

Abstract
Growing evidence suggests that close appositions between the endoplasmic reticulum (ER) and other membranes, including appositions with the plasma membrane (PM), mediate exchange of lipids between the two bilayers. The mechanisms of such exchange, which allows lipid transfer independently of vesicular transport, remain poorly understood. The presence of an SMP (synaptotagmin-like-mitochondrial-lipid binding protein) domain, a proposed lipid binding module, in several proteins localized at membrane contact sites raised the possibility that such domains may be implicated in lipid transport1,2. SMP-containing proteins include components of the ERMES complex, an ER-mitochondrial tether3, and the Extended-Synaptotagmins/tricalbins, which are ER-PM tethers4-6. Here we present at 2.44 Å resolution the crystal structure of a fragment of Extended-Synaptotagmin 2 (E-Syt2), including an SMP domain and two adjacent C2 domains. The SMP domain has a beta-barrel structure like protein modules in the TULIP superfamily. It dimerizes to form a ~90 Å long cylinder traversed by a channel lined entirely with hydrophobic residues, with the two C2A-C2B fragments forming arched structures flexibly linked to the SMP domain. Importantly, structural analysis complemented by mass spectrometry revealed the presence of glycerophospholipids in the E-Syt2 SMP channel, indicating a direct role for E-Syts in lipid transport. These findings provide strong evidence for a role of SMP domain containing proteins in the control of lipid transfer at membrane contact sites and have broad implication beyond the field of ER to PM appositions.
ER-PM contact sites present in all eukaryotes are thought to have multiple functions7-9, including an evolutionarily conserved role in the regulation of lipid composition and metabolism at the PM8-11. The Extended-Synaptotagmins (E-Syts) are ER-resident proteins that act as tethers linking the ER and PM membranes into close apposition4-6. There are three E-Syts (1/2/3) in mammalian cells, which homo- or heterodimerize6. Each E-Syt has an N-terminal ER-membrane anchor, followed by a short linker region, an SMP domain, and three or more C2 domains (Fig. 1a)6. The ER-membrane anchor and C2 domains are required for tethering, whereas the role of the SMP domain has been unclear6. Bioinformatics analyses suggesting that SMP domains belong to the tubular-lipid-binding (TULIP) superfamily were therefore intriguing because several TULIP proteins are known to bind lipids and participate in lipid transfer1,2.
Figure 1.
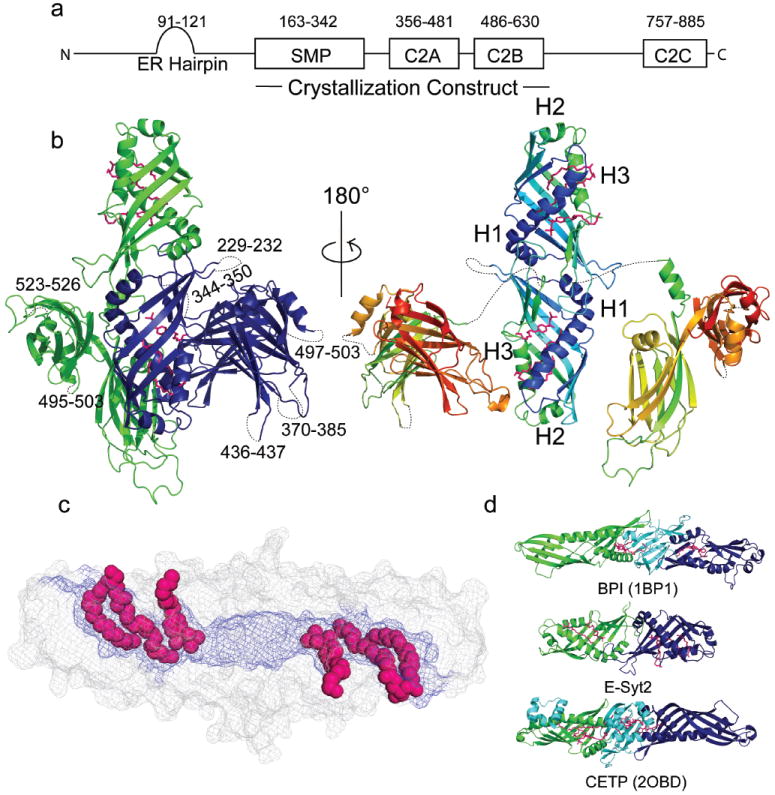
E-Syt2 Structure. (a) E-Syt2 domain architecture. Domain boundaries indicated as residue numbers. (b) Left: Monomers in the Esyt2 dimer are green or blue. C2A-C2B arches (see also Fig. 3) contact the SMP barrel as in the crystal. Right: Esyt2 is colored from blue (N-terminus) to red (C-terminus), and the C2A-C2B arches are pulled away from the SMP dimer. The “seam” along the side of the SMP dimer is partially visible. Detergent and lipid molecules are pink. (c) Surface representation of the SMP dimer. Hydrophobic residues (blue) line the channel. Lipid fatty acid moieties are in space-filling representation. (d) Ribbon diagrams for BPI, E-Syt2, and CTEP. Structural elements in BPI or CTEP not present in Esyt2 are cyan.
To obtain insights regarding E-Syt function, we have determined the crystal structure for a fragment of human E-Syt2 (residues 163-634) comprising the SMP, C2A and C2B domains (Table ED1). The C-terminal portion of E-Syt2, which consists of an extended linker region followed by a third C2 domain (C2C) that interacts with PI(4,5)P2 at the plasma membrane6, was not in the construct.
This E-Syt2 fragment dimerizes both in solution, as assessed by size exclusion chromatography, and in the crystal. Each SMP domain is a beta barrel (Fig. 1b), comprising a highly twisted beta-sheet and two alpha helices, H1and H3. A third helix, H2, partially caps one end of the barrel. The other end of the barrel associates with the corresponding end of the second SMP domain to form a 90 Å long cylinder. The residues at the interface of the two SMP domains are among the most highly conserved (Fig. ED1), consistent with the physiological relevance of the dimer, and they most likely mediate homo- and heterodimerization of the E-Syts 6. A ~10 Å diameter channel lined exclusively with hydrophobic residues runs through the cylinder (Fig. 1c). It connects with solvent at both ends and via a narrow seam along the length of the cylinder.
There is no significant primary sequence similarity between the E-Syt2 SMP domain and proteins of known fold. Nevertheless, as predicted by bioinformatics studies1,2, our structure firmly establishes the SMP domain as a member of the TULIP superfamily. In previously characterized TULIP proteins like bactericidal/permeability-increasing protein (BPI) and cholesteryl ester transfer protein (CETP) (Fig. 1d), SMP-like modules also form tubular structures harboring an elongated hydrophobic channel12,13. BPI and CETP do not dimerize, however, as each monomer comprises two SMP-like domains rather than one, with the two modules arranged in tandem and connected by a beta-sheet not present in the E-Syts.
Within the SMP channel, the electron density maps show clear densities for two lipid-like molecules per E-Syt2 monomer (Fig. 2a, b). One density is consistent with a diacylglycerol lipid, whereas the other is reminiscent of Triton X100, a detergent used in protein purification. Triton was modeled into this density with its 4-(1,1,3,3 tetramethylbutyl)-phenyl group buried within the hydrophobic channel and its hydrophilic polyethylene chain extended through the seam into solvent (Fig. 2). A non-denaturing MS analysis14 of recombinant E-Syt2 purified in the absence of Triton shows that ligand-bound proteins are present and that two lipid molecules can bind per monomer (Fig. ED2), so most likely a second lipid molecule binds in place of the detergent in a physiological context. Denaturing negative ionization high resolution MS and MSMS analysis further identified ligands of the recombinant E-Syt2 as two major components of the bacterial membrane, phosphatidylglycerol and phosphatidylethanolamine (PE) (Fig. ED3, Table ED2).
Figure 2.
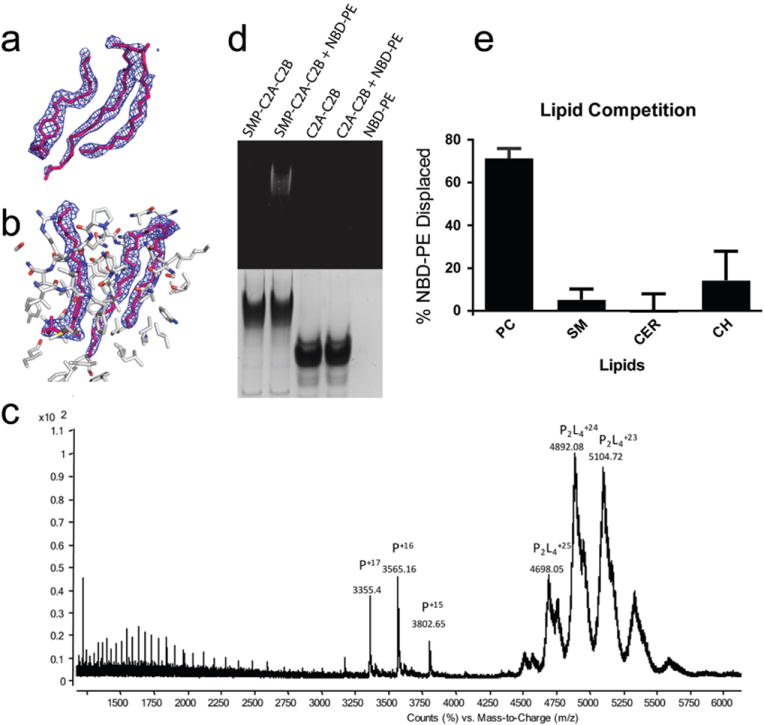
E-Syt2 binds lipid-like molecules in the SMP hydrophobic channel. (a) 2Fo-Fc electron density (1.0 σ) into which the Triton X100 detergent and phosphoglycerol lipid were modeled; models after refinement are superimposed. Lipid headgroups were not modeled. (b) Final 2Fo-Fc electron density for lipid-like molecules. Protein atoms within 8 Å of lipid are indicated. (c) Positive mode, non-denaturing, ESI mass spectrum of ESyt-2 expressed in mammalian cells. In native conditions (20mM acetic acid, pH 6.9) a conformation of apo ESyt-2 centered around the +16 charge state is the most abundant monomeric state (indicated as P), MW= 57032.4±1.6 Da. The most represented species in native conditions are dimers containing at least 4 lipid molecules (P2L4) with a charge state centered around +24. (d) An E-Syt2 construct that includes the SMP domain binds fluorescent NBD-PE, whereas the C2A-C2B arch alone does not. Proteins were incubated with fluorescent lipid, then analyzed by native PAGE, which was examined for fluorescence (top) and stained by Coomassie (bottom). The band with the SMP-containing protein co-migrates with fluorescent PE. (e) E-Syt2 preloaded with NBD-PE, then further purified, was incubated with phosphatidylcholine (PC), sphingomyelin (SM), ceramide (CER) or cholesterol (CH). Samples were visualized on native gels by fluorescence and Coomassie staining. Fluorescence signal was normalized to the amount of protein in each lane, which was assessed by Coomassie staining. The experiment was performed in quintiplicate; error bars represent standard deviation. PC, but not any of the sterols, displaces NBD-PE. (See also Fig. ED7.)
The electron density maps show that the lipid fatty acid moieties lie in the hydrophobic channel, whereas the polar head group protrudes through the seam and is solvated. There is no density for the head group, implying that it does not make extensive contacts with E-Syt2 and is disordered. This suggests that the SMP domain could bind a variety of glycerophospholipids. BPI and CETP bind diverse lipids in a similar manner, with hydrophobic portions buried in the hydrophobic channel and the hydrophilic groups exposed to solvent along the seam12,13.
To identify lipids bound to E-Syt2 in mammalian cells, a 3xFLAG-tagged E-Syt2 fragment comprising the SMP and C2A-C2B domains was expressed in Expi293 cells and purified by anti-FLAG immunoprecipitation. Denaturing MS of this construct and quantitative lipidomic analysis revealed the presence of the glycerophospholipids phosphatidylcholine (PC, 67%), PE (21%), phosphatidylinositol (7%), and phosphatidylserine (3%) (Fig. ED4-ED5, Table ED3). These relative amounts of the major phospholipidsroughly reflect their relative abundance in animal cell membranes15, suggesting a lack of specificity with respect to particular glycerophospholipid ligands. We inspected for but did not detect other lipid species such as LysoPC, sphingosine or sphingosine-1-phosphate, sphingomyelin, any phosphoinositides, or significant amounts of ceramide. Nor were cholesterol esters, which are bound by CETP 13, or cholesterol detected.
When analyzed by non-denaturing MS, ESyt-2 was found to occur both as monomer and dimer (Fig. 2c). The charge state distribution centered around +16 represents monomers mainly in the apo form, corresponding to a molecular weight of 57030 Da after deconvolution (Fig. ED6A). Only a low percentage of the monomers (~10%) was bound to 1 or 2 lipid molecules, with masses corresponding to the main lipid species identified previously. Most of the protein was dimeric with a charge state distribution centered around +24. After deconvolution of this spectrum (Fig. ED6b-c, ED7), the MW of the dimer is ~117500 Da, suggesting the presence of at least 4 bound lipid molecules (assuming an average mass of 750-800 Da per lipid). Almost no dimers were detected in the apo form. These findings indicate that dimerization is required for stable binding of the lipids.
Binding to glycerophospholipids was also demonstrated in vitro by testing the ability of purified lipids to displace preloaded fluorescent NBD-PE from E-Syt2 (Fig. 2d,e). Incubation of preloaded E-Syt2 with PC displaced NBD-PE, as revealed by loss of fluorescence in the band corresponding to E-Syt2 in native PAGE (Fig. 2e). In contrast, consistent with the MS results, cholesterol, sphingomyelin, and ceramide did not displace PE, suggesting that they are not bound by E-Syt2 even when abundantly present. The greater rigidity of these lipids as compared to glycerophospholipids may prevent their entry into the E-Syt2 lipid binding site.
C2A and C2B both fold into type II C2 domains and are rigidly linked into an arch spanning ~70 Å across (Fig. 3 shows best view of arch), as recently described16. In the crystal structure, both C2A-C2B modules of the E-Syt2 dimer contact the same SMP domain, with different C2A-C2B residues interacting with different patches on the SMP (Fig. 1a, left). Residues at the interfaces are not conserved (Fig. ED1), suggesting that the C2-SMP interactions are crystal contacts and that C2A-C2B does not interact with the SMP dimer insolution (Fig. 1b, right). Rather, the E-Syt2 C2A-C2B fragment interacts with lipid bilayers in a Ca2+-dependent manner17. Accordingly, calcium-coordinating residues were identified in loops at the end of the arch structure in C2A16.
Figure 3.
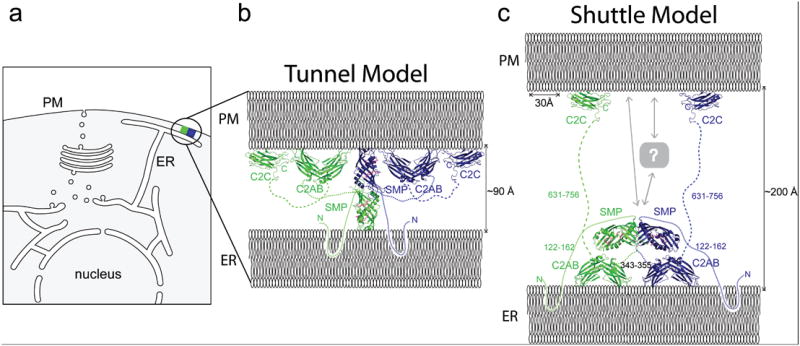
ER-PM contact site and a model of E-Syt2. (a) Schematic of a cell with a ER-PM contact site. (b) “Tunnel model” of E-Syt2, where E-Syt2 bridges the ER and PM to transfer lipids between them. The E-Syt2 SMP dimer is ~90 Å long, so the ER and PM cannot be further apart if E-Syt2 acts as a tunnel. E-Syt2 is anchored to the ER via its N-terminal region and to the PM via the C2C domain (PDBID 2DMG). C2A-C2B interacts with membrane via loops at the ends of the arch and could interact with the ER, PM or either, depending on the inter-membrane distance. The model is to scale. (c) “Shuttle model” of E-Syt2 tethering the ER and PM and drawn to scale. Length of unstructured linker regions is indicated in residues. At larger ER-PM distances as shown here, C2A-C2B cannot reach the PM. The SMP dimer may shuttle lipids between the ER and PM, potentially together with partner proteins (labeled “?”).
The distance between the ER and PM membranes at contact sites is estimated at 100-300 Å8,18,19. E-Syt2 spans this space with an N-terminal membrane anchor inserted into the ER and its C2C domain bound to the PM via interaction with PI(4,5)P2, a phosphoinositide enriched there6 (Fig. 3). While C2C, when expressed alone, is recruited to the PM6,17, we and others17 did not observe any association of C2A-C2B with a particular organelle in living cells. Given geometric restraints imposed by domain dimensions and linker lengths, an interaction of C2A-C2B with the cytosolic surface of the ER is plausible and consistent with acidic phospholipid-independent membrane interactions observed for it17. An interaction of C2A-C2B with the PM at very narrow ER-PM contacts (<~100 Å), where it can reach the PM, is also plausible, however.
The structure of the E-Syt2 SMP domain and identification of lipids in its hydrophobic channel conclusively establishes this domain as a lipid-binding module. The structural similarity of E-Syt2 to proteins in the TULIP superfamily, several of which function in lipid transfer extra-cytosolically2,12,13, further suggests a role in lipid transport. Moreover, the presence of this module in proteins that localize to contacts between the ER and other membranes4-6,20 strongly indicates a role in the transfer of lipids from their site of synthesis in the ER to these membranes, such as the PM in the case of E-Syt2. In answer to how such transfer may take place, SMP dimers could in principle form a bridge between apposed membranes along which lipids are transferred (“tunnel model”, Fig. 3), with their hydrophobic moieties sliding along the hydrophobic channel and their hydrophilic headgroups solvent exposed at the seam. CETP was proposed to function in exactly this manner to channel cholesterol esters between lipoprotein particles21. However, as it is only 90 Å long, the E-Syt2 SMP dimer may be too short to span the inter-membrane space. Another possibility, plausible for ER-PM distances under ~200 Å, is that the SMP dimer shuttles between membranes, extracting lipid from one and delivering it to the second (“shuttle model”, Fig. 3). In a third, not mutually exclusive model, E-Syt2 functions in concert with other lipid transfer proteins to effect transport. This model is appealing because it does not require E-Syts to preferentially bind specific lipids, which could instead be selected by the partner proteins. Phosphatidylinositol transfer proteins (PITPs) and proteins in the OSH family are interesting candidates because they can interact with specific lipids22,23 and are thought to function in lipid transfer at contact sites8,9.
The identification of E-Syts as lipid transfer proteins represents an important step forward in dissecting the biology of ER-PM contact sites. Nevertheless, our work is also more generally applicable to other contact sites, such as ER-mitochondrial contacts, where the ERMES tethering complex is predicted to contain three SMP modules1,2.We suspect that these hetero-dimerize in a head-to-head fashion, as observed in the E-Syt2 SMP homodimer, to form one or more lipid carrier modules with hydrophobic lipid-binding channels.
Methods
Cloning, protein expression, purification
Human E-Syt2 (residues 163-634) was cloned into a modified pCDF vector, which codes for an N-terminal GST-tag and a Precision protease cleavage site. The plasmid was transformed into Escherichia coli BL21(DE3) cells. Cells were grown to an OD600 0.7, then shifted to 20 °C, and protein expression was induced by adding 1 mM IPTG. Cells were harvested 20 h after induction. Selenomethionine-substituted E-Syt2 was produced similarly, according to ref 24. Cells were collected by centrifugation, resuspended in lysis buffer (20 mM Tris [pH 8.0], 500 mM NaCl, 5 mm DTT, 10 mM EDTA) supplemented with protease inhibitors (Complete EDTA-free, from Roche) and lysed using a cell disruptor (Avestin). E-Syt2 was isolated by Glutathione Sepharose resin, washed once with lysis buffer supplemented with 1% Triton X-100 and twice in lysis buffer, except without EDTA. The GST tag was cleaved off using Precision protease and the hexahistidine-tagged protease removed by passing the protein solution over Ni-NTA resin. The protein was further purified by gel filtration (Superdex 200, GE Healthcare), eluted in buffer containing 20 mM Tris (pH 8.0), 300 mM NaCl, and 5 mM DTT, and concentrated to approximately 8 mg/mL.
For purification from mammalian cells, human E-Syt2 (residues 163-634) was cloned into pCDNA 3.1 with an N-terminal 3XFLAG tag. Expi293 cells were transfected for 3 days before harvesting. Cells were lysed in buffer (20mM Tris [pH 8.0], 250mM NaCl, 10mM EDTA) supplemented with protease inhibitors (Complete EDTA-free, from Roche) by freeze-thaw method using liquid nitrogen for 5 times. Proteins were purified by anti-FLAG M2 resin (Sigma) and eluted in buffer (20mM Tris [pH 8.0], 250mM NaCl) containing 100ug/ml 3XFLAG peptide. Further purification was by size exclusion chromatography, as described above.
Crystallization, Data Collection, and Structure Determination
Crystals of native protein were grown at room temperature by the hanging drop method. Equal volumes ofprotein solution and mother liquor (1.3 ul) were mixed and suspended over mother liquor (100 mM malonic acid-imidazole-boric acid (MIB) buffer (Qiagen) at pH 4.3, 12.5% PEG 1500). The crystals belong to spacegroup P212121, with a=74.3, b=88.4, c=167.2 Å. Selenomethionine substituted crystals were grown under similar conditions, except that the mother liquor additionally contained sodium iodide (100 mM MIB (Qiagen) at pH 4.6, 14% PEG1500, 0.1 M NaI). Crystals were transferred to mother liquor supplemented with 16% ethylene glycol, loop-mounted and flash frozen in liquid nitrogen. For the selenium substituted crystals, data were collected at two wavelengths chosen to maximize the selenium and iodine anomalous signals. All data were collected at NE-CAT beamline 24-ID-C at the APS and processed using XDS25. Phases were calculated in Phenix26 by the multi-wavelength anomalous dispersion method using the data from the selenium-substituted crystals, and electron density maps were further improved by solvent flattening and non-crystallographic two-fold averaging. We used COOT27 for model building. As model building progressed, we iteratively combined phases calculated from partial models with experimental phases to improve the maps in still unbuilt regions. The model was refined to 2.44 Å against data from native crystals using Phenix26. Initial rounds of refinement were carried out using least squares, real space, individual B-factor and torsion angle annealing options and were alternated with manual rebuilding in COOT27. Detergent and lipid molecules as well as waters were modeled in the final stages of refinement, for which we used least-squares, individual B-factor, and TLS refinement options. Figures were prepared using PyMol software28. The model has good Ramachandran geometry (96%, 4%, and 0.1% of the residues are in the allowed, generously allowed and disallowed regions, respectively).
Mass Spectrometry analyses
Purified protein samples were desalted with a Vivaspin 10,000 MWCO device (GE Healthcare) to exchange the initial buffer with 20 mM ammonium acetate, pH 6.8, and to eliminate eventually present contaminating small molecules. The sample was then directly infused at a flow of 1 μl/min and a concentration of 0.4 mg/ml into a quadrupole time-of-flight (QTOF) 6550 Agilent (Santa Clara, USA) mass spectrometer equipped with an Agilent 1200 series system. Nondenaturing analyses were performed in positive ion mode with nanoelectrospray voltage set to 1400 V (Vcap), temperature 280°C, drying gas flow 11 l/min and fragmentor 175 V. The samples were injected in 20 mM ammonium acetate, pH 6.8, and spectra acquired at a scan rate of 1 spectra/sec. Data were collected over a mass range of 1000-10000 m/z and deconvoluted using Bioconfirm software (Agilent) and the Maximum Entropy deconvolution algorithm. Denaturing mass spectrometry, to identify the possible ligands and to confirm the theoretical mass of apo ESyt-2, was performed in positive and negative ion mode using conditions that could break the protein-ligand complex (high vapor acidic solutions containing 5% acetic acid in 20mM ammonium acetate). The same acidic conditions were also used to dissociate native dimers and to confirm that their presence was not an instrumental artifact. Data were acquired between 100 and 1000 m/z(both positive and negative mode) to identify the ligands and between 1000 and 10000 m/z (positive mode) to monitor the protein conformational states. The structural identity of ligands was confirmed by collision-induced dissociation (CID) in positive and negative ion mode after selection of precursor ions in MSMS mode and fragmentation with a collision energy value of 25 eV.
To improve lipid identification, a lipid extract obtained by sonicating (10 minutes) the protein (4μg) in 100 μl of methanol (or chloroform/methanol for extraction of neutral lipids) was collected after centrifugation of the sample and protein precipitation. The sample was then resuspended in solvent B (see below) and analysed by LC-MSMS with the same mass spectrometer equipped with a ChipLC system and a HILIC chip (Amide 80, Agilent) for the chromatographic separation of the isolated lipids. The Agilent 1200 series HPLC used a capillary pump for sample injection onto the Enrichment column and a Nano pump for separation. Solvents used for HILIC HPLC: 50% acetonitrile in water containing 25mM ammonium formate pH 4.6 adjusted with formic acid (solvent A), 95% acetonitrile containing 25mM ammonium formate pH 4.6 adjusted with formic acid (solvent B). LC gradient used: 100% B from 0 to 1.5 mins, 100% to 85% B from 1.5 to 3 mins, 85 to 80% B from 3 to 11.5 mins, 100% B from 11.5 to 13.5 mins, 100% B from 13.6 to19 mins. The Agilent 6550 quadrupole time-of-flight (QTOF) mass spectrometer was operated in positive ion mode and negative ion mode; electrospray voltage was set to 1580 V (Vcap), temperature 185°C, drying gas 14 L/min, fragmentor voltage 175 V. The instrument was operated in auto MS/MS mode with MS acquisition rate of 2 spectra/sec and MS/MS acquisition rate of 4 spectra/sec.
Quantification of the lipid species released from ESyt-2 expressed in mammalian cells was performed by LC-MSMS (MRM mode) after spiking lipid standards in the sample before the extraction and using a 6460 QQQ (Agilent) mass spectrometer, equipped with a 1290 series chromatographic system. Lipid standards: 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE), 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine (DMPS), 1,2-dimyristoyl-sn-glycero-3-phospho-(1’-rac-glycerol)(DMPG), 1,2-dihexadecanoyl-sn-glycero-3-phosphate (DPPA), 1-heptadecanoyl-2-hydroxy-sn-glycero-3-phosphate (LPA), N-heptadecanoyl-D-erythro-sphingosine (Cer) were purchased from Avanti polar lipids (Alabaster, AL, USA), Dioctanoyl Phosphatidylinositol (PI) was purchased from Echelon Biosciences (USA). The experimental conditions and the transitions monitored to quantify the lipid species are reported together in the Supplementary methods Excel file.
In vitro lipid binding assay
Lipid loading. Gel filtrated SMP-C2A-C2B (19μl at ~2mg/ml) or C2A-C2B (at ~3 mg/ml) in 20mM Tris HCl pH 8, 150 mM NaCl, and 5mM DTT were mixed with 1 μl NBD-PE 16:0 at 1mg/ml in methanol. The lipid control was 19μl of the above buffer with 1 μl NBD-PE 16:0. They were incubated on ice for one hour and visualized on a DTT containing native PAGE gel. Fluorescence was visualized using Imagequant LAS4000 and protein via Coomassie staining. Only the SMP-containing construct binds NBD-PE.
Lipid competition assay
NBD-PE preloaded protein was obtained by incubating GST-SMP-C2A-C2B with NBD-PE as described in the lipid loading section. To separate E-Syt2 from excess PE, GST-SMP-C2A-C2B was rebound to Glutathione resin, washed, and cleaved off using Precision protease.
Lipids were dried under nitrogen and resuspended at 6.4mM in methanol. Ceramide was heated to 42 °C to dissolve the lipid. Lipid (1μl) was added to SMP-C2A-C2B preloaded with NBD-PE (19 μl at ~1mg/ml). Samples were incubated for one hour at room temperature, then visualized on native gels as described above. Gel images were quantified using imageJ29. Fluorescent signal was normalized to the amount of protein in each lane, which was assessed by Coomassie staining.
Extended Data
Fig. ED1. E-Syt2 structure analysis.
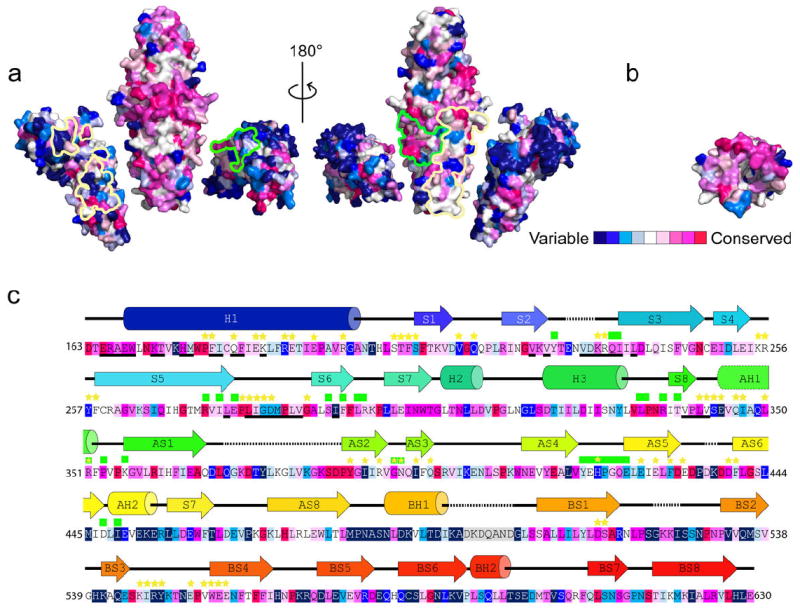
(a) E-Syt2 dimer colored according to sequence conservation in E-Syt and tricalbin proteins1. C2A-C2B is pulled away from the SMP dimer (as in Fig 1B, right). Residues at the C2A-C2B:SMP interface are outlined in green or yellow. The interfaces correspond to crystal contacts but likely are not physiologically significant as these residues are not well conserved. Note the band of conserved residues at the SMP dimerization interface. (b) A cross-section of the SMP barrel, exposing residues at the dimerization interface. The surface is colored according to sequence conservation. (c) E-Syt2 sequence. Secondary structure elements are colored as in Fig. 1, right, and annotated, as is sequence conservation. Residues at dimerization interface are underlined bold. Yellow Stars and Green Squares indicate residues at the C2A-C2B:SMP interfaces and match their respective outline color as in panel A. (1Ashkenazy, H., Erez, E., Martz, E., Pupko, T. Consurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucl. Acids Res. 38, W529-533 (2010).)
Fig. ED2. Binding of phospholipids by Esyt-2 expressed in bacteria.
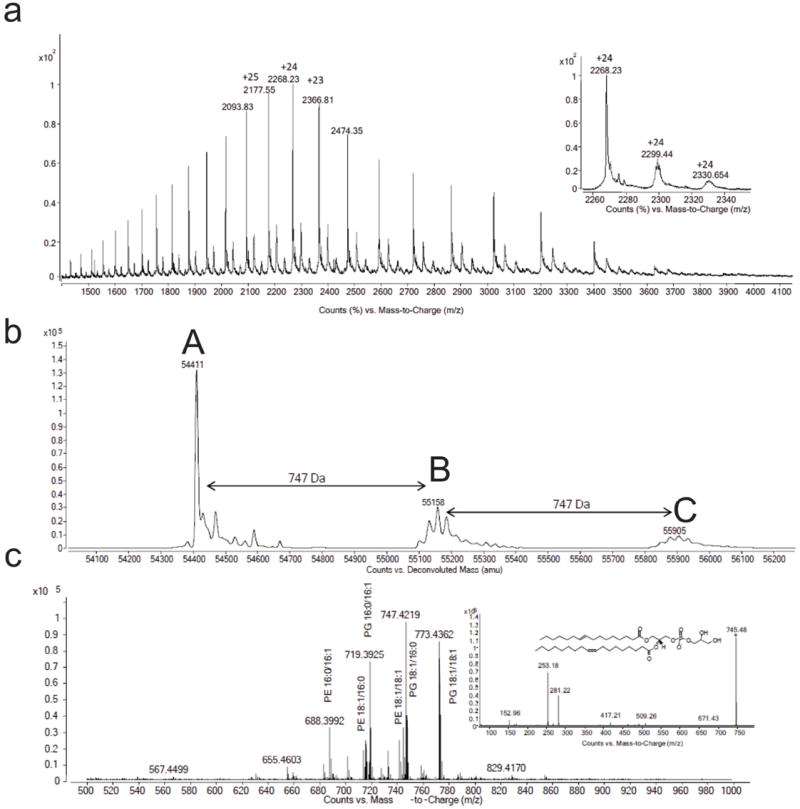
Positive mode non denaturing MS was carried out on Esyt-2 protein expressed in E. coli. Spectra are shown before (a) and after (b) deconvolution with the apo (A, MW= 54414.2±1.5 Da) and the bound species (B and C, with one and two lipid molecules respectively) represented. The average molecular weight difference between the different forms is shown and can correspond to a phosphatidylglycerol (PG) molecular mass (see c). (c) When exposed to denaturing conditions but, partly, also in non denaturing ones, ESyt-2 releases lipids that can be monitored in negative mode MS. (Inset) Negative mode production scan on m/z 745.48 released from Esyt-2 protein confirmed the identity of one ligands as PG 16:1/18:1.
Fig. ED3. MSMS fragmentation spectra in negative mode of two representative abundant glycerophospholipids released from bacterially expressed ESyt-2 under denaturing MS conditions.
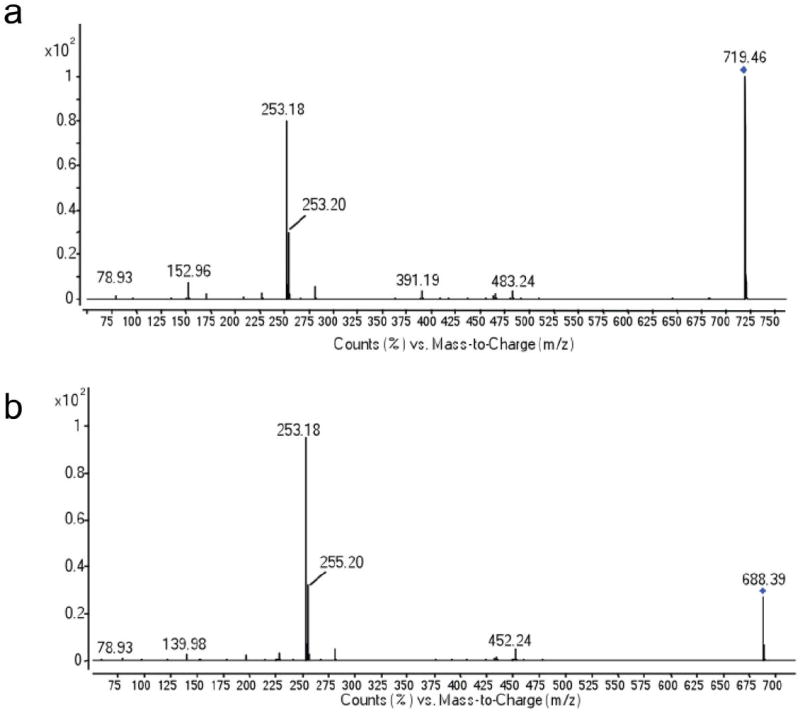
(a) Fragmentation of PG 16:0/16:1 (719.4648 m/z). (b) Fragmentation of PE 16:0/16:1 (688.3992 m/z). All the other possible lipid ligands released from the protein and detected by MS were fragmented in the same way and the identity confirmed.
Fig. ED4. Negative mode MS spectra of glycerophospholipids released from ESyt-2, expressed in mammalian cells, under denaturing MS conditions.
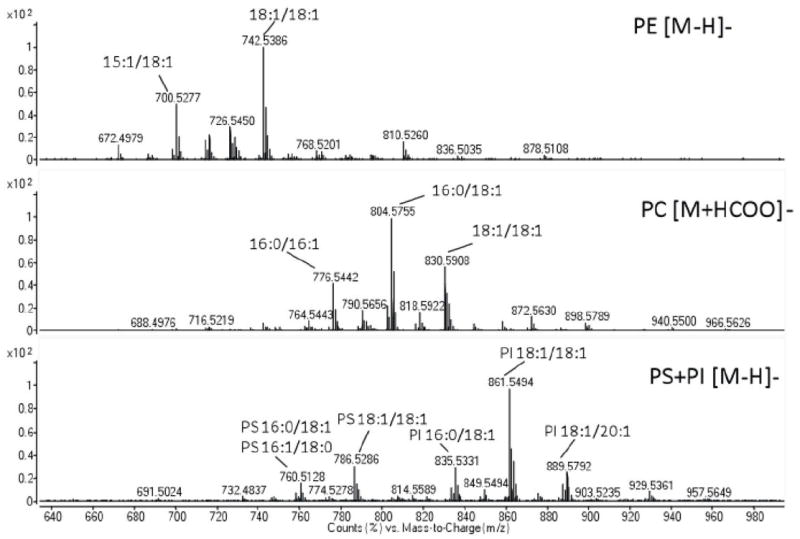
The four most abundant glycerophospholipid classes (PE, PC, PS, PI) are shown. The acyl chains compositions of the most represented molecular species in each class are also reported.
Fig. ED5. Positive and negative mode MSMS fragmentation spectra of representative abundant glycerophospholipids released from ESyt-2 expressed in mammalian cells, under denaturing MS conditions.
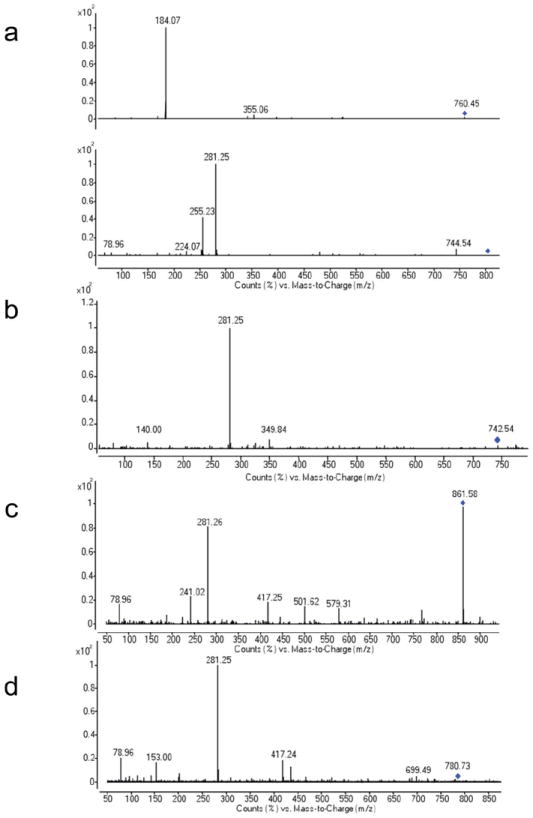
(a) Fragmentation of PC 16:0/18:1 (760.45 m/z). (b) Fragmentation of PE 18:1/18:1 (742.53 m/z). (c) Fragmentation of PI 18:1/18:1 (861.58 m/z). (d) Fragmentation of PS 18:1/18:1 (786.53 m/z).
Fig. ED6. Deconvoluted spectra of ESyt-2.
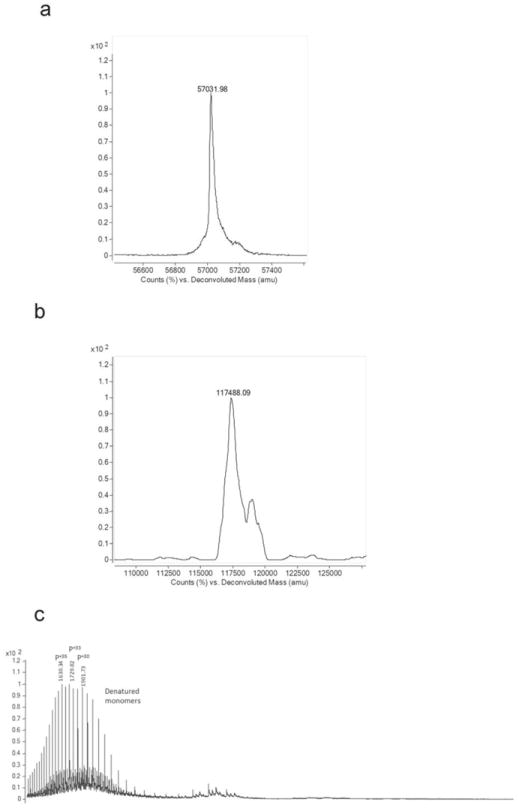
Deconvoluted spectra of the monomeric ESyt-2 form indicated as P (a) and of the lipid-bound dimeric form indicated as P2L4 (b) in figure 2c. (c) Positive mode ESI MS spectrum in denaturing conditions (5% acetic acid in 20mM ammonium acetate) of ESyt-2 expressed in mammalian cells. To exclude the possibility that the dimer-associated MS peaks in Fig. 2c and ED6b could be an artifact generated during ionization, the sample was denatured in acidic conditions. Only a charge distribution around +33 was detected (indicated as P), probably representing a more open and denatured monomeric apo protein conformation, confirmed by the deconvoluted spectrum (not shown). In these experimental conditions, no peaks corresponding to dimeric structures could be detected.
Fig. ED7. Lipid Competition Assay.
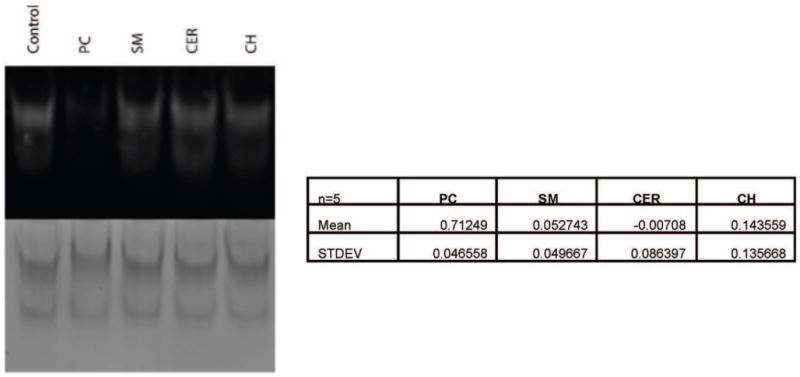
NBD-PE-preloaded SMP-C2A-C2B mixed with methanol (control), phosphatidylcholine (PC), Sphingomyelin (SM), Ceramide (CER), or Cholesterol (CH). Quantification of the 5 trials is shown.
Table ED1.
Data Collection and Refinement Statistics.
| Data collection | E-Syt2(163-634) | |||
| Crystal | Native | SeMet, Iodide | ||
| Space group | P212121 | P212121 | ||
| Unit cell dimensions | ||||
| 74.33, 88.38, 167.19 | 73.54, 88.33, 166.24 | 73.39, 88.17, 165.87 | ||
| a, b, c (Å) | ||||
| Peak | Remote | |||
|
|
||||
| Wavelength (Å) | 0.9792 | 0.9792 | 1.9074 | |
| Resolution (Å) | 47.14–2.44 (2.57-2.44) | 46.94–2.8 (2.95-2.8) | 46.84-2.8 (2.95-2.8) | |
| Rmerge(%) | 8.9 (60.5) | 10.8 (62.9) | 6.7(36.9) | |
| I/σ | 13.6 (2.4) | 14.1(2.5) | 20.8(4.3) | |
| Completeness (%) | 99.9 (99.8) | 99.9(99.8) | 99.6(99.4) | |
| Redundancy | 6.3 (6.4) | 6.4(5.9) | 6.2(6.1) | |
| Refinement | ||||
| Resolution (Å) | 47.14–2.44 | |||
| No. unique reflections | 41769 | |||
| Rwork/Rfree% | 21.02/25.56 | |||
| No. atoms | ||||
| Protein | 7143 | |||
| Water | 212 | |||
| Ligand | 138 | |||
| Average B (Å2) | ||||
| Protein | 50.79 | |||
| Water | 45.54 | |||
| Ligand | 53.59 | |||
| R.m.s deviations | ||||
| Bond lengths (Å) | 0.003 | |||
| Bond angles (°) | 0.846 | |||
Numbers in parenthesis refer to values in highest resolution bin.
Table ED2. Phospholipids present as ligands in E-Syt2 purified from E. coli and observed after denaturing mass spectrometry in negative ionization mode.
Some precursors are a mixture of isobaric forms and cy refers to a cyclopropyl group. In bold the most abundant species.
| [M-H]- | lipid species |
|---|---|
|
| |
| 660 | PE 14:0/16.1 |
|
| |
| 662 | PE 14:0/16:0 |
|
| |
| 686 | PE 16:1/16:1 |
|
| |
| 688 | PE 16:0/16:1 |
|
| |
| 691 | PG 14:0/16:1 |
|
| |
| 693 | PG 16:0/14:0 |
|
| |
| 702 | PE 16:0/cy17:0 |
|
| |
| 714 | PE 18:1/16:1 |
| PE cy17:0/cy17:0 | |
|
| |
| 716 | PE 16:0/18:1 |
|
| |
| 719 | PG 16:0/16:1 |
| PG 14:0/18:1 | |
|
| |
| 728 | PE 18:1/cy17:0 |
|
| |
| 730 | PE 16:0/cy19:0 |
| PE 18:0/cy17:0 | |
| PE 17:0/18:1 | |
|
| |
| 733 | PG 16:0/cy17:0 |
|
| |
| 742 | PE 18:1/18:1 |
| PE cy19:0/cy17:0 | |
|
| |
| 745 | PG 18:1/16:1 |
|
| |
| 747 | PG 16:0/18:1 |
|
| |
| 759 | PG 18:1/cy17:0 |
|
| |
| 773 | PG 18:1/18:1 |
Table ED3. A targeted quantitative lipidomic approach based on LC-MSMS was used to quantify the different lipid species released by ESyt-2 after organic solvent extraction.
Here only the total amount of each lipid class was reported. See supplementary methods for experimental details about the different lipids that were monitored.
| ng/4μg of protein | ||||||
|---|---|---|---|---|---|---|
| Cer | PC | PE | PG | PI | PS | |
| Mean | 1.71 | 145.97 | 44.68 | 0.3 | 15.64 | 4.92 |
| SD | 0.02 | 3.3 | 0 | 0.01 | 4.39 | 0.24 |
Supplementary Material
Acknowledgments
We are grateful to the staff at NE-CAT beamline 24ID-C at the APS for assistance with data collection. We thank Hongying Shen and Nikit Kumar for discussion. This work was supported by the NIH (GM080616 to KMR, R37NS36251 and DK082700 to PDC). MRW and FT are funded by grants from the National University of Singapore via the Life Sciences Institute (LSI)and the Biochemical Research Council (BMRC-SERC Diag-034). CMS is recipient of an NSF Graduate Research Fellowship (DGE-1122492). YS was supported by the Uehara Memorial Foundation and the Japanese Society for Promotion of Science.
Footnotes
Author Contributions: All authors participated in the design of the experiments, data analysis, and manuscript editing. CMS, XW, PN, FT, and YS carried out the experiments. FT, MRW, PDC and KMR wrote this manuscript.
Author Information:Coordinates and structure factors have been deposited in the PDB (ID 4P42).
The authors have no competing financial interests.
References
- 1.Kopec KO, Alva V, Lupas AN. Homology of SMP domains to the TULIP superfamily of lipid-binding proteins provides a structural basis for lipid exchange between ER and mitochondria. Bioinformatics. 2010;26:1927–1931. doi: 10.1093/bioinformatics/btq326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kopec KO, Alva V, Lupas AN. Bioinformatics of the TULIP domain superfamily. Biochem Soc Trans. 2011;39:1033–1038. doi: 10.1042/BST0391033. [DOI] [PubMed] [Google Scholar]
- 3.Kornmann B, et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toulmay A, Prinz WA. A conserved membrane binding domain targets proteins to organelle contact sites. J Cell Sci. 2012;125:49–58. doi: 10.1242/jcs.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manford AG, Stefan CJ, Yuan HL, MacGurn JA, Emr SD. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev Cell. 2012;23:1129–1140. doi: 10.1016/j.devcel.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Giordano F, et al. PI(4,5)P2-Dependent and Ca2+-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 2013;153:1494–1509. doi: 10.1016/j.cell.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine T, Loewen CJ. Inter-organelle membrane contact sites: through a glass, darkly. Curr Opin Cell Biol. 2006;18:371–378. doi: 10.1016/j.ceb.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Toulmay A, Prinz WA. Lipid transfer and signaling at organelle contact sites: the tip of the iceberg. Current Opinion in Cell Biology. 2011;23:458–463. doi: 10.1016/j.ceb.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stefan CJ, Manford AG, Emr SD. ER-PM connections: sites of information transfer and inter-organelle communication. Curr Opin Cell Biol. 2013;25:434–442. doi: 10.1016/j.ceb.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefan CJ, et al. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell. 2011;144:389–401. doi: 10.1016/j.cell.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 11.Tavassoli S, et al. Plasma membrane-endoplasmic reticulum contact sites regulate phosphatidylcholine synthesis. EMBO Rep. 2013;14:434–440. doi: 10.1038/embor.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beamer LJ, Carroll SF, Eisenberg D. Crystal structure of human BPI and two bound phospholipids at 2.4 Angstrom resolution. Science. 1997;276:1861–1864. doi: 10.1126/science.276.5320.1861. [DOI] [PubMed] [Google Scholar]
- 13.Qiu X, et al. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. NSMB. 2007;14:106–113. doi: 10.1038/nsmb1197. [DOI] [PubMed] [Google Scholar]
- 14.Krylova IN, et al. Structural Analyses Reveal Phosphatidyl Inositols as Ligands for the NR5 Orphan Receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 15.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Reviews Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J, et al. Structure and Ca2+-binding properties of the Tandem C2 Domains of E-Syt2. Structure. 2013 doi: 10.1016/j.str.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Min S-W, Chang W-P, Südhof TC. E-Syts, a family of membranous Ca2+-sensor proteins with multiple C2 domains. PNAS. 2007;104:3823–3828. doi: 10.1073/pnas.0611725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orci L, et al. From the Cover: STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. PNAS. 2009;106:19358–19362. doi: 10.1073/pnas.0911280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West M, Zurek N, Hoenger A, Voeltz GK. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J Cell Biol. 2011;193:333–346. doi: 10.1083/jcb.201011039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helle SCJ, et al. Organization and function of membrane contact sites. Biochimica et Biophysica Acta. 2013;1833:2526–2541. doi: 10.1016/j.bbamcr.2013.01.028. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, et al. Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat Chem Biol. 2012;8:342–349. doi: 10.1038/nchembio.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Saint-Jean M, et al. Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J Cell Biol. 2011;195:965–978. doi: 10.1083/jcb.201104062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulz TA, et al. Lipid-regulated sterol transfer between closely apposed membranes by oxysterol-binding protein homologues. 2009;187:889–903. doi: 10.1083/jcb.200905007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doublié S. In: Methods Enzymol. Carter CW, Sweet RM, editors. Vol. 276. Academic Press; 1997. pp. 523–530. [Google Scholar]
- 25.Kabsch W. XDS Acta Cryst D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott wG, Cowtan K. Features and Development of Coot. Acta Cryst D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; [Google Scholar]
- 29.Collins TJ. ImageJ for microscopy. Biotechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.