphenylglycine, MCPG; 1 mM; −40.2 ± 4.9%), of the endoplasmic reticulum Ca2+-ATPase (cyclopiazonic acid, CPA; 30 μM; −28.1 ± 6.8%) and of Rho kinase (Y-27632, 10 μM; −45.8 ± 8.3%) (Fig. 3e,f and Supplementary Fig. 5b,c), suggesting that it required neither intracellular Ca2+ mobilization nor Rho kinase activation. However, the drug FK506 abolished shrinkage (Fig. 3e,f; 0.86 ± 3.4%), indicating that the Ca2+-dependent phosphatase calcineurin was involved in the detection of Ca2+ transients. Calcineurin promotes dephosphorylation and the activation of actin-depolymerizing factors (ADF/cofilin)29, which can be blocked by a phospho-cofilin (P-cofilin) peptide with a phosphorylated Ser3 (refs. 7,29). We found that the P-cofilin peptide abolished spine shrinkage (Fig. 3e,f; 1.0 ± 8.4%), suggesting that cofilin was involved in the shrinkage, which is consistent with a report that cofilin diffuses in the cytosol30 and accumulates in spines undergoing LTD31.
Heterosynaptic competition of spines along a dendrite
Spine enlargement can compete against spreading shrinkage and induce heterosynaptic competition along a dendrite. To demonstrate this, we challenged two neighboring spines with repetitive pairings of spiking and glutamate uncaging: one with LTP and the other with the LTD protocol in the presence of muscimol (Fig. 4a,b). Spine shrinkage spread to neighboring spines with the LTD protocol except for the specific spine that was exposed to the LTP protocol (Fig. 4b–d). Thus, spine enlargement outcompeted shrinkage without affecting the shrinkage of surrounding spines (Fig. 4b–d). These data indicate that the individual modifiability of spines was preserved even when competitive enlargement and shrinkage stimuli were applied along the dendrite. This is consistent with an in vivo imaging study32 in which spine elimination was shown to occur nearly independent of the surrounding spines, assuming that the spines are competing along a dendrite. Moreover, structural plasticity was associated with parallel changes in glutamate-induced currents for both enlargement and shrinkage during heterosynaptic interactions (Fig. 4e).
Figure 4. Competition of the enlargement and shrinkage of neighboring spines.

(a) Spike timing protocol for the heterosynaptic competition of spine enlargement and shrinkage with 200 nM muscimol. Glutamate uncaging was applied 5 ms before the spike and 10 ms after the spike, and repetitive pairing was performed 80 times (5 Hz). (b) Fluorescence images of a dendrite subjected to the LTP (e) and LTD protocols (s). Scale bar, 2 μm. The soma was located to the right. (c) Volume changes in the spines shown in b subjected to the LTD protocol (s) and its neighbors (n) or to the LTP protocol (e) and its neighbors distal to spines with LTD stimuli (ne). The duration of glutamate uncaging in the LTP protocol was 3 ms. (d) Volume changes in spines subjected to the LTD protocol (14 spines, 14 dendrites, 14 slices, 13 rats) and their neighbors (34 spines, 14 dendrites, 14 slices, 13 rats) and to the LTP protocol (14 spines, 14 dendrites, 14 dendrites, 13 rats) and its distal neighbors (12 spines, 12 dendrites, 11 slices, 11 rats). The duration of glutamate uncaging in the LTP protocol was 1.8 ms (8 spines, 8 dendrites, 8 slices, 7 rats) or 3 ms (6 spines, 6 dendrites, 6 slices, 6 rats). (e) Peak amplitude changes in glutamate-induced currents (2pEPSCs) in spines subjected to the LTP protocol (5 spines, 5 dendrites, 5 slices, 5 rats), LTD protocol (5 spines, 5 dendrites, 5 slices, 5 rats), neighboring spines (6 spines, 5 dendrites, 5 slices, 5 rats) and distant ones (4 spines, 4 dendrites, 4 slices, 4 rats) ≥15 μm from stimulated ones. Glutamate-induced currents were recorded with voltage clamp at a holding potential of −65 mV. (f) Competition of spine enlargement against the spreading shrinkage induced by neighboring spine stimulation with the LTD protocol (muscimol, 200 nM or 50 nM; Supplementary Fig. 7a,b). Enlargement outcompeted shrinkage when spines were subjected to LTP protocols as in a with glutamate uncagings of 1.2 ms, 1.8 ms or 3 ms with 200 nM muscimol and 0.6 ms or 1.2 ms with 50 nM muscimol (Supplementary Fig. 7a,b). *P < 0.05, **P < 0.01 by Steel’s test versus 0 ms. The curves were drawn by eye. (g) The volume changes in spines subjected to the LTP protocol (5 spines, 5 dendrites, 5 slices, 3 rats, 3 ms), the LTD protocol and neighboring spines (23 spines, 5 dendrites, 5 slices, 3 rats) in the presence of dephosphorylated (dp)-cofilin peptide in the patch pipette and 200 nM muscimol. Data are presented as mean ± s.e.m.
Spine shrinkage spread gave rise to competition between spine enlargement and shrinkage (Fig. 4f and Supplementary Figs. 5c and 7a,b). Spine enlargement required longer uncaging (1.2–3 ms) when the optimal LTD protocol (200 nM muscimol) was applied to the neighboring spines (Fig. 4b), whereas enlargement was induced with a short duration of glutamate uncaging (0.6 ms), which mimicked miniature excitatory postsynaptic currents, when spine shrinkage was weakly induced with 50 nM muscimol (Figs. 3e,4f). The spine subjected to the LTP protocol instead exhibited shrinkage when cofilin phosphorylation was blocked by a dephosphorylated cofilin peptide (Fig. 4f,g)29, supporting the finding that the competitive interactions of spines involved ADF/cofilin.
Ca2+ nanodomain effects of NMDAR receptors
Our data indicate that GABAA receptors promoted spine shrinkage by suppressing bAP-triggered bulk increases in [Ca2+]i. In fact, GABA uncaging suppressed the bAP-triggered Ca2+ transients within 20 μm of the uncaging site (Fig. 5), consistent with the effects of GABA uncaging on spine shrinkage (Fig. 2f). GABA uncaging also suppressed Ca2+ transients by bAP and glutamate uncaging in the LTD timing (Fig. 6a and Supplementary Fig. 8), similarly to muscimol (Fig. 6b and Supplementary Fig. 9). This reduction was mainly due to bAP-triggered Ca2+ influx and depended less on NMDA receptors (Fig. 6a), possibly reflecting the far steeper voltage dependence of voltage-gated Ca2+ channels relative to NMDA receptors. Moreover, spine shrinkage was induced in the presence of 2–3 mM ethylene glycol tetraacetic acid (EGTA; −38.8 ± 5.8%) in the patch pipette, even in the absence of muscimol (Fig. 6c–e and Supplementary Fig. 7c), but not in the presence of 10 mM EGTA (Fig. 6e and Supplementary Fig. 7c; 11.9 ± 13.1%), indicating that shrinkage required moderate increases in bulk [Ca2+]i (Fig. 6f,g and Supplementary Fig. 10), as proposed for LTD33, 34, 35.
Figure 5. The spatial profile of GABA uncaging-mediated inhibition of bAP-induced dendritic Ca2+ transients.

(a) mCherry fluorescence image of a dendrite to which a combination of bAPs and GABA uncaging was applied at the 0-μm location. The soma was located to the bottom. (b,c) GCaMP6s (see Online Methods) images of the dendrite shown in a. The images were normalized by the images before bAP, and the relative increases in the fluorescence were pseudocolor coded. Each image is an average of 6–8 frames. (d) Time courses of fluorescence changes without (black) or with (blue) GABA uncaging at each dendritic location. The traces are averages of 4–13 dendrites. (e) Relative reductions in the peak amplitudes of bAP-induced Ca2+ transient along the dendrite. Kruskal–Wallis test, P < 0.0055. Steel’s test, *P < 0.05, **P < 0.01 (4–13 dendrites, 4–10 slices, 2–5 rats). Data are presented as mean ± s.e.m.
Figure 6. Spine Ca2+ signaling and effects of cytosolic EGTA.

(a) Effects of GABA uncaging (blue) on peak fluorescence ratios between the low-affinity (KD = 2.3 μM) Ca2+ indicator Fluo5F and Alexa 594 (G/R) for spine [Ca2+]i increases (Supplementary Fig. 8) by a combination of bAP and glutamate uncaging in LTD (22 spines, 6 dendrites, 3 slices, 3 rats), bAP (25 spines, 9 dendrites, 5 slices, 5 rats) and glutamate uncaging (17 spines, 6 dendrites, 5 slices, 4 rats). Two-sided paired t-test versus without GABA uncaging. (b) Effects of muscimol on G/R ratios between Fluo5F and Alexa 594 (Supplementary Fig. 9) by a combination of bAP and glutamate uncaging in LTD at 0 nM (51 spines, 13 dendrites, 13 slices, 8 spines), 50 nM (33 spines, 6 dendrites, 6 slices, 6 rats) and 200 nM (18 spines, 7 dendrites, 7 slices, 2 rats). Two-sided paired t-test versus 0 nM. (c) LTD protocol with 3 mM EGTA in the patch pipette instead of muscimol in the extracellular solution. (d) Spine shrinkage by the LTD protocol with 3 mM EGTA in the patch pipette. Arrow indicates spine in which glutamate was uncaged. Scale bar, 2 μm. (e) Dependence of shrinkage on EGTA concentration (Supplementary Fig. 7c). (f) Dependence of G/R ratio on EGTA concentration (Supplementary Fig. 10). Averaged fluorescence ratios with 0.5 mM EGTA were normalized to 100%. (g) Spine shrinkage dependence on G/R ratio. Data are presented as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001 versus 0% by Wilcoxon signed-rank test.
Notably, spine shrinkage was never induced when the laser strength was reduced (3–4 mW) during glutamate uncaging (Fig. 7a and Supplementary Fig. 7e), even though the spine [Ca2+]i increases were similarly suppressed (Fig. 7b and Supplementary Fig. 9e–i) as in the presence of muscimol (Fig. 6b). This suggests that spine shrinkage required sufficient activation of glutamate receptors and possibly involved the Ca2+ nanodomain of NMDA receptors36. To test this hypothesis, we compared the effects of whole-cell applications of two Ca2+-buffering compounds, EGTA and 1,2-bis(o-aminophenoxy)ethane-N,N,N′N′-tetraacetic acid (BAPTA). These have similar affinities for Ca2+ (0.2 μM), but BAPTA binds Ca2+ 160 times faster (kon = 4.0 × 108 M−1 s−1)37, 38.
Figure 7. Spine Ca2+ signaling and effects of cytosolic BAPTA.
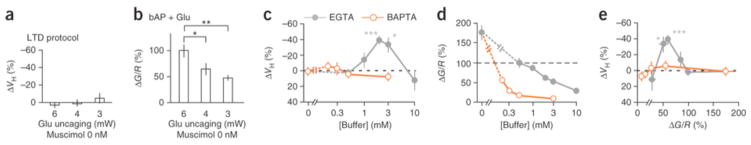
(a) Effect on spine shrinkage of laser irradiation power used for glutamate uncaging, with 6 mW (24 spines, 15 dendrites, 8 slices, 5 rats), 4 mW (28 spines, 6 dendrites, 2 slices, 2 rats) or 3 mW (15 spines, 6 dendrites, 2 slices, 2 rats) (Supplementary Fig. 7e). (b) Effect on fluorescence ratio, for the same spines, of laser power used for glutamate uncaging (30 spines, 5 dendrites, 5 slices, 3 rats), with 6 mW, 4 mW or 3 mW (Supplementary Fig. 9e). P = 0.014 by ANOVA. P = 0.016 and 0.0016 by Scheffe test for 4 mW and 3 mW, respectively, versus 6 mW. (c) Dependence of shrinkage on the BAPTA concentration (Supplementary Fig. 7d), relative to that of EGTA (same as in Fig. 6e). (d) Dependence of peak fluorescence ratio (G/R) on BAPTA concentration (Supplementary Fig. 10), relative to that of EGTA (same as in Fig. 6f). Averaged fluorescence ratios with 0.5 mM EGTA were normalized as 100%. (e) Spine shrinkage dependence on G/R ratio in the presence of BAPTA and EGTA (same as Fig. 6g). Data are presented as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001 versus 0% by Wilcoxon signed-rank test.
Spine shrinkage was not induced with BAPTA (0.2 mM) (Fig. 7), which generated bulk [Ca2+]i increases similar to those with EGTA (2–3 mM) (Fig. 7d and Supplementary Fig. 10c). The BAPTA concentration, which was ten times lower (0.2 mM), was as effective for global Ca2+ transients as EGTA (2 mM) (Fig. 7d and Supplementary Fig. 10c) because of its large kon value39. In support of this, the decay of Ca2+ transients was consistently slowed with BAPTA (Supplementary Fig. 10c)39. Even considering the difference in the concentration, the predicted length constants of the Ca2+ domain ( , where DCa represents the Ca2+ diffusion constant of 220 μm2 s−1), were smaller with BAPTA (50 nm) than with EGTA (200 nm)36. Thus, in addition to the moderate increases in bulk [Ca2+]i, spine shrinkage required a Ca2+ nanodomain of NMDA receptors that was larger than 50 nm. Therefore, it was dependent on the sufficient activation of glutamate receptors.
Discussion
Promotion of spine shrinkage by GABA
We established conditions that induced the marked shrinkage and even the elimination of dendritic spines when both glutamate and GABA receptors were activated in synchrony with action potentials. The shrinkage was markedly augmented by the activation of GABAA receptors, which suppressed the global [Ca2+]i increases that are seen with the LTD protocol. Reduction in bulk increases in global [Ca2+]i was able to replace the effects of GABA, as the cytosolic application of EGTA (2–3 mM) mimicked the effects of GABA. This is consistent with reports of moderate increases in [Ca2+]i proposed for LTD33, 34, 35. Thus, GABAA receptors themselves were not required for spine shrinkage, as long as [Ca2+]i increases were in the appropriate range. This may resolve the controversy concerning the role of GABA in LTD induction among different experimental conditions21, 22, 40, 41. Under our conditions, the [Ca2+]i increase had to be high enough to prohibit spine shrinkage and the [Ca2+]i increase needed to be suppressed by GABAergic inhibition or EGTA to induce spine shrinkage (Supplementary Figs. 5c and 9i).
Our data also indicated that weak activation of glutamate receptors was not able to replace the effects of GABA and that sufficient activation of NMDA receptors was necessary for spine shrinkage to produce the Ca2+ nanodomain of NMDA receptors with a size over 50 nm. GABAergic inhibition enhanced spine shrinkage because it effectively suppressed global increases in [Ca2+]i, although it less efficiently suppressed Ca2+ entry through NMDA receptors owing to the weaker voltage dependence of NMDA receptors. The Ca2+ domain of NMDA receptors was sensed by downstream signaling molecules to detect the sufficient activation of glutamatergic receptors in a spine. If this mechanism were not in place, moderate increases in dendritic [Ca2+]i readily induced by spikes could result in synaptic plasticity even when there was no excitatory synaptic input. Calcineurin was necessary for spine shrinkage, and it is a good candidate for sensing both global and nanodomain [Ca2+]i increases because it has high-affinity EF-hand Ca2+-binding sites, in addition to a binding site for calmodulin42, whose Ca2+ affinity is low and which often acts as a sensor for the Ca2+ nanodomain19.
The promotion of synapse elimination and LTD by GABAergic inhibition may be a general phenomenon in the cortex. In fact, competitive elimination of spine synapses is greatly enhanced during the critical period of sensory cortices, and such synaptic plasticity is facilitated by maturation of GABAergic neurons or even application of GABAA agonists1, 16, 43. This is consistent with our finding that spine shrinkage can be promoted either by uncaging of a caged GABA compound that mimics IPSCs or by tonic application of a GABAA agonist, muscimol. Spine elimination is infrequent in the adult cortex in vivo9, 11, 32 possibly because plasticity is reduced after closure of the critical period1, the exact conditions for spine shrinkage may be only infrequently met and the drive toward spine enlargement caused by reverberating network activities outcompetes the drive toward spine shrinkage in the cell assembly.
GABAergic inhibition during a short period (<50 ms) before action potentials was sufficient to induce spine shrinkage. This is, to our knowledge, the first demonstration of a narrow time window for GABA action, and the precise spike-timing dependence of GABA influence on spine shrinkage likely assists the formation of neuronal networks that operate synchronously, with oscillatory firing patterns generated by inhibitory input44. Because GABA uncaging induced rapid currents as IPSCs, the same effects on Ca2+ signaling and synaptic plasticity would be expected for IPSCs. Apical dendritic branches of CA1 pyramidal neurons are innervated by specific inhibitory neurons—for example, bistratified cells that are involved in theta and gamma oscillations45. Because GABA uncaging in our experiments was local and did not suppress somatic Na+ spikes, local GABAergic circuits can fine-tune the selection of synapses by suppression of local Ca2+ transients.
Competitive interactions of dendritic spines
The spread of spine shrinkage contrasted clearly with enlargement, which was restricted to stimulated spines5, 18, 19, although enlargement was triggered by stronger stimuli (LTP protocol or high-frequency stimulation), which must be associated with larger increases in [Ca2+]i. Therefore, if Ca2+ were responsible for the spread of spine shrinkage, spine shrinkage should have been induced in the spines surrounding the enlarged spines, which was not the case. Thus, Ca2+ cannot simply account for the spread of spine shrinkage. Another candidate for a spreading factor is dephosphorylated ADF/cofilin, which is known to be diffusible in the cytosol30, to accumulate with β-arrestin in spines during LTD31 and to sever and depolymerize actin fibers and thereby destabilize the postsynaptic density. The precise molecular mechanisms as to how cofilin induces long-term spreading spine shrinkage require further investigation.
Recently, input-specific spine shrinkage has been reported following low-frequency stimulation of a single spine with glutamate uncaging46, in which the shrinkage occurred to a smaller extent (25%) than in our study under a protocol in which extracellular Ca2+ concentrations were lowered. In our conditions, which used physiological Ca2+ concentrations and action potentials, the extent of spine shrinkage often exceeded 50% and even included spine elimination. Thus, there may be two forms of spine shrinkage. The spreading form of spine shrinkage has been detected in both cultured and acute slice preparations, consistent with the spreading LTD reported in many studies23, 24, 25.
STDP has been thought to mediate the competitive modification in the functional strengths of synaptic connections in an entire cell15. We have demonstrated that both LTP and LTD that were induced with STDP protocols can be associated with the structural plasticity of spines that involves enlargement and shrinkage, respectively, establishing that STDP can structurally select synapses. In addition, we here report a new form of heterosynaptic competition that depends on the local spread of spine shrinkage. The spread of spine shrinkage triggered competitive interactions of spines along a dendrite, and such shrinkage strengthened the competition among synapses and even eliminated spines that were left unstimulated. This non-Hebbian plasticity is necessary in theory for the effective formation of neuronal networks47 because unused synapses should be removed for optimization of the neuronal circuit. Because shrinkage spread only up to 15 μm, this competition may be involved in the local optimization of synaptic connections along a dendrite48, 49, unlike the homeostatic plasticity that globally affects synapses in a cell.
Our study suggests that spine shrinkage is induced by a signaling cascade involving calcineurin and actin depolymerizing factor, ADF/cofilin7. In contrast, spine enlargement involves the activation of calcium/calmodulin-dependent protein kinase II (CaMKII), which eventually causes the phosphorylation of cofilin29. We found that the competitive interactions between the enlargement and shrinkage of spines critically involved cofilin phosphorylation and dephosphorylation, such that if phosphorylation dominated, spines enlarged. As CaMKII and calcineurin deactivate when [Ca2+]i is restored, the long-term structural effects are likely mediated by downstream effectors, such as cofilin. The cofilin-dependent local competition is likely used in the brain in vivo, as knockout mice lacking cofilin-1 show severe deficits in LTP and LTD, as well as in many learning tasks50.
In summary, we have established conditions for the reliable induction of spine shrinkage and elimination and found a new mechanism by which GABAA receptors modulate synaptic plasticity through local dendritic Ca2+ signaling. Thus, the excitation-inhibition balance affects synaptic contacts not only by the modulation of spike activities but also more directly by the GABAergic inhibition of local dendritic Ca2+ signaling.
Methods
Slice culture preparation
Hippocampal slices (350 μm thick) were prepared from 6- to 8-d-old Sprague–Dawley rats (both male and female), mounted onto 0.4-μm culture plate inserts (EMD Millipore) and incubated at 35 °C under 5% CO2 in medium comprising 50% minimum essential media (Gibco), 25% Hanks’ balanced salt solution (Gibco), 25% horse serum (Gibco) and glucose (6.5 g/l). After 8–13 d in vitro, slices were transferred individually to recording chambers and superfused with artificial cerebral spinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3 and 20 mM glucose, which was bubbled with 95% O2 and 5% CO2. Bathing solutions also contained 200 μM Trolox (Sigma). Hippocampal CA3 regions were removed in order to reduce burst firing. All physiological experiments were performed at 30–32 °C.
In (2R)-amino-5-phosphonovaleric acid (APV; 50 μM, R&D Systems), CGP55845 (1 μM, Tocris), MCPG (1 mM, R&D Systems), cyclopiazonic acid (CPA; 30 μM, Sigma) and Y-27632 (10 μM, R&D Systems) experiments, slices were superfused for at least 30 min with each inhibitor before glutamate uncaging. In FK506 (1 μM, Sigma) experiments, slices were incubated with FK506 for 1–3 h before being transferred to recording chambers and perfused with normal ACSF. CPA and FK506 stock solutions were dissolved in dimethylsulfoxide (DMSO; final concentration 0.005–0.03%), which did not affect spine shrinkage (mean ± s.e.m.: control, −48.6 ± 4.1%, n = 21; DMSO alone, −48.1 ± 4.8%, n = 9). MK-801, P-cofilin peptide (MA(pS)GVAVSDGVIKVFN, 0.5 mM, BEX) or dephosphorylated cofilin peptide (MASGVAVSDGVIKVFN, 0.5 mM, BEX) were dissolved in pipette solutions. The experimental protocols were approved by the Animal Experimental Committee of the Faculty of Medicine, University of Tokyo.
Acute slice preparation
Acute hippocampal slices (350 μm thick) were prepared from 19- to 28-d-old Sprague-Dawley rats, using a chilled solution containing 120 mM choline chloride, 3 mM KCl, 8 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, 1 mM sodium pyruvate, 5 mM sodium ascorbate and 25 mM glucose, which was bubbled with 95% O2 and 5% CO2. The slices were kept in ACSF and incubated at 33 °C for 1 h, and then placed at room temperature until use. Electrophysiological experiments were conducted as the cultured-slice experiments, except that 0.5 mM 4-carboxymethoxy-5,7-dinitroindolinyl-glutamate (CDNI-glutamate) and 400 nM muscimol were included in the bathing solution. The irradiation of the 720 nm laser was 2.4 ms duration, 80 times at 1 Hz. A longer glutamate uncaging was used in acute preparations than in cultured ones because intact dendrites were found in deeper layers (50 μm) of acute slices, and stronger irradiation was required to generate EPSC-like currents with similar amplitudes5. We also used a slightly higher concentration of muscimol (400 nM) than in slice culture preparations (200 nM), as it appeared more effective in inducing spine shrinkage in acute slices (data not shown).
Two-photon excitation imaging
Two-color imaging and uncaging were performed as follows51. Two-photon imaging of dendritic spines was performed using an upright microscope (BX61WI; Olympus) equipped with a FV1000 laser-scanning microscope system (FV1000; Olympus) and a water-immersion objective lens (LUMPlanFI/IR; 60×; numerical aperture, 0.9). The system included two mode-locked, femtosecond-pulse Ti:sapphire lasers (MaiTai, Spectra Physics) set at 720 nm and 830 nm for glutamate uncaging and Alexa 594, respectively. Each laser was connected to the microscope through an independent scan head and gated using an acousto-optic modulator for two-photon imaging and uncaging of caged glutamate with 2 mM CDNI-glutamate. For two-color uncaging, a blue argon laser (458 nm; Showa Optronics) was directed into one of the scan heads for one-photon uncaging of the caged-GABA compound ruthenium-based GABA (RuBi-GABA, 50 μM, approximately 0.2 mW).
Second or third dendritic branches were used for the imaging and uncaging experiments. Three-dimensional dendritic reconstructions were generated by summing the pixel fluorescence values in the 17–29 xy images separated by 0.5 μm. Dendritic fluorescence, which increased gradually, even 20 min after whole-cell perfusion, was corrected by the fluorescence of an entire dendritic region. Spine head volumes were estimated from total fluorescence intensity. Only one spine per dendrite was stimulated for shrinkage because of the spread. Neighboring spines were within 3 μm of stimulated spines unless otherwise stated. Shrinkage amplitudes were averaged among neighboring spines (2–5 spines) in dendrites within 3 μm of stimulated spines. Spines that changed volume by ≥30% before uncaging were excluded from data analyses (13.6%, 48 of 352 spines). Long-term spine shrinkages were measured by ratio of the average before induction to the average between 60 and 80 min after induction.
We estimated spine-head volume (V) from the total fluorescence intensity (F) in the stacked images of spines. A conversion coefficient, V/F, was obtained from the fluorescence profile of the largest, roundest spine on each dendrite4, 5, which was subsequently applied to all other spines on the dendrite. Two-photon images were analyzed with IPLab (Scanalytics Inc.), ImageJ (NIH; http://rsb.info.nih.gov/ij/) and MATLAB (MathWorks) software.
Electrophysiology and two-color uncaging
For normal whole-cell recordings, patch-clamp electrodes (open-tip resistance, 4–7 MΩ) were filled with 138 mM potassium gluconate, 4 mM MgCl2, 10 mM disodium phosphocreatine, 50 μM Alexa 594 (Life Technologies Corporation), 4 mM ATP (sodium salt), 0.4 mM GTP (sodium salt), 10 mM HEPES (pH 7.2 with KOH), 0.5 mM EGTA (potassium salt) and 5 μM β-actin (human platelet; Cytoskeleton). Series resistance was 19.5 ± 4.8 MΩ (mean ± s.d.), and the resting membrane potential was −59.2 ± 2.7 mV (mean ± s.d.). Cells were voltage clamped at −65 mV, except during spike and uncaging (Axopatch 200B, Molecular Devices). Cells with resting potentials more than −53 mV at uncaging were excluded from the data analyses. Small hyperpolarizing holding currents were applied to maintain voltages near −65 mV during spike and uncaging. Action potentials were induced by depolarizing currents of 1–2 nA for 2 ms. AMPA receptor–mediated currents were obtained by voltage-clamping cells at −65 mV during two-photon uncaging at the spine tip with 6-mW laser power. Currents were evoked 3–5 times at each time point, low-pass filtered at 2 kHz, sampled at 10 kHz and averaged.
CDNI-glutamate (2 mM)52, 53 and RuBi-GABA (0.05 mM, R&D Systems)54 were puffed locally from glass pipettes near selected dendrites. Selective photolysis of CDNI-glutamate and RuBi-GABA was performed with femtosecond lasers at 720 nm (0.6 ms, unless otherwise stated) and argon lasers at 458 nm, respectively. CDNI-glutamate was not photolyzed by the blue laser. RuBi-GABA, which has a low two-photon cross-section, did not induce measurable currents by femtosecond lasers at 720 nm with 3–6.5 mW (Supplementary Fig. 1). Photoreleased glutamate levels were adjusted by changing laser powers (approximately 6 mW) to evoke currents through AMPA-sensitive glutamate receptors with amplitudes (approximately 5–20 pA) similar to those of miniature excitatory postsynaptic currents. Photoreleased GABA levels were adjusted by changing laser powers (0.15–0.2 mW) to evoke currents through GABAA receptors with amplitudes similar to those of miniature IPSCs. We uncaged GABA for 1 ms at each of four points that flanked the dendritic shaft in order to alleviate possible damage from laser irradiation (Fig. 1g). The damage was small, as laser irradiation without caged compounds resulted in no increase in [Ca2+]i.
In the experiments shown in Figures 3 and 4, GABA uncaging was replaced with 200 nM or 50 nM muscimol (Sigma) in the puffing solution, and repetitive pairing was performed 80 times (5 Hz). For the enlargement and shrinkage competition of spines with spike-timing protocols with 200 nM muscimol, glutamate uncaging was applied 5 ms before the spikes for spine enlargement and 10 ms after the spikes for spine shrinkage. The puff application of muscimol covered more than a 200 μm radius because the opening of the glass application pipette was larger than 100–200 μm.
For the whole-cell recordings of tonic GABA-mediated inhibitory currents (Supplementary Fig. 2), patch-clamp electrodes contained 120 mM CsCl, 2 mM NaCl, 10 mM disodium phosphocreatine, 50 μM Alexa 594, 2 mM ATP (sodium salt), 0.5 mM GTP (sodium salt), 10 mM HEPES (pH 7.2 with CsOH), 10 mM EGTA (cesium salt) and 4 mM MgCl2, whereas the external solutions contained 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM; Sigma), APV (50 μM) and tetrodotoxin (1 μM). The cells were voltage-clamped at −65 mV.
Ca2+ imaging
For the Ca2+ measurements, the pipette solutions contained 300 μM of a low-affinity Ca2+ indicator, Fluo-5F (KCa, 2.3 μM; Life Technologies Corporation) and 20 μM Alexa 594, and the fluorescence intensities were measured at 490–540 nm (G) and 570–630 nm (R), respectively. We obtained the ratio, ΔG(t)/R = (G(t) − G0)/R, where G(t) is the fluorescence intensity at time t, G0 is the initial intensity and R is the red-channel averaged intensity. Spine ratio time courses were obtained three times and averaged. To test GABA uncaging or muscimol effects, 5–9 spines were selected from the same dendrites and their mean amplitudes compared in the presence of 0.5 mM EGTA in the internal solution. When Ca2+ buffers were systematically altered (Figs. 6 and 7) with 1, 2, 3 or 10 mM EGTA or 0.2, 0.3, 0.5 or 3 mM BAPTA instead of 0.5 mM EGTA, 16.7% of CaCl2 relative to the buffer was added to set the baseline Ca2+ concentration to 30 nM. The peak values of ΔG(t)/R were obtained from 5–9 spines from the same dendrite, and the mean values were compared across dendrites (Figs. 6a,b,f and 7b,d). They were normalized to 100% according to the mean value for 0.5 mM EGTA.
For the imaging of bAP-induced Ca2+ transient along a dendrite, neurons were transfected with mCherry and GCaMP6s (Addgene)55 constructs using a gene gun (PDS-1000; Bio-Rad) at P14 and used for the experiments between P17 and P22. Two-photon excitation was performed at 970 nm, and the fluorescence intensities were measured at 490–540 nm for GCaMP6s and 570–630 nm for mCherry. The pyramidal neurons were whole-cell clamped to generate action potentials. RuBi-GABA (50–300 μM) was uncaged with a 468-nm laser (0.1–0.2 mW) at 5–10 ms before action potentials. The Ca2+ images were smoothed by a Gaussian spatial filter with an s.d. of 13.8 μm (Fig. 5b).
Statistical analysis
Spine shrinkage, which is presented as mean ± s.e.m. (n = number of dendrites), was analyzed by Wilcoxon signed-rank tests in Figures 1,2,3, 6e and 7c and Supplementary Figure 6d. The data in Figures 3e and 4f were significant by Kruskal–Wallis tests (P < 0.00001 and P = 0.004) and further analyzed by Steel tests. For the Ca2+ imaging data, the averaged [Ca2+]i values were obtained from several spines in a dendrite and compared across dendrites with parametric tests (Figs. 6a,b and 7b): the data in Figure 6a,b were tested by two-sided paired t-tests; those in Figure 7b were significant by ANOVA (P = 0.014), and a post hoc analysis was performed with a Scheffe test. P < 0.05 was considered statistically significant. Data collection and analysis were not performed blind to the conditions of the experiments. Data were not randomized for analysis. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications in the field5, 6, 19.
Supplementary Material
Acknowledgments
We thank K. Mizuno, K. Ohashi, G.J. Augustine, C. O’Donnell, S. Okabe and J. Eilers for discussions and C. Maeda, M. Ogasawara, H. Ohno, M. Ishikawa and K. Tamura for technical assistance. This work was supported by Grants-in-Aid for Specially Promoted Area (no. 21000009 to H.K.), Scientific Research (C) (no. 21500367 to J.N.), Scientific Research on Priority Areas (‘Elucidation of neural network function in the brain’, no. 20021008 to M.M.), Young Scientist (A) (no. 19680020 to M.M.), Scientific Research on Innovative Areas ‘Mesoscopic Neurocircuitry’ (no. 22115005 to M.M.), the Global COE Program (‘Integrative Life Science based on the study of Biosignaling Mechanisms’ to H.K.) and the Strategic Research Program for Brain Sciences (‘Neuroinformatics of Emotion’ to H.K.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by the US National Institutes of Health (grants GM53395 and NS69720 to G.C.R.E.-D.). In addition, this work was supported by a Mitsubishi Foundation grant to M.M. and a Research Grant from the Human Frontier Science Program to H.K.
Contributor Information
Tatsuya Hayama, Laboratory of Structural Physiology, Center for Disease Biology and Integrative Medicine, Faculty of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, Japan. CREST, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan.
Jun Noguchi, Laboratory of Structural Physiology, Center for Disease Biology and Integrative Medicine, Faculty of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, Japan. CREST, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan.
Satoshi Watanabe, Laboratory of Structural Physiology, Center for Disease Biology and Integrative Medicine, Faculty of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, Japan. CREST, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan.
Noriko Takahashi, Laboratory of Structural Physiology, Center for Disease Biology and Integrative Medicine, Faculty of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, Japan. CREST, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan.
Akiko Hayashi-Takagi, Laboratory of Structural Physiology, Center for Disease Biology and Integrative Medicine, Faculty of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, Japan. CREST, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan. PRESTO, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan.
Graham C R Ellis-Davies, Department of Neuroscience, Mount Sinai School of Medicine, New York, New York, USA.
Masanori Matsuzaki, CREST, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan. PRESTO, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan. Division of Brain Circuits, National Institute for Basic Biology, The Graduate University of Advanced Studies (Sokendai), Okazaki, Japan.
Haruo Kasai, Laboratory of Structural Physiology, Center for Disease Biology and Integrative Medicine, Faculty of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, Japan. CREST, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan.
References
- 1.Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- 2.Nakayama H, et al. GABAergic inhibition regulates developmental synapse elimination in the cerebellum. Neuron. 2012;74:384–396. doi: 10.1016/j.neuron.2012.02.032. [DOI] [PubMed] [Google Scholar]
- 3.Matsuzaki M, et al. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noguchi J, et al. In vivo two-photon uncaging of glutamate revealing the structure-function relationships of dendritic spines in the neocortex of adult mice. J Physiol (Lond ) 2011;589:2447–2457. doi: 10.1113/jphysiol.2011.207100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuzaki M, Honkura N, Ellis-Davies G, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 8.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 9.Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Yang G, Pan F, Gan W. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 12.Mori H, Mishina M. Structure and function of the NMDA receptor channel. Neuropharmacology. 1995;34:1219–1237. doi: 10.1016/0028-3908(95)00109-j. [DOI] [PubMed] [Google Scholar]
- 13.Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- 14.Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 15.Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- 16.Hensch TK, Fagiolini M. Excitatory-inhibitory balance and critical period plasticity in developing visual cortex. Prog Brain Res. 2005;147:115–124. doi: 10.1016/S0079-6123(04)47009-5. [DOI] [PubMed] [Google Scholar]
- 17.Mataga N, Mizuguchi Y, Hensch TK. Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron. 2004;44:1031–1041. doi: 10.1016/j.neuron.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka J, et al. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanemoto Y, et al. Spatial distributions of GABA receptors and local inhibition of Ca2+ transients studied with GABA uncaging in the dendrites of CA1 pyramidal neurons. PLoS ONE. 2011;6:e22652. doi: 10.1371/journal.pone.0022652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steele PM, Mauk MD. Inhibitory control of LTP and LTD: stability of synapse strength. J Neurophysiol. 1999;81:1559–1566. doi: 10.1152/jn.1999.81.4.1559. [DOI] [PubMed] [Google Scholar]
- 22.Nishiyama M, Togashi K, Aihara T, Hong K. GABAergic activities control spike timing- and frequency-dependent long-term depression at hippocampal excitatory synapses. Front Synaptic Neurosci. 2010;2:22. doi: 10.3389/fnsyn.2010.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abraham WC, Goddard GV. Asymmetric relationships between homosynaptic long-term potentiation and heterosynaptic long-term depression. Nature. 1983;305:717–719. doi: 10.1038/305717a0. [DOI] [PubMed] [Google Scholar]
- 24.Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- 25.Wang SS, Khiroug L, Augustine GJ. Quantification of spread of cerebellar long-term depression with chemical two-photon uncaging of glutamate. Proc Natl Acad Sci USA. 2000;97:8635–8640. doi: 10.1073/pnas.130414597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fino E, et al. RuBi-glutamate: two-photon and visible-light photoactivation of neurons and dendritic spines. Front Neural Circuits. 2009;3:2. doi: 10.3389/neuro.04.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuzaki M, Hayama T, Kasai H, Ellis-Davies GCR. Two-photon uncaging of gamma-aminobutyric acid in intact brain tissue. Nat Chem Biol. 2010;6:255–257. doi: 10.1038/nchembio.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai D, et al. Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by gamma-aminobutyric acid(A) receptors in hippocampal neurons. Mol Pharmacol. 2001;59:814–824. doi: 10.1124/mol.59.4.814. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein BW, Bamburg JR. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20:187–195. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai FP, et al. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 2008;27:982–992. doi: 10.1038/emboj.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pontrello CG, et al. Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci USA. 2012;109:E442–E451. doi: 10.1073/pnas.1118803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai CS, Franke TF, Gan WB. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- 33.Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- 36.Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- 38.Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markram H, Roth A, Helmchen F. Competitive calcium binding: implications for dendritic calcium signaling. J Comput Neurosci. 1998;5:331–348. doi: 10.1023/a:1008891229546. [DOI] [PubMed] [Google Scholar]
- 40.Wittenberg GM, Wang SS. Malleability of spike-timing-dependent plasticity at the CA3–CA1 synapse. J Neurosci. 2006;26:6610–6617. doi: 10.1523/JNEUROSCI.5388-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abraham WC, Wickens JR. Heterosynaptic long-term depression is facilitated by blockade of inhibition in area CA1 of the hippocampus. Brain Res. 1991;546:336–340. doi: 10.1016/0006-8993(91)91498-p. [DOI] [PubMed] [Google Scholar]
- 42.Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 43.Yang EJ, Lin EW, Hensch TK. Critical period for acoustic preference in mice. Proc Natl Acad Sci USA. 2012;109:17213–17220. doi: 10.1073/pnas.1200705109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paulsen O, Moser EI. A model of hippocampal memory encoding and retrieval: GABAergic control of synaptic plasticity. Trends Neurosci. 1998;21:273–278. doi: 10.1016/s0166-2236(97)01205-8. [DOI] [PubMed] [Google Scholar]
- 45.Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oh WC, Hill TC, Zito K. Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc Natl Acad Sci USA. 2013;110:E305–E312. doi: 10.1073/pnas.1214705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiete IR, Senn W, Wang CZ, Hahnloser RH. Spike-time-dependent plasticity and heterosynaptic competition organize networks to produce long scale-free sequences of neural activity. Neuron. 2010;65:563–576. doi: 10.1016/j.neuron.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 48.Govindarajan A, Kelleher RJ, Tonegawa S. A clustered plasticity model of long-term memory engrams. Nat Rev Neurosci. 2006;7:575–583. doi: 10.1038/nrn1937. [DOI] [PubMed] [Google Scholar]
- 49.Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:291–307. doi: 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 50.Rust MB, et al. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010;29:1889–1902. doi: 10.1038/emboj.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuzaki M, Kasai H. Two-photon uncaging microscopy. Cold Spring Harbor Protocols. 2011 doi: 10.1101/pdb.prot5620. pdb prot5620. [DOI] [PubMed] [Google Scholar]
- 52.Ellis-Davies GCR. A practical guide to the synthesis of dinitroindolinyl-caged neurotransmitters. Nat Protoc. 2011;6:314–326. doi: 10.1038/nprot.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellis-Davies GCR, Matsuzaki M, Paukert M, Kasai H, Bergles D. 4-Carboxymethoxy-5,7-dinitroindolinyl-Glu: an improved caged glutamate for expeditious ultraviolet and two-photon photolysis in brain slices. J Neurosci. 2007;27:6601–6604. doi: 10.1523/JNEUROSCI.1519-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rial Verde EM, Zayat L, Etchenique R, Yuste R. Photorelease of GABA with visible light using an inorganic caging group. Front Neural Circuits. 2008;2:2. doi: 10.3389/neuro.04.002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.