

Abstract
Histatins are salivary cationic peptides that provide the first line of defense against oral candidiasis caused by Candida albicans. This minireview presents a critical evaluation of our knowledge of the candidacidal mechanism of histatin 5 (Hst 5). Hst 5 is the most potent among all histatin family members with regard to its antifungal activity. The mode of action of Hst 5 has been a subject of intense debate. Unlike other classical host innate immune proteins, pore formation or membrane lysis by Hst 5 has largely been disproven, and it is now known that all targets of Hst 5 are intracellular. Hst 5 binds C. albicans cell wall proteins (Ssa1/2) and glycans and is taken up by the cells through fungal polyamine transporters in an energy-dependent manner. Once inside the fungal cells, Hst 5 may affect mitochondrial functions and cause oxidative stress; however, the ultimate cause of cell death is by volume dysregulation and ion imbalance triggered by osmotic stress. Besides these diverse targets, a novel mechanism based on the metal binding abilities of Hst 5 is discussed. Finally, translational approaches for Hst 5, based on peptide design and synergy with other known drugs, are considered a step forward for bench-to-bed application of Hst 5.
INTRODUCTION
Histatins (Hsts) were first described as basic components of the secretions of human salivary glands based on their electrophoretic mobility in cationic gel systems (1). The mean concentration of total Hsts secreted from unstimulated parotid glands was shown to be approximately 53 μg/ml, with Hst 5 being a major component (2) at an estimated range of 15 to 30 μM in whole saliva (3). The family of salivary Hsts comprises a group of 12 structurally related members of which histatin 1 (Hst 1) and Hst 3 are full-length proteins encoded by closely related loci of two distinct genes, HIS1 (encoding Hst 1) and HIS2 (encoding Hst 3) (4). The smaller proteins, Hst 2 (derived from Hst 1) and Hsts 4 to 12 (derived from Hst 3), are generated by proteolytic cleavage of parent Hsts by salivary proteases during secretion (5; reviewed in reference 6).
Early attempts to purify these Hsts from whole saliva, using ion-exchange chromatography or gel filtration, yielded groups of proteins rich in the amino acid histidine; therefore, they were initially referred to as histidine-rich peptides (HRPs) (reviewed in reference 1). Further purification efforts using gel filtration followed by reverse-phase high-performance liquid chromatography finally isolated three major distinct purified components consisting of Hsts 1, 3, and 5 (1). Subsequently, a novel and less labor-intensive purification method that exploited the metal binding ability of Hsts, using zinc precipitation under alkaline conditions, was developed (7).
Hsts emerged as important components of the human innate immune system with the discovery of their candidacidal and to a lesser extent bactericidal effects. This coincided with the AIDS epidemic of the 1980s that was accompanied by emergence of oral thrush caused by Candida species in HIV-infected immunocompromised individuals, leading to a great interest in Hsts as potential therapeutics. Their role in preventing oral candidiasis in this subpopulation was further bolstered in a recent study by Khan et al. (8), showing that reduction in Hst 5 levels in HIV-positive individuals contributes to enhanced prevalence of Candida albicans among these patients. Besides their microbicidal properties, Hsts 1, 3, and 5 participate in the formation of a protective layer (pellicle) on smooth tooth surfaces, where they prevent microbial colonization and stabilize mineral-solute interactions (9, 10). Recently, it has also been suggested that Hsts 1 and 2 may be the primary reason for efficient wound healing in the oral cavity, based on their ability to promote reepithelialization by stimulating epithelial cell migration in a human full-skin wound model (11, 12).
Of the many proposed functions of Hsts, the most significant and well researched one is their strong antifungal activity. Among all Hsts, Hst 5 has the most potent fungicidal activity against pathogenic fungi, including Candida albicans (1, 13) and other medically important Candida species, such as Candida kefyr, Candida krusei, and Candida parapsilosis (MIC50, 10 to 20 μg/ml), as well as Cryptococcus neoformans and Aspergillus fumigatus (MIC50, 5 to 6 μg/ml) (14). Hst 5 potentially inhibits almost 100% of the C. albicans cells from germinating into more-virulent hyphae (13, 15).
Although over a decade of studies have been devoted toward understanding the mechanism of fungicidal activity of Hsts, the crucial toxic event is still incompletely defined. Earlier studies pointed toward membranolytic mechanisms for Hst 5 in causing fungal cell death; however, more-recent studies have shown that multiple intracellular targets are the more likely sites of action. This minireview will primarily focus on the evolution of our understanding of the candidacidal mechanism of Hst 5.
STRUCTURE-BASED CLUES TO THE MEMBRANE LYSIS HYPOTHESIS
Domain-based analyses of the complete Hst 5 protein (Asp-Ser-His-Ala-Lys-Arg-His-His-Gly-Tyr-Lys-Arg-Lys-Phe-His-Glu-Lys-His-His-Ser-His-Arg-Gly-Tyr) found that a minimum of 12 residues at the C terminus (“functional domain”) were required for killing of fungal cells (16). Fungicidal activity was shown to increase proportionally with an increase in length from 12 to 16 residues from the C terminus (16). Specifically, Lys13, Arg12, and Glu16 were identified as important residues by mutational analyses (17, 18). Extensive structural studies of Hst 5 revealed that these peptides lack any secondary structure and are unordered in aqueous solutions (19–21). However, when placed in organic solutions such as trifluoroethylene or in membrane-mimetic solutions like dimethyl sulfoxide (DMSO)-water, they assumed α-helical structures. Furthermore, the C-terminal functional domain that was shown to be important for biological activity also was needed for α-helicity (16), thus suggesting a role for this structural requirement in fungicidal activity. Adoption of α-helicity in membrane-mimetic solutions by Hsts, together with the observations that Hst-mediated killing of C. albicans was accompanied with release of small molecules like K+ (22) and ATP (23), led researchers to conclude that Hsts kill cells by forming pores in the cellular membrane. Thus, early workers identified the plasma membrane as the primary cellular target.
CELL MEMBRANE: THE MISIDENTIFIED TARGET OF Hst 5
Comparison of Hst 5 and a peptide comprised of its functional domain (dh5), with substitution analogs (dhvar1/4) designed to increase amphipathic character and membrane insertion, revealed significant differences among these peptides (24, 25). Unlike Hst 5, analogs with highly stabilized α-helical structures had higher fungicidal activity, dissipated cytoplasmic transmembrane potential, and were hemolytic. In contrast, hydrophobicity analyses of dh5 and Hst 5 showed similarities with primary sequences predominantly found in globular regions of a protein rather than membrane seeking regions, leading to the conclusion that membrane insertion would be unlikely (24, 25). Subsequently, other workers replaced selected Hst 5 residues with proline to prevent α-helix formation in order to reduce its ability to insert into fungal membranes and found that these Hst derivatives were equally effective in their fungicidal activity (26). Collectively, these studies concluded that membrane insertion leading to lysis is not the mechanism involved in Hst 5-mediated killing.
Further abundant evidence has emerged supporting the concept that membrane lysis is an effect, rather than the cause, of Hst 5-mediated killing. Lack of immediate membrane lytic activity by Hst 5 was demonstrated in elegant studies measuring the release and dequenching of the fluorescent dye calcein from dye-loaded C. albicans cells (6). Cells treated for 60 min with Hst 5 (50 and 200 μM) exhibited 86% loss of viability, while only 10% of intracellular calcein was released, clearly refuting membrane lysis as the cause of cell death (6). Also, it was shown that in spite of the cellular loss of ATP upon Hst 5 exposure, cells continued to maintain their membrane potential and remained alive for up to 80 min after ATP efflux (23). ATP release and killing of C. albicans cells from endogenously expressed chromosomally encoded Hst 5 provided further strong evidence that Hst 5 targets are intracellular (27).
Another report still emphasized on single pore formation in the membrane as an important step in uptake of Hst 5 at a concentration of 50 μM (28). Interestingly, energy-depleted cells had longer cell wall retention times, and a small number of such cells had simultaneous uptake of Hst 5 and propidium iodine (PI) (indicative of membrane permeabilization or pore formation) (29). This led to two important observations: Hst 5 uptake is energy dependent, and more importantly, Hst 5 may indeed have direct membrane effects, albeit when applied to cells at sufficiently higher concentrations (greater than the maximal physiological concentration of 30 μM) over a longer time span. Thus, the membrane lytic effects of Hst 5 reported previously may be due to use of higher concentrations of this peptide, although no evidence of a “single” pore as reported in that study (28) has been corroborated. Lack of permanent pores has also been demonstrated previously by immunogold labeling and freeze fracturing of the plasma membrane of Hst 5-treated C. albicans cells (30, 31).
Another model for uptake that involves the plasma membrane has also been described. This model is based on exploitation of the transmembrane potential of the plasma membrane for direct transfer of an appropriately charged Hst 5 into the cytosol (32). The basis for this conclusion is the fact that disruption of mitochondrial function that can affect transmembrane potential offers protection against Hst 5 uptake and subsequent toxicity (32). However, the mitochondrion itself is the probable target for Hst 5-mediated toxicity, and this is discussed in the subsequent sections.
BINDING AND UPTAKE OF Hst 5
Besides the above-mentioned uptake mechanisms based on direct interactions with the plasma membrane, a transporter-mediated model and a model involving the endocytic pathway have also been proposed; these models offer explanation for passage of Hst 5 through the complex structure of the C. albicans cell wall. A basal layer (comprised of β1-3 and β1-6 glucans and chitin) covered by a network of glycosylated glycosylphosphatidylinositol (GPI)-anchored proteins comprises this complex wall structure (reviewed in reference 33). Transporter proteins and energy expenditure in the form of ATP utilization are needed to mediate passage of Hst 5 through this thick cell wall. Hst 5 binds both cell wall β-glucans (29) as well as fungal Ssa2 and Ssa1 proteins in the cell wall (6, 34–37) in order to stabilize and perhaps localize itself with a transport complex. Ultimately, it is transported into the cell through the fungal polyamine transporters Dur3 and Dur31 in an energy-dependent process (38). Charge similarity between Hst 5 and polyamines is likely the reason why both of these cationic molecules can share a transporter. It has recently been shown that cellular uptake is enhanced by conjugation of the active fragment of Hst 5 (shown outlined by a broken line in Fig. 1) with the polyamine spermidine at either the N or C terminus (39). These conjugates were highly effective as topical therapeutics in a murine model of oral candidiasis compared to the original peptide. In fact, lack of transporter-mediated intracellular transport is the basis for Hst 5 resistance in Candida glabrata, and expression of C. albicans Dur3 and Dur31 in C. glabrata resulted in both higher killing and uptake of Hst 5 (40), underscoring the key role of these transporters in Hst 5 translocation. Hst 5 uptake and killing are lower in C. albicans ssa2Δ/Δ cells (35), showing that binding to the cell wall is an important step in allowing for localized retention before energy-dependent uptake. However, surface binding, unlike uptake, is energy independent, since energy-depleted cells (cells treated with sodium azide, which reduces ATP synthesis) still had abundant Hst 5 binding but not intracellular uptake (29).
FIG 1.
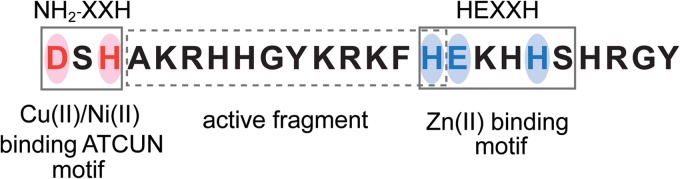
Peptide sequence of Hst 5 with its functional domains. Hst 5 has a Cu/Ni binding ATCUN motif at the C terminus, while a Zn binding motif is present toward the N terminus. The active fragment of Hst 5 is shown outlined by a broken line.
It is important to point out here that energy-dependent cytosolic translocation is not the only mechanism that allows Hst 5 entry into the cells. Vacuolar localization of Hst 5 occurred in Hst 5-treated C. albicans cells and this localization was reduced in endocytic mutants (28, 29), indicating a role for the endocytic pathway in uptake. However, endocytic uptake is insignificant in terms of Hst 5 toxicity, since cells defective in the endocytic pathway or cells lacking vacuoles had no differences in Hst 5 susceptibilities compared to the wild-type (WT) cells (28, 29). In contrast, C. albicans cells lacking genes encoding Rvs161 and Rvs167 Bin-Amphiphysin-Rvs homology domain proteins (that were shown to play a significant role in endocytosis) exhibited resistance toward Hst 5, leading the authors to conclude that endocytosis may play some role in the candidacidal mechanism of Hst 5 (41). Alternatively, this resistance may be an indirect result of changes in lipid composition or the physical properties of the plasma membrane in these mutants. Thus, uptake of Hst 5 may involve all three uptake pathways (transporter-mediated uptake, direct transfer across the membrane, and receptor-mediated endocytosis). The actual uptake potentially is a combination of one or more these processes, the choice of which is seemingly dependent on Hst 5 concentration (28, 29).
CELL DEATH BY IONIC IMBALANCE AND VOLUME DYSREGULATION
Release of small molecules like potassium ions (42) and ATP (23) were among the first observations made by researchers trying to understand the mechanism of killing by Hst 5. K+ release upon Hst 5 treatment offers one of the primary mechanisms by which Hst 5 interferes with cellular metabolism and ultimately leads to cell death. Cells lose volume, concomitantly with loss in viability, when treated with Hst 5 (22). Pharmacological inhibitors of anion transporters significantly prevented volume reduction and killing, suggesting that volume loss is a result of ion efflux (22). Subsequently, Trk1 was identified as an ion transporter responsible for potassium loss, since cells lacking the protein were resistant to Hst 5 (43). Another observation, in line with such volume dysregulation, is that vacuolar expansion was also observed along with cytosolic translocation, but in either case, PI uptake (indicative of cell lysis) always followed but never preceded vacuolar expansion (29). Interestingly, extracellular NaCl (100 mM), but not sorbitol, prevented vacuolar expansion and PI entry in cells that already contained cytosolic Hst 5 (29), thus showing a critical role for ionic balance in Hst 5 toxicity.
ATP AND MITOCHONDRION-MEDIATED MECHANISM
Evidence also exists for the role of released cellular ATP in Hst 5-mediated killing. Treatment with Hst 5 in the presence of an ATP scavenger, apyrase, led to reduction in killing, suggesting a direct involvement for released ATP in cell death (44). Interestingly, anaerobically grown cells were less sensitive to Hst 5, showing that it is not entirely reduction in intracellular ATP content that is responsible for killing; rather, ATP efflux may present a signal for cytotoxicity (45). It is known that released ATP signals cellular toxicity in higher eukaryotes by binding to membrane nucleotide P2X receptors (reviewed in reference 44). The presence of such purinergic receptors in the C. albicans cell wall has been suggested by cross-reactivity with rat anti-P2X receptor antibody, so that released ATP itself may mediate Hst 5-induced killing (44).
Because of the involvement of ATP in the Hst 5-mediated killing, much attention was paid to the ATP-generating organelle, the mitochondrion, as a potential target. Treatment of cells with classical mitochondrial inhibitors, prior to Hst 5 exposure, inhibited cell killing (32, 45, 46). However, similar results were obtained with generalized metabolic inhibitors and with high-salt conditions that disrupt ionic interactions, such as those potentially occurring between Hst 5 and the cell wall (32, 37). Thus, mitochondrial involvement is likely to be primarily due to its role in maintenance of membrane potential that is important for Hst 5 internalization. Fluorescein isothiocyanate (FITC)-labeled Hst 5 was taken up by fungal cells only in metabolically active states, whereas metabolic inhibition of any sort, whether induced by perturbing agents or due to slowing down of active respiration, resulted in loss of peptide uptake and killing (45, 46). However, inhibition of mitochondrial transmembrane potential (47) along with localization of Hst 5 in the mitochondria (32) made it tempting to conclude that Hst 5-mediated mitochondrial dysfunction is an important mechanism in killing. Furthermore, Hst 5 inhibited respiration in isolated mitochondria (47) as well as in intact yeast cells, and this inhibition correlated with cell killing (32). Interestingly, energy depletion itself induces changes in the physical state of the lipid bilayer, and this was offered as an alternative explanation for Hst 5 resistance of nonrespiring cells, independent of the involvement of mitochondria (48).
Interference with mitochondrial respiratory machinery can lead to generation of reactive oxygen species (ROS), and their role in Hst 5-mediated killing was also studied. Use of the oxygen radical-sensitive probe dihydroethidium (HEt) indeed revealed ROS generation in Hst 5-treated C. albicans cells (49). In the presence of the ROS scavenger l-cysteine, both ROS production and cell killing were prevented (49), establishing ROS generation as a mechanism for Hst 5-mediated killing. It is, however, imperative to point out that no direct interaction between Hst 5 and the respiratory chain enzymes was shown. Nonetheless, changes in the mitochondrial proteome have been shown in Hst 5-treated C. albicans cells (50), including reduction of ubiquinol cytochrome c (cyt-c) reductase core protein 2, ATP synthase gamma chain, and malate dehydrogenase proteins, indicative of mitochondrial dysfunction.
Hst 5-induced ROS generation as a cause of cell death has since been challenged. First, none of the classical markers of ROS-mediated oxidative damage, including protein carbonylation and chromosome laddering, were observed as a result of Hst 5 treatment of C. albicans (51). Second, Veerman et al. showed that TEMPO (2,2,66,6-tetramethylpiperidine-N-oxyl), a cell-permeant ROS scavenger, did not inhibit cell killing nor reduce the fluorescence of HEt (52). Furthermore, these authors showed that HEt-induced fluorescence upon Hst 5 treatment was in fact caused by redistribution of preexisting ethidium over the whole cell and not a result of ROS generation in the mitochondria. However, Hst 5-induced ROS generation can potentially be both transient and localized (the possibility of which is proposed and discussed in the following sections) and therefore more difficult to detect.
INVOLVEMENT OF MAPK SIGNALING
Mitogen-activated protein kinase (MAPK) pathways are signaling pathways that respond to environmental signals, and antifungal-mediated insult to the cell is no exception (reviewed in reference 53). Many antifungal drugs are known to activate these pathways in C. albicans. Hst 5-mediated oxidative stress as well as osmotic stress (as caused by ion loss) can therefore explain the induction of stress MAPK Hog1 in Hst 5-treated C. albicans cells. Hst 5 treatment of C. albicans caused Hog1 phosphorylation, and hog1Δ/Δ mutants were found to be more susceptible to Hst 5 (54). Hog1-induced glycerol production was enhanced in cells exposed to Hst 5 (54). All this corroborated with the proposed induction of osmotic stress as an important mechanistic mediator in Hst 5 candidacidal activity. As mentioned above, the Hog1 pathway is also activated in response to oxidative stress, and therefore, Hst 5-induced oxidative stress cannot be completely ruled out.
Recently, we found that the cell wall integrity MAPK Cek1 also plays a role in the fungicidal mechanism of Hst 5 (55). We observed that cells are more susceptible to Hst 5 under environmental conditions that activate Cek1 phosphorylation, such as temperature shift to 37°C and use of N-acetylglucosamine (GlcNAc) as the sole carbon source. This is not surprising, since Hst 5 binds to Candida cell wall components, including β-glucans, whose levels are maintained by the Cek1 pathway (56). Interestingly, Ssa2, the cell wall binding partner of Hst 5, is a heat shock protein whose expression increases with a temperature shift from 30°C to 37°C (35), which incidentally is also a cue for Cek1 induction. Furthermore, Hog1 represses activation of Cek1 MAPK (57); this potentially allows for the integration of various environmental signals with the outcome of Hst 5 treatment of C. albicans. This interplay of MAPK activity in C. albicans in relationship to Hst 5 activity is depicted in Fig. 2.
FIG 2.

Interplay of Hst 5 treatment with MAPK pathways of C. albicans. Activation of Cek1 MAPK by environmental conditions such as GlcNAc as a carbon source or growth temperature of 37°C may potentially enhance Hst 5 susceptibility by Cek1-mediated cell wall modifications (left). On the other hand, Hst 5 treatment itself causes oxidative stress, leading to the activation of Hog1 stress MAPK in C. albicans (right). Inhibition of Cek1 pathway by Hog1 activation offers another layer of complexity to this interplay between Hst 5 treatment and cellular MAPK pathways. Proteins that are phosphorylated are indicated by a blue P.
However, C. albicans response to environmental conditions may also reduce Hst 5 activity. Two such examples involve the Cek1 MAPK pathway: (i) C. albicans secreted aspartyl proteases (Saps) with a role in activation of the pathway (58) potentially degrade Hst 5 (59) and (ii) the interaction of the shed domain of Cek1 head sensor Msb2 with Hst 5 potentially offers protection against Hst 5 (60), although it still remains elusive whether the exact concentrations of shed Msb2 in vivo can functionally inhibit Hst 5 activity in the oral cavity. The roles of shed Msb2 and Saps in Hst 5 activity are discussed in detail in the accompanying minireview (by Marc Swidergall and Joachim F. Ernst [61]).
Hst 5 METAL BINDING ABILITIES AS A POTENTIAL CANDIDACIDAL MECHANISM
Perplexingly, purified Hst 5 exhibits only 10 to 15% of its in vitro fungicidal activity when added into whole saliva (62, 63). Two major processes are likely to account for these differences: (i) the dynamic turnover of salivary proteins balancing secretion with proteolytic degradation (64, 65) and (ii) the phenomenon first described as “masking” (62) that is likely due to Hsts binding with salts and possibly with various metals, including Ca (66), Cu, and Zn in saliva. A similar phenomenon was also observed for the candidacidal activity of another antimicrobial salivary protein, lactoferrin (67). Studies have established that various transitional metals, such as Zn, Ni, Cu, and Fe, are intrinsically present in the saliva (68–70). However, the metal content of healthy or control subjects showed significant variation between individuals and experimental conditions. Because of the potential peptide-metal interactions in saliva, the metal binding abilities of Hst 5 have been extensively studied. Hst 5 binds Zn and Cu (71) and possesses definitive metal binding motifs (Fig. 1) for Cu and Ni (ACTUN motif, encompassing the first 3 N-terminal amino acids, Asp-Ser-His) as well as for Zn (HEXXH motif at the C terminus [72, 73]). Multiple strong binding sites within the binding motif are believed to occur for Cu, while Zn has only one strong putative metal binding site (74).
Although binding to Cu, Zn, or Ni did not stabilize the α-helical structure of Hst 5 (74), important consequences of metal binding have been observed. Zinc binding was shown to allow Hst 5 to catalyze fusion of lipid vesicles (73), and some increase in the bactericidal activity for Hst 5 has been reported for Zn-Hst 5 (75). However, effects of Cu binding provide unique mechanistic insights into novel potential candidacidal mechanisms for Hst 5. A high level of hydrogen peroxide production was observed in solutions with Hst 5 and Cu, in the presence of a reductant such as ascorbate (76–78). This raises an interesting possibility for ROS generation by a Cu-Hst 5 complex. Yeast mitochondria have been shown to contain detectable levels of nonprotein copper or “free” copper (79), and this, together with the fact that Hst 5 is shown to localize to the mitochondria, again raises ROS production as a possible mechanism involved in killing. The lack of global effects of ROS production in Hst 5-treated cells could be explained if production of ROS by Hst 5 is a transient effect that is localized only to the mitochondria. Interestingly, C. albicans mitochondrial superoxide dismutase (Sod5) is a Cu/Zn-containing protein that is upregulated during the yeast-to-hypha transition and induced during oxidative stress, while Hst 5 inhibits germination and Hst 5-mediated chelation of Cu and Zn could potentially interfere with the ability of Sod5 function.
Surprisingly, Hst 5 interactions with Fe, an important component of human diet and saliva, have not been investigated previously. Iron is known to effectively coordinate with histidine residues that are abundantly found in Hst 5. Our recent data show that iron does indeed bind to Hst 5 as evidenced by a change in Hst 5 structure when measured by circular dichroism (CD) upon titration of Hst 5 with iron (63). Although the most immediate effect of iron binding was a decrease in the in vitro candidacidal activity of Hst 5, we found altered expression levels in large numbers of C. albicans iron uptake genes in response to Hst 5 treatment (63). This suggests that iron chelation, and metal chelation in general (as depicted in Fig. 3), may be one of the primary and yet unexplored candidacidal mechanisms of Hst 5. Iron is an essential nutrient for all living organisms, since it serves as a cofactor in proteins involved in electron transfer and is a component of iron-sulfur cluster proteins or other iron-containing proteins that function as enzymes in major metabolic processes in the cell. Chelation of iron and other important metals that serve as micronutrients can create nutrient limiting conditions, negatively affecting pathogen growth. On the other hand, intracellular metal chelation by Hst 5 can potentially disrupt important fungal cellular functions. For example, mitochondria are the most susceptible to changes in cellular iron levels, owing to various iron-containing proteins in their organellar protein machinery. This suggests that the previously proposed mitochondrial dysfunction may instead be a response to Hst 5-induced disruption of cellular iron homeostasis that can be an important potential mechanism for Hst 5-mediated killing of C. albicans. Lastly, like Cu, Hst 5-Fe interactions can very well present a source for generation of ROS that could oxidize important proteins in the mitochondria, leading to cell death.
FIG 3.

Hst 5-mediated metal chelation as a potential candidacidal mechanism. Both extracellular and intracellular metal chelation can potentially contribute toward cell death, either by scavenging away important transition metals needed for growth (left), or by dysregulation of metal homeostasis that affects cell organellar functions such as mitochondrial functions (right).
SUMMARY
Much progress has been made in our understanding of the mechanism of Hst 5. It is clear that Hst 5 affects multiple intracellular targets. However, the ultimate cause of cell death seems to be counterintuitively simple, the loss of potassium ions leading to osmotic imbalance. Nevertheless, there is a possibility that additional mechanisms are either simultaneously in play within the cell or that certain environmental conditions may favor one mechanism over the other. For example, metal binding abilities of Hst 5 would assume greater significance under nutrient-limited conditions, since metal binding by Hst 5 could potentially further limit the essential metals needed for growth. However, metal binding would not be of major significance if nutrients were abundant in the environment. Similarly, environmental conditions leading to Cek1 MAPK activation in C. albicans would make cells more susceptible to Hst 5, while MAPK Hog1 activation would potentially mitigate Hst 5-mediated oxidative and osmotic stress. Thus, Hst 5 may have evolved to utilize multiple environmentally induced physiological states of C. albicans to maximize its killing potential.
The past 2 decades have highlighted the candidacidal potential of Hst 5. With all the considerable knowledge gained about the mechanism of action of this peptide, we are at an interesting juncture with regard to bench-to-bed translational breakthrough. This breakthrough will most likely involve either an intelligent design of novel Hst 5 derivatives for enhanced killing potential (as observed with the spermidine conjugates) or use of Hst 5 in conjunction with other drugs or compounds that would potentiate Hst 5 activity. Examples of this are coadministration of Hst 5 with drugs like caspofungin that activate Cek1 MAPK to make C. albicans cells more susceptible, or coadministration with iron chelator drugs that can potentially “protect” Hst 5 activity from iron binding-mediated loss in the killing potential of Hst 5. However, effective routes of administration and treatment efficacies will need to be determined in appropriate experimental models for various forms of candidiasis, including topical use against mucosal candidiasis (oral and dermal) and systemic use for more widespread disease.
ACKNOWLEDGMENT
We thank John Nyquist for the illustrations in this article.
Footnotes
Published ahead of print 20 June 2014
REFERENCES
- 1.Oppenheim FG, Xu T, McMillian FM, Levitz SM, Diamond RD, Offner GD, Troxler RF. 1988. Histatins, a novel family of histidine-rich proteins in human parotid secretion. Isolation, characterization, primary structure, and fungistatic effects on Candida albicans. J. Biol. Chem. 263:7472–7477 [PubMed] [Google Scholar]
- 2.Johnson DA, Yeh CK, Dodds MW. 2000. Effect of donor age on the concentrations of histatins in human parotid and submandibular/sublingual saliva. Arch. Oral Biol. 45:731–740. 10.1016/S0003-9969(00)00047-9 [DOI] [PubMed] [Google Scholar]
- 3.Campese M, Sun X, Bosch JA, Oppenheim FG, Helmerhorst EJ. 2009. Concentration and fate of histatins and acidic proline-rich proteins in the oral environment. Arch. Oral Biol. 54:345–353. 10.1016/j.archoralbio.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabatini LM, Azen EA. 1989. Histatins, a family of salivary histidine-rich proteins, are encoded by at least two loci (HIS1 and HIS2). Biochem. Biophys. Res. Commun. 160:495–502. 10.1016/0006-291X(89)92460-1 [DOI] [PubMed] [Google Scholar]
- 5.Castagnola M, Inzitari R, Rossetti DV, Olmi C, Cabras T, Piras V, Nicolussi P, Sanna MT, Pellegrini M, Giardina B, Messana I. 2004. A cascade of 24 histatins (histatin 3 fragments) in human saliva. Suggestions for a pre-secretory sequential cleavage pathway. J. Biol. Chem. 279:41436–41443 [DOI] [PubMed] [Google Scholar]
- 6.Edgerton M, Koshlukova SE, Lo TE, Chrzan BG, Straubinger RM, Raj PA. 1998. Candidacidal activity of salivary histatins. Identification of a histatin 5-binding protein on Candida albicans. J. Biol. Chem. 273:20438–20447 [DOI] [PubMed] [Google Scholar]
- 7.Flora B, Gusman H, Helmerhorst EJ, Troxler RF, Oppenheim FG. 2001. A new method for the isolation of histatins 1, 3, and 5 from parotid secretion using zinc precipitation. Protein Expr. Purif. 23:198–206. 10.1006/prep.2001.1493 [DOI] [PubMed] [Google Scholar]
- 8.Khan SA, Fidel PL, Jr, Thunayyan AA, Varlotta S, Meiller TF, Jabra-Rizk MA. 2013. Impaired histatin-5 levels and salivary antimicrobial activity against in HIV infected individuals. J. AIDS Clin. Res. 4:pii=1000193. 10.4172/2155-6113.1000193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen JL, Lamkin MS, Oppenheim FG. 1992. Adsorption of human salivary proteins to hydroxyapatite: a comparison between whole saliva and glandular salivary secretions. J. Dent. Res. 71:1569–1576. 10.1177/00220345920710090501 [DOI] [PubMed] [Google Scholar]
- 10.Siqueira WL, Margolis HC, Helmerhorst EJ, Mendes FM, Oppenheim FG. 2010. Evidence of intact histatins in the in vivo acquired enamel pellicle. J. Dent. Res. 89:626–630. 10.1177/0022034510363384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oudhoff MJ, Blaauboer ME, Nazmi K, Scheres N, Bolscher JG, Veerman EC. 2010. The role of salivary histatin and the human cathelicidin LL-37 in wound healing and innate immunity. Biol. Chem. 391:541–548 [DOI] [PubMed] [Google Scholar]
- 12.Oudhoff MJ, Bolscher JG, Nazmi K, Kalay H, van't Hof W, Amerongen AV, Veerman EC. 2008. Histatins are the major wound-closure stimulating factors in human saliva as identified in a cell culture assay. FASEB J. 22:3805–3812. 10.1096/fj.08-112003 [DOI] [PubMed] [Google Scholar]
- 13.Xu T, Levitz SM, Diamond RD, Oppenheim FG. 1991. Anticandidal activity of major human salivary histatins. Infect. Immun. 59:2549–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helmerhorst EJ, Reijnders IM, van't Hof W, Simoons-Smit I, Veerman EC, Amerongen AV. 1999. Amphotericin B- and fluconazole-resistant Candida spp., Aspergillus fumigatus, and other newly emerging pathogenic fungi are susceptible to basic antifungal peptides. Antimicrob. Agents Chemother. 43:702–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin AL, Shi Q, Johnson DA, Patterson TF, Rinaldi MG, Yeh CK. 1999. Further characterization of human salivary anticandidal activities in a human immunodeficiency virus-positive cohort by use of microassays. Clin. Diagn. Lab. Immun. 6:851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raj PA, Edgerton M, Levine MJ. 1990. Salivary histatin 5: dependence of sequence, chain length, and helical conformation for candidacidal activity. J. Biol. Chem. 265:3898–3905 [PubMed] [Google Scholar]
- 17.Driscoll J, Duan C, Zuo Y, Xu T, Troxler R, Oppenheim FG. 1996. Candidacidal activity of human salivary histatin recombinant variants produced by site-directed mutagenesis. Gene 177:29–34. 10.1016/0378-1119(96)00265-X [DOI] [PubMed] [Google Scholar]
- 18.Tsai H, Raj PA, Bobek LA. 1996. Candidacidal activity of recombinant human salivary histatin-5 and variants. Infect. Immun. 64:5000–5007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raj PA, Soni SD, Levine MJ. 1994. Membrane-induced helical conformation of an active candidacidal fragment of salivary histatins. J. Biol. Chem. 269:9610–9619 [PubMed] [Google Scholar]
- 20.Brewer D, Hunter H, Lajoie G. 1998. NMR studies of the antimicrobial salivary peptides histatin 3 and histatin 5 in aqueous and nonaqueous solutions. Biochem. Cell Biol. 76:247–256. 10.1139/o98-066 [DOI] [PubMed] [Google Scholar]
- 21.Raj PA, Marcus E, Sukumaran DK. 1998. Structure of human salivary histatin 5 in aqueous and nonaqueous solutions. Biopolymers 45:51–67. 10.1002/(SICI)1097-0282(199801)45:1<51::AID-BIP5>3.0.CO;2-Y [DOI] [PubMed] [Google Scholar]
- 22.Baev D, Li XS, Dong J, Keng P, Edgerton M. 2002. Human salivary histatin 5 causes disordered volume regulation and cell cycle arrest in Candida albicans. Infect. Immun. 70:4777–4784. 10.1128/IAI.70.9.4777-4784.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koshlukova SE, Lloyd TL, Araujo MW, Edgerton M. 1999. Salivary histatin 5 induces non-lytic release of ATP from Candida albicans leading to cell death. J. Biol. Chem. 274:18872–18879. 10.1074/jbc.274.27.18872 [DOI] [PubMed] [Google Scholar]
- 24.Helmerhorst EJ, van't Hof W, Breeuwer P, Veerman EC, Abee T, Troxler RF, Amerongen AV, Oppenheim FG. 2001. Characterization of histatin 5 with respect to amphipathicity, hydrophobicity, and effects on cell and mitochondrial membrane integrity excludes a candidacidal mechanism of pore formation. J. Biol. Chem. 276:5643–5649. 10.1074/jbc.M008229200 [DOI] [PubMed] [Google Scholar]
- 25.Helmerhorst EJ, Van't Hof W, Veerman EC, Simoons-Smit I, Nieuw Amerongen AV. 1997. Synthetic histatin analogues with broad-spectrum antimicrobial activity. Biochem. J. 326(Part 1):39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Situ H, Balasubramanian SV, Bobek LA. 2000. Role of alpha-helical conformation of histatin-5 in candidacidal activity examined by proline variants. Biochim. Biophys. Acta 1475:377–382. 10.1016/S0304-4165(00)00096-9 [DOI] [PubMed] [Google Scholar]
- 27.Baev D, Lil XW, Edgerton M. 2001. Genetically engineered human salivary histatin genes are functional in Candida albicans: development of a new system for studying histatin candidacidal activity. Microbiology 147:3323–3334 [DOI] [PubMed] [Google Scholar]
- 28.Mochon AB, Liu H. 2008. The antimicrobial peptide histatin-5 causes a spatially restricted disruption on the Candida albicans surface, allowing rapid entry of the peptide into the cytoplasm. PLoS Pathog. 4:e1000190. 10.1371/journal.ppat.1000190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang WS, Bajwa JS, Sun JN, Edgerton M. 2010. Salivary histatin 5 internalization by translocation, but not endocytosis, is required for fungicidal activity in Candida albicans. Mol. Microbiol. 77:354–370. 10.1111/j.1365-2958.2010.07210.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruissen AL, Groenink J, Van't Hof W, Walgreen-Weterings E, van Marle J, van Veen HA, Voorhout WF, Veerman EC, Nieuw Amerongen AV. 2002. Histatin 5 and derivatives. Their localization and effects on the ultra-structural level. Peptides 23:1391–1399. 10.1016/S0196-9781(02)00076-1 [DOI] [PubMed] [Google Scholar]
- 31.den Hertog AL, van Marle J, van Veen HA, Van't Hof W, Bolscher JG, Veerman EC, Nieuw Amerongen AV. 2005. Candidacidal effects of two antimicrobial peptides: histatin 5 causes small membrane defects, but LL-37 causes massive disruption of the cell membrane. Biochem. J. 388:689–695. 10.1042/BJ20042099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helmerhorst EJ, Breeuwer P, van't Hof W, Walgreen-Weterings E, Oomen LC, Veerman EC, Amerongen AV, Abee T. 1999. The cellular target of histatin 5 on Candida albicans is the energized mitochondrion. J. Biol. Chem. 274:7286–7291. 10.1074/jbc.274.11.7286 [DOI] [PubMed] [Google Scholar]
- 33.Chaffin WL, Lopez-Ribot JL, Casanova M, Gozalbo D, Martinez JP. 1998. Cell wall and secreted proteins of Candida albicans: identification, function, and expression. Microbiol. Mol. Biol. Rev. 62:130–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li XS, Reddy MS, Baev D, Edgerton M. 2003. Candida albicans Ssa1/2p is the cell envelope binding protein for human salivary histatin 5. J. Biol. Chem. 278:28553–28561. 10.1074/jbc.M300680200 [DOI] [PubMed] [Google Scholar]
- 35.Li XS, Sun JN, Okamoto-Shibayama K, Edgerton M. 2006. Candida albicans cell wall ssa proteins bind and facilitate import of salivary histatin 5 required for toxicity. J. Biol. Chem. 281:22453–22463. 10.1074/jbc.M604064200 [DOI] [PubMed] [Google Scholar]
- 36.Sun JN, Li W, Jang WS, Nayyar N, Sutton MD, Edgerton M. 2008. Uptake of the antifungal cationic peptide histatin 5 by Candida albicans Ssa2p requires binding to non-conventional sites within the ATPase domain. Mol. Microbiol. 70:1246–1260. 10.1111/j.1365-2958.2008.06480.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang WS, Li XS, Sun JN, Edgerton M. 2008. The P-113 fragment of histatin 5 requires a specific peptide sequence for intracellular translocation in Candida albicans, which is independent of cell wall binding. Antimicrob. Agents Chemother. 52:497–504. 10.1128/AAC.01199-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar R, Chadha S, Saraswat D, Bajwa JS, Li RA, Conti HR, Edgerton M. 2011. Histatin 5 uptake by Candida albicans utilizes polyamine transporters Dur3 and Dur31 proteins. J. Biol. Chem. 286:43748–43758. 10.1074/jbc.M111.311175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tati S, Li R, Puri S, Kumar R, Davidow P, Edgerton M. 2014. Histatin 5-spermidine conjugates have enhanced fungicidal activity and efficacy as a topical therapeutic for oral candidiasis. Antimicrob. Agents Chemother. 58:756–766. 10.1128/AAC.01851-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tati S, Jang WS, Li R, Kumar R, Puri S, Edgerton M. 2013. Histatin 5 resistance of Candida glabrata can be reversed by insertion of Candida albicans polyamine transporter-encoding genes DUR3 and DUR31. PLoS One 8:e61480. 10.1371/journal.pone.0061480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Douglas LM, Martin SW, Konopka JB. 2009. BAR domain proteins Rvs161 and Rvs167 contribute to Candida albicans endocytosis, morphogenesis, and virulence. Infect. Immun. 77:4150–4160. 10.1128/IAI.00683-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pollock JJ, Denepitiya L, MacKay BJ, Iacono VJ. 1984. Fungistatic and fungicidal activity of human parotid salivary histidine-rich polypeptides on Candida albicans. Infect. Immun. 44:702–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baev D, Rivetta A, Vylkova S, Sun JNN, Zeng GF, Slayman CL, Edgerton M. 2004. The TRK1 potassium transporter is the critical effector for killing of Candida albicans by the cationic protein, histatin 5. J. Biol. Chem. 279:55060–55072. 10.1074/jbc.M411031200 [DOI] [PubMed] [Google Scholar]
- 44.Koshlukova SE, Araujo MW, Baev D, Edgerton M. 2000. Released ATP is an extracellular cytotoxic mediator in salivary histatin 5-induced killing of Candida albicans. Infect. Immun. 68:6848–6856. 10.1128/IAI.68.12.6848-6856.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gyurko C, Lendenmann U, Troxler RF, Oppenheim FG. 2000. Candida albicans mutants deficient in respiration are resistant to the small cationic salivary antimicrobial peptide histatin 5. Antimicrob. Agents Chemother. 44:348–354. 10.1128/AAC.44.2.348-354.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gyurko C, Lendenmann U, Helmerhorst EJ, Troxler RF, Oppenheim FG. 2001. Killing of Candida albicans by histatin 5: cellular uptake and energy requirement. Antonie Van Leeuwenhoek 79:297–309. 10.1023/A:1012070600340 [DOI] [PubMed] [Google Scholar]
- 47.Petruzzelli R, Clementi ME, Marini S, Coletta M, Di Stasio E, Giardina B, Misiti F. 2003. Respiratory inhibition of isolated mammalian mitochondria by salivary antifungal peptide histatin-5. Biochem. Biophys. Res. Commun. 311:1034–1040. 10.1016/j.bbrc.2003.10.104 [DOI] [PubMed] [Google Scholar]
- 48.Veerman EC, Valentijn-Benz M, Nazmi K, Ruissen AL, Walgreen-Weterings E, van Marle J, Doust AB, van't Hof W, Bolscher JG, Amerongen AV. 2007. Energy depletion protects Candida albicans against antimicrobial peptides by rigidifying its cell membrane. J. Biol. Chem. 282:18831–18841. 10.1074/jbc.M610555200 [DOI] [PubMed] [Google Scholar]
- 49.Helmerhorst EJ, Troxler RF, Oppenheim FG. 2001. The human salivary peptide histatin 5 exerts its antifungal activity through the formation of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A. 98:14637–14642. 10.1073/pnas.141366998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Komatsu T, Salih E, Helmerhorst EJ, Offner GD, Oppenheim FG. 2011. Influence of histatin 5 on Candida albicans mitochondrial protein expression assessed by quantitative mass spectrometry. J. Proteome Res. 10:646–655. 10.1021/pr100861k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wunder D, Dong J, Baev D, Edgerton M. 2004. Human salivary histatin 5 fungicidal action does not induce programmed cell death pathways in Candida albicans. Antimicrob. Agents Chemother. 48:110–115. 10.1128/AAC.48.1.110-115.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Veerman EC, Nazmi K, Van't Hof W, Bolscher JG, Den Hertog AL, Nieuw Amerongen AV. 2004. Reactive oxygen species play no role in the candidacidal activity of the salivary antimicrobial peptide histatin 5. Biochem. J. 381:447–452. 10.1042/BJ20040208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hayes BM, Anderson MA, Traven A, van der Weerden NL, Bleackley MR. 14 February 2014. Activation of stress signalling pathways enhances tolerance of fungi to chemical fungicides and antifungal proteins. Cell. Mol. Life Sci. 10.1007/s00018-014-1573-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vylkova S, Jang WS, Li W, Nayyar N, Edgerton M. 2007. Histatin 5 initiates osmotic stress response in Candida albicans via activation of the Hog1 mitogen-activated protein kinase pathway. Eukaryot. Cell 6:1876–1888. 10.1128/EC.00039-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li R, Puri S, Tati S, Edgerton M. 2014. Cek1 phosphorylation facilitates antifungal activity of Hst 5, poster 97A, p 146 Final Prog. Abstr. 12th ASM Conf. Candida candidiasis. American Society for Microbiology, Washington, DC [Google Scholar]
- 56.Galan-Diez M, Arana DM, Serrano-Gomez D, Kremer L, Casasnovas JM, Ortega M, Cuesta-Dominguez A, Corbi AL, Pla J, Fernandez-Ruiz E. 2010. Candida albicans beta-glucan exposure is controlled by the fungal CEK1-mediated mitogen-activated protein kinase pathway that modulates immune responses triggered through dectin-1. Infect. Immun. 78:1426–1436. 10.1128/IAI.00989-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roman E, Nombela C, Pla J. 2005. The Sho1 adaptor protein links oxidative stress to morphogenesis and cell wall biosynthesis in the fungal pathogen Candida albicans. Mol. Cell. Biol. 25:10611–10627. 10.1128/MCB.25.23.10611-10627.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Puri S, Kumar R, Chadha S, Tati S, Conti HR, Hube B, Cullen PJ, Edgerton M. 2012. Secreted aspartic protease cleavage of Candida albicans Msb2 activates Cek1 MAPK signaling affecting biofilm formation and oropharyngeal candidiasis. PLoS One 7:e46020. 10.1371/journal.pone.0046020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meiller TF, Hube B, Schild L, Shirtliff ME, Scheper MA, Winkler R, Ton A, Jabra-Rizk MA. 2009. A novel immune evasion strategy of Candida albicans: proteolytic cleavage of a salivary antimicrobial peptide. PLoS One 4:e5039. 10.1371/journal.pone.0005039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szafranski-Schneider E, Swidergall M, Cottier F, Tielker D, Roman E, Pla J, Ernst JF. 2012. Msb2 shedding protects Candida albicans against antimicrobial peptides. PLoS Pathog. 8:e1002501. 10.1371/journal.ppat.1002501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swidergall M, Ernst JF. 2014. Interplay between Candida albicans and the antimicrobial peptide armory. Eukaryot. Cell 13:950–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Helmerhorst EJ, Flora B, Troxler RF, Oppenheim FG. 2004. Dialysis unmasks the fungicidal properties of glandular salivary secretions. Infect. Immun. 72:2703–2709. 10.1128/IAI.72.5.2703-2709.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Puri S, Li R, Edgerton M. 2014. Iron binding negatively impacts the fungicidal activity of salivary protein histatin 5 against Candida albicans, abstr S10C:6, p 77 Final Prog. 12th ASM Conf. Candida candidiasis. American Society for Microbiology, Washington, DC [Google Scholar]
- 64.Helmerhorst EJ, Alagl AS, Siqueira WL, Oppenheim FG. 2006. Oral fluid proteolytic effects on histatin 5 structure and function. Arch. Oral Biol. 51:1061–1070. 10.1016/j.archoralbio.2006.06.005 [DOI] [PubMed] [Google Scholar]
- 65.Xu L, Lal K, Santarpia RP, III, Pollock JJ. 1993. Salivary proteolysis of histidine-rich polypeptides and the antifungal activity of peptide degradation products. Arch. Oral Biol. 38:277–283. 10.1016/0003-9969(93)90133-7 [DOI] [PubMed] [Google Scholar]
- 66.Dong J, Vylkova S, Li XS, Edgerton M. 2003. Calcium blocks fungicidal activity of human salivary histatin 5 through disruption of binding with Candida albicans. J. Dent. Res. 82:748–752. 10.1177/154405910308200917 [DOI] [PubMed] [Google Scholar]
- 67.Viejo-Diaz M, Andres MT, Fierro JF. 2004. Modulation of in vitro fungicidal activity of human lactoferrin against Candida albicans by extracellular cation concentration and target cell metabolic activity. Antimicrob. Agents Chemother. 48:1242–1248. 10.1128/AAC.48.4.1242-1248.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garhammer P, Hiller KA, Reitinger T, Schmalz G. 2004. Metal content of saliva of patients with and without metal restorations. Clin. Oral Invest. 8:238–242. 10.1007/s00784-004-0281-4 [DOI] [PubMed] [Google Scholar]
- 69.Matos de Souza R, Macedo de Menezes L. 2008. Nickel, chromium and iron levels in the saliva of patients with simulated fixed orthodontic appliances. Angle Orthod. 78:345–350. 10.2319/111806-466.1 [DOI] [PubMed] [Google Scholar]
- 70.Mikulewicz M, Chojnacka K, Wozniak B, Downarowicz P. 2012. Release of metal ions from orthodontic appliances: an in vitro study. Biol. Trace Elem. Res. 146:272–280. 10.1007/s12011-011-9233-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gusman H, Lendenmann U, Grogan J, Troxler RF, Oppenheim FG. 2001. Is salivary histatin 5 a metallopeptide? Biochim. Biophys. Acta 1545:86–95. 10.1016/S0167-4838(00)00265-X [DOI] [PubMed] [Google Scholar]
- 72.Grogan J, McKnight CJ, Troxler RF, Oppenheim FG. 2001. Zinc and copper bind to unique sites of histatin 5. FEBS Lett. 491:76–80. 10.1016/S0014-5793(01)02157-3 [DOI] [PubMed] [Google Scholar]
- 73.Melino S, Rufini S, Sette M, Morero R, Grottesi A, Paci M, Petruzzelli R. 1999. Zn(2+) ions selectively induce antimicrobial salivary peptide histatin-5 to fuse negatively charged vesicles. Identification and characterization of a zinc-binding motif present in the functional domain. Biochemistry 38:9626–9633 [DOI] [PubMed] [Google Scholar]
- 74.Brewer D, Lajoie G. 2000. Evaluation of the metal binding properties of the histidine-rich antimicrobial peptides histatin 3 and 5 by electrospray ionization mass spectrometry. Rapid Commun. Mass. Spectrom. 14:1736–1745. 10.1002/1097-0231(20001015)14:19<1736::AID-RCM86>3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- 75.Rydengard V, Andersson Nordahl E, Schmidtchen A. 2006. Zinc potentiates the antibacterial effects of histidine-rich peptides against Enterococcus faecalis. FEBS J. 273:2399–2406. 10.1111/j.1742-4658.2006.05246.x [DOI] [PubMed] [Google Scholar]
- 76.Cabras T, Patamia M, Melino S, Inzitari R, Messana I, Castagnola M, Petruzzelli R. 2007. Pro-oxidant activity of histatin 5 related Cu(II)-model peptide probed by mass spectrometry. Biochem. Biophys. Res. Commun. 358:277–284. 10.1016/j.bbrc.2007.04.121 [DOI] [PubMed] [Google Scholar]
- 77.Houghton EA, Nicholas KM. 2009. In vitro reactive oxygen species production by histatins and copper(I,II). J. Biol. Inorg. Chem. 14:243–251. 10.1007/s00775-008-0444-x [DOI] [PubMed] [Google Scholar]
- 78.Tay WM, Hanafy AI, Angerhofer A, Ming LJ. 2009. A plausible role of salivary copper in antimicrobial activity of histatin-5–metal binding and oxidative activity of its copper complex. Bioorg. Med. Chem. Lett. 19:6709–6712. 10.1016/j.bmcl.2009.09.119 [DOI] [PubMed] [Google Scholar]
- 79.Cobine PA, Ojeda LD, Rigby KM, Winge DR. 2004. Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix. J. Biol. Chem. 279:14447–14455. 10.1074/jbc.M312693200 [DOI] [PubMed] [Google Scholar]