

Abstract
Glucosamine and N-acetylglucosamine are among the most abundant sugars on the planet, and their introduction into the oral cavity via the diet and host secretions, and through bacterial biosynthesis, provides oral biofilm bacteria with a source of carbon, nitrogen, and energy. In this study, we demonstrated that the dental caries pathogen Streptococcus mutans possesses an inducible system for the metabolism of N-acetylglucosamine and glucosamine. These amino sugars are transported by the phosphoenolpyruvate:sugar phosphotransferase system (PTS), with the glucose/mannose enzyme II permease encoded by manLMN playing a dominant role. Additionally, a previously uncharacterized gene product encoded downstream of the manLMN operon, ManO, was shown to influence the efficiency of uptake and growth on N-acetylglucosamine and, to a lesser extent, glucosamine. A transcriptional regulator, designated NagR, was able to bind the promoter regions in vitro, and repress the expression in vivo, of the nagA and nagB genes, encoding N-acetylglucosamine-6-phosphate deacetylase and glucosamine-6-phosphate deaminase, respectively. The binding activity of NagR could be inhibited by glucosamine-6-phosphate in vitro. Importantly, in contrast to the case with certain other Firmicutes, the gene for de novo synthesis of glucosamine-6-phosphate in S. mutans, glmS, was also shown to be regulated by NagR, and NagR could bind the glmS promoter region in vitro. Finally, metabolism of these amino sugars by S. mutans resulted in the production of significant quantities of ammonia, which can neutralize cytoplasmic pH and increase acid tolerance, thus contributing to enhanced persistence and pathogenic potential.
INTRODUCTION
Dental caries is caused by the accumulation of polymicrobial biofilms that produce organic acids from dietary carbohydrates, leading to demineralization of the tooth. Streptococcus mutans is one of the primary organisms associated with the development of dental caries (1). Not only are the levels of S. mutans strongly correlated with the initiation and progression of carious lesions (2), but also the organism is particularly well adapted to contribute to the caries process by virtue of its ability to produce a strongly adhesive biofilm matrix, its capacity to transport an array of carbohydrates, its ability to generate organic acid end products from many carbohydrate sources, and its capacity to grow and continue to metabolize sugars at low pH (3–5).
The oral cavity is a unique environment in which microorganisms are periodically presented with large quantities of carbohydrate-rich foodstuffs but must survive on nutrients obtained from host secretions, sloughed host cells, or metabolic products of other microbes during fasting by the host. The inhabitants of the oral cavity have evolved specialized mechanisms for addressing the nutrient fluctuations presented by this “feast or famine” lifestyle (6). For saccharolytic organisms, including S. mutans and other caries pathogens, the intermittent introduction of carbohydrates in the diet of the host presents an array of energy sources, ranging from monosaccharides and disaccharides to more complex oligosaccharides and polysaccharides. Not surprisingly, many oral bacteria possess glycosidases and transport systems that provide them with access to the spectrum of carbohydrates released into oral biofilms. Bacteria use a variety of mechanisms for internalizing these carbohydrates from the environment, including ABC transporters. However, the phosphoenolpyruvate:sugar phosphotransferase system (PTS) is the dominant carbohydrate transport pathway in many bacteria, including some of the most abundant bacteria in dental biofilms (7–9).
A functional PTS consists of enzyme I (EI), which transfers a phosphoryl group from phosphoenolpyruvate (PEP) to the HPr protein at histidine residue 15 (HPr∼P). HPr∼P can then donate its phosphoryl group to various sugar-specific enzyme II (EII) permeases. The EII permeases are composed of cytoplasmically localized EIIA and EIIB domains and membrane-associated EIIC, and in some cases EIID, domains. The EII proteins catalyze the concomitant internalization and phosphorylation of their cognate sugar(s). The genome of S. mutans contains the genes for as many as 15 unique EII PTS permeases, most of which are found in the majority of strains, although some are restricted to specific isolates (7, 10, 11). Of relevance to this study, the glucose/mannose-specific EII permease (ManLMN) of S. mutans is capable of transporting an array of carbohydrates, including glucose, mannose, galactose, and 2-deoxyglucose (12, 13). In addition to its role in carbohydrate transport, EIIABMan (ManL) has also been identified as having a dominant influence on carbohydrate catabolite repression (CCR), although a complete understanding of how ManL exerts CCR has not yet been realized (12, 14).
N-Acetylglucosamine (GlcNAc) is commonly utilized as a monomeric unit in many naturally occurring polymers, such as the chitin that forms the outer covering of arthropods and the cell walls of many fungi. Glucosamine (GlcN) and GlcNAc are also important building blocks for the peptidoglycan of bacterial cell walls and certain lipopolysaccharides (15–17). In addition, GlcN and GlcNAc catabolism provides organisms with a carbon and a nitrogen source. Due to their importance in catabolic and anabolic processes, the metabolism of these amino sugars has been studied in detail in many bacterial species. Some of the most prominent early studies involving the metabolism of these amino sugars were conducted in Escherichia coli and described GlcNAc as a highly preferred sugar in the hierarchy of catabolite repression (18). It is now well understood that GlcNAc enters E. coli as GlcNAc-6-phosphate (GlcNAc-6-P) predominately through a GlcNAc-specific PTS system encoded by nagE and also through the mannose PTS transport system encoded by manXYZ (19, 20). Catabolism of GlcNAc-6-P begins with deacetylation by the gene product of nagA, GlcNAc-6-P deacetylase (21). GlcN-6-P can then be converted directly to UDP-GlcNAc through the activity of GlmM and GlmU (22–24) and used for cell wall biosynthesis or other anabolic processes. Alternatively, GlcN-6-P can be acted on by NagB, a GlcN-6-P deaminase (21), to generate fructose-6-phosphate (fructose-6-P), which can enter the Embden-Meyerhof-Parnas pathway. Like GlcNAc, GlcN can also be internalized and phosphorylated by the ManXYZ PTS and subsequently deaminated by NagB to yield fructose-6-P (25, 26). Of note, GlcNAc-6-P also serves as an important allosteric effector for downstream metabolic processes (27, 28).
In certain Firmicutes, transport of amino sugars also occurs via sugar-specific PTS permeases. For example, Bacillus subtilis transports GlcNAc via an EIICB protein (NagP), and homologs of key amino sugar metabolic enzymes, including NagA (29) and NagB (30), subsequently catabolize GlcNAc-6-P to fructose-6-P. Glucosamine is imported through an EIICBA protein (GamP) and also via the major glucose transporter PtsG (31, 32), and the protein encoded by the gamA gene subsequently metabolizes GlcN-6-P to fructose-6-P (32). Notably, GamA also serves as a major GlcN-6-P deaminase during GlcNAc metabolism in B. subtilis (32). The transcription of genes associated with the metabolism of GlcNAc, and GlcN in E. coli, is controlled primarily by the activity of a ROK (repressor, open reading frame [ORF], and kinase) family transcriptional repressor, NagC (33). Studies with E. coli have demonstrated that GlcNAc-6-P binds to NagC and releases the repressor to allow transcription of metabolic genes (27). In B. subtilis, the transcriptional repressor NagR, formerly known as YvoA, regulates the metabolism of GlcNAc by binding near nag gene promoters and exerting control over the transcription of metabolic genes, including nagA, nagB, and nagP (34), whereas the genes for GlcN metabolism are controlled by another homologous regulator, GamR (YbgA) (35). Functional studies conducted on NagR have suggested that either GlcNAc-6-P (36) or GlcN-6-P (34) could serve as an allosteric regulator that disengages NagR from nag operon promoters, although the precise mechanism for how these and other potential allosteric effectors regulate NagR binding has not been established (35). It is also noteworthy that GlcN-6-P is an allosteric regulator of GamR (35).
When amino sugars are required for cell wall biosynthesis and unavailable via exogenous sources, cells utilize fructose-6-P and glutamine to produce GlcN-6-P via a GlcN-6-P synthase encoded by glmS. The production of amino sugars is an important process in bacteria because of the requirements for these carbohydrates to generate a cell wall and other polysaccharides, as noted above. In the model Gram-negative bacterium E. coli, GlmS levels are controlled by the presence of a strong hairpin at the 5′ end of the transcript, which leads to occlusion of the ribosome binding site (37). A regulatory small RNA (sRNA), GlmZ, binds to and stabilizes the 5′ untranslated region (UTR) of glmS, and this interaction allows for efficient translation of glmS mRNA (38). GlmZ is subject to processing, which removes necessary residues for binding to glmS, and a second sRNA, GlmY, is required to prevent cleavage of these residues from GlmZ (39). When cells experience a shortage of GlcN-6-P, production of GlmY increases, preventing the processing of GlmZ (40). The regulation of GlmS production via trans-acting sRNAs appears to be conserved among other members of Enterobacteriaceae, including Yersinia pseudotuberculosis and Salmonella enterica serovar Typhimurium (41).
The mechanisms regulating amino sugar anabolism by Gram-positive bacteria differ in many ways from those in Gram-negative bacteria. For example, in some Firmicutes, glmS is regulated by a cis-acting ribozyme capable of sensing metabolites directly (42). Residues contained in the 5′ UTR of the glmS mRNA form a ribozyme structure (42). In the presence of GlcN-6-P, the ribozyme initiates self-cleavage of the glmS transcript from a larger mRNA structure, preventing translation and resulting in decreased amounts of GlmS within the cell (42). Apparent homologs of the glmS ribozyme have been identified in multiple bacterial species, but the vast majority of these homologous structures are present in Gram-positive bacteria (43). Notably, the presence of a canonical ribozyme sequence was not predicted upstream of the glmS promoter in species of the mutans streptococci, including S. mutans (43, 44).
Relatively few studies have been conducted on amino sugar metabolism in S. mutans. S. mutans strain GS-5 was shown to possess both an inducible GlcNAc uptake system and a second, lower-affinity system, based in part on the observation that GlcNAc-transporting strains were more sensitive to inhibition when grown in the presence of the GlcNAc analog streptozotocin (45). The ability to ferment GlcNAc may also confer a selective advantage on S. mutans. In particular, the fermentation of GlcNAc has been used as one method for differentiating S. mutans from its close cariogenic relative Streptococcus sobrinus (46, 47). In fact, S. mutans strain NCTC 10449 was capable of outcompeting S. sobrinus SL-1 in mixed cultures when glucose and GlcNAc were provided as the carbohydrate sources, whereas S. sobrinus was the dominant organism when glucose was the sole carbohydrate (48). S. sobrinus SL-1 was incapable of growing with GlcNAc as the primary carbohydrate source, and studies comparing the GlcNAc-6-P deacetylase and GlcN-6-P deaminase activities of the strains revealed that S. mutans NCTC 10449 consistently produced higher levels of these enzymes than S. sobrinus SL-1 in cells grown on GlcN or on a combination of glucose and the individual amino sugars (48).
Recent progress has been made dissecting the gene products responsible for GlcNAc metabolism in S. mutans UA159. A strain containing a deletion of glmS required GlcNAc for growth, and nagB mutants could not grow in the presence of GlcNAc. Further, a decrease in the levels of the secreted glucosyltransferase enzymes GtfB and GtfC, which produce exopolysaccharides from sucrose, as well as the cell surface protein antigen Pac was noted in the nagB mutant strain and was associated with a decrease in biofilm formation. In contrast, a glmS mutant displayed the opposite phenotypes, showing elevated production of GtfB, GtfC, and Pac and an increase in biofilm formation compared to those of the wild-type strain (44). While the levels of nagB and glmS mRNA in response to growth carbohydrate have been measured in S. mutans, the mechanisms by which these genes are regulated in response to carbohydrate source and other factors have not been investigated, nor have the primary transport pathways for GlcNAc and GlcN been established. In this study, we examined the route by which the amino sugars GlcNAc and GlcN enter S. mutans and the mechanisms for the induction and repression of the nagA, nagB, and glmS genes. Finally, we began to investigate the contribution of amino sugar metabolism to physiologic homeostasis in S. mutans.
MATERIALS AND METHODS
Bacterial growth and genetic manipulation.
Escherichia coli strain DH10B was grown in Luria-Bertani medium (L broth) at 37°C in air. Streptococcus mutans strains, including UA159 and five clinical isolates, were maintained using brain heart infusion (BHI; Difco Laboratories, Detroit, MI) medium and routinely cultured in a tryptone-vitamin (TV) base medium (49) supplemented with various concentrations of carbohydrate. When desired, strains were cultured in a modified version of the chemically defined medium FMC (50), by substituting the carbohydrates for glucose. N-Acetyl-d-glucosamine (GlcNAc) and glucosamine (GlcN, as GlcN-HCl) were purchased from Sigma-Aldrich (St. Louis, MO) and used at final concentration of 20 mM, unless otherwise noted. GlcN-HCl was prepared fresh and added to media, due to its instability with prolonged exposure to neutral or alkaline conditions. To monitor growth, bacteria were cultivated overnight in BHI, unless specified otherwise, diluted 1:150 into fresh TV or FMC base medium containing the desired carbohydrates and incubated at 37°C in a Bioscreen C Lab system (Helsinki, Finland). All samples were covered with an oil overlay (approximately 60 μl) to reduce exposure to air, and the optical densities at 600 nm (OD600) were measured every 30 min.
Genetic manipulation of the bacterial genome was performed primarily using an allelic-exchange method described in previous reports (51, 52), and mutants were maintained on the media detailed above supplemented with erythromycin (10 μg/ml), kanamycin (1 mg/ml), or spectinomycin (1 mg/ml) (Sigma), when needed. All primers used in this study for the engineering of mutants or plasmids, quantitative reverse transcription-PCR (qRT-PCR), etc., are listed in Table 1. To complement an existing manL mutant (ΔmanL) (53), a promoterless copy of manL was introduced into the chromosome using the integration vector pBGE (51), which allows for the introduction of foreign DNA downstream of the gtfA promoter, to create the ΔmanL pBGE-manL strain. The manO mutant of S. mutans was complemented by providing the manO gene in trans on plasmid pIB184 (54). The complementing plasmid was constructed by first amplifying the manO structural gene and its ribosome binding site using primers manOcomBm5′ and manOcomXh3′ (Table 1) to introduce BamHI and XhoI sites, respectively. The PCR product was then directionally cloned into BamHI and XhoI sites in the expression vector pIB184 to create plasmid pIB184-manO (54). Similarly, the plasmid pIB184-nagR was used to complement a nagR deletion mutant and was constructed by amplifying the nagR gene using primers nagR-5′Bm and nagR-3′RI (Table 1) to include BamHI and EcoRI restriction sites for inserting the gene into pIB184 (54). The integrity of manL in pBGE and manO and nagR in pIB184 was confirmed by DNA sequencing, and the plasmids were introduced into S. mutans strains via natural transformation (55).
TABLE 1.
Primers used in this studya
| Primer name | Sequence | Purpose |
|---|---|---|
| nagR-1 | 5′-GGCAGCATAAGGGCAGTCGCT-3′ | nagR deletion |
| nagR-2Bm | 5′-TTTGATCATGGATCCGAATATAAGCC-3′ | nagR deletion |
| nagR-3Bm | 5′-GGCTCAAGGATCCTATTTTGAAAATGG-3′ | nagR deletion |
| busR-4 | 5′-GCCCCATAATAACAAAGACTTCTCCT-3′ | nagR deletion |
| nagR-5′Bm | 5′-CCATTAGTAGGATCCAATATTTTGTGGTTA-3′ | pIB184-nagR creation |
| nagR-3′RI | 5′-GCTAATCATTCTTTCTTTGAATTCCATTACTT-3′ | pIB184-nagR creation |
| nagA-S | 5′-ATATGAAATTGCGCCTGGAC-3′ | nagA qRT-PCR |
| nagA-AS | 5′-CCGAGAGCAATTCTCGACTC-3′ | nagA qRT-PCR |
| nagB-S | 5′-TGTCAGCCGATGATAAG AG 3′ | nagB qRT-PCR |
| nagB-AS | 5′-GCTAAATCTGCTGCCAGTCC-3′ | nagB qRT-PCR |
| glmS-S | 5′-TGCTGATTTGCGAGCTAAGA-3′ | glmS qRT-PCR |
| glmS-AS | 5′-TGCATGTGGATGAGCATTTT-3′ | glmS qRT-PCR |
| gyrA-S | 5′-TTGCGACTATCTGCTATGTG-3′ | gyrA qRT-PCR |
| gyrA-AS | 5′-CCAAGAATCTGCTGTCCG-3′ | gyrA qRT-PCR |
| ManLMNA-15′ | 5′-GCAATCGCCCACAAAATAGT-3′ | manLMN deletion |
| ManLMNA-13′Xb | 5′-TTCGCCATGTCTAGAGATAACGATTCCGATAGC-3′ | manLMN deletion |
| ManLMNA+15′RI | 5′-CTTATATAGAATTCCCCAAAGACAGCGAAGTTA-3′ | manLMN deletion |
| ManLMNA + 13′ | 5′-ATTTTGGATGAAGCCACCAG-3′ | manLMN deletion |
| manL-comp-5′Bm | 5′-GAGAATAACAGGATCCTTTTTATCTGAGA-3′ | pBGE-manL creation |
| manL-comp-3′Xh | 5′-GACATTATTCTCGAGCCTTTCGTTGTTT-3′ | pBGE-manL creation |
| manO-km5′-1 | 5′-TATTTCTCTCTTTCTATTTTTGCACACTAG-3′ | manO deletion |
| manO-Bm5′-2 | 5′-ATACACCGGATCCTGTGTATTCAACTTTTGT-3′ | manO deletion |
| manO-Bm3′-3 | 5′-AAAATTCCTCGGATCCTCATCTGATTCTGGTAA-3′ | manO deletion |
| manO-km3′-4 | 5′-TCTTGATAACAGTTTGATTTTGATGAAGTTG-3′ | manO deletion |
| manOcomBm5′ | 5′-ATAATAGGATCCATTTATTCAGGAGTAAATTT-3′ | pIB184-manO creation |
| manOcomXh3′ | 5′-TACATTTTCTCGAGCTAACCCTTCTTTTTTCGT-3′ | pIB184-manO creation |
| nagR-His/MBP-5′Bm | 5′-GGTTAAGGAGGATCCATGTTACCGGCTT-3′ | MBP-NagR fusion |
| nagR-His/MBP-3′HIII | 5′-CTTTTGTTTCCAAAGCTTACTTATTTTCT-3′ | MBP-NagR fusion |
| PnagA-5′ | 5′-GAAGAGAACAGGAATTAATTTGCTTGGT-3′ | nagA probe for EMSA |
| PnagA-3′bio | 5′-CATTATACTCTTATTAATAACAGTTGTAAATGGT-3′ | nagA probe for EMSA |
| PnagB-5′ | 5′-AGTTTTTGAAGAAGTTAGATCAGACAGT-3′ | nagB probe for EMSA |
| PnagB-3′bio | 5′-AACGCCCTCCTAATCGGTCTATACA-3′ | nagB probe for EMSA |
| PglmS-5′ | 5′-AAGAGTAGTACAGCTACTCTTTTT-3′ | glmS probe for EMSA |
| PglmS-3′bio | 5′-CTACTCTTTTTTTAAATGTCAAAAAGACTATAC-3′ | glmS probe for EMSA |
Underlined are restriction enzyme sites engineered to facilitate cloning. Bm, BamHI; RI, EcoRI; HIII, HindIII; Xb, XbaI; Xh, XhoI.
Recombinant NagR protein was engineered by cloning the entire coding sequence of nagR into pMal-p2X (New England BioLabs, Ipswich, MA) in E. coli DH10B to create a maltose-binding protein (MBP) fusion to NagR. Overexpression of the MBP-tagged NagR protein was induced by treating exponentially growing cells (OD600 ≈ 0.5) in L broth with 0.05 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h. After harvesting of cells, the MBP-NagR protein was purified from cell lysates using an amylose resin (New England BioLabs), and then NagR was cleaved from MBP using factor Xa (New England BioLabs) and purified according to instructions from the supplier.
PTS assay.
PTS-dependent transport of sugars was assayed as described elsewhere (56, 57), with minor modifications. Cells were cultured under the desired conditions and collected by centrifugation, and then the pellets were washed, snap-frozen in a dry ice-ethanol bath, and stored at −80°C. Frozen cell pellets were thawed on ice, washed twice with 0.1 M sodium-potassium phosphate buffer (pH 7.2) containing 5 mM MgCl2 (KPB), and resuspended in 0.1 volume of the same buffer. Permeabilization of cells was achieved by the addition of 0.05 volume of toluene-acetone (1:9, vol/vol) and 2 min of vortexing, twice. The reaction mixture included permeabilized cells, 100 μM NADH, 10 mM NaF, a 10 mM concentration of the desired carbohydrate, and approximately 10 U of a lactate dehydrogenase solution (Sigma) in 0.1 M KPB. The reaction mixture was equilibrated at 37°C, and the reaction was initiated by the addition of 5 mM PEP. Reaction mixtures were incubated at 37°C, and the rates of oxidation of NADH were monitored over time. PTS activity was calculated from the rate of PEP-dependent oxidation of NADH and normalized to the concentration of protein determined with a bicinchoninic acid (BCA) assay (Thermo Scientific, Rockford, IL).
Immunoblotting.
S. mutans UA159 or its derivatives were grown in TV medium supplemented with sugars to mid-exponential phase (OD600 ≈ 0.5). Cells were harvested, resuspended in 750 μl of PBS with 4% SDS, and lysed with a Bead Beater (Biospec, Bartlesville, OK) in the presence of 0.5 ml of glass beads (average diameter, 0.1 mm). Whole-cell lysates were centrifuged at 16,000 × g at 4°C for 10 min, and the BCA assay (Thermo Scientific) was utilized to determine the protein concentrations of the clarified cell lysates. A portion of each cell lysate (20 μg) was placed in a solution of SDS sample buffer (58) containing a final concentration of 2% 2-mercaptoethanol. Samples were immersed in a boiling water bath for 5 min and centrifuged at 16,000 × g for 5 min, and then proteins in the clarified supernatants were separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane using a Trans-Blot SD (Bio-Rad, Hercules, CA). A solution of Tris-buffered saline containing Tween 20 (TBST; 25 mM Tris-HCl [pH 7.4], 2.5 mM KCl, 150 mM NaCl, 0.05% Tween 20) and 5% nonfat dried milk was used to block the membranes. A primary antiserum elicited to a purified recombinant ManL protein from S. mutans was incubated with membranes (53). Membranes were then washed in TBST and incubated with a goat anti-rabbit peroxidase-conjugated antibody (1:4,000 dilution; KPL, Gaithersburg, MD). After a final washing with TBST, protein bands were detected using the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
pH drop assay.
Bacterial cultures were subjected to pH drop experiments as previously described (59, 60). Briefly, cells were grown in TV base medium supplemented with sugar(s) to an OD600 of ≈0.5 and centrifuged at 4°C. Cells were washed with ice-cold deionized water and resuspended in 5 ml of 50 mM KCl–1 mM MgCl2. The OD600 of the suspension was adjusted to 4.5 to normalize the cell density across samples, and 4.75 ml of the cell suspension was placed in a beaker and stirred. A solution of 0.1 M KOH was used to adjust the pH of the cell suspension to 7.2. After a stable pH was achieved, 0.25 ml of a 1 M solution of the desired sugar was added, and the pH was recorded every 30 s for 30 min by a pH meter with a computer interface.
Ammonia assay.
Cells were grown overnight in TV base medium supplemented with the desired sugar and subcultured 1:20 into the same medium. When the cultures reached mid-exponential phase (OD600 ≈ 0.5), they were centrifuged at 4°C and washed once with ice-cold deionized water. Cells were resuspended in 5 ml of 50 mM KCl–1 mM MgCl2, and the OD600 of the suspension was adjusted to 4.5. A portion of the cell suspension (4.75 ml) was removed, and the pH was adjusted to 7.2 using 0.1 M KOH. Once a stable pH was established, the pH of the solution was recorded, and 0.25 ml of a 1 M solution of the desired carbohydrate was added. Cells were incubated for 1 h with occasional agitation at 37°C and 5% CO2, at which point the pH of the solution was again recorded, and the supernatant fluids were collected after centrifugation. Ammonia concentrations in the supernatants were measured using an ammonia detection assay (Sigma) according to the supplier's instructions. Negative controls included 50 mM KCl–1 mM MgCl2 alone or containing each of the tested carbohydrates. Concentrations of ammonia were determined using a standard test solution provided by the supplier.
RNA methods.
Total bacterial RNA was extracted from exponentially growing cells using the RNeasy minikit (Qiagen, Germantown, MD) as described elsewhere (61), with some modifications. Briefly, 10 ml of each bacterial culture was pelleted, resuspended in 1 ml of RNAprotect Bacteria Reagent (Qiagen), and incubated at room temperature for 5 min. Cells were then spun down and resuspended in 280 μl of TE-SDS buffer (50 mM Tris, 10 mM EDTA, 0.4% SDS [pH 8.0]) and mixed with 300 μl of acidic phenol (Sigma) in a screw-cap tube containing 0.25 ml of glass beads. Cells were homogenized by bead beating followed by centrifugation for 10 min at 16,000 × g, and 200 μl of the aqueous phase was removed and processed using the RNeasy minikit. DNA was removed using 2 consecutive on-column treatments with RNase-free DNase I (Qiagen). RNA was eluted in 30 μl of nuclease-free water.
For real-time quantitative RT-PCR, 1 μg of total RNA was subjected to reverse transcription using the SuperScript III reverse transcriptase kit and random hexamers, according to protocols provided by the supplier (Life Technologies, Carlsbad, CA). To probe the transcript levels of various genes, real-time PCR was performed on first-strand cDNA using gene-specific primers. The transcript levels of the housekeeping gene gyrA (62) were used for normalization of the data.
EMSA.
DNA fragments of approximately 100 bp that contained the promoter regions of nagA, nagB, and glmS were amplified by PCR using primers labeled with biotin at the 5′ end and then were gel purified. A 10-μl reaction mixture contained 10 fmol of biotin-labeled probe, 5 mM MgCl2, and 2 μg of poly(dI-dC) in binding buffer (50 mM KCl, 1 mM EDTA, 5 mM dithiothreitol, 10 mM HEPES [pH 7.9], and 4% glycerol). Recombinant MBP-NagR protein or NagR released by factor Xa treatment was added to the reaction, along with glucosamine-6-phosphate (GlcN-6-P) or N-acetylglucosamine-6-phosphate (GlcNAc-6-P), when desired. The reaction mixtures were incubated at room temperature for 20 min before being resolved on a 4% nondenaturing polyacrylamide gel in 0.5× Tris-borate-EDTA (TBE) buffer (63). After blotting to a nylon membrane (GeneScreen Plus; PerkinElmer, Waltham, MA), biotin-labeled DNA probes were detected using a LightShift chemiluminescent electrophoretic mobility shift assay (EMSA) kit (Thermo Scientific). It was also noted that both MBP-NagR and NagR tended to form aggregates and failed to enter the polyacrylamide gel once bound to DNA probes. To mitigate this problem, various concentrations (0.05% to 2%) of the nonionic detergent NP-40 (Thermo Scientific) were added to some of the reaction mixtures. While NP-40 did decrease the aggregation and reveal more discrete shifted products, addition of NP-40 at concentrations as high as 2% still displayed a substantial amount of product remaining in the wells of the gel (see Fig. S14 in the supplemental material). Since the presence of NP-40 at such high levels might interfere with the normal function of the protein, the data presented in this report are those obtained in the absence of NP-40.
RESULTS
Involvement of EIIMan in N-acetylglucosamine and glucosamine metabolism.
It has been demonstrated that Streptococcus mutans is capable of metabolizing GlcNAc (N-acetylglucosamine) and GlcN (glucosamine) (45, 46), although it was not defined how these carbohydrates were transported. In this work, we tested whether the glucose/mannose EII permease (ManLMN) could be responsible for the internalization of GlcNAc or GlcN, since similar PTS permeases have been shown to, or have been predicted to, function in this capacity in certain other microorganisms (19, 25, 26, 64). An allelic-exchange mutation of the entire man operon, manLMN, was created in S. mutans UA159 by replacing the genes with a nonpolar erythromycin cassette. The parental and manLMN mutant strains were cultivated in TV medium supplemented with 20 mM glucose, GlcNAc, or GlcN. Growth of the manLMN mutant on glucose was comparable to that of other EIIMan mutants in previous studies (12) (data not shown). The manLMN mutant and a mutant lacking only ManL (EIIABMan) (53) displayed essentially no growth when GlcNAc was used as the primary carbohydrate source (Fig. 1A) and slower growth with lower final yields than for wild-type cells growing on GlcN (Fig. 1B). Attempts were made to complement the manLMN mutant using the integration vector pBGE (51), but we were not able to obtain stable clones of the operon. Since similar defects were observed in the manL mutant growing on GlcNAc or GlcN (Fig. 1A and B), a wild-type copy of manL was inserted into the chromosome of the ΔmanL strain using the integration vector pBGE, which allows for integration of foreign DNA into the gtfA locus with expression of the gene of interest driven from the gtfA promoter (51). The complemented ΔmanL pBGE-manL strain showed partial restoration of growth on GlcNAc (Fig. 1A), with substantially lower growth rates and a lower final yield than for the wild-type strain. Additionally, growth on GlcN (Fig. 1B) and glucose (data not shown) showed very little improvement as a result of the complementation. We attribute these observations to the fact that the expression levels of manL from the gtfA promoter were lower than from its cognate promoter (see Fig. S2 in the supplemental material), which resulted in lower levels of ManL protein (see Fig. S3 in the supplemental material).
FIG 1.

Growth curves of S. mutans UA159 and derivatives on TV media supplemented with various carbohydrates. (A and B) UA159, a manLMN deletion mutant, a manL deletion mutant (ΔmanL), a manL deletion mutant containing the empty pBGE vector (ΔmanL pBGE), and a manL deletion mutant complemented with pBGE-manL (ΔmanL pBGE-manL) were grown in TV medium supplemented with 20 mM GlcNAc (A) or 20 mM GlcN (B); (C) UA159 (light shapes) and a PTS enzyme I deletion mutant (dark shapes) were grown in TV medium supplemented with 20 mM glucose (triangles), 20 mM GlcNAc (squares), or 20 mM GlcN (circles).
We recently conducted a study in which the genomes of 57 clinical isolates of S. mutans were sequenced and compared (10, 65). In an attempt to demonstrate the importance of the man operon in amino sugar transport across multiple strains of the same species, similar allelic-replacement mutations of the man operon were generated in five additional isolates. The five isolates of S. mutans and their corresponding man operon mutants were inoculated into TV with GlcNAc or GlcN. The man operon mutants all displayed near complete loss of growth on GlcNAc (see Fig. S4A in the supplemental material). Also similar to the case with S. mutans UA159, growth on GlcN was negatively impacted by man deletion, and the severities of this impact varied among isolates (see Fig. S4B). In addition, cells of Streptococcus gordonii strain DL1 and an isogenic mutant bearing a deletion of the manL homolog (66) were grown on TV medium containing 20 mM GlcNAc or GlcN. In this case, the mutant strain displayed an extended lag phase and slower growth in GlcNAc (see Fig. S4C), as well as a modestly longer lag phase and slightly slower growth in GlcN (see Fig. S4D). Notably, the mutant displayed a higher final yield on both amino sugars. Thus, the ManLMN PTS permease contributes significantly to the ability of S. mutans and at least one other abundant oral streptococcus to grow with GlcNAc or GlcN as the primary carbohydrate and energy source.
To determine if the amino sugars were transported primarily by the PTS, cells bearing an allelic-exchange mutation of the ptsI gene encoding the general PTS enzyme EI of S. mutans UA159 (labeled ΔEI) (67), which displayed a phenotype similar to that of a previously characterized ptsI mutant (68), were inoculated into media with GlcNAc or GlcN as the primary growth carbohydrate. As shown in Fig. 1C, the ΔEI strain was not capable of growing on either GlcNAc or GlcN. We hypothesized that additional PTS transporters may be responsible for the residual growth observed in manLMN mutants on GlcN (Fig. 1B). To address this, previously engineered mutants harboring deletions in other known PTS permeases were tested for their growth on GlcNAc and GlcN. Compared to S. mutans UA159, the mutant strains lacking the cellobiose or fructose PTS EIIs each showed slightly reduced growth on GlcN, but the impact was not as pronounced as that of ManL (see Fig. S5 in the supplemental material). At the same time, mutants bearing deletions in the EII permease of the lactose PTS (SMU.1492) (13) or maltose PTS (SMU.2047) (69, 70) showed little or no defect on growth on GlcN (data not shown). In addition, when GlcN was the primary carbohydrate source, strains lacking the cellobiose or fructose PTS permease components and manL displayed poorer growth than did strains bearing only the manL deletion (see Fig. S5). When grown with GlcNAc, strains with single deletions of the cellobiose or fructose PTS showed defects in growth similar to those seen when GlcN was the primary carbohydrate source, while strains with a manL deletion in combination with the cellobiose PTS or fructose PTS mutations showed little to no growth (data not shown), similar to results seen with a manL deletion alone (Fig. 1A).
Transport of N-acetylglucosamine and glucosamine is mediated through EIIMan.
To further demonstrate the role of the man operon in the transport of amino sugars, cells of S. mutans UA159 and the manLMN mutant were grown in TV supplemented with 0.5% glucose, and PEP-dependent carbohydrate transport was assayed. Compared to the wild-type strain, cells lacking the manLMN operon displayed almost no detectable PTS-dependent uptake of GlcNAc and only a very low level of transport of GlcN (Fig. 2A). To determine if growth in the presence of GlcNAc or GlcN would result in enhanced transport of these sugars via the PTS, strain UA159 was grown in media containing glucose, GlcN, or a combination of glucose and GlcN. These cells displayed significantly higher (∼60% increase) transport of GlcN when GlcN was the primary carbohydrate source than did cells grown with glucose alone or a combination of glucose and GlcN (see Fig. S6A in the supplemental material). We noted that the manLMN deletion mutant strain was capable of slower but significant growth when GlcN was the primary carbohydrate source in TV base medium. However, when a PTS assay was performed using cells grown on GlcN, the manLMN deletion strain again displayed significantly lower PTS-dependent transport of GlcN than did the wild-type strain (see Fig. S6B). This low level of PTS-dependent transport provides further evidence that other PTS enzymes may play a secondary role in transport of GlcN (see Fig. S5). As the manLMN mutant could not be grown in the presence of GlcNAc as the primary carbohydrate source, the wild-type and manLMN mutant strains were grown on a combination of glucose and GlcNAc to determine if the presence of GlcNAc in the media could stimulate PTS transport via permeases other than the EIIMan complex. However, under these growth conditions the manLMN mutant strain was unable to transport GlcNAc via the PTS (see Fig. S6C).
FIG 2.
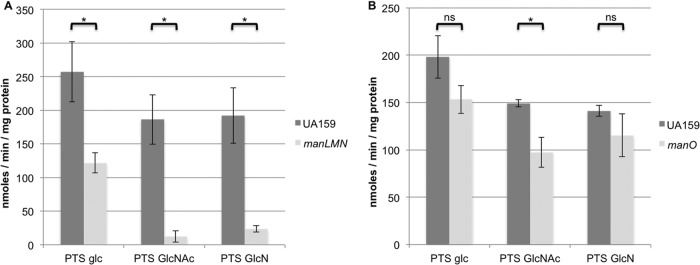
PTS-dependent sugar:phosphotransferase activity. S. mutans UA159 and a manLMN deletion mutant (A) or a manO deletion mutant (B) were grown in TV base medium supplemented with glucose. PTS-dependent transport of glucose (glc), GlcNAc, or GlcN was determined using permeabilized cells as described in Materials and Methods. Each bar represents the average of results of three independent experiments, with error bars indicating the standard deviation. *, P < 0.05 (by the Student t test). ns, not significant.
A manO mutant displayed lower growth rates on, and PTS-dependent transport of, N-acetylglucosamine and glucosamine.
In some streptococci, including Streptococcus bovis and Streptococcus salivarius, the man operon contains an additional transcript, manO, located downstream of manLMN (12, 71, 72). In S. mutans UA159 and over 50% of the clinical isolates sequenced in a previous study (10), an apparent homolog of manO (SMU.1883), which encodes a 122-amino-acid protein, is located 3.85 kbp downstream of manN. However, in the remaining sequenced clinical isolates, manO is about 300 bp downstream of manN. It has been suggested that ManO may be capable of binding DNA and influencing transcription of the man operon in S. salivarius (72), although there has not yet been any experimental evidence generated to support this idea. To explore whether ManO could impact the transport of amino sugars, we created an allelic-exchange mutant of manO with a nonpolar kanamycin marker. Cells lacking ManO that were grown in GlcNAc displayed the most significant increase in doubling time. However, the final yield for the manO mutant on GlcNAc was comparable to that of the wild-type strain, as was the case for the other sugars tested. Loss of ManO also negatively impacted growth on glucose and GlcN (Fig. 3). To complement the manO mutation, a wild-type copy of the manO gene was inserted into the expression vector pIB184 (54), and the resulting plasmid, pIB184-manO, was used to transform the manO mutant. When grown with GlcNAc, GlcN, or glucose as the primary carbohydrate source, the complemented strain displayed growth that was comparable to that of the wild-type strain containing the empty vector (see Fig. S7 in the supplemental material).
FIG 3.
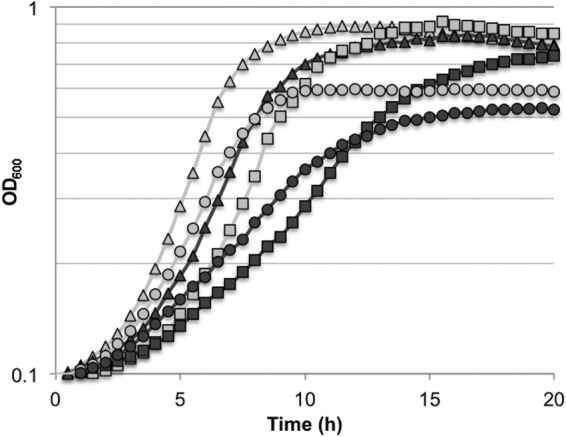
Growth curves of S. mutans UA159 (light shapes) and a manO deletion mutant (dark shapes) grown in TV base medium supplemented with 20 mM glucose (triangles), 20 mM GlcNAc (squares), or 20 mM GlcN (circles).
To determine if deletion of manO could impact PTS-dependent transport of the carbohydrates of interest, the manO mutant and wild-type strain were grown in TV base medium supplemented with 20 mM glucose. The manO mutant displayed a significant defect in PTS-dependent transport of GlcNAc, and transport of glucose and GlcN was consistently lower than for the wild-type strain, though the differences were not statistically significant (Fig. 2B). To explore the potential impact of loss of ManO on the amount of the EIIAB component of the EIIMan complex, ManL, S. mutans UA159 and the manO mutant were grown on TV media containing 20 mM glucose, and ManL levels were compared by immunoblotting. The levels of ManL in the two cell types were comparable (see Fig. S8 in the supplemental material).
The metabolism of N-acetylglucosamine impacts acid tolerance in S. mutans.
The impact of metabolism of GlcNAc on acid tolerance by S. mutans UA159 grown in glucose, GlcNAc, or a combination of the two sugars was assessed as detailed in Materials and Methods. When GlcNAc was used as the substrate in a pH drop assay, cells grown on a combination of glucose and GlcNAc reached a terminal pH after 30 min of 4.29 ± 0.06, which was significantly lower than the terminal pH achieved by cells grown on the sugars individually (Fig. 4A). Additionally, cells grown in GlcNAc alone were capable of achieving a terminal pH of 4.62 ± 0.09, which was significantly lower than that achieved by cells grown on glucose (4.97 ± 0.07) and, as noted above, significantly higher than for cells grown on a combination of glucose and GlcNAc (Fig. 4A). Cells grown on a combination of these sugars were also capable of lowering the pH at a significantly higher rate than for cells grown on individual sugars. Specifically, cells grown with both glucose and GlcNAc reached a pH of 6.05 ± 0.18 2 min after provision of exogenous GlcNAc, whereas cells grown on glucose or GlcNAc alone reached a pH of 6.73 ± 0.004 and 6.64 ± 0.12, respectively, in the same time interval (Fig. 4A). Of note, when S. mutans strain UA159 was grown with a combination of glucose and GlcNAc, a statistically significant increase in PTS-dependent transport of GlcNAc was seen compared to that for cells grown with glucose or GlcNAc as the primary carbohydrate source (see Fig. S9 in the supplemental material). These results would seem to imply that the faster drop in pH observed in cells grown with a combination of GlcNAc and glucose as the primary carbohydrate source may in part be due to enhanced carbohydrate transport capacity. As EIIMan is the primary transporter of both GlcNAc and glucose, we also performed an immunoblot to determine if the growth carbohydrate influenced the amount of ManL in the cells, but ManL protein levels were generally consistent in cells grown with glucose, GlcNAc, or a combination of glucose and GlcNAc (see Fig. S10 in the supplemental material). Protein levels for other components of the EIIMan complex were not analyzed because antibodies against these proteins are not available.
FIG 4.
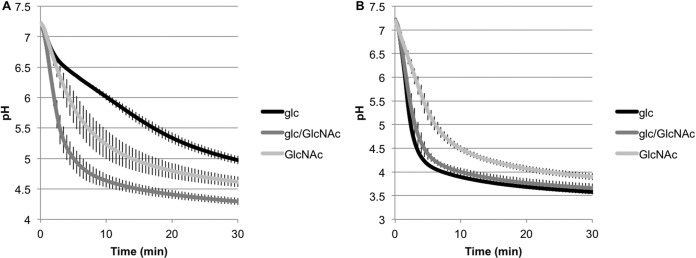
pH drop assays. Cells of S. mutans UA159 were grown to mid-exponential phase in TV base medium supplemented with 20 mM glucose (glc), 20 mM glc and 20 mM GlcNAc (glc/GlcNAc), or 20 mM GlcNAc (GlcNAc) before being subjected to pH drop assays as described in Materials and Methods. The assay was initiated by the addition of 50 mM (final concentration) of either GlcNAc (A) or glucose (B), and the change in the pH was monitored at 30-s intervals for 30 min. The data represent the averages of results of three independent experiments, with error bars indicating the standard deviation.
In a similar experiment using glucose as the substrate for pH drops, cells grown in glucose or a combination of glucose and GlcNAc were capable of lowering the pH at comparable rates and reached roughly the same terminal pH after 30 min. However, cells grown on GlcNAc alone yielded significantly higher terminal pH values than did cells grown on glucose or a combination of the sugars (Fig. 4B). Additionally, cells grown on GlcNAc alone lowered the pH at a significantly lower rate (6.42 ± 0.14 after 2 min), whereas cells grown on glucose or a combination of the sugars reached pH values of 5.40 ± 0.15 and 5.74 ± 0.25, respectively, in the same amount of time (Fig. 4B). Due to its instability under neutral or alkaline pH conditions, we determined that GlcN was not suited to serve as a substrate in pH drop assays, in which carbohydrates are usually added as neutral pH solutions.
The metabolism of N-acetylglucosamine and glucosamine produces substantial quantities of ammonia.
One advantage of metabolizing amino sugars is the production of ammonia, which can neutralize the environment produced as a result of rapid carbohydrate catabolism. Additionally, the production of ammonia inside cells has the dual benefit of neutralizing the cytoplasm and providing a nitrogen source, the latter eliminating the need to devote energy to transport ammonium ion or other nitrogen sources from the environment. To determine if significant quantities of ammonia were produced during metabolism of amino sugars, we grew S. mutans UA159 to mid-exponential phase with glucose, GlcNAc, or GlcN as the primary carbohydrate source and resuspended equal densities of cells in the same solution as used for pH drop assays. Cells were rapidly neutralized and incubated with the original growth carbohydrate, namely, glucose, GlcNAc, or GlcN. After 1 h of incubation, cells incubated in GlcNAc or GlcN produced significantly more ammonia than cells incubated with glucose, and cells incubated with GlcN produced the most ammonia of the three carbohydrates tested (Fig. 5). Interestingly, we also noted that the average final pH of the solution for cells incubated with glucose, GlcNAc, or GlcN correlated with the production of ammonia (data not shown), with the highest terminal pH being associated with the carbohydrate that yielded the most ammonia. Clearly, then, ammonia production from amino sugars can create a more alkaline environment, allowing cells to metabolize carbohydrates for a longer period by protecting acid-sensitive enzymes.
FIG 5.
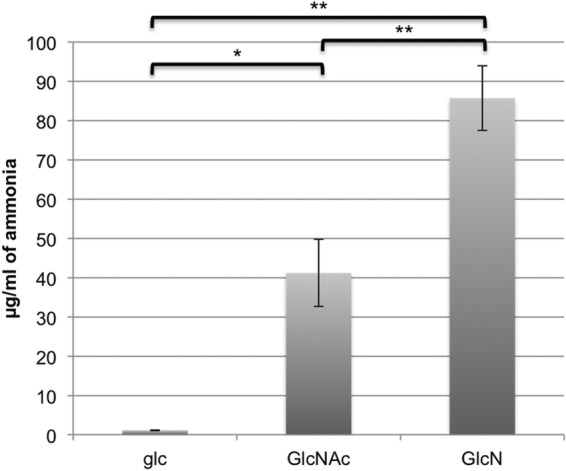
Measurements of ammonia in supernatant fluids of UA159 incubated with 50 mM glucose (glc), 50 mM GlcNAc, or 50 mM GlcN. The data are averages of results of three independent experiments, with error bars indicating the standard deviation. *, P < 0.05; **, P < 0.005 (by the Student t test).
NagR is the major regulator of glucosamine and N-acetylglucosamine metabolism.
In many bacteria, metabolism of GlcNAc and GlcN is governed by the binding of a GntR/HutC-type regulator, NagR, to the promoter regions of amino sugar catabolism genes, including nagA and nagB (34, 73, 74). A gene with substantial homology to confirmed nagR genes is present in S. mutans UA159, SMU.1065c, hereafter designated nagR. Of note, nagR is immediately upstream (separated by 3 bp) of a predicted osmolarity-responsive transcriptional regulator gene, busR (SMU.1064c) (see Fig. S1A). An allelic-exchange mutant of nagR was created by replacing SMU.1065c with a nonpolar kanamycin cassette. When S. mutans UA159 and the nagR mutant were grown in TV supplemented with 20 mM GlcN or GlcNAc, there was no significant difference in growth rate or final yield due to the loss of NagR (data not shown). To determine if contaminants from the tryptone base media were influencing our results, cells were also grown using the chemically defined medium FMC (50), but GlcNAc or GlcN was substituted for glucose at a final concentration of 20 mM. There was also little change in the growth rate of the nagR mutant in the chemically defined medium FMC with GlcN as the sole carbohydrate source, although there was a small increase in final yield (Fig. 6A). However, the nagR mutant grew significantly faster than the wild-type strain on FMC constituted with 20 mM GlcNAc as the sole carbohydrate source and displayed a markedly shorter lag phase (Fig. 6B). In contrast, the wild-type cells growing in FMC with GlcN did not show the same lag phase as in FMC with GlcNAc. Thus, the absence of NagR allowed for more rapid adaptation of the mutant strain when cells were transferred from BHI to FMC containing GlcNAc but not when transferred from BHI to FMC with GlcN. The different phenotypes of the nagR mutant in TV- and FMC-based media lead us to suspect that trace amounts of GlcN or other inducing reagents may be present in the tryptone in TV base medium, which stimulated the transcription of the nag genes in the wild-type strain, leading to a less pronounced effect of the loss of NagR.
FIG 6.
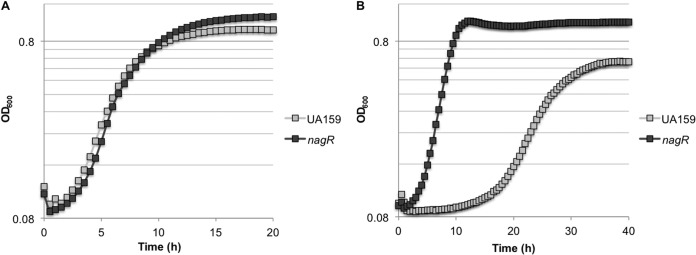
Growth curves of S. mutans UA159 and a nagR mutant grown in FMC medium formulated with 20 mM GlcN (A) or 20 mM GlcNAc (B) as the sole carbohydrate source.
In order to better understand the phenotypes of the nagR mutant, real-time quantitative RT-PCR was employed to probe the mRNA levels of three genes critical to GlcNAc and GlcN metabolism, nagA, nagB, and glmS, which are not genetically linked in S. mutans (see Fig. S1A in the supplemental material). To avoid the potentially confounding effects of trace carbohydrates in tryptone, FMC lacking glucose (50) was used as the base medium and glucose, GlcNAc, or GlcN was added to a final concentration of 20 mM. As presented in Table 2, there were profoundly altered expression levels of all three genes in the nagR mutant compared to the parental strain. In the wild-type genetic background, the presence of GlcNAc or GlcN resulted in significant increases in nagA and nagB transcripts relative to that measured in glucose-grown cells. Notably, glmS mRNA levels were significantly decreased when UA159 cells were grown in GlcNAc or GlcN rather than glucose. These results are consistent with previous findings with S. mutans (44). In contrast to the wild-type strain, the nagR mutant presented markedly higher levels of nagA and nagB transcripts when grown in glucose, and similarly high levels with GlcNAc and GlcN, consistent with the prediction that NagR functions as a negative regulator of these genes. Surprisingly, glmS message levels were also found to be significantly higher in the nagR mutant than in the parental strain, regardless of the growth carbohydrate. Of note, expression levels of the putative osmoregulator gene busR were not significantly altered in the nagR mutant grown on GlcNAc or GlcN. However, when growing on glucose, the nagR mutant yielded modestly higher levels (about 2-fold) of busR transcript than the wild type (data not shown).
TABLE 2.
Relative mRNA levels of nagA, nagB, and glmS as determined by qRT-PCRa
| Carbohydrate | Strain | Ratio of indicated mRNA level to gyrA mRNA level (avg ± SD)c |
||
|---|---|---|---|---|
| nagA | nagB | glmS | ||
| Glucose | UA159 | 0.24 ± 0.08 | 1.64 ± 0.13 | 0.18 ± 0.03 |
| nagR mutant | 3.29 ± 0.98* | 16.64 ± 1.28† | 2.23 ± 0.50‡ | |
| nagR/pIB184 strain | 3.14 ± 0.07 | 32.63 ± 3.48 | 4.13 ± 0.19 | |
| nagR/pIB184-nagR strain | 0.32 ± 0.08d | 4.25 ± 1.38d | 0.82 ± 0.11d | |
| GlcNAc | UA159 | 2.21 ± 0.27b | 19.24 ± 1.36b | 0.11 ± 0.01b |
| nagR mutant | 3.02 ± 0.51* | 25.60 ± 2.77† | 2.53 ± 0.28‡ | |
| GlcN | UA159 | 1.40 ± 0.30b | 10.91 ± 2.48b | 0.05 ± 0.01b |
| nagR mutant | 1.66 ± 0.29* | 10.48 ± 1.03† | 1.54 ± 0.23‡ | |
| nagR/pIB184 strain | 1.71 ± 0.20 | 12.02 ± 1.41 | 1.45 ± 0.33 | |
| nagR/pIB184-nagR strain | 0.37 ± 0.14d | 4.38 ± 1.23d | 0.11 ± 0.05d | |
Cells were grown in FMC containing 20 mM glucose, GlcNAc, or GlcN as the primary carbohydrate source. Gene expression relative to that of the constitutively expressed gyrA gene is reported as a ratio.
The transcript level was significantly different from that in the same strain grown with glucose (one-way analysis of variance [ANOVA]; P < 0.05).
A two-way ANOVA was performed individually on nagA (*), nagB (†), and glmS (‡) transcript data, and each mRNA level was significantly affected as a result of nagR deletion (P < 0.0001), change in carbohydrate source (PnagA = 0.0064, PnagB < 0.0001, and PglmS = 0.0088), and the interaction of these two factors (PnagA = 0.0009, PnagB < 0.0001, and PglmS = 0.0232).
A two-way ANOVA was performed individually on nagA, nagB, and glmS transcript data in comparison to that of nagR/pIB184, and each mRNA level was significantly affected as a result of nagR complementation (P < 0.0001), change in carbohydrate source (P < 0.0001), and the interaction of these two factors (P < 0.0001).
To confirm that the observed changes in growth phenotype and gene regulation were due exclusively to loss of NagR, a complemented strain was constructed by introducing a wild-type copy of nagR into the nagR mutant strain on the expression plasmid pIB184 (54), resulting in high-level, constitutive transcription of nagR (approximately 40-fold above wild-type levels). When tested for growth, the complemented nagR mutant displayed reduced growth on GlcN and especially on GlcNAc (see Fig. S11 in the supplemental material), likely as a result of nagR overexpression. When gene transcription was assayed in cells grown under glucose or GlcN conditions, mRNA levels of nagA, nagB, and glmS in the complemented strain were found to be comparable to, or in some cases lower than, those measured in the wild-type strain (Table 2). Notably, busR mRNA levels in glucose-grown, nagR-overexpressing cells remained the same as in the wild-type background (data not shown). These data provide further evidence that NagR is a dominant transcriptional regulator of the genes for both catabolism and anabolism of GlcN and GlcNAc in S. mutans UA159, in stark contrast to how these genes are regulated in other Gram-positive bacteria in which, to our knowledge, anabolic processes are coordinated by NagR-independent mechanisms.
Recombinant NagR protein binds to the promoter regions of nagA, nagB, and glmS in vitro.
To further explore the mechanisms by which NagR regulates expression of nagA, nagB, and glmS, we tested the ability of recombinant NagR protein to interact with DNA fragments carrying these promoter sequences, as an online database (http://regprecise.lbl.gov/RegPrecise/collections.jsp) (75) indicated that conserved putative NagR-binding elements exist in the regions tested (see Fig. S1B in the supplemental material). Using electrophoretic mobility shift assay (EMSA) with a NagR protein fused to a maltose-binding protein (MBP-NagR), all three DNA probes containing the promoter regions of nagA, nagB, and glmS genes were shifted by the recombinant protein (data not shown). Interestingly, this shift did not result in distinct bands migrating more slowly in the gel. Rather, complexes of DNA and protein appeared to form but failed to enter the gel, with a corresponding loss of the unbound probe migrating freely into the gel. Given the possibility that the presence of the MBP tag could be interfering with the interaction of NagR with its cognate binding sites, recombinant MBP-NagR was treated with factor Xa, followed by adsorption with an amylose resin to deplete MBP and undigested MBP-NagR. When used in EMSAs, the released NagR retarded the progression of all three probes through the gel (Fig. 7). However, the complexes formed by the interaction of NagR with the probes again failed to enter the gel and form distinctive bands (Fig. 7), even more so than when the MBP-NagR fusion protein was used.
FIG 7.

EMSAs performed using the NagR protein and biotinylated DNA fragments containing regions upstream of the nagB (A), nagA (B), or glmS (C) coding sequences. DNA probes and purified protein were generated as described in Materials and Methods. All EMSAs are representative examples of at least three independent experiments showing similar results.
In trying to reduce the formation of protein-DNA aggregates, the nonionic detergent NP-40 was tested in some of the reaction mixtures, but only a modest decrease in the formation of aggregates and an increase in discrete shifted products could be realized (see Fig. S14 in the supplemental material). Importantly, the loss of nagB probe signal due to binding to NagR was readily reversed by the addition of small amounts of proteinase K into the binding reaction (Fig. 7A), and similar results were seen for nagA and glmS probes (data not shown). Notably, high-molecular-weight complexes similar to those formed by S. mutans NagR and the three promoter probes have been observed in previous studies of NagR/NagC (27, 34), and the complexes from these studies also appeared to settle on the top of the gel instead of migrating as clear bands. As controls, we performed gel shift experiments using purified maltose-binding protein with all three probes and saw no change in migration of the probes or loss of signal in any case (Fig. 7A; see also Fig. S12 in the supplemental material). Additionally, the migration of two unrelated probes, corresponding to the promoters of nlmD and pdhD (M. Watts and R. A. Burne, unpublished data), was not influenced by the presence of released NagR or MBP-NagR (see Fig. S13), indicating that contamination of the NagR protein preparation by nucleases did not account for the results.
Because GlcNAc and GlcN appear to be internalized exclusively by the PTS, we explored whether the phosphorylated forms of GlcNAc and GlcN, GlcNAc-6-P and GlcN-6-P, could alter the binding of NagR to its targets. As shown in Fig. 7, the presence of 20 mM GlcN-6-P, but not GlcNAc-6-P, inhibited the binding of NagR to probes containing promoter sequences of nagA, nagB, and glmS. Similar results were obtained with the recombinant MBP-NagR protein (data not shown). Other common carbohydrates and intermediates, including GlcNAc, GlcN, fructose-6-P, and fructose-1,6-bisphosphate, were also tested, but none of these compounds affected the binding of NagR to its targets (data not shown). These results support the idea that GlcN-6-P may serve in vivo as the allosteric effector that alleviates repression of nagA, nagB, and possibly glmS by NagR. This model would be consistent with previous results obtained with B. subtilis showing that the binding activity of the NagR homolog, formerly YvoA, to the nagA promoter was inhibited by GlcN-6-P (34). However, since transcription of glmS and the genes for catabolism, nagA and nagB, are oppositely regulated in the presence of glucose or GlcN and GlcNAc (Table 2) (44), the apparent affinity of NagR for the glmS promoter region and the effects of GlcN-6-P on binding of NagR to the glmS probe suggest a novel mechanism for transcriptional regulation of glmS by S. mutans in response to the carbohydrate source.
DISCUSSION
This study provides genetic and biochemical evidence that EIIMan is the primary transporter for GlcNAc and GlcN in S. mutans and that both amino sugars are imported exclusively via the PTS (Fig. 1C). We would also predict that multiple species of the genus Streptococcus utilize homologs of the glucose/mannose-specific PTS for internalizing these amino sugars since ManLMN are fairly well conserved, and we showed here that manL of S. gordonii is also required for efficient GlcNAc and GlcN metabolism (see Fig. S4C and D in the supplemental material). These results are not completely unexpected, as the mannose PTS permease is capable of transporting both sugars in E. coli (19, 26), and ManLMN of S. mutans contributes to the uptake of a number of sugars besides mannose (12, 13). However, a novel discovery from this study is that a previously uncharacterized protein, ManO, which is encoded within or near the manLMN operons of multiple streptococci (12, 71, 72) could influence the transport and metabolism of GlcNAc and GlcN.
The presence of a manO homolog has been described for other species of streptococci, and it has been suggested that ManO of Streptococcus salivarius could serve as a transcriptional regulator (72). Unpublished data collected by previous members of our laboratory seem to indicate that a manO mutant bears a slight defect in PTS-dependent uptake of a variety of sugars other than GlcNAc and GlcN (J. Abranches and R. A. Burne, unpublished data). These data led us to test whether ManO could influence amino sugar transport, perhaps by modifying the substrate specificity of the ManLMN permease. It is also noteworthy that there appear to be two distinct populations of S. mutans strains that show evidence of divergence in the organization of the EIIMan gene cluster (see Fig. S1A) (10). One of these, represented by strain UA159, carries a 3.85-kbp region between manN and manO that encodes a putative ABC transporter and a small hypothetical protein. The other group of S. mutans strains does not contain the large intergenic region encoding these two proteins but instead has a 0.3-kbp intergenic region with no apparent open reading frames (ORFs). Also notable is the substantial difference in DNA sequences of manM and manN between these two groups of S. mutans strains. For example, ManM and ManN of strain SMU33 (10), which is missing the large intergenic region, are 73% and 77% identical to ManM and ManN of S. mutans UA159. However, when ManM and ManN of strain UA159 are compared to strains that have the 3.85-kbp insertion, it is found that these proteins are nearly 100% identical (data not shown). In contrast, the degree of conservation of ManL, ManO, and the region upstream of manL across all 57 strains appears to be near 100%. These findings could suggest the existence of two lineages of S. mutans that arose from evolution in distinct host genetic backgrounds or host microenvironments that provided differential access to particular carbohydrates. Experiments are under way to probe potential interactions between ManO and the other components of the EIIMan complex, though initial studies of the association between ManO and ManL via a two-hybrid assay have failed to demonstrate an interaction (data not shown). The finding that ManL protein levels remained relatively unchanged in a manO mutant (see Fig. S8 in the supplemental material) is consistent with observations made in our laboratory that the activity of the manL promoter was not influenced by the deletion of manO in S. mutans UA159 (Abranches and Burne, unpublished). Our working hypothesis is that ManO is a structural component of the EIIMan complex required for efficient carbohydrate transport. Future experiments are under way to fully elucidate the role of ManO in the EIIMan complex.
Reintroduction of manL onto the chromosome of the manL deletion strain, under the control of the gtfA promoter, restored the ability of the organism to grow on GlcNAc as the primary carbohydrate source and enhanced growth on GlcN compared to that of the manL mutant. However, complementation of the manL mutant in this manner did not fully restore growth to wild-type levels, a finding that we attribute to lower levels of manL mRNA and ManL protein when expression of manL was driven by the gtfA promoter (see Fig. S2 and S3 in the supplemental material). It must also be considered that the defect in GlcN transport in the manL mutant could be partially compensated for by other EII permeases, e.g., the cellobiose-specific PTS (see Fig. S5A) (celD strain), as some of these transporters appear to be inducible by the presence of GlcN (see Fig. S6A, UA159 glc versus UA159 GlcN). Further, the expression of the cellobiose PTS operon (cel) is subject to EIIMan-dependent inducer exclusion (51). Therefore, in the absence of ManL, expression of the cel operon should increase and would mask to some extent the effects of loss of ManL on amino sugar metabolism. Conversely, complementation of ΔmanL by pBGE-manL should reduce the expression of the cel operon by restoring EIIMan-mediated repression of cel genes. Thus, the GlcN-related growth and PTS phenotypes of these strains could reflect contributions of the EIIMan, EIICel, and possibly minor effects from other PTS permeases.
The NagR protein of S. mutans shares a high degree of similarity to GntR/HutC-type transcription regulators of other bacteria. For example, NagR of S. mutans is 36% identical to NagR/YvoA of B. subtilis, including conservation in the N-terminal helix-turn-helix and C-terminal effector-binding domains. Similar to what has been established in E. coli and B. subtilis, expression of genes that are essential for GlcNAc and GlcN catabolism, nagA and nagB, was clearly inducible in S. mutans when amino sugars were added to the culture media (Table 2). Further, by using recombinant MBP-NagR or NagR in EMSAs, we have demonstrated the ability of NagR to bind to presumptive target promoter regions upstream of nagA, nagB, and glmS. Importantly, this binding activity was disrupted by the addition of GlcN-6-P but not GlcNAc-6-P. On the basis of these findings, we propose that GlcN-6-P is likely the inducing signal detected by NagR in S. mutans. In support of this hypothesis, we have noted that the metabolism of GlcNAc by S. mutans requires a period of adaptation, as evidenced by the prolonged lag phase after transferring strain UA159 grown in BHI broth into FMC with GlcNAc as the sole carbohydrate source (Fig. 6B), although the long lag phase was not observed when transitioning onto FMC with GlcN (Fig. 6A). The duration of adaptation is likely determined by two factors. First, the induction of nagA and nagB gene expression may be delayed in cells inoculated into FMC containing GlcNAc because GlcNAc-6-P must be converted to GlcN-6-P and accumulate in sufficient quantities to relieve NagR repression. Second, an extended lag phase may arise as a result of growth-inhibitory effects of GlcNAc-6-P that could accumulate rapidly in cells via ManLMN-dependent uptake. Interestingly, it has been reported that GlcNAc is preferred over GlcN in E. coli and that the inducing signal for nag gene expression is GlcNAc-6-P (20, 27, 76). Further, inactivation of the nagA gene in E. coli results in significant accumulation of GlcNAc-6-P due to the turnover of peptidoglycan and recycling of its components (20, 77). While this can lead to elevated expression of nag genes in E. coli, due to GlcNAc-6-P binding to NagC, we have noted that for S. mutans, a nagA mutant grown on glucose does not produce enhanced levels of the nagB transcript (data not shown). Thus, in spite of similarities in the repressor of nag gene expression in E. coli and S. mutans, there are fundamental differences in the way in which the metabolic intermediates of GlcN and GlcNAc modulate induction of the nag genes through these transcriptional regulators. Lastly, we have noted that the levels of nagB transcripts are considerably higher than that of nagA in all strains and under various growth conditions (Table 2). Setting aside possible posttranscriptional regulation of NagA and NagB levels or activity, the differences in the absolute levels of mRNAs for these genes could be associated with the need for cells to remain responsive to changes in carbohydrate source while maintaining GlcN-6-P pools at levels that are optimal for growth.
One surprising finding was that nagA, nagB, and glmS, which are not genetically linked, are under direct control of NagR. Further, the binding of NagR to all three promoter regions was inhibited by GlcN-6-P. However, as shown in Table 2 and reported previously (44), expression of glmS is inversely related to that of nagA and nagB in the wild-type background, consistent with the fact that GlmS is required for biosynthesis of the amino sugars and NagA and NagB for their catabolism. Thus, the mechanism by which NagR interacts with the glmS promoter and controls glmS expression must be different than for its control of nagA and nagB in vivo. Analysis of the 5′ UTR of glmS in S. mutans has not identified canonical residues for the formation of a ribozyme complex (44), which is the primary mode of regulation for glmS in multiple Gram-positive bacteria. Likewise, bioinformatic studies of the diversity and distribution of ribozymes in bacteria failed to detect the presence of a glmS ribozyme sequence in S. mutans, in any other streptococcus, or in most lactobacilli (43, 44). However, regulation of glmS by additional transcriptional regulators or regulatory RNAs cannot yet be excluded. We have noted in a transcriptome sequencing (RNA-Seq) analysis performed on S. mutans UA159 that the glmS transcript was upregulated in galactose relative to glucose, and this effect appears to be independent of CcpA (78). Further, it has been demonstrated in studies with E. coli that one of the galactose permeases, galP, is under the direct control of the NagR homolog NagC, suggesting that coordinated regulation of galactose and amino sugar metabolism could be common in bacteria (79). In addition, we have identified two putative sRNAs, one upstream and one downstream of the glmS gene, which could act to differentially regulate glmS in response to carbohydrate source and availability (data not shown). Alternatively, it has been demonstrated that NagR could play a role as both an activator and a repressor of transcription in E. coli and Vibrio cholerae (80, 81). Therefore, it is possible that NagR of S. mutans can activate glmS in response to changes in the pools of metabolic intermediates, including, but not limited to, GlcN-6-P. In a recent study subjecting the B. subtilis genome to DNase I protection assays probed with NagR, it was suggested that NagR may be capable of binding specifically to the operator of nag operon genes and nonspecifically to neighboring DNA at higher NagR concentrations, perhaps through protein-protein interaction (35). Further, GlcNAc-6-P was shown to disrupt this nonspecific binding, but specific binding of NagR to the operator was preserved. It should be considered, then, that in regions of the S. mutans genome such as the glmS and nagB promoters, where two putative NagR-binding sequences are predicted by the RegPrecise database (see Fig. S1B in the supplemental material), NagR protein may form bridges between operator sequences to serve as another level of regulation. We are currently exploring how glmS transcription is controlled in the mutans streptococci.
Another layer of potential complexity in NagR function in S. mutans is its tight genetic linkage to busR, which is predicted to encode an osmolarity-responsive transcriptional regulator. As mentioned above, nagR and busR are located only 3 nucleotides apart, and examination of previously generated RNA-Seq data (78) indicates that nagR and busR transcript levels are nearly identical and that these genes are probably cotranscribed under the conditions tested. In addition, qRT-PCR results have shown that transcript levels of both nagR and busR were significantly lower in UA159 when cells were grown on FMC containing GlcN rather than GlcNAc or glucose (data not shown). Subsequent qRT-PCR studies have revealed that an allelic-exchange replacement of the nagR gene with a nonpolar antibiotic resistance marker did not negatively influence the expression of busR, and a wild-type copy of the nagR gene expressed on a plasmid from a strong promoter successfully reversed the growth defect of the nagR mutant without influencing the expression of busR. Interestingly, these two transcriptional regulators are immediately followed by a pair of ABC transporter genes, opuAa and opuAb, that are likely required for bacterial resistance to osmotic stress (82). When opuAa mRNA levels were assayed by qRT-PCR, we found that expression increased 2- to 3-fold in a busR deletion mutant grown in FMC containing glucose as the sole carbohydrate source (data not shown), while the levels remained unchanged in a nagR deletion mutant. The elucidation of the exact nature of this differential regulation is beyond the scope of this communication, although we plan to investigate the functions of NagR and BusR in regulating amino sugar metabolism and other phenotypic traits that may be important for establishment, persistence, or virulence of S. mutans.
The virulence of cariogenic bacteria, including S. mutans, is predominately associated with acidogenicity and acid tolerance (1, 4). It has been demonstrated previously that factors promoting a more alkaline internal pH benefit the organism and perhaps increase its pathogenicity (83). Our study provides evidence that the metabolism of GlcN and GlcNAc, albeit at relatively high concentrations, clearly increases the total concentration of ammonia in the environment and presumably within the cytoplasm (Fig. 5). The effect of GlcN and GlcNAc metabolism also impacted the final pH of the culture, illustrating that the metabolism of these amino sugars may provide cariogenic species with a mechanism for maintaining a more alkaline internal pH. This benefit was demonstrated in our pH drop assay in which UA159 cells were grown in glucose, GlcNAc, or the combination of both and then tested for the ability to lower the environmental pH through utilization of GlcNAc (Fig. 4A). Among all these growth conditions, cells grown in the combination of glucose and GlcNAc dropped the pH to the lowest point and did so at the highest rate; these cells also presented the highest levels of GlcNAc transport when tested in a PTS assay (see Fig. S9 in the supplemental material). The enhanced capacity of cells grown with glucose alone or a combination of glucose and GlcNAc to lower the pH when exogenous glucose was provided may be explained as follows. Previous studies have demonstrated that exposure to a moderately acidic environment (e.g., pH 5.5 for 2 h) is correlated with enhanced acid tolerance, attributable to the induction of the adaptive acid tolerance response (ATR) (60, 84–87). Two hallmarks of induction of the ATR in S. mutans are more rapid metabolism of carbohydrate and attainment of a lower final pH in pH drop assays. Cells grown with glucose or a combination of glucose and GlcNAc as the primary carbohydrate source routinely experienced a lower pH by mid-exponential phase (data not shown) than cells grown only on GlcNAc. Therefore, we posit that cells grown with GlcNAc alone likely maintained a more alkaline cytoplasmic pH due to ammonia production (Fig. 5) from deamination of GlcN-6-P, so the ATR was not induced. It must also be considered that the metabolism of amino sugars provides a competitive advantage for some species. As mentioned above, the metabolism of GlcNAc favors S. mutans over S. sobrinus in mixed cultures due mostly to higher levels of GlcNAc-metabolizing enzymes in S. mutans (48). The production of ammonia via amino sugar metabolism could serve to moderate cytoplasmic pH and supply nitrogen to metabolizing cells, which could contribute to the caries process by allowing cells to continue to make acid at low pH. Further studies are needed to explore this hypothesis and to elucidate the impact of amino sugar metabolism by pathogens and commensals on the initiation and progression of dental caries.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grant DE12236 from the National Institute of Dental and Craniofacial Research. Z.D.M. was supported by a University of Florida Alumni Fellowship.
We thank Ariana Rosa-Alberty for assistance during mutant construction and growth studies.
Footnotes
Published ahead of print 13 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00820-14.
REFERENCES
- 1.Loesche WJ. 1986. Role of Streptococcus mutans in human dental decay. Microbiol. Rev. 50:353–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamada S, Slade HD. 1980. Biology, immunology, and cariogenicity of Streptococcus mutans. Microbiol. Rev. 44:331–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemos JA, Burne RA. 2008. A model of efficiency: stress tolerance by Streptococcus mutans. Microbiology 154:3247–3255. 10.1099/mic.0.2008/023770-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burne RA. 1998. Oral streptococci. Products of their environment. J. Dent. Res. 77:445–452. 10.1177/00220345980770030301 [DOI] [PubMed] [Google Scholar]
- 5.Banas JA. 2004. Virulence properties of Streptococcus mutans. Front. Biosci. 9:1267–1277. 10.2741/1305 [DOI] [PubMed] [Google Scholar]
- 6.Carlsson J. 1984. Regulation of sugar metabolism in relation to the “feast-and-famine” existence of plaque, p 205–211 In Guggenheim B. (ed), Cariology. Karger, Basel, Switzerland [Google Scholar]
- 7.Ajdić D, McShan WM, McLaughlin RE, Savić G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. U. S. A. 99:14434–14439. 10.1073/pnas.172501299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vadeboncoeur C, Pelletier M. 1997. The phosphoenolpyruvate:sugar phosphotransferase system of oral streptococci and its role in the control of sugar metabolism. FEMS Microbiol. Rev. 19:187–207. 10.1111/j.1574-6976.1997.tb00297.x [DOI] [PubMed] [Google Scholar]
- 9.Ajdić D, Pham VT. 2007. Global transcriptional analysis of Streptococcus mutans sugar transporters using microarrays. J. Bacteriol. 189:5049–5059. 10.1128/JB.00338-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornejo OE, Lefébure T, Pavinski Bitar PD, Lang P, Richards VP, Eilertson K, Do T, Beighton D, Zeng L, Ahn SJ, Burne RA, Siepel A, Bustamante CD, Stanhope MJ. 2013. Evolutionary and population genomics of the cavity causing bacteria Streptococcus mutans. Mol. Biol. Evol. 30:881–893. 10.1093/molbev/mss278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeng L, Xue P, Stanhope MJ, Burne RA. 2013. A galactose-specific sugar: phosphotransferase permease is prevalent in the non-core genome of Streptococcus mutans. Mol. Oral Microbiol. 28:292–301. 10.1111/omi.12025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abranches J, Chen YY, Burne RA. 2003. Characterization of Streptococcus mutans strains deficient in EIIABMan of the sugar phosphotransferase system. Appl. Environ. Microbiol. 69:4760–4769. 10.1128/AEM.69.8.4760-4769.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng L, Das S, Burne RA. 2010. Utilization of lactose and galactose by Streptococcus mutans: transport, toxicity, and carbon catabolite repression. J. Bacteriol. 192:2434–2444. 10.1128/JB.01624-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng L, Burne RA. 2008. Multiple sugar: phosphotransferase system permeases participate in catabolite modification of gene expression in Streptococcus mutans. Mol. Microbiol. 70:197–208. 10.1111/j.1365-2958.2008.06403.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobrogosz WJ. 1968. N-Acetylglucosamine assimilation in Escherichia coli and its relation to catabolite repression. J. Bacteriol. 95:585–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schäffer C, Messner P. 2005. The structure of secondary cell wall polymers: how Gram-positive bacteria stick their cell walls together. Microbiology 151:643–651. 10.1099/mic.0.27749-0 [DOI] [PubMed] [Google Scholar]
- 17.Trent MS. 2004. Biosynthesis, transport, and modification of lipid A. Biochem. Cell Biol. 82:71–86. 10.1139/o03-070 [DOI] [PubMed] [Google Scholar]
- 18.Dobrogosz WJ. 1968. Effect of amino sugars on catabolite repression in Escherichia coli. J. Bacteriol. 95:578–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White RJ. 1970. The role of the phosphoenolpyruvate phosphotransferase system in the transport of N-acetyl-D-glucosamine by Escherichia coli. Biochem. J. 118:89–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Álvarez-Añorve LI, Bustos-Jaimes I, Calcagno ML, Plumbridge J. 2009. Allosteric regulation of glucosamine-6-phosphate deaminase (NagB) and growth of Escherichia coli on glucosamine. J. Bacteriol. 191:6401–6407. 10.1128/JB.00633-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White RJ. 1968. Control of amino sugar metabolism in Escherichia coli and isolation of mutants unable to degrade amino sugars. Biochem. J. 106:847–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mengin-Lecreulx D, van Heijenoort J. 1996. Characterization of the essential gene glmM encoding phosphoglucosamine mutase in Escherichia coli. J. Biol. Chem. 271:32–39. 10.1074/jbc.271.1.32 [DOI] [PubMed] [Google Scholar]
- 23.Mengin-Lecreulx D, van Heijenoort J. 1993. Identification of the glmU gene encoding N-acetylglucosamine-1-phosphate uridyltransferase in Escherichia coli. J. Bacteriol. 175:6150–6157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mengin-Lecreulx D, van Heijenoort J. 1994. Copurification of glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridyltransferase activities of Escherichia coli: characterization of the glmU gene product as a bifunctional enzyme catalyzing two subsequent steps in the pathway for UDP-N-acetylglucosamine synthesis. J. Bacteriol. 176:5788–5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones-Mortimer MC, Kornberg HL. 1980. Amino-sugar transport systems of Escherichia coli K12. J. Gen. Microbiol. 117:369–376 [DOI] [PubMed] [Google Scholar]
- 26.Curtis SJ, Epstein W. 1975. Phosphorylation of D-glucose in Escherichia coli mutants defective in glucosephosphotransferase, mannosephosphotransferase, and glucokinase. J. Bacteriol. 122:1189–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plumbridge JA. 1991. Repression and induction of the nag regulon of Escherichia coli K-12: the roles of nagC and nagA in maintenance of the uninduced state. Mol. Microbiol. 5:2053–2062. 10.1111/j.1365-2958.1991.tb00828.x [DOI] [PubMed] [Google Scholar]
- 28.Calcagno M, Campos PJ, Mulliert G, Suástegui J. 1984. Purification, molecular and kinetic properties of glucosamine-6-phosphate isomerase (deaminase) from Escherichia coli. Biochim. Biophys. Acta 787:165–173. 10.1016/0167-4838(84)90076-1 [DOI] [PubMed] [Google Scholar]
- 29.Vincent F, Yates D, Garman E, Davies GJ, Brannigan JA. 2004. The three-dimensional structure of the N-acetylglucosamine-6-phosphate deacetylase, NagA, from Bacillus subtilis: a member of the urease superfamily. J. Biol. Chem. 279:2809–2816. 10.1074/jbc.M310165200 [DOI] [PubMed] [Google Scholar]
- 30.Vincent F, Davies GJ, Brannigan JA. 2005. Structure and kinetics of a monomeric glucosamine 6-phosphate deaminase: missing link of the NagB superfamily? J. Biol. Chem. 280:19649–19655. 10.1074/jbc.M502131200 [DOI] [PubMed] [Google Scholar]
- 31.Reizer J, Bachem S, Reizer A, Arnaud M, Saier MH, Jr, Stülke J. 1999. Novel phosphotransferase system genes revealed by genome analysis—the complete complement of PTS proteins encoded within the genome of Bacillus subtilis. Microbiology 145(Part 12):3419–3429 [DOI] [PubMed] [Google Scholar]
- 32.Gaugué I, Oberto J, Putzer H, Plumbridge J. 2013. The use of amino sugars by Bacillus subtilis: presence of a unique operon for the catabolism of glucosamine. PLoS One 8:e63025. 10.1371/journal.pone.0063025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Titgemeyer F, Reizer J, Reizer A, Saier MH., Jr 1994. Evolutionary relationships between sugar kinases and transcriptional repressors in bacteria. Microbiology 140(Part 9):2349–2354. 10.1099/13500872-140-9-2349 [DOI] [PubMed] [Google Scholar]
- 34.Bertram R, Rigali S, Wood N, Lulko AT, Kuipers OP, Titgemeyer F. 2011. Regulon of the N-acetylglucosamine utilization regulator NagR in Bacillus subtilis. J. Bacteriol. 193:3525–3536. 10.1128/JB.00264-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaugué I, Oberto J, Plumbridge J. 2014. Regulation of amino sugar utilization in Bacillus subtilis by the GntR family regulators, NagR and GamR. Mol. Microbiol. 92:100–115. 10.1111/mmi.12544 [DOI] [PubMed] [Google Scholar]
- 36.Resch M, Schiltz E, Titgemeyer F, Muller YA. 2010. Insight into the induction mechanism of the GntR/HutC bacterial transcription regulator YvoA. Nucleic Acids Res. 38:2485–2497. 10.1093/nar/gkp1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Görke B, Vogel J. 2008. Noncoding RNA control of the making and breaking of sugars. Genes Dev. 22:2914–2925. 10.1101/gad.1717808 [DOI] [PubMed] [Google Scholar]
- 38.Kalamorz F, Reichenbach B, März W, Rak B, Görke B. 2007. Feedback control of glucosamine-6-phosphate synthase GlmS expression depends on the small RNA GlmZ and involves the novel protein YhbJ in Escherichia coli. Mol. Microbiol. 65:1518–1533. 10.1111/j.1365-2958.2007.05888.x [DOI] [PubMed] [Google Scholar]
- 39.Urban JH, Vogel J. 2008. Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol. 6:e64. 10.1371/journal.pbio.0060064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reichenbach B, Maes A, Kalamorz F, Hajnsdorf E, Görke B. 2008. The small RNA GlmY acts upstream of the sRNA GlmZ in the activation of glmS expression and is subject to regulation by polyadenylation in Escherichia coli. Nucleic Acids Res. 36:2570–2580. 10.1093/nar/gkn091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Göpel Y, Lüttmann D, Heroven AK, Reichenbach B, Dersch P, Görke B. 2011. Common and divergent features in transcriptional control of the homologous small RNAs GlmY and GlmZ in Enterobacteriaceae. Nucleic Acids Res. 39:1294–1309. 10.1093/nar/gkq986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. 2004. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428:281–286. 10.1038/nature02362 [DOI] [PubMed] [Google Scholar]
- 43.McCown PJ, Roth A, Breaker RR. 2011. An expanded collection and refined consensus model of glmS ribozymes. RNA 17:728–736. 10.1261/rna.2590811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawada-Matsuo M, Mazda Y, Oogai Y, Kajiya M, Kawai T, Yamada S, Miyawaki S, Oho T, Komatsuzawa H. 2012. GlmS and NagB regulate amino sugar metabolism in opposing directions and affect Streptococcus mutans virulence. PLoS One 7:e33382. 10.1371/journal.pone.0033382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacobson GR, Poy F, Lengeler JW. 1990. Inhibition of Streptococcus mutans by the antibiotic streptozotocin: mechanisms of uptake and the selection of carbohydrate-negative mutants. Infect. Immun. 58:543–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beighton D, Russell RR, Whiley RA. 1991. A simple biochemical scheme for the differentiation of Streptococcus mutans and Streptococcus sobrinus. Caries Res. 25:174–178. 10.1159/000261363 [DOI] [PubMed] [Google Scholar]
- 47.Kral TA, Daneo-Moore L. 1981. Biochemical differentiation of certain oral streptococci. J. Dent. Res. 60:1713–1718. 10.1177/00220345810600091301 [DOI] [PubMed] [Google Scholar]
- 48.Homer KA, Patel R, Beighton D. 1993. Effects of N-acetylglucosamine on carbohydrate fermentation by Streptococcus mutans NCTC 10449 and Streptococcus sobrinus SL-1. Infect. Immun. 61:295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burne RA, Wen ZT, Chen YY, Penders JE. 1999. Regulation of expression of the fructan hydrolase gene of Streptococcus mutans GS-5 by induction and carbon catabolite repression. J. Bacteriol. 181:2863–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terleckyj B, Willett NP, Shockman GD. 1975. Growth of several cariogenic strains of oral streptococci in a chemically defined medium. Infect. Immun. 11:649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng L, Burne RA. 2009. Transcriptional regulation of the cellobiose operon of Streptococcus mutans. J. Bacteriol. 191:2153–2162. 10.1128/JB.01641-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lau PC, Sung CK, Lee JH, Morrison DA, Cvitkovitch DG. 2002. PCR ligation mutagenesis in transformable streptococci: application and efficiency. J. Microbiol. Methods 49:193–205. 10.1016/S0167-7012(01)00369-4 [DOI] [PubMed] [Google Scholar]
- 53.Moye ZD, Zeng L, Burne RA. 2014. Modification of gene expression and virulence traits in Streptococcus mutans in response to carbohydrate availability. Appl. Environ. Microbiol. 80:972–985. 10.1128/AEM.03579-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biswas I, Jha JK, Fromm N. 2008. Shuttle expression plasmids for genetic studies in Streptococcus mutans. Microbiology 154:2275–2282. 10.1099/mic.0.2008/019265-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petersen FC, Scheie AA. 2010. Natural transformation of oral streptococci. Methods Mol. Biol. 666:167–180. 10.1007/978-1-60761-820-1_12 [DOI] [PubMed] [Google Scholar]
- 56.Kornberg HL, Reeves RE. 1972. Inducible phosphoenolpyruvate-dependent hexose phosphotransferase activities in Escherichia coli. Biochem. J. 128:1339–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.LeBlanc DJ, Crow VL, Lee LN, Garon CF. 1979. Influence of the lactose plasmid on the metabolism of galactose by Streptococcus lactis. J. Bacteriol. 137:878–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 59.Abranches J, Nascimento MM, Zeng L, Browngardt CM, Wen ZT, Rivera MF, Burne RA. 2008. CcpA regulates central metabolism and virulence gene expression in Streptococcus mutans. J. Bacteriol. 190:2340–2349. 10.1128/JB.01237-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belli WA, Marquis RE. 1991. Adaptation of Streptococcus mutans and Enterococcus hirae to acid stress in continuous culture. Appl. Environ. Microbiol. 57:1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahn SJ, Lemos JA, Burne RA. 2005. Role of HtrA in growth and competence of Streptococcus mutans UA159. J. Bacteriol. 187:3028–3038. 10.1128/JB.187.9.3028-3038.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakano K, Lapirattanakul J, Nomura R, Nemoto H, Alaluusua S, Grönroos L, Vaara M, Hamada S, Ooshima T, Nakagawa I. 2007. Streptococcus mutans clonal variation revealed by multilocus sequence typing. J. Clin. Microbiol. 45:2616–2625. 10.1128/JCM.02343-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 64.Carvalho SM, Farshchi Andisi V, Gradstedt H, Neef J, Kuipers OP, Neves AR, Bijlsma JJ. 2013. Pyruvate oxidase influences the sugar utilization pattern and capsule production in Streptococcus pneumoniae. PLoS One 8:e68277. 10.1371/journal.pone.0068277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palmer SR, Miller JH, Abranches J, Zeng L, Lefébure T, Richards VP, Lemos JA, Stanhope MJ, Burne RA. 2013. Phenotypic heterogeneity of genomically-diverse isolates of Streptococcus mutans. PLoS One 8:e61358. 10.1371/journal.pone.0061358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tong H, Zeng L, Burne RA. 2011. The EIIABMan phosphotransferase system permease regulates carbohydrate catabolite repression in Streptococcus gordonii. Appl. Environ. Microbiol. 77:1957–1965. 10.1128/AEM.02385-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zeng L, Burne RA. 2010. Seryl-phosphorylated HPr regulates CcpA-independent carbon catabolite repression in conjunction with PTS permeases in Streptococcus mutans. Mol. Microbiol. 75:1145–1158. 10.1111/j.1365-2958.2009.07029.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cvitkovitch DG, Boyd DA, Thevenot T, Hamilton IR. 1995. Glucose transport by a mutant of Streptococcus mutans unable to accumulate sugars via the phosphoenolpyruvate phosphotransferase system. J. Bacteriol. 177:2251–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abranches J, Candella MM, Wen ZT, Baker HV, Burne RA. 2006. Different roles of EIIABMan and EIIGlc in regulation of energy metabolism, biofilm development, and competence in Streptococcus mutans. J. Bacteriol. 188:3748–3756. 10.1128/JB.00169-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Webb AJ, Homer KA, Hosie AH. 2007. A phosphoenolpyruvate-dependent phosphotransferase system is the principal maltose transporter in Streptococcus mutans. J. Bacteriol. 189:3322–3327. 10.1128/JB.01633-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Asanuma N, Yoshii T, Hino T. 2004. Molecular characteristics of phosphoenolpyruvate: mannose phosphotransferase system in Streptococcus bovis. Curr. Microbiol. 49:4–9. 10.1007/s00284-003-4232-0 [DOI] [PubMed] [Google Scholar]
- 72.Lortie LA, Pelletier M, Vadeboncoeur C, Frenette M. 2000. The gene encoding IIABLMan in Streptococcus salivarius is part of a tetracistronic operon encoding a phosphoenolpyruvate: mannose/glucose phosphotransferase system. Microbiology 146(Part 3):677–685 [DOI] [PubMed] [Google Scholar]
- 73.Rigali S, Nothaft H, Noens EE, Schlicht M, Colson S, Müller M, Joris B, Koerten HK, Hopwood DA, Titgemeyer F, van Wezel GP. 2006. The sugar phosphotransferase system of Streptomyces coelicolor is regulated by the GntR-family regulator DasR and links N-acetylglucosamine metabolism to the control of development. Mol. Microbiol. 61:1237–1251. 10.1111/j.1365-2958.2006.05319.x [DOI] [PubMed] [Google Scholar]
- 74.Świątek MA, Tenconi E, Rigali S, van Wezel GP. 2012. Functional analysis of the N-acetylglucosamine metabolic genes of Streptomyces coelicolor and role in control of development and antibiotic production. J. Bacteriol. 194:1136–1144. 10.1128/JB.06370-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Novichkov PS, Kazakov AE, Ravcheev DA, Leyn SA, Kovaleva GY, Sutormin RA, Kazanov MD, Riehl W, Arkin AP, Dubchak I, Rodionov DA. 2013. RegPrecise 3.0—a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics 14:745. 10.1186/1471-2164-14-745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Álvarez-Añorve LI, Calcagno ML, Plumbridge J. 2005. Why does Escherichia coli grow more slowly on glucosamine than on N-acetylglucosamine? Effects of enzyme levels and allosteric activation of GlcN6P deaminase (NagB) on growth rates. J. Bacteriol. 187:2974–2982. 10.1128/JB.187.9.2974-2982.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Plumbridge J. 2009. An alternative route for recycling of N-acetylglucosamine from peptidoglycan involves the N-acetylglucosamine phosphotransferase system in Escherichia coli. J. Bacteriol. 191:5641–5647. 10.1128/JB.00448-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zeng L, Choi SC, Danko CG, Siepel A, Stanhope MJ, Burne RA. 2013. Gene regulation by CcpA and catabolite repression explored by RNA-Seq in Streptococcus mutans. PLoS One 8:e60465. 10.1371/journal.pone.0060465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.El Qaidi S, Allemand F, Oberto J, Plumbridge J. 2009. Repression of galP, the galactose transporter in Escherichia coli, requires the specific regulator of N-acetylglucosamine metabolism. Mol. Microbiol. 71:146–157. 10.1111/j.1365-2958.2008.06515.x [DOI] [PubMed] [Google Scholar]
- 80.Ghosh S, Rao KH, Sengupta M, Bhattacharya SK, Datta A. 2011. Two gene clusters co-ordinate for a functional N-acetylglucosamine catabolic pathway in Vibrio cholerae. Mol. Microbiol. 80:1549–1560. 10.1111/j.1365-2958.2011.07664.x [DOI] [PubMed] [Google Scholar]
- 81.Plumbridge J. 1995. Co-ordinated regulation of amino sugar biosynthesis and degradation: the NagC repressor acts as both an activator and a repressor for the transcription of the glmUS operon and requires two separated NagC binding sites. EMBO J. 14:3958–3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abranches J, Lemos JA, Burne RA. 2006. Osmotic stress responses of Streptococcus mutans UA159. FEMS Microbiol. Lett. 255:240–246. 10.1111/j.1574-6968.2005.00076.x [DOI] [PubMed] [Google Scholar]
- 83.Burne RA, Zeng L, Ahn SJ, Palmer SR, Liu Y, Lefebure T, Stanhope MJ, Nascimento MM. 2012. Progress dissecting the oral microbiome in caries and health. Adv. Dent. Res. 24:77–80. 10.1177/0022034512449462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bender GR, Sutton SV, Marquis RE. 1986. Acid tolerance, proton permeabilities, and membrane ATPases of oral streptococci. Infect. Immun. 53:331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bender GR, Thibodeau EA, Marquis RE. 1985. Reduction of acidurance of streptococcal growth and glycolysis by fluoride and gramicidin. J. Dent. Res. 64:90–95. 10.1177/00220345850640021701 [DOI] [PubMed] [Google Scholar]
- 86.Bowden GH, Hamilton IR. 1987. Environmental pH as a factor in the competition between strains of the oral streptococci Streptococcus mutans, S. sanguis, and “S. mitior” growing in continuous culture. Can. J. Microbiol. 33:824–827 [DOI] [PubMed] [Google Scholar]
- 87.Svensäter G, Larsson UB, Greif EC, Cvitkovitch DG, Hamilton IR. 1997. Acid tolerance response and survival by oral bacteria. Oral Microbiol. Immunol. 12:266–273. 10.1111/j.1399-302X.1997.tb00390.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.