

Abstract
Thermoanaerobacterium saccharolyticum, a Gram-positive thermophilic anaerobic bacterium, grows robustly on insoluble hemicellulose, which requires a specialized suite of secreted and transmembrane proteins. We report here the characterization of proteins secreted by this organism. Cultures were grown on hemicellulose, glucose, xylose, starch, and xylan in pH-controlled bioreactors, and samples were analyzed via spotted microarrays and liquid chromatography-mass spectrometry. Key hydrolases and transporters employed by T. saccharolyticum for growth on hemicellulose were, for the most part, hitherto uncharacterized and existed in two clusters (Tsac_1445 through Tsac_1464 for xylan/xylose and Tsac_1344 through Tsac_1349 for starch). A phosphotransferase system subunit, Tsac_0032, also appeared to be exclusive to growth on glucose. Previously identified hydrolases that showed strong conditional expression changes included XynA (Tsac_1459), XynC (Tsac_0897), and a pullulanase, Apu (Tsac_1342). An omnipresent transcript and protein making up a large percentage of the overall secretome, Tsac_0361, was tentatively identified as the primary S-layer component in T. saccharolyticum, and deletion of the Tsac_0361 gene resulted in gross morphological changes to the cells. The view of hemicellulose degradation revealed here will be enabling for metabolic engineering efforts in biofuel-producing organisms that degrade cellulose well but lack the ability to catabolize C5 sugars.
INTRODUCTION
Bacterial conversion of plant biomass plays a central role in the global carbon cycle and agricultural processes and is of interest for new industrial processes (1). However, due to the recalcitrance of these substrates (2), their solubilization presents a significant challenge in both fundamental and applied contexts (3). The three primary components of plant biomass are cellulose (a β-1-4-linked chain of glucose), hemicellulose (a branched heteropolymer composed of several sugars with variable acetylation and sulfation), and lignin (4). Cellulose and hemicellulose are typically present in a ratio of about 3:1 in woody materials, with lower ratios being common in grasses (5). Microbially mediated solubilization of both cellulose and hemicellulose requires specialized suites of enzymes, readily occurs under anoxic as well as oxic conditions, and is often carried out by specialized microorganisms and communities. Anaerobic fermentation of cellulose and hemicellulose is of interest because of the production of reduced organic compounds with potential utility as fuels or chemicals.
Recently, in order to characterize the enzymatic and secretory pathways of hemicellulose-utilizing anaerobes, researchers have begun to employ transcriptomics, proteomics, and a combination of the two (6–8) as higher-throughput methods of determining the enzymes involved in solubilization. In these reports, many genes and their products previously annotated as hypothetical or putative could be assigned predicted functions. In addition, this type of approach allows the elucidation of complex regulatory networks involving posttranscription control (9–12).
Among the secreted proteins, the S layer is an important part of the cell envelopes of most bacteria (including those involved in hemicellulose deconstruction) and almost all archaea (13). This is a protein latticework that serves to protect cells from a number of environmental factors, such as predation by prokaryotic parasites, high calcium and sulfate ion concentrations (14), shear stress, and possibly, solvents (15). In addition, the S layer acts as an anchor point for surface glycosylation (16, 17), can serve as a molecular sieve (18–20), and is involved with protein anchoring. For example, the S layer produced by Bacillus stearothermophilus DSM 2358 anchors a surface-associated amylase (21).
Thermoanaerobacterium saccharolyticum is a hemicellulolytic, anaerobic, thermophilic, Gram-positive bacterium and an important community member of consortia responsible for cellulose degradation (22) and was first isolated from a hydrothermal spring (23). In addition to its role in the environment, T. saccharolyticum also has the potential to be used as an industrial organism, given its amenability to genetic engineering and fermentation (24–28). It grows rapidly on most hemicellulose components of plant cell walls, in addition to the hydrolysis products of cellulose (i.e., glucose and cellobiose) (23, 27, 29). While a number of studies involving T. saccharolyticum have looked at individual or small groups of biomass-active enzymes (23, 29–37), none have examined the transcriptome and proteome as a whole.
In this study, we sought to characterize the collection of proteins that T. saccharolyticum localizes to its membrane or secretes during growth on various soluble and insoluble hemicellulose-related substrates and to thereby elucidate various aspects of the organism's physiology.
MATERIALS AND METHODS
Strains and media.
Wild-type strain T. saccharolyticum JW/SL-YS485 (DSM 8691), obtained from DSMZ (the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Germany), was grown exclusively in TSC1 medium (26), which consists of (liter−1) 2.0 g sodium citrate tribasic dihydrate, 1.9 g (NH4)2SO4, 0.1 g FeSO4·7H2O, 1.0 g MgSO4, 1.0 g KH2PO4, 0.1 g CaCl2·2H2O, and 8.5 g yeast extract, with 5 g/liter of one of the following: glucose, starch from potatoes, xylose, or xylan from birch. All growth, unless otherwise specified, was performed in a Coy anaerobic chamber.
Reactor inoculation, growth, and sampling.
Reactor starter cultures consisted of 50 ml of TSC1 medium with the appropriate carbon source in 115-ml nitrogen-flushed anaerobic serum bottles which was inoculated with T. saccharolyticum, and the starter cultures were subcultured twice to ensure acclimation to the medium prior to inoculation into the reactor. Batch fermentations in TSC1 medium with the appropriate carbon source were performed in Sartorius Biostat Qplus fermentors with a 1-liter working volume (Sartorius-Stedim, Bohemia, NY), which were constantly purged with a 95% nitrogen–5% CO2 gas mixture. The fermentors were maintained at a pH of 6.1 via the addition of 5 M KOH, the temperature was held at 55°C, and the cultures were agitated at 150 rpm. The reactors were sampled at 1/3 the maximum optical density at 600 nm (OD600; which was an OD600 of 0.33) or 1/3 the maximum base addition (total base addition was calibrated on the basis of repeated test runs with a given substrate) when OD600 readings were impossible to obtain due to the presence of insoluble substrates (i.e., starch and xylan). A second set of samples was taken 1 h after the cessation of base addition and the increase in the OD for soluble substrates ceased. For both time points, samples were taken from the same reactors for transcriptomic and proteomic analyses. Samples for transcriptomic and proteomic analyses taken at the time point of 1/3 the maximum optical density are referred to as mid-log-phase samples, and those taken at 1 h after the cessation of growth are referred to as stationary-phase samples.
For transcriptomic analysis, 15-ml samples were mixed with 30 ml RNAprotect Bacteria reagent (Qiagen Corp., Valencia, CA) and incubated at room temperature for 5 min, followed by centrifugation at 4,000 × g for 10 min at 4°C. The pellets were then resuspended in 1 ml SET (50 mM Tris-HCl, pH 8.0, 50 mM EDTA, 20% [wt/vol] sucrose) buffer and stored at −80°C. For proteomic analysis, 250 ml of culture was removed and centrifuged at 8,000 × g, and the pellets and cleared supernatants were stored separately at −80°C. Biological duplicates were used for all experiments. Due to the scale of the experiments described herein, budgetary constraints limited us to two biological replicates.
Prediction of transmembrane and secreted proteins.
To predict which proteins were secreted via the Sec system (the only identifiable secretion system present in the T. saccharolyticum genome, as assessed by BLAST analysis) and which were present in the membrane, two prediction packages were employed. Sec tags were predicted using the SignalP (v. 4.1) program (38). Transmembrane domain-containing proteins were predicted using the TMHMM server (v. 2.0) (39), followed by the removal of all proteins containing only 1 transmembrane domain, which was predicted to be a Sec tag.
Microarrays.
The T. saccharolyticum JW/SL-YS485 2.88-Mb genome has a 35.1% G+C content and consists of a chromosome (GenBank accession number CP003184.1), phage sequence (GenBank accession number CP003186.1), and plasmid pMU3262 (GenBank accession number CP003185.1), and the genome carries 2,840 candidate protein-coding models. Oligonucleotide probes that were 70 nucleotides in length for a whole-genome DNA microarray were designed using CommOligo software (40), as described previously (41), and commercially synthesized without modification in 96-well plates. Probe concentrations were adjusted to 100 pmol/μl, and the probes were transferred to 384-well printing plates in dimethyl sulfoxide at a final concentration of 50% using a BioMek FX liquid-handling robot (Beckman Coulter, Fullerton, CA) and then spotted onto UltraGAPS glass slides (Corning Life Sciences, Corning, NY) using a BioRobotics Microgrid II microarrayer (Genomic Solutions, Ann Arbor, MI) in a dust-free clean room maintained at 21°C and 50% relative humidity. The spotted DNA was stabilized on slides by UV cross-linking using a UV 1800 Stratalinker cross-linker (Stratagene, La Jolla, CA) according to the slide manufacturer's instructions (Corning Life Sciences). Total RNA was purified using an RNeasy Plus minikit (Qiagen Corp.), which was used as the template to generate cDNA copies labeled with Cy5-dUTP (Amersham Biosciences, Piscataway, NJ), and then the purified cDNA was dried using an SDP1010 SpeedVac system (ThermoSavant, Holbrook, NY). The hybridization and washing conditions for the oligonucleotide microarrays have been described elsewhere (42), except that in this study hybridizations were conducted using a 12-bay hybridization station (BioMicro Systems, Inc., Salt Lake City, UT), and the arrays were dried using a Maui wash system (BioMicro Systems, Inc.). DNA microarray outlier detection and statistical analyses were conducted essentially as described previously (43), except that JMP Genomics (v. 4.1) software (SAS Institute, Cary, NC) was used for statistical analyses. Data were normalized using the LOWESS normalization algorithm within JMP Genomics software. An analysis of variance (ANOVA) was performed to determine differential expression levels between conditions and time points using the false discovery rate (FDR) testing method (P < 0.05).
Analysis of peptides by multidimensional LC-MS/MS analysis.
Both whole-cell secretome samples and cell-free secretome samples (which were centrifuged, after which the supernatant was filtered through a 0.22-μm-pore-size filter) were prepared for liquid chromatography (LC)-tandem mass spectrometry (MS/MS) analysis as follows: proteins were denatured and reduced by addition of SDS lysis buffer (4% SDS in 100 mM Tris-HCl, pH 8.0, with 10 mM dithiothreitol [DTT]) at a 1:1 (vol/vol) ratio, and the mixture was boiled and sonicated with a Branson sonic disruptor (20% amplitude, 2 min; 10-s pulse; 10-s pause). Trichloroacetic acid was added to a concentration of 20% (wt/vol) to precipitate sample proteins from the detergent and solutes. Ice-cold, acetone-washed pelleted proteins were resuspended with 8 M urea in 100 mM Tris-HCl, pH 8.0. The amount of recovered protein was measured using a bicinchoninic acid (BCA) assay (Thermo-Pierce, Rockford, IL). Proteins were reduced with 5 mM DTT, alkylated with 10 mM iodoacetamide, and digested with two separate and sequential aliquots of sequencing-grade trypsin (Promega) at a 1:100 (wt/wt) enzyme-to-protein ratio. As 8 M urea inhibits trypsin, samples were diluted to 4 M for an overnight digestion, followed by dilution with 2 M urea for a 4-h digestion. Samples were then adjusted with 150 mM NaCl and 0.1% formic acid and filtered through a 500-μl 10-kDa-cutoff spin column filter (VWR brand). Peptide concentrations were then measured using the BCA assay.
For the LC-MS/MS measurements, a 25-μg aliquot of peptides was bomb loaded onto a biphasic MudPIT back column as described previously (44, 45). Loaded peptides were then washed with solvent A (5% acetonitrile, 95% high-pressure liquid chromatography [HPLC]-grade water, 0.1% formic acid) for 20 min, followed by a 25-min gradient to solvent B (70% acetonitrile, 30% HPLC-grade water, 0.1% formic acid) offline. Desalted peptides were then placed in-line with an in-house-pulled, reverse-phase-packed nanospray emitter and analyzed by a two-dimensional liquid chromatographic method (11 strong cation-exchange steps, each followed by a 2-h reversed-phase elution) in which the liquid chromatograph was interfaced on-line with a hybrid LTQ-Velos/Orbitrap mass spectrometer (Thermo Fisher, Waltham, MA) operating in a data-dependent fashion. Each full scan (2 microscans) generated by the Orbitrap mass analyzer (30,000 resolution) was followed by 10 MS/MS events (2 microscans) in the LTQ-Velos/Orbitrap mass spectrometer. Two replicate measurements were obtained for each sample.
MS data analysis and evaluation.
Acquired MS/MS spectra were assigned to specific peptide sequences, along with common contaminant protein entries as well as reversed decoy entries to assess protein-level false discovery rates, using the SEQUEST search algorithm (46) with a FASTA proteome database specific to T. saccharolyticum. SEQUEST-scored peptide sequence data were filtered and assembled into protein loci using the DTASelect software package (47) with the following conservative criteria: for XCorr, +1 = 1.8, +2 = 2.5, and +3 = 3.5; DeltCN = 0.08; and 2 unique peptides per protein identification. Prior to semiquantitative analysis, spectral counts were rebalanced to properly distribute nonunique/shared peptides between their potential parent proteins solely on the basis of their unique spectral counts. Normalized spectral abundance factor (NSAF) values were then calculated for each protein (48).
NSAF values were imported into the JMP Genomics software package (v. 4.1; SAS Institute) for statistical analyses (49). After transformation with the natural logarithm and standardization to correct for signal intensity, one-way ANOVA was used to identify proteins that showed significant differences in abundance under various growth conditions (P < 0.01).
The predicted protein sequence of T. saccharolyticum was submitted to the SignalP server (v. 3.0) (50) to predict the presence of signal peptides. Hidden Markov model analysis with parameters for Gram-positive bacteria was used to identify signal peptidase I cleavage sites within the first 70 residues of each protein. Probability scores (SProb) were used to distinguish proteins that could be translocated by the Sec-dependent pathway (51). This analysis did not identify proteins translocated by alternative, less common secretory pathways.
Deletion of Tsac_0361.
The deletion of Tsac_0361 was performed via nonreplicative vector-driven replacement of the open reading frame with a kanamycin resistance cassette using two regions of homology of roughly 1,000 bp each on either side of Tsac_0361. The suicide vector pALKO1-2 was constructed using Gibson cloning (52, 53), the primers for which can be found in Table 1. Transformation was performed as previously described (25). Selection was performed on TSC1 medium supplemented with 200 μg/ml kanamycin. Deletions were confirmed by PCR using primers kanregion_F and kanregion_R (Table 1).
TABLE 1.
Primers used in construction of pALKO1-2
| Primer namea | Sequence |
|---|---|
| PMC500BACK_F | TCTATCAGCTGTCCCTCCTGTTC |
| PMC500BACK_R | CCAGGCATCAAATAAAACGAAAG |
| 0361upstreamF | CAGTCTTTCGACTGAGCCTTTCGTTTTATTTGATGCCTGGCATCTTATAAGATAGTGTACGAGGAAAGTG |
| 0361KOupWT_R | GTATAATCTTACCTATCACCTCAAATGGTTCGCTGGGTTTGGAATAAATCCTCCTCCTTAATATATAGTG |
| 0361KOdownWT_F | CTAGATAGGGGTCCCGAGCGCCTACGAGGAATTTGTATCGCACAAGCCTAACAAAAAAGTCCTC |
| 0361downstreamR | CCACCCCGTCAGTAGCTGAACAGGAGGGACAGCTGATAGAGCAAGTGATCTAAATCTAAATATCTTCTTC |
| kanregion_F | AAACCCAGCGAACCATTTGAGG |
| kanregion_R | CGATACAAATTCCTCGTAGGCGC |
F, forward; R, reverse.
Microscopy.
Cultures for microscopy were grown in TSC1 medium lacking antibiotic selection. Phase-contrast microscopy was performed on overnight cultures of wild-type and T. saccharolyticum ΔTsac_0361 strains. Preparation of samples for transmission electron microscopy (TEM) was carried out at room temperature, unless otherwise noted. All cell types were washed with 3% glutaraldehyde (GTA), 1% paraformaldehyde (PF) in 0.1 M Na cacodylate buffer (pH 7.2 to 7.4) (NaCac). Cells were then fixed in 3% GTA, 1% PF, 0.2% tannic acid (TA) in 0.1 M NaCac for 2 h. Fresh fixative was then added prior to a further 1-h incubation. Finally, cells were placed on a rotator for 24 h at 40°C. Cells were washed with 0.1 M NaCac. Samples were postfixed in 1% osmium tetroxide (OsO4) and 1,000 ppm ruthenium red (RR) in 0.1 M NaCac for 2 h. Cells were then rinsed twice in distilled H2O. Dehydration via ethanol was performed in a 30%, 50%, and 70% series for 30 min each in tubes in a rotor at room temperature. A final 24-h incubation in 70% ethanol was performed in a rotor at 40°C. En bloc staining was done with 1% uranyl acetate in 70% ethanol for 3 h at 4°C on a rotator in the dark. Samples were then further dehydrated in 85% and 95% ethanol for 1 h each on a rotator at room temperature. A final dehydration was performed in 100% ethanol over the course of 6 h on a rotator at room temperature. Samples were then embedded in LX112 resin. Thin sections were stained with 2% uranyl acetate (UA) in methanol for 15 min and Reynold's lead citrate for 3 min. TEM images were taken at 100 kV on a FEI Tecnai F20ST FEG TEM equipped with a digital camera (XR-41B; Advanced Microscopy Techniques, Woburn, MA).
Data analysis.
Data were analyzed using the R statistical software package (v.2.15.2; the R Foundation) (54), unless otherwise specified.
RESULTS
Prediction of secreted and transmembrane domain-containing proteins.
Using the SignalP server, a total of 131 proteins with secretory sequences were found in the genome sequence of T. saccharolyticum strain JW/SL-YS485 (see Table S1 in the supplemental material). Of these, 33 are annotated as hypothetical proteins and 15 are predicted to be carbohydrate-active enzymes by the Carbohydrate Active Enzymes database (55). All of the predicted secreted proteins use the Sec system, with no evidence for other secretory systems (e.g., the Tat system [56]) being found, and this finding is also true for other related organisms, for example, Clostridium thermocellum and Clostridium acetobutylicum (57). Another 649 proteins contain transmembrane sequences (see Table S2 in the supplemental material). Most of the proteins containing transmembrane domains have multiple membrane-spanning domains, with a large portion of these being either phosphotransferase (PTS) or ABC transporters.
Substrate-specific gene expression revealed by transcriptomics.
To examine the transcriptional response to different growth substrates, microarrays were used during growth on glucose, starch, xylose, or xylan in mid-log or stationary phase (see File S2 in the supplemental material). The data were normalized, and genes coding for proteins with Sec tags or transmembrane domains were compared (Fig. 1). mRNA levels from stationary-phase samples were found to be quite variable and were excluded from our analysis.
FIG 1.

Comparison of microarray results between different conditions during mid-log growth (log2 expression values) with strongly differentially expressed genes. The sugars studied were glucose (which has 6 carbons), starch (which is made up of glucose monomers), xylose (which has 5 carbons), and xylan (which is made up of xylose monomers). (A) Expression with growth on glucose at mid-log phase versus expression with growth on starch at mid-log phase; (B) expression with growth on xylose at mid-log phase versus expression with growth on xylan at mid-log phase; (C) expression with growth on glucose at mid-log phase versus expression with growth on xylose at mid-log phase; (D) expression with growth on starch at mid-log phase versus expression with growth on xylan at mid-log phase.
One of the most noticeable trends was the increased number of upregulated substrate-specific genes as the substrate complexity increased. This tendency can best be seen in Fig. 1C and D. Whereas growth on glucose versus xylose should require a shift in only the transporter complement (Fig. 1C), the difference between growth on starch and xylan required a host of changes, including transporters, sensory genes, and hydrolases (Fig. 1D). One of the genes upregulated during growth on xylan which is of particular interest is Tsac_0898, which will be further discussed below.
Surprisingly, much of the diversity between substrates in the secreted and transmembrane proteins came in the form of transporters rather than hydrolases. Of these changes, many either occurred in putative operons or were localized to specific regions of the chromosome (Fig. 1).
Characterization of the proteome.
Unlike for the microarray experiments, we chose to investigate those proteins with Sec tag or transmembrane domains as well as the unsorted, whole-cell results (see File S2 in the supplemental material). ANOVAs were performed on proteins predicted to contain either a Sec or a transmembrane domain to choose those with significant differences between conditions. The 50 proteins with the lowest ANOVA P values (which fell between 0.008 and 1.97E−14; see Table S3 in the supplemental material) were ordered by hierarchical clustering (Fig. 2).
FIG 2.

Hierarchical clustered heat map of the genes for 50 proteins that contain transmembrane or Sec tag domains that were the most significantly expressed, as detected by LC-MS/MS. The data were normalized, and the results were analyzed by ANOVA, which was performed to determine differences in expression between growth substrates. FAD, flavin adenine dinucleotide; Stat_Av, average value for stationary-phase growth; Mid_Av, average value for mid-log-phase growth.
Clustering primarily occurred with sugar type (i.e., 5 versus 6 carbons), with additional clustering between mid-log-phase and stationary-phase cultures being found. While the glucose and starch cultures clustered quite strongly, the stationary-phase xylose cultures appeared to be more similar to stationary-phase xylan cultures and thus complicate the dendrogram. Cultures grown on xylose and xylan both had a relatively large number of proteins which responded strongly to their presence. Conversely, cultures grown on glucose and starch had relatively few proteins that were strongly associated with these substrates alone. It should be noted that we had hoped to use the supernatant fractions that we collected to interrogate the T. saccharolyticum secretome. Unfortunately, due to the strong cell surface tethering of a number of important proteins, compounded by mild contamination of the supernatants by cytosolic proteins, we abandoned further analysis of these data (see Fig. S1 in the supplemental material).
The same analysis was also performed on the full set of LC-MS/MS results, regardless of the predicted localization (Fig. 3). When whole-cell proteins were included in the analysis, cluster differentiation became much stronger, with clear boundaries and smaller ANOVA P values (see Table S4 in the supplemental material). This suggests that while the cytosolic enzyme complement is closely tied to substrate type, the expression of those proteins which are destined for the membrane or extracellular environment is less tightly regulated.
FIG 3.

Hierarchical clustered heat map of the genes for 50 total proteins in the pellet that were the most significantly expressed, as detected by LC-MS/MS. The data were normalized, and the results were analyzed by ANOVA, which was performed to determine differences in expression between growth substrates.
Of those proteins that showed almost no change in response to substrate, Tsac_0361 stood out. In the pellet, for those proteins which contain transmembrane or Sec tag domains, Tsac_0361 comprised approximately 24% of total protein length-normalized LC-MS/MS peptide detections. In addition, Tsac_0361 was the 4th most frequently detected protein in the pellets, regardless of localization. While these numbers cannot be taken as absolute quantities, given the limitations and caveats of LC-MS/MS, they do tend to correlate with abundance. Given that Tsac_0361, unlike the other frequently detected proteins, is unidentified and is poorly annotated (with the exception of a predicted S-layer homology [SLH] domain), we considered a test for this protein to be reasonable to determine the relevance of our data set for the identification of novel, physiologically important proteins.
Tsac_0361 is the S-layer structural protein.
BLASTP searches revealed that the Tsac_0361 protein is unique to T. saccharolyticum, and the only region of the protein that shared homology to other proteins was an N-terminal S-layer homology domain. This suggests that whatever the function of Tsac_0361, it would most likely be cell surface associated, as many cell surface-tethered proteins, including XynA, contain SLH domains.
To experimentally explore its function, we deleted Tsac_0361 from a wild-type T. saccharolyticum strain. While the strain was capable of growth, maximum achievable cell densities were low (roughly 20% of the density for the parent strain) and the medium seemed to become increasingly viscous during growth. To determine if there was a morphological effect, we observed the ΔTsac_0361 mutant using phase-contrast microscopy (Fig. 4). The cells showed aberrant and diverse morphologies. Some cells appeared to fail to divide correctly or produced large blebs. These blebs appeared to completely lack a cell wall and will be discussed in more detail below.
FIG 4.
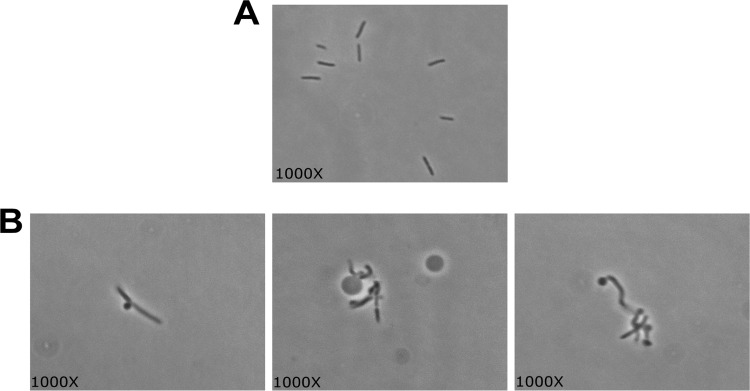
Phase-contrast microscopy of wild-type T. saccharolyticum (A) and T. saccharolyticum ΔTsac_0361 (B). Additional panes in panel B demonstrate the variety of morphologies observed.
This poor growth phenotype, combined with the presence of the SLH domain and the abundance of Tsac_0361, led us to hypothesize that this protein may be an important structural component of the cell envelope. Specifically, we suspected that Tsac_0361 was the primary component of the S layer in T. saccharolyticum. In order to test this, we chose to visualize the cell envelope of the two strains via transmission electron microscopy (TEM).
The results of the comparison of the wild-type and ΔTsac_0361 strains by TEM are shown in Fig. 5. The wild-type cells have recognizable S layers, seen as a second white band on top of the peptidoglycan, which is further decorated (S layers are heavily glycosylated and act as the anchor point for some extracellular proteins [58, 59]). In contrast, while the deletion mutant maintains normal membrane and peptidoglycan layers, it lacks the S layer and its accompanying decoration. Additionally, the previously mentioned blebs did, indeed, appear to lack peptidoglycan (Fig. 6).
FIG 5.
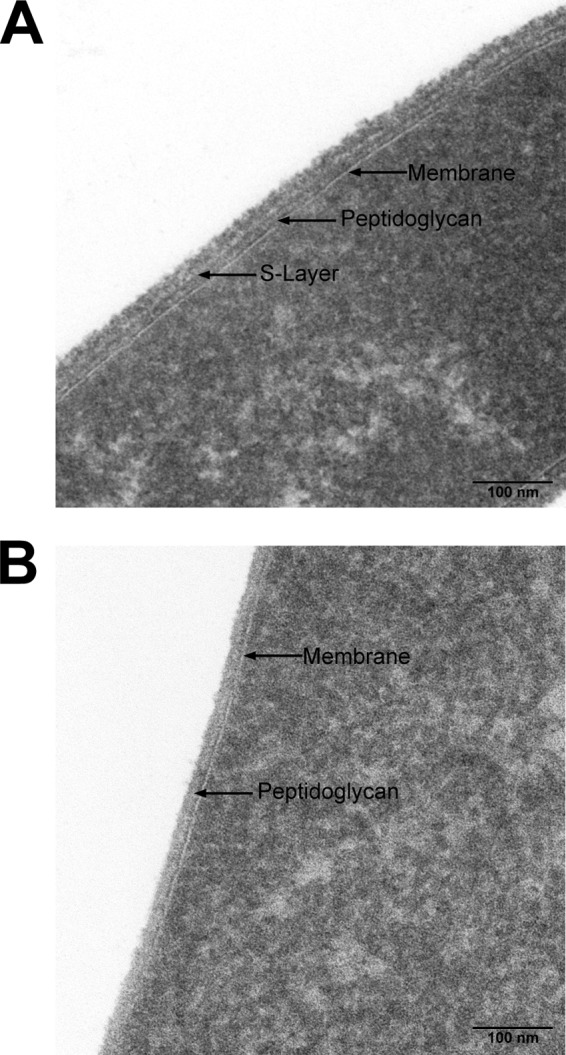
Transmission electron micrographs of wild-type T. saccharolyticum (A) and T. saccharolyticum ΔTsac_0361 (B). The membrane, peptidoglycan, and S layer are present in the wild type and are indicated, whereas only the membrane and peptidoglycan are present in the strain with the Tsac_0361 deletion.
FIG 6.
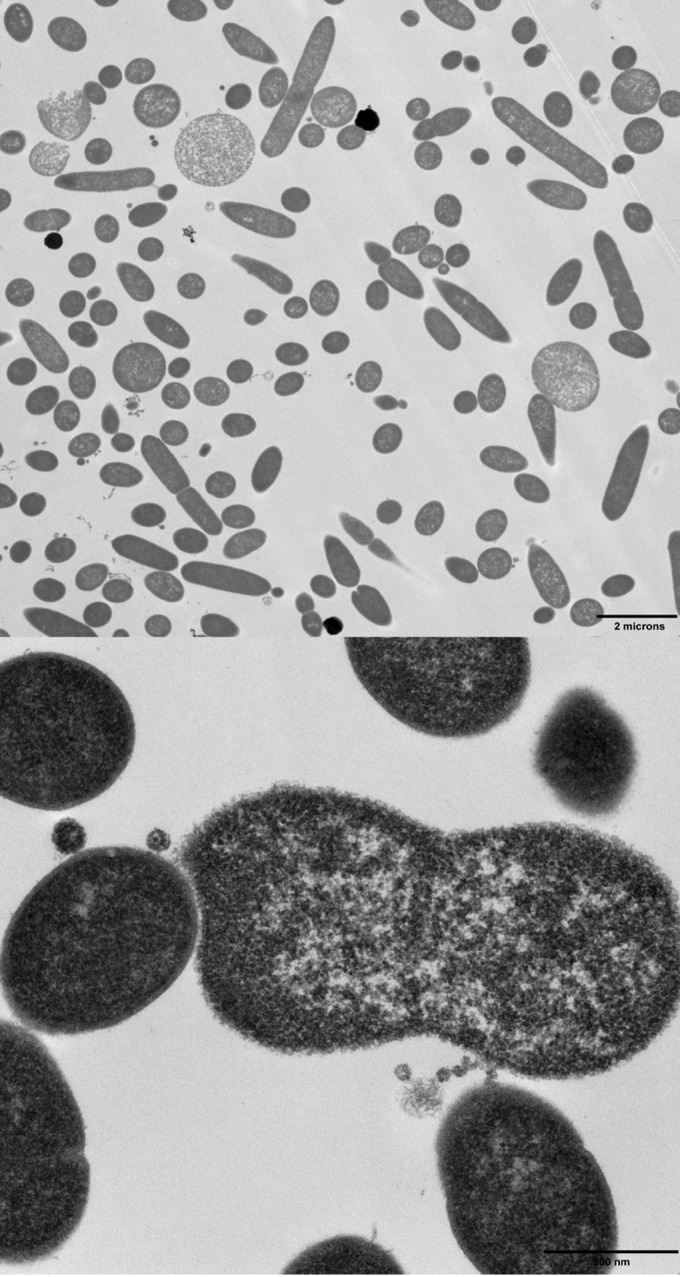
Transmission electron micrograph showing the production of blebs during growth of the ΔTsac_0361 mutant. The blebs both are larger than normal cells and lack a discernible cell wall. Additionally, they appear to be flexible and bend around other cells, further supporting a lack of a cell wall.
We attempted to complement the deletion of the chromosomal copy of Tsac_0361 via the expression of Tsac_0361 driven by the constitutive C. thermocellum cellobiose phosphorylase (cbp; Cthe_0275) promoter on the replicative plasmid pMC805, which is based on pIKM1 (60). While a clear increase in the number of cells with a wild-type morphology was observed, the complementation appeared to be incomplete when we used the cbp promoter to drive Tsac_0361 expression (data not shown).
DISCUSSION
Here we report the results of the first analysis of the transcriptome and proteome of T. saccharolyticum when the organism is growing on various carbon sources as well as the identification of an important component of the cell envelope.
A large number of proteomic and transcriptomic experiments have been performed to analyze different aspects of bacterial physiology. We, along with a growing number of researchers (9–12), consider the combined transcriptomic and proteomic approach to paint a more complete picture than any single method. One example of this is in the form of the heat shock sigma factor SigH. It has been shown in other systems that there is substantial posttranscriptional regulation (61). We see this reflected in our data as well, with significant levels of transcript being present but no protein being detected. This finding is not surprising, as a heat shock event would be highly unlikely in our controlled-fermentation environment. Conversely, not all proteins make ideal substrates for mass spectrometry. Tsac_1420, the ATP synthase subunit c, although essential for generating the proton motive force, is quite small and is cut only 3 times by trypsin. This produces both very small and very large fragments, but such fragments are not ideal for MS. The result is that this protein is missed by MS but not by the microarrays. With a multipronged approach, additional layers of regulation that would have previously gone unnoticed can be extracted.
Many of the key hydrolases present in T. saccharolyticum have been identified previously (29, 30, 33, 37). These hydrolases include XynA (Tsac_1459), XynC (Tsac_0897), and a pullulanase, Apu (Tsac_1342). A number of uncharacterized enzymes were also identified. During growth on starch, a glucan, 1,4-alpha-glucosidase (Tsac_1365), was strongly upregulated, as was Tsac_1346, a putative alpha-amylase. Most of the major enzymes involved with xylan utilization have been previously described (37), but one gene, Tsac_0898, which is annotated as a sulfatase-modifying factor protein, has not been studied. We show that this protein's expression, as determined by both microarray analysis and LC-MS/MS, is linked to mid-log growth on xylan and stationary-phase growth on xylose or xylan. Sulfatase-modifying proteins activate sulfatases by altering either a cysteine or a serine residue to form C-α-formylglycine (FGly) at the short motif C/SXPXR in the active site of the enzyme (62–64). Once active, sulfatases hydrolyze sulfate esters in a wide variety of processes (65, 66). These proteins and their modifying factors are conserved across all domains (67). Although the role of the as yet unidentified T. saccharolyticum sulfatase is unknown, other sulfatases have been shown to be active on chondroitin sulfate and heparin, both of which are polysaccharides (66, 68), suggesting that Tsac_0898 may be involved in desulfurization of sulfated hemicelluloses.
The transporters employed by T. saccharolyticum for growth on hemicellulose have not been previously characterized. Here we show that these transporters exist in two major gene clusters (Tsac_1445 through Tsac_1464 for xylan/xylose and Tsac_1344 through Tsac_1349 for starch). A PTS system subunit, Tsac_0032, also appeared to be exclusive to growth on glucose.
It is of note that the transcriptomic and proteomic profiles of the stationary-phase xylose cultures more closely resemble those of mid-log-phase and stationary-phase xylan cultures than those of mid-log-phase xylose cultures. This makes intuitive sense, given that once it is solubilized, xylan (i.e., xylose) is depleted and the organism would attempt to liberate more from an expected environmental source (e.g., a blade of grass). This trend in the upregulation of hemicellulases did not appear to be the simple result of starvation or the onset of stationary phase, as no such shift in the glucose- or starch-grown cultures was observed. The implication is that the regulation of hemicellulase expression involves a degree of memory. This type of regulation has been seen in other bacteria and is beginning to be appreciated as an important component of gene regulation (69–71). This phenotype, combined with the ease of genetic manipulation in this organism, suggests that T. saccharolyticum might be useful as a potential model organism for second-order gene regulation within thermophilic bacteria.
Contrary to our predictions, the cell protein complement as a whole, rather than just proteins involved with interfacing with the environment (transmembrane and secreted proteins), showed the largest changes in response to substrate. This was surprising, as we expected relatively subtle differences in cytosolic proteins between growth substrates. One anticipated exception to this prediction were those enzymes participating in the pentose phosphate pathway, for example, xylulokinase, which we show to be strongly differentially regulated.
Of those secreted or transmembrane proteins (and their mRNAs) that did change, most are poorly annotated transporters which deserve further study. Similar transcriptomic experiments have been shown to be particularly useful in predicting the function of various mono- and polysaccharide transporters (6, 7, 72, 73). These proteins also have potential applications in biotechnology. The heterologous expression of some or all of these transporters could increase the catabolic repertoire of industrially useful microbes. One such organism that would benefit from this approach is the well-characterized thermophilic bacterium Clostridium thermocellum. While it has the ability to hydrolyze hemicellulose, it cannot catabolize the liberated sugars. Expression of the transporters identified here, along with components of the pentose phosphate pathway, could allow C. thermocellum access to an additional 20 to 40% of the plant biomass that is comprised of hemicellulose (5). This task is made easier by the chromosomal arrangement of these genes, which are generally clustered on the chromosome (in both an operon-dependent and an operon-independent fashion), which would ease cloning and heterologous expression. Studies of other hemicellulolytic bacteria have also shown the same general trend in gene clustering (6, 7, 72, 73).
Some of the first evidence for bacterial protein glycosylation came from the study of glycosylated S-layer proteins in Thermoanaerobacterium thermosaccharolyticum in 1976 (16). While this pioneering work on bacterial glycosylation set the stage for work in many other microbes, the gene coding for the S-layer protein in T. thermosaccharolyticum has remained unidentified. On the basis of homology to Tsac_0361, the T. thermosaccharolyticum S-layer protein is likely Thethe_02622, which shares 92% amino acid identity with Tsac_0361.
The large number of LC-MS/MS detections of Tsac_0361 suggested that this protein was generally important within the T. saccharolyticum pool of secreted proteins. Although no characterized homologs were identified, the basic structure (two amino-terminal SLH domains), followed by a long run of seemingly degenerate sequence, was reminiscent of that of the S-layer proteins from other Firmicutes. The importance for normal growth but the nonessentiality further supported this hypothesis. Finally, via TEM, we have shown the distinct lack of an S layer and the accompanying decoration in the deletion strain. Further work will be required to complete the characterization of Tsac_0361 and determine the cause of the incomplete complementation, which we suspect is the result of insufficient expression to consistently assemble a complete S layer. We propose the renaming of Tsac_0361 to SlpA (S-layer protein A). The identification of slpA and its deletion will facilitate the study of S layers and their role in glycosylation, protein anchoring, and general cell physiology within the genus Thermoanaerobacterium.
Supplementary Material
ACKNOWLEDGMENTS
We thank Louisa Howard of the Rippell Electron Microscopy facility at Dartmouth College for assistance with the EM studies and Alicia Eve Ballok for critical reading of the manuscript.
This research was supported by Mascoma Corporation, Lebanon, NH, and a grant from the BioEnergy Science Center (BESC), Oak Ridge National Laboratory, a U.S. Department of Energy (DOE) Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science.
Footnotes
Published ahead of print 6 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00998-14.
REFERENCES
- 1.Falkowski P, Scholes RJ, Boyle E, Canadell J, Canfield D, Elser J, Gruber N, Hibbard K, Hogberg P, Linder S, Mackenzie FT, Moore B, III, Pedersen T, Rosenthal Y, Seitzinger S, Smetacek V, Steffen W. 2000. The global carbon cycle: a test of our knowledge of earth as a system. Science 290:291–296. 10.1126/science.290.5490.291 [DOI] [PubMed] [Google Scholar]
- 2.Klemm D, Heublein B, Fink HP, Bohn A. 2005. Cellulose: fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 44:3358–3393. 10.1002/anie.200460587 [DOI] [PubMed] [Google Scholar]
- 3.Wilson DB. 2011. Microbial diversity of cellulose hydrolysis. Curr. Opin. Microbiol. 14:259–263. 10.1016/j.mib.2011.04.004 [DOI] [PubMed] [Google Scholar]
- 4.Popper ZA. 2008. Evolution and diversity of green plant cell walls. Curr. Opin. Plant Biol. 11:286–292. 10.1016/j.pbi.2008.02.012 [DOI] [PubMed] [Google Scholar]
- 5.McKendry P. 2002. Energy production from biomass (part 1): overview of biomass. Bioresour. Technol. 83:37–46. 10.1016/S0960-8524(01)00118-3 [DOI] [PubMed] [Google Scholar]
- 6.Conners SB, Montero CI, Comfort DA, Shockley KR, Johnson MR, Chhabra SR, Kelly RM. 2005. An expression-driven approach to the prediction of carbohydrate transport and utilization regulons in the hyperthermophilic bacterium Thermotoga maritima. J. Bacteriol. 187:7267–7282. 10.1128/JB.187.21.7267-7282.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dodd D, Moon YH, Swaminathan K, Mackie RI, Cann IKO. 2010. Transcriptomic analyses of xylan degradation by Prevotella bryantii and insights into energy acquisition by xylanolytic Bacteroidetes. J. Biol. Chem. 285:30261–30273. 10.1074/jbc.M110.141788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunne JC, Li D, Kelly WJ, Leahy SC, Bond JJ, Attwood GT, Jordan TW. 2012. Extracellular polysaccharide-degrading proteome of Butyrivibrio proteoclasticus. J. Proteome Res. 11:131–142. 10.1021/pr200864j [DOI] [PubMed] [Google Scholar]
- 9.Gilad O, Jacobsen S, Stuer-Lauridsen B, Pedersen MB, Garrigues C, Svensson B. 2010. Combined transcriptome and proteome analysis of Bifidobacterium animalis subsp. lactis BB-12 grown on xylo-oligosaccharides and a model of their utilization. Appl. Environ. Microbiol. 76:7285–7291. 10.1128/AEM.00738-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raberg M, Peplinski K, Heiss S, Ehrenreich A, Voigt B, Doring C, Bomeke M, Hecker M, Steinbuchel A. 2011. Proteomic and transcriptomic elucidation of the mutant Ralstonia eutropha G(+)1 with regard to glucose utilization. Appl. Environ. Microbiol. 77:2058–2070. 10.1128/AEM.02015-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer A, Yang SJ, Bayer AS, Vaezzadeh AR, Herzig S, Stenz L, Girard M, Sakoulas G, Scherl A, Yeaman MR, Proctor RA, Schrenzel J, Francois P. 2011. Daptomycin resistance mechanisms in clinically derived Staphylococcus aureus strains assessed by a combined transcriptomics and proteomics approach. J. Antimicrob. Chemother. 66:1696–1711. 10.1093/jac/dkr195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pohl S, Tu WY, Aldridge PD, Gillespie C, Hahne H, Mader U, Read TD, Harwood CR. 2011. Combined proteomic and transcriptomic analysis of the response of Bacillus anthracis to oxidative stress. Proteomics 11:3036–3055. 10.1002/pmic.201100085 [DOI] [PubMed] [Google Scholar]
- 13.Sleytr UB, Sara M. 1997. Bacterial and archaeal S-layer proteins: structure-function relationships and their biotechnological applications. Trends Biotechnol. 15:20–26. 10.1016/S0167-7799(96)10063-9 [DOI] [PubMed] [Google Scholar]
- 14.Sara M, Sleytr UB. 2000. S-layer proteins. J. Bacteriol. 182:859–868. 10.1128/JB.182.4.859-868.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rothfuss H, Lara JC, Schmid AK, Lidstrom ME. 2006. Involvement of the S-layer proteins Hpi and SlpA in the maintenance of cell envelope integrity in Deinococcus radiodurans R1. Microbiology 152:2779–2787. 10.1099/mic.0.28971-0 [DOI] [PubMed] [Google Scholar]
- 16.Sleytr UB, Thorne KJ. 1976. Chemical characterization of the regularly arranged surface layers of Clostridium thermosaccharolyticum and Clostridium thermohydrosulfuricum. J. Bacteriol. 126:377–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaffer C, Wugeditsch T, Kahlig H, Scheberl A, Zayni S, Messner P. 2002. The surface layer (S-layer) glycoprotein of Geobacillus stearothermophilus NRS 2004/3a—analysis of its glycosylation. J. Biol. Chem. 277:6230–6239. 10.1074/jbc.M108873200 [DOI] [PubMed] [Google Scholar]
- 18.Sara M, Sleytr UB. 1987. Molecular-sieving through S layers of Bacillus stearothermophilus strains. J. Bacteriol. 169:4092–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sara M, Sleytr UB. 1987. Production and characteristics of ultrafiltration membranes with uniform pores from two-dimensional arrays of proteins. J. Membr. Sci. 33:27–49. 10.1016/S0376-7388(00)80050-2 [DOI] [Google Scholar]
- 20.Sara M, Pum D, Sleytr UB. 1992. Permeability and charge-dependent adsorption properties of the S-layer lattice from Bacillus coagulans E38-66. J. Bacteriol. 174:3487–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egelseer E, Schocher I, Sara M, Sleytr UB. 1995. The S-layer from Bacillus stearothermophilus DSM-2358 functions as an adhesion site for a high-molecular-weight amylase. J. Bacteriol. 177:1444–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Izquierdo JA, Sizova MV, Lynd LR. 2010. Diversity of bacteria and glycosyl hydrolase family 48 genes in cellulolytic consortia enriched from thermophilic biocompost. Appl. Environ. Microbiol. 76:3545–3553. 10.1128/AEM.02689-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee YE, Jain MK, Lee CY, Lowe SE, Zeikus JG. 1993. Taxonomic distinction of saccharolytic thermophilic anaerobes: description of Thermoanaerobacterium xylanolyticum gen. nov., sp. nov., and Thermoanaerobacterium saccharolyticum gen. nov., sp. nov., reclassification of Thermoanaerobium brockii, Clostridium thermosulfurogenes, and Clostridium thermohydrosulfuricum E100-69 as Thermoanaerobacter brockii comb. nov., Thermoanaerobacterium thermosulfurigenes comb. nov., and Thermoanaerobacter thermohydrosulfuricus comb. nov., respectively; and transfer of Clostridium thermohydrosulfuricum 39E to Thermoanaerobacter ethanolicus. Int. J. Syst. Bacteriol. 43:41–51. 10.1099/00207713-43-1-41 [DOI] [Google Scholar]
- 24.Shaw AJ, Podkaminer KK, Desai SG, Bardsley JS, Rogers SR, Thorne PG, Hogsett DA, Lynd LR. 2008. Metabolic engineering of a thermophilic bacterium to produce ethanol at high yield. Proc. Natl. Acad. Sci. U. S. A. 105:13769–13774. 10.1073/pnas.0801266105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw AJ, Hogsett DA, Lynd LR. 2010. Natural competence in Thermoanaerobacter and Thermoanaerobacterium species. Appl. Environ. Microbiol. 76:4713–4719. 10.1128/AEM.00402-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaw AJ, Covalla SF, Hogsett DA, Herring CD. 2011. Marker removal system for Thermoanaerobacterium saccharolyticum and development of a markerless ethanologen. Appl. Environ. Microbiol. 77:2534–2536. 10.1128/AEM.01731-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Argyros DA, Tripathi SA, Barrett TF, Rogers SR, Feinberg LF, Olson DG, Foden JM, Miller BB, Lynd LR, Hogsett DA, Caiazza NC. 2011. High ethanol titers from cellulose by using metabolically engineered thermophilic, anaerobic microbes. Appl. Environ. Microbiol. 77:8288–8294. 10.1128/AEM.00646-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Podkaminer KK, Shao XJ, Hogsett DA, Lynd LR. 2011. Enzyme inactivation by ethanol and development of a kinetic model for thermophilic simultaneous saccharification and fermentation at 50 degrees C with Thermoanaerobacterium saccharolyticum ALK2. Biotechnol. Bioeng. 108:1268–1278. 10.1002/bit.23050 [DOI] [PubMed] [Google Scholar]
- 29.Lee YE, Lowe SE, Zeikus JG. 1993. Regulation and characterization of xylanolytic enzymes of Thermoanaerobacterium saccharolyticum B6A-RI. Appl. Environ. Microbiol. 59:763–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee YE, Lowe SE, Zeikus JG. 1993. Gene cloning, sequencing, and biochemical characterization of endoxylanase from Thermoanaerobacterium saccharolyticum B6A-RI. Appl. Environ. Microbiol. 59:3134–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee YE, Zeikus JG. 1993. Genetic organization, sequence and biochemical characterization of recombinant beta-xylosidase from Thermoanaerobacterium saccharolyticum strain B6A-RI. J. Gen. Microbiol. 139(Pt 6):1235–1243. 10.1099/00221287-139-6-1235 [DOI] [PubMed] [Google Scholar]
- 32.Lee YE, Ramesh MV, Zeikus JG. 1993. Cloning, sequencing and biochemical characterization of xylose isomerase from Thermoanaerobacterium saccharolyticum strain B6A-RI. J. Gen. Microbiol. 139(Pt 6):1227–1234. 10.1099/00221287-139-6-1227 [DOI] [PubMed] [Google Scholar]
- 33.Ramesh MV, Podkovyrov SM, Lowe SE, Zeikus JG. 1994. Cloning and sequencing of the Thermoanaerobacterium saccharolyticum B6A-RI apu gene and purification and characterization of the amylopullulanase from Escherichia coli. Appl. Environ. Microbiol. 60:94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bronnenmeier K, Meissner H, Stocker S, Staudenbauer WL. 1995. Alpha-d-glucuronidases from the xylanolytic thermophiles Clostridium stercorarium and Thermoanaerobacterium saccharolyticum. Microbiology 141(Pt 9):2033–2040. 10.1099/13500872-141-9-2033 [DOI] [PubMed] [Google Scholar]
- 35.Liu SY, Wiegel J, Gherardini FC. 1996. Purification and cloning of a thermostable xylose (glucose) isomerase with an acidic pH optimum from Thermoanaerobacterium strain JW/SL-YS 489. J. Bacteriol. 178:5938–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lorenz WW, Wiegel J. 1997. Isolation, analysis, and expression of two genes from Thermoanaerobacterium sp. strain JW/SL YS485: a beta-xylosidase and a novel acetyl xylan esterase with cephalosporin C deacetylase activity. J. Bacteriol. 179:5436–5441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Podkaminer KK, Guss AM, Trajano HL, Hogsett DA, Lynd LR. 2012. Characterization of xylan utilization and discovery of a new endoxylanase in Thermoanaerobacterium saccharolyticum through targeted gene deletions. Appl. Environ. Microbiol. 78:8441–8447. 10.1128/AEM.02130-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 39.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580. 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 40.Li X, He Z, Zhou J. 2005. Selection of optimal oligonucleotide probes for microarrays using multiple criteria, global alignment and parameter estimation. Nucleic Acids Res. 33:6114–6123. 10.1093/nar/gki914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown SD, Raman B, McKeown CK, Kale SP, He ZL, Mielenz JR. 2007. Construction and evaluation of a Clostridium thermocellum ATCC 27405 whole-genome oligonucleotide microarray. Appl. Biochem. Biotechnol. 137:663–674. 10.1007/s12010-007-9087-6 [DOI] [PubMed] [Google Scholar]
- 42.Chhabra SR, He Q, Huang KH, Gaucher SP, Alm EJ, He Z, Hadi MZ, Hazen TC, Wall JD, Zhou J, Arkin AP, Singh AK. 2006. Global analysis of heat shock response in Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 188:1817–1828. 10.1128/JB.188.5.1817-1828.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown SD, Martin M, Deshpande S, Seal S, Huang K, Alm E, Yang YF, Wu LY, Yan TF, Liu XD, Arkin A, Chourey K, Zhou JZ, Thompson DK. 2006. Cellular response of Shewanella oneidensis to strontium stress. Appl. Environ. Microbiol. 72:890–900. 10.1128/AEM.72.1.890-900.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Washburn MP, Wolters D, Yates JR. 2001. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19:242–247. 10.1038/85686 [DOI] [PubMed] [Google Scholar]
- 45.McDonald WH, Ohi R, Miyamoto DT, Mitchison TJ, Yates JR. 2002. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. Int. J. Mass Spectrom. 219:245–251. 10.1016/S1387-3806(02)00563-8 [DOI] [Google Scholar]
- 46.Eng JK, McCormack AL, Yates JR. 1994. An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989. 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]
- 47.Tabb DL, McDonald WH, Yates JR. 2002. DTASelect and contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1:21–26. 10.1021/pr015504q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. 2006. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5:2339–2347. 10.1021/pr060161n [DOI] [PubMed] [Google Scholar]
- 49.Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE. 2009. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat. Biotechnol. 28:83–89. 10.1038/nbt.1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2:953–971. 10.1038/nprot.2007.131 [DOI] [PubMed] [Google Scholar]
- 51.Erickson BK, Mueller RS, VerBerkmoes NC, Shah M, Singer SW, Thelen MP, Banfield JF, Hettich RL. 2010. Computational prediction and experimental validation of signal peptide cleavages in the extracellular proteome of a natural microbial community. J. Proteome Res. 9:2148–2159. 10.1021/pr900877a [DOI] [PubMed] [Google Scholar]
- 52.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6:343–345. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 53.Gibson DG, Smith HO, Hutchison CA, Venter JC, Merryman C. 2010. Chemical synthesis of the mouse mitochondrial genome. Nat. Methods 7:901–903. 10.1038/nmeth.1515 [DOI] [PubMed] [Google Scholar]
- 54.Core Team R. 2012. R: a language and environment for statistical computing. Core Team R, Vienna, Austria [Google Scholar]
- 55.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42:D490–D495. 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freudl R. 2013. Leaving home ain't easy: protein export systems in Gram-positive bacteria. Res. Microbiol. 164:664–674. 10.1016/j.resmic.2013.03.014 [DOI] [PubMed] [Google Scholar]
- 57.Desvaux M, Khan A, Scott-Tucker A, Chaudhuri RR, Pallen MJ, Henderson IR. 2005. Genomic analysis of the protein secretion systems in Clostridium acetobutylicum ATCC 824. Biochim. Biophys. Acta 1745:223–253. 10.1016/j.bbamcr.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 58.Messner P, Steiner K, Zarschler K, Schaffer C. 2008. S-layer nanoglycobiology of bacteria. Carbohydr. Res. 343:1934–1951. 10.1016/j.carres.2007.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schneewind O, Missiakas DM. 2012. Protein secretion and surface display in Gram-positive bacteria. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367:1123–1139. 10.1098/rstb.2011.0210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mai V, Lorenz WW, Wiegel J. 1997. Transformation of Thermoanaerobacterium sp. strain JW/SL-YS485 with plasmid pIKM1 conferring kanamycin resistance. FEMS Microbiol. Lett. 148:163–167. 10.1111/j.1574-6968.1997.tb10283.x [DOI] [Google Scholar]
- 61.Viollier PH, Weihofen A, Folcher M, Thompson CJ. 2003. Post-transcriptional regulation of the Streptomyces coelicolor stress responsive sigma factor, SigH, involves translational control, proteolytic processing, and an anti-sigma factor homolog. J. Mol. Biol. 325:637–649. 10.1016/S0022-2836(02)01280-9 [DOI] [PubMed] [Google Scholar]
- 62.Dierks T, Miech C, Hummerjohann J, Schmidt B, Kertesz MA, von Figura K. 1998. Posttranslational formation of formylglycine in prokaryotic sulfatases by modification of either cysteine or serine. J. Biol. Chem. 273:25560–25564. 10.1074/jbc.273.40.25560 [DOI] [PubMed] [Google Scholar]
- 63.Dierks T, Lecca MR, Schlotterhose P, Schmidt B, von Figura K. 1999. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J. 18:2084–2091. 10.1093/emboj/18.8.2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sardiello M, Annunziata I, Roma G, Ballabio A. 2005. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum. Mol. Genet. 14:3203–3217. 10.1093/hmg/ddi351 [DOI] [PubMed] [Google Scholar]
- 65.Parenti G, Meroni G, Ballabio A. 1997. The sulfatase gene family. Curr. Opin. Genet. Dev. 7:386–391. 10.1016/S0959-437X(97)80153-0 [DOI] [PubMed] [Google Scholar]
- 66.Hanson SR, Best MD, Wong CH. 2004. Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew. Chem. Int. Ed. Engl. 43:5736–5763. 10.1002/anie.200300632 [DOI] [PubMed] [Google Scholar]
- 67.Landgrebe J, Dierks T, Schmidt B, von Figura K. 2003. The human SUMF1 gene, required for posttranslational sulfatase modification, defines a new gene family which is conserved from pro- to eukaryotes. Gene 316:47–56. 10.1016/S0378-1119(03)00746-7 [DOI] [PubMed] [Google Scholar]
- 68.Salyers AA, Vercellotti JR, West SE, Wilkins TD. 1977. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl. Environ. Microbiol. 33:319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolf DM, Fontaine-Bodin L, Bischofs I, Price G, Keasling J, Arkin AP. 2008. Memory in microbes: quantifying history-dependent behavior in a bacterium. PLoS One 3:e1700. 10.1371/journal.pone.0001700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gawande PV, Griffiths MW. 2005. Growth history influences starvation-induced expression of uspA, grpE, and rpoS and subsequent cryotolerance in Escherichia coli O157:H7. J. Food Prot. 68:1154–1158 [DOI] [PubMed] [Google Scholar]
- 71.Rozen Y, Belkin S. 2001. Survival of enteric bacteria in seawater. FEMS Microbiol. Rev. 25:513–529. 10.1111/j.1574-6976.2001.tb00589.x [DOI] [PubMed] [Google Scholar]
- 72.Song YJ, Xue YF, Ma YH. 2013. Global microarray analysis of carbohydrate use in alkaliphilic hemicellulolytic bacterium Bacillus sp. N16-5. PLoS One 8:e54090. 10.1371/journal.pone.0054090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hottes AK, Meewan M, Yang D, Arana N, Romero P, McAdams HH, Stephens C. 2004. Transcriptional profiling of Caulobacter crescentus during growth on complex and minimal media. J. Bacteriol. 186:1448–1461. 10.1128/JB.186.5.1448-1461.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.