

Abstract
Scald caused by Rhynchosporium commune is an important foliar disease of barley. Insertion mutagenesis of R. commune generated a nonpathogenic fungal mutant which carries the inserted plasmid in the upstream region of a gene named PFP1. The characteristic feature of the gene product is an Epc-N domain. This motif is also found in homologous proteins shown to be components of histone acetyltransferase (HAT) complexes of fungi and animals. Therefore, PFP1 is suggested to be the subunit of a HAT complex in R. commune with an essential role in the epigenetic control of fungal pathogenicity. Targeted PFP1 disruption also yielded nonpathogenic mutants which showed wild-type-like growth ex planta, except for the occurrence of hyphal swellings. Complementation of the deletion mutants with the wild-type gene reestablished pathogenicity and suppressed the hyphal swellings. However, despite wild-type-level PFP1 expression, the complementation mutants did not reach wild-type-level virulence. This indicates that the function of the protein complex and, thus, fungal virulence are influenced by a position-affected long-range control of PFP1 expression.
INTRODUCTION
Rhynchosporium commune (formerly Rhynchosporium secalis) is a haploid imperfect fungus causing scald, an important foliar disease of barley in all growing areas around the world (1–4). Despite the lack of a sexual stage, the fungus is classified as an Ascomycete on the basis of nucleotide sequence comparisons (5). It is characterized by unusual hemibiotrophic (6), entirely intercellular development, which is almost exclusively restricted to the subcuticular region of host leaves. The disease process begins on the surface of barley leaves with the formation of germ tubes from the two-celled conidiospores. After penetrating the cuticle, frequently without the formation of distinct appressoria, the hyphae spread mainly longitudinally along the leaf. Initially, thin hyphae with distantly spaced septae grow predominantly in the pectin-rich layer of the outer epidermis cell walls (7). Later, thick short-septate hyphae appear, and they eventually form a dense stroma that supports the formation of new spores (8–11). The typical disease symptoms, necrotic lesions, occur after a latent period lasting from a few days up to several weeks, when mesophyll cells in leaf regions that are heavily colonized by the fungus collapse (12, 13). Hence, fungal growth in planta can be divided into 4 phases: germination and penetration, early development of thin hyphae with a very slow gain of fungal biomass, exponential growth of mainly thick hyphae with a massive increase in biomass, and late stationary phase with sporulation (13).
The genetic and biochemical basis of pathogenicity is poorly understood to date. Early reports described the putative involvement of low-molecular-weight toxins (14). Later, a small group of secreted effector proteins (NIP1, NIP2, NIP3) was identified (15). No biochemical activity has been able to be attributed to NIP2 to date, whereas NIP1 and NIP3 have been found to stimulate the plant plasma membrane-localized H+-ATPase (16). The three NIP genes show strong expression during the early fungal growth stage, followed by a decline during the period of rapid fungal growth, and they contribute quantitatively to fungal virulence (13). Furthermore, NIP1 was shown to be a dual-function effector (17): on top of its nonspecific virulence activity, the protein is the avirulence factor corresponding to barley resistance gene Rrs1 (18, 19). Integrating structural information (20) with results from binding studies (21) revealed that both functions of NIP1 appear to be mediated through the same plant membrane-localized NIP1-binding site, which is most likely not encoded by the resistance gene.
Many fungal plant pathogens have been studied using either reverse genetics or bioinformatics tools with genomics, transcriptomics, or proteomics data, followed by functional analysis via gene disruption to learn more about the molecular processes that control pathogen development on the host. Of particular interest are pathogenicity genes, which are classically defined as “necessary for disease development, but not essential for the pathogen to complete its life cycle in vitro” (22). The products of these genes are therefore potential targets for fungicides. Disruption of pathogenicity genes can result in a loss of fungal pathogenicity or a strong reduction in disease development, which is a less dramatic outcome. The functions of pathogenicity genes are very diverse and can be associated with host recognition, formation of infection structures, penetration of the cuticle and host cell wall, suppression of host defense, nutrient uptake, and further colonization of the host (for a review, see references 23 and 24).
Several of the described pathogenicity genes have pleiotropic effects and do not strictly obey the definition. For instance, C-type cyclin genes in Fusarium spp. (25, 26) and in Mycosphaerella graminicola (27) have important roles in fungal development, secondary metabolite production, and sporulation, in addition to their impact on pathogenicity. Furthermore, proteins modulating chromatin structure have been shown to be involved in controlling pathogenicity. The FTL1 gene of Fusarium graminearum (28) and the TIG1 gene of Magnaporthe oryzae (29), both of which encode transducin β-like components of a histone deacetylation (HDAC) complex, are defective in plant infection and conidiogenesis. Moreover, deletion of the HDC1 gene in Cochliobolus carbonum, which is related to Saccharomyces cerevisiae HOS2 HDAC, yielded mutants with reduced penetration efficiency and strongly reduced virulence (30).
Gene disruption by insertion mutagenesis has been very effective in investigating fungal pathogenesis (31). Restriction enzyme-mediated integration (REMI) (32–34) is a method to disrupt genes by nonhomologous integration of transforming plasmid DNA (35). Therefore, REMI mutants of R. commune were generated and screened for the loss of pathogenicity on formerly susceptible plants. In this study, we describe the identification and characterization of the PFP1 gene encoding a subunit of a histone acetyltransferase (HAT) complex.
MATERIALS AND METHODS
Fungal and plant culture conditions.
Culture of fungal isolate UK7 and of susceptible barley cultivar ‘Ingrid' and inoculations was as described previously (9, 11, 36).
REMI mutagenesis.
Fungal protoplasts were isolated as described previously (36) using a mixture of glucanase (5 mg/ml) and driselase (5 mg/ml; Interspex Products, San Mateo, CA) instead of Novozyme 234 for cell wall degradation. Plasmid pAN7-1 (37) was linearized using the restriction enzyme BamHI or HindIII. Fungal protoplasts were transformed with 10 μg of prelinearized or circular plasmid DNA in the presence of BamHI (20 units) or HindIII (50 units) using the polyethylene glycol-CaCl2 technique (36). Individual transformants were transferred twice onto selective agar plates before being tested for pathogenicity on barley primary leaves.
Targeted gene disruption.
Fungal protoplasts were prepared by incubating homogenized mycelia (36) for 6 h at 29°C on a laboratory shaker (60 rpm) in a solution of 125 mg of lysing enzymes from Trichoderma harzianum (Sigma-Aldrich, Steinheim, Germany) in 10 ml of protoplast buffer. After cellular debris was removed, the protoplast suspension was adjusted to 5 × 107/ml and used for transformation. Two gene disruption constructs were generated by PCR-based fusion of a resistance cassette derived from plasmid pAN8-1 (38) comprising the ble gene from Streptoalloteichus hindustanus under the control of the glyceraldehyde-3-phosphate dehydrogenase (GPD) gene promoter and the TrpC terminator from Aspergillus nidulans (39) with the 5′ and 3′ regions of the PFP1 gene (40). The constructs combined 1 kb of sequence upstream of the translation start codon (primers Fusion-primer1s and Fusion-primer2as; see Table S1 and Fig. S1 in the supplemental material) and either 1.1 kb covering most of exon 2 (primers Fusion-primer3s and Fusion-primer4as) or 0.9 kb downstream of the translation stop codon (primers Fusion-primer7s and Fusion-primer8as) with the ble gene (primers Fusion-primer5s and Fusion-primer6as-2). The constructs were amplified when needed using primer Fusion-primer1s in combination with Fusion-primer4as or Fusion-primer8as, excised from an agarose gel after size fractionation, and used at a concentration of 20 μg per transformation assay. DNA from all transformants was tested by PCR for the absence of the PFP1 gene (primer combinations PFP3s/PFP2as and PFP27s/Fusion-primer4as) and for the presence of the ble gene (primers pSS2-ble3s and pSS2-ble4as). Successful deletion was confirmed by a third PCR analysis, in which the 5′ and 3′ integration sites were amplified (primers 5′-Int1s/5′-Int3s and 5′-Int2as and primers 3′-Int1s/3′-Int3s and 3′-Int4as), and by Southern hybridization. Before further characterization, fungal mutants were grown on barley cultivar ‘Ingrid' and reisolated (13).
Complementation of REMI and deletion mutants.
For REMI complementation, a 4,902-bp fragment of the PFP1 sequence was amplified by PCR using the primers LB2057WT16bis and LB2057WT17 and cloned into the pGEM-T Easy vector (Promega). The complementation vector was generated by inserting the PFP1-containing EcoRI fragment into plasmid pAN8-1 (which carries a gene for phleomycin resistance). After transformation, four independent mutants were grown on lima bean agar for inoculation experiments. For complementation of the deletion mutants, a 2,200-bp hygromycin B resistance cassette from plasmid pAN7-1 and the 5,977-bp PFP1 sequence were amplified by PCR using primers GFP-Fus-HPH1-2s, GFP-Fus-HPH2as, PFP48s, and PFP49as. A complementation vector was generated by cloning both amplification products into pCR2.1 (Invitrogen). Vectors lacking the PFP1 sequence were used as controls. After transformation of protoplasts from the mutants, the presence of the transferred PFP1 gene was verified via PCR (primers PFP3s and PFP2as).
Pathogenicity assays.
Fungal REMI transformants were first subjected to two rounds of a rapid cotton swab assay (41). All mutants that failed to cause disease symptoms in these preliminary assays were further grown for spore isolation. Standard spray inoculation (9) was carried out using 7-day-old plants and 104 to 5 × 105 spores/10 plants for the REMI mutants and 105 spores/10 plants for the deletion and complementation mutants. Lesion development was evaluated visually at 2 to 3 weeks postinoculation.
Quantification of fungal development.
Relative quantification of fungal biomass in planta was achieved by quantitative PCR (qPCR). By application of the efficiency calibrated model (42), the GPD gene (primers GPD-RTs and GPD-RTas) served as the fungal target gene and a gene encoding a putative member of the high-temperature-requirement A (HTRA) family of serine proteases (GenBank accession no. AK359241; primers HO05J24F and HO05J24R) served as the barley reference gene (13). To quantify fungal growth in liquid culture, fungal cultures were initiated with 103 or 104 spores in sucrose-supplemented Fries medium no. 3 (36) in 50-ml Falcon tubes and incubated under mild shaking at 17°C in the dark. After 14 and 21 days, the media were removed by filtration and the mycelia were lyophilized and weighed. To estimate fungal growth on solid lima bean agar (11), colonies were initiated with 5,000 conidia on agar-coated microscope slides and grown at 17°C in the dark. The germination frequencies at 24 and 48 h and the number of germ tubes at 48 h were counted under a microscope (AxioVision LE; Carl Zeiss MicroImaging, Jena, Germany) using 3 groups of 100 spores each in 4 biological replicates. In addition, the lengths of 30 to 40 germ tubes after 48 h were measured in each of 4 biological replicates.
Quantification of fungal gene expression.
To quantify fungal gene expression, total RNA was extracted from inoculated barley leaves or from fungal mycelia grown ex planta either manually (mycelia were ground with a mortar and pestle) or using a cryogenic grinder (Labman Automation Ltd., Stokesley, North Yorkshire, United Kingdom). Transcripts of the PFP1 target gene (primers PFP-RTs and PFP-RTas) were quantified relative to those of the constitutively expressed GPD gene (primers GPD-RTs and GPD-RTas) by quantitative reverse transcription-PCR (qRT-PCR) (13, 42). The absence of genomic DNA was routinely tested by amplifying the GPD gene sequence. Primers GPD-cDNA-test-s and GPD-cDNA-test-as yielded a product of 861 bp with cDNA and one of 1,115 bp with genomic DNA as the template (see Fig. S2 in the supplemental material).
DNA techniques.
The regions flanking the plasmid integration site in the DNA from the REMI mutant were sequenced after (i) PCR walking using the blunt end-producing restriction enzymes PvuII and EcoRV (43) and (ii) thermal asymmetric interlaced PCR (TAIL-PCR) (44). To obtain the cDNA sequence, 5′ rapid amplification of cDNA ends (RACE) and 3′ RACE (45, 46) were carried out using a SMART RACE cDNA amplification kit (Clontech Laboratories) according to the manufacturer's instructions. For 5′ RACE, gene-specific primer PFP21as was used. The 5′ cDNA sequence was amplified using nested universal primer A and primer PFP23as and cloned into plasmid pCR2.1 for sequencing. For 3′ RACE, an oligo(dT) primer with a gene-specific extension [oligo(dT)-spec] and primer PFP3s were used for first-strand cDNA synthesis. Nested primer oligo(dT)-out containing the specific extension of oligo(dT)-spec was used along with primer PFP19s to generate the second cDNA strand. The 3′ cDNA sequence was amplified using primer PFP13s and the nested primer oligo(dT)-in and cloned in pCR2.1 for sequencing.
Proteome analysis.
For proteome analysis, digestion of proteins and assessment of spectral data were performed as described in reference 47 using a self-made database. Mass spectrometry data were achieved as described in reference 47 with a 300-min gradient and as described in reference 48 with minor changes of the solvent and gradient.
Statistical analysis.
All quantitative data were subjected to an unpaired t test (confidence interval of 95% using GraphPad Prism software, version 4.0 [49]).
Nucleotide sequence accession number.
The PFP1 gene has been deposited in the GenBank database under accession number KJ410022.
RESULTS
Identification of a nonpathogenic fungal insertion mutant.
R. commune mutants were generated by restriction enzyme-mediated integration (REMI) through transformation of fungal protoplasts with plasmid pAN7-1 (37). Of ∼1,300 REMI mutants subjected to pathogenicity testing, 21 lost the ability to cause disease symptoms on barley. To identify the genes affected by the insertion, the DNA sequences flanking the plasmid integration sites were obtained by PCR walking (43) and by TAIL-PCR (44) of genomic mutant DNA. In this way, a total sequence of 5,977 bp was generated for one of the REMI mutants. The plasmid had integrated into a BamHI site (between nucleotide positions 1255 and 1256) 274 bp upstream of a long putative open reading frame (ORF) (Fig. 1). Comparison of genomic and cDNA sequences revealed a gene containing two exons of 2,612 bp (nucleotide positions 1529 to 4140) and 1,018 bp (nucleotide positions 4186 to 5203) that are interrupted by a 45-bp intron. 3′ RACE confirmed that position 5203, which is followed by three stop codons within the next 24 bp, constitutes the end of the coding sequence. The gene was named putative function in pathogenicity 1 (PFP1). In a preliminary complementation experiment, the REMI mutant was retransformed with the PFP1 gene. Inoculation of a susceptible barley cultivar with mutant conidia produced symptoms considerably weaker than those produced by wild-type strain UK7, suggesting that the mutants had regained pathogenicity but failed to express full virulence (see Fig. S3 in the supplemental material).
FIG 1.
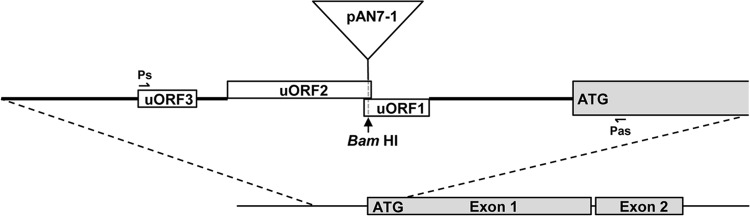
Structure of the PFP1 genomic region (5,977 bp) showing the site of pAN7-1 integration. Exons are marked in gray. uORF, upstream open reading frame; Ps, sense primer PFP-ESTs; Pas, antisense primer PFP23as.
Plasmid integration had occurred as a perfect REMI event, preserving BamHI sites on both sides of the insertion. However, Southern analysis using the hph selective marker gene and a 1,020-bp fragment 5′ of the first exon as probes revealed that two copies of pAN7-1 had integrated in tandem in a head-to-tail pattern (see Fig. S4 in the supplemental material). Closer inspection revealed three short ORFs upstream of the PFP1 coding sequence. RT-PCR using primers located at the 5′ end of the distal ORF (PFP1 EST s) and inside the first exon of PFP1 (PFP23as) demonstrated that the PFP1 mRNA contains all three putative upstream ORFs (uORFs). Interestingly, plasmid integration had occurred at a BamHI site in the 12-bp overlap between the two proximal uORFs (Fig. 1).
RT-PCR using primers located upstream of uORF3 (Fusion-primer1s) and at the 3′ end of exon 2 (Fusion-primer4as) demonstrated a wild-type PFP1 mRNA length of >4,600 nucleotides (see Fig. S2 in the supplemental material). The transcript start site is not known, but it must be located >430 bp upstream of uORF3.
Characterization of PFP1 gene product.
The deduced 1,210-amino-acid protein showed the highest similarity to hypothetical PHD finger motif-containing proteins from different fungi. More detailed analysis of the primary structure revealed the presence of an Epc-N (enhancer of polycomb N terminus) domain at amino acid positions 347 to 787 (Fig. 2). This domain was described as a protein-protein interaction module found in chromatin-associated proteins, notably, as components of histone acetyltransferase (HAT) complexes (50). The Epc-N domain is interrupted by a 5-center Zn-binding motif containing a PHD domain, an additional potentially Zn-binding domain called Zn knuckle, and a PHD-like motif (PZPM; amino acids 457 to 642). On the basis of the absence or presence of this motif, Epc-N proteins are grouped into two families (families 1 and 2), both of which are further divided into two subfamilies (subfamilies I and II and subfamilies III and IV, respectively). The absence of a bromodomain, a protein module specifically recognizing ε-N-acetylation of lysine and characterizing subfamily III proteins, makes PFP1 a member of subfamily IV. A putative AT hook, a structural motif found in many DNA-binding proteins (51), is located between the PFP1 N terminus and the Epc-N domain in amino acid positions 235 to 239. Furthermore, the EMBOSS epestfind algorithm located three putative protein-destabilizing motifs with PEST scores of >+5 (52) near the C terminus in the region from amino acids 1031 to 1173. Finally, a potential nuclear localization signal at the C terminus (amino acids 1197 to 1210) suggests the nuclear localization of PFP1.
FIG 2.
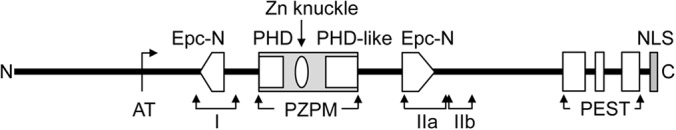
Structure of the PFP1 protein. AT, AT hook (amino acids RPGRP); Epc-N, Epc-N domain interrupted by PZPM domain; PHD (C4HC3); Zn knuckle (C2H2); PHD-like domain (C5HC2H); I, IIa, and IIb, domains I, IIa, and IIb, respectively; PEST, putative destabilizing motifs; NLS, putative nuclear localization signal.
PFP1 expression during pathogenesis.
Expression of PFP1 during the growth of fungal wild-type strain UK7 on susceptible barley cultivar ‘Ingrid' was quantified relative to the constitutive expression of the fungal glyceraldehyde-3-phosphate dehydrogenase (GPD) gene using qRT-PCR (Fig. 3) (42). The latter was selected as the reference gene on the basis of results from a preliminary experiment, in which the expression profiles of the fungal GPD gene and the ubiquitin and actin genes were compared during pathogenesis on barley (not shown). Use of the actin gene was abandoned due to the difficulty with finding primers that reliably discriminate between the fungal gene and the plant gene. Ubiquitin gene expression increased drastically after 10 days postinoculation (dpi). In contrast, GPD transcript abundance rose only slightly after 10 dpi. The highest PFP1 transcript levels were found at 0 dpi (less than 1 h after inoculation), followed by a continuous decrease during the first two stages of fungal development in planta and a stable but very low expression from about 6 dpi on.
FIG 3.
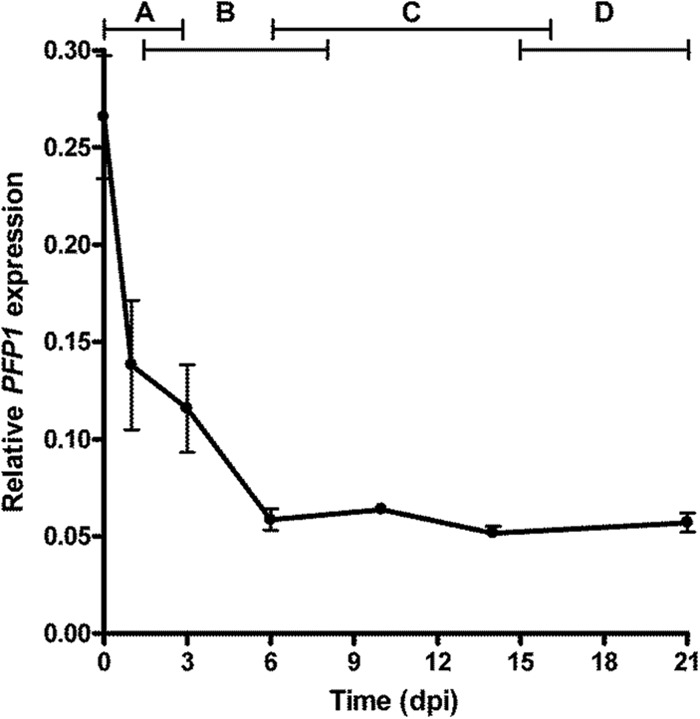
PFP1 expression during development of fungal wild-type strain UK7 on barley cultivar ‘Ingrid'. PFP1 expression relative to the expression of the GPD gene was quantified by qRT-PCR (n = 4). The four phases of fungal development are indicated at the top. A, germination and penetration; B, formation of thin hyphae; C, rapid growth with thick hyphae; D, stationary phase.
Generation and phenotypic characterization of a targeted PFP1 deletion mutant.
The REMI mutation did not impair the integrity of the PFP1 coding sequence. The presence of an mRNA comprising the sequences coding for both HPH and PFP1, as expected by the genomic structure, was shown by RT-PCR using primers in the hph gene (primers HPH1s and HPH200-f) and at the 3′ end of PFP1 exon 2 (primer Fusion-primer4as; see Fig. S4 in the supplemental material). This provoked the question whether plasmid integration into the PFP1 promoter affects PFP1 expression. Therefore, relative PFP1 transcript abundance in fungal mycelia growing ex planta in liquid medium was quantified by qRT-PCR (42) using primers in the sequence of exon 2. Relative PFP1 transcript levels turned out to be very low, although they were 8.6-fold higher in the REMI mutant than in wild-type strain UK7 (Fig. 4). It remains to be shown whether the 5′ uORFs are of any regulatory relevance in this context. However, plasmid integration had occurred such that the hph and PFP1 ORFs are separated by a stretch of 295 bp containing 8 stop codons and several short ORFs. It is therefore unlikely that the PFP1 ORF is translated either as a read-through of the hph gene or by reinitiation of translation. Despite the occurrence of the PFP1 transcript, the REMI mutant may therefore be a null mutant. However, the very low PFP1 mRNA abundance in the wild type and the REMI mutant, possibly aggravated by the short half-life of the protein, which is suggested by the presence of PEST motifs, precluded the detection of the PFP1 protein in extracts from wild-type and mutant mycelia by liquid chromatography/mass spectrometry (LC/MS)-based analysis.
FIG 4.
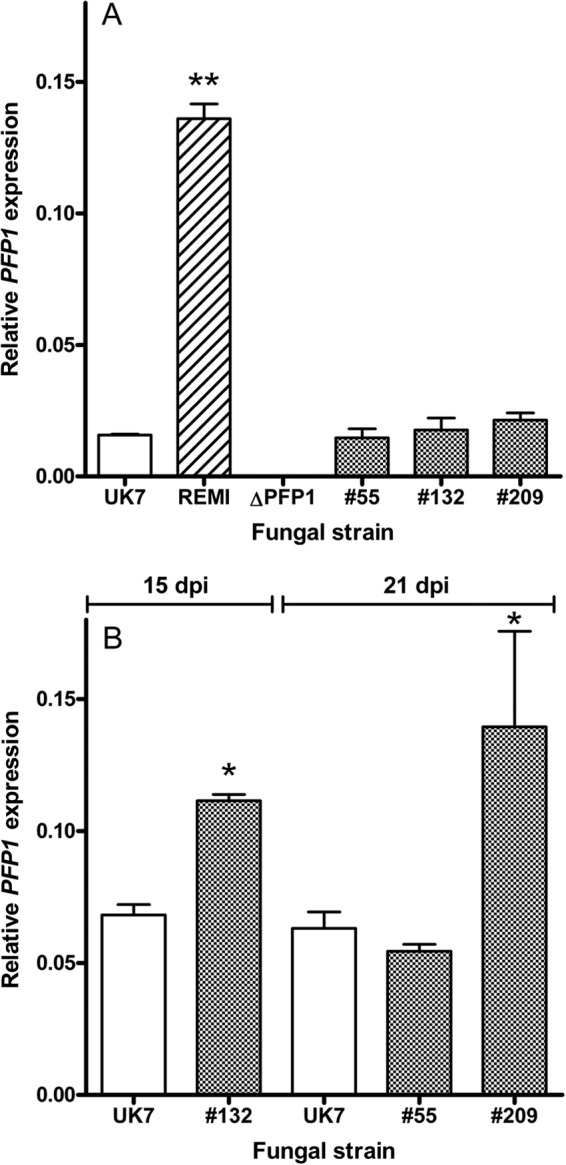
PFP1 expression after 10 days of culture ex planta (n = 4) (A) and at 15 or 21 dpi in planta (n = 4) (B) in wild-type strain UK7, the REMI mutant, the deletion mutant UK7ΔPFP1 (ΔPFP1), and 3 independent UK7ΔPFP1 complementation mutants (mutants 55, 132, and 209). *, P < 0.05 compared with the results for UK7; **, P < 0.01 compared with the results for UK7.
For further functional analysis, deletion mutants were generated by targeted disruption of the PFP1 coding sequence. To this end, two gene constructs that contained the phleomycin resistance cassette between 5′ and 3′ genomic fragments flanking either exon 1 or the entire PFP1 coding sequence were amplified by fusion PCR (40). Transformation of fungal protoplasts from strain UK7 using these constructs yielded two exon 1 deletion mutants, UK7ΔPFP1-ex1.1 and UK7ΔPFP1-ex1.2, and one total deletion mutant, UK7ΔPFP1. The latter mutant, which failed to accumulate the PFP1 transcript (Fig. 4A) and, hence, is a true null mutant, was used in all following experiments. In addition, some experiments were carried out using the partial deletion mutants.
Inoculation of susceptible barley cultivar ‘Ingrid' with spores of deletion mutant UK7ΔPFP1 resulted in a strongly attenuated disease phenotype compared to that of wild-type strain UK7. Necrotic areas remained very small and mostly restricted to the leaf margins (Fig. 5; see also Fig. S5 in the supplemental material). This phenotype was also observed with the exon 1 deletion mutants UK7ΔPFP1-ex1.1 and UK7ΔPFP1-ex1.2 (not shown). In addition, microscopic analysis revealed that the mutant mycelia developed much slower than wild-type mycelia and failed to produce the typical stroma (Fig. 6). As a consequence, at 21 dpi wild-type strain UK7 had generated >20 times more biomass than the deletion mutant, UK7ΔPFP1 (Fig. 7).
FIG 5.

Disease phenotype on barley cultivar ‘Ingrid' of fungal wild-type strain UK7, deletion mutant UK7ΔPFP1 (ΔPFP1), and 3 UK7ΔPFP1 complementation mutants (mutants 55, 132, and 209). C, noninoculated control leaf.
FIG 6.
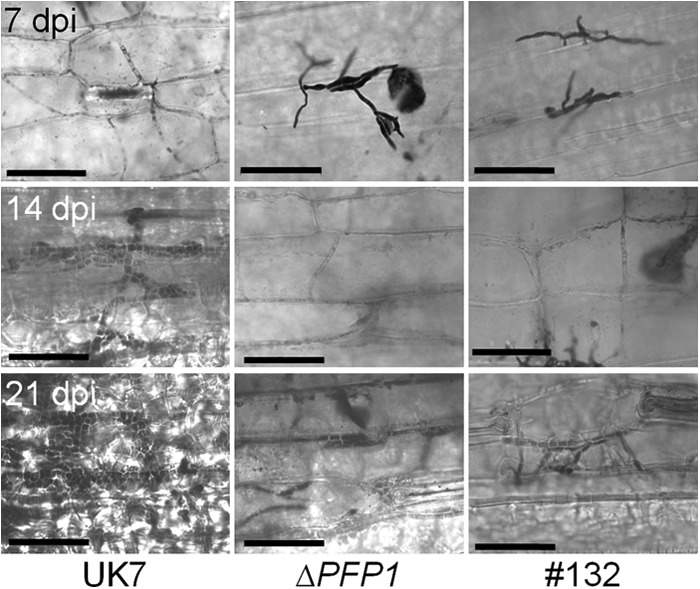
Development of fungal mycelia in planta. At 7, 14, and 21 dpi, infected leaves were harvested and stained with trypan blue and fungal structures were analyzed under a microscope. Wild-type strain UK7 grows normally and establishes a dense subcuticular stroma. In contrast, the deletion mutant ΔPFP1 can be found in only a very few small lesions, where the fungus starts to develop thick, short-septate hyphae only very late upon inoculation. The complementation mutant 132 also grows slower than the wild type but succeeds with the production of many more symptoms than the deletion mutant. Bars = 50 μm.
FIG 7.
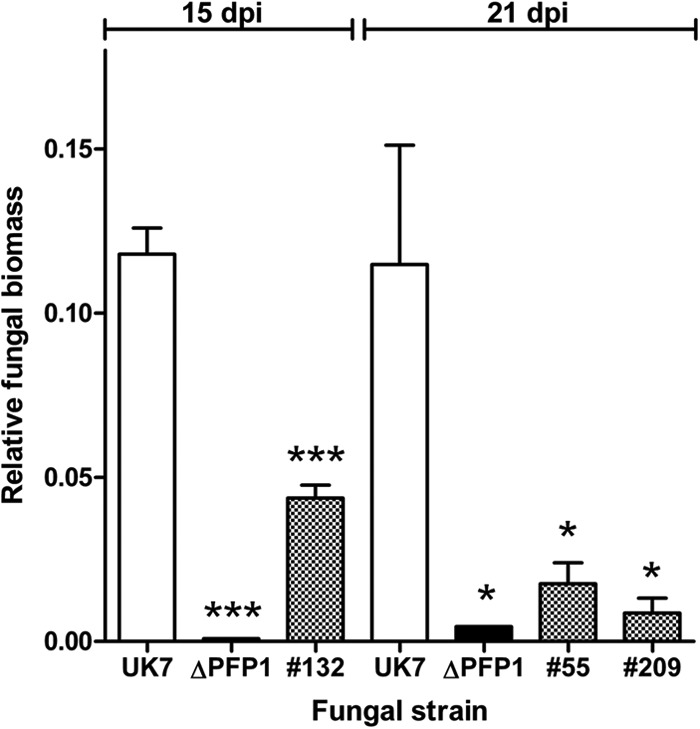
Fungal growth in planta. Fungal biomass was quantified by qPCR via amplification of the GPD gene and compared to that obtained via amplification of a plant HTRA-like protease gene. Due to the differences in growth of the complementation mutants, DNA was isolated 15 and 21 dpi of barley cultivar ‘Ingrid' (n = 4). ***, P < 0.00101 compared with the results for UK7; **, P < 0.0101 compared with the results for UK7; *, P < 0.0501 compared with the results for UK7.
When grown ex planta in liquid medium, germ tube formation and early hyphal development were delayed in the mutant compared to the times of germ tube formation and early hyphal development for the wild type. Nevertheless, after 21 days of liquid culture, the dry weights of mutant and wild-type mycelia did not deviate substantially (Fig. 8). When grown on agar, the germination frequency of the mutant was slightly but significantly reduced at 24 h (20 to 30%) and at 48 h (5 to 12%) compared to that of the wild type. Mutant spores formed about two germ tubes within 48 h, whereas wild-type spores formed an average of about four. At 48 h, the germ tubes were about 40% shorter in the mutant than in the wild type (not shown). Nevertheless, after 20 days of culture, mutant and wild-type mycelia covered comparable areas on agar. However, the mycelia differed in their morphology, with the wild-type mycelia appearing very uniform, whereas mutant mycelia were inhomogeneous, with single colonies of very different sizes being seen (Fig. 8). The most notable characteristic of the mutant when grown both in liquid culture and on lima bean agar was the formation of chlamydospore-like structures/hyphal swellings, which never occurred in cultures of wild-type strain UK7 (Fig. 9).
FIG 8.
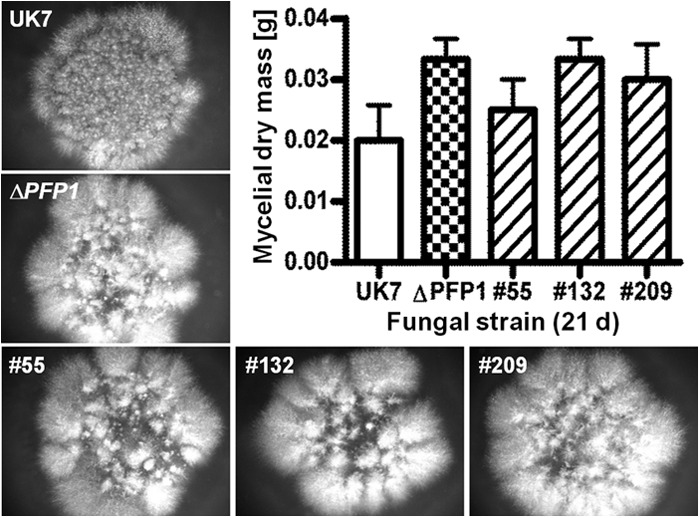
Fungal mycelia growing ex planta. The colonies (diameter, ≈13 mm) of wild-type UK7, the PFP1 deletion mutant, and complementation mutants 55, 132, and 209 looked similar after growing for 21 days on lima bean agar. When grown for 21 days in liquid culture, the fungal dry masses did not differ substantially (n = 3). P values were as follows: for ΔPFP1, 0.1161; for mutant 55, 0.5908; for mutant 132, 0.1161; and for mutant 209, 0.2879.
FIG 9.
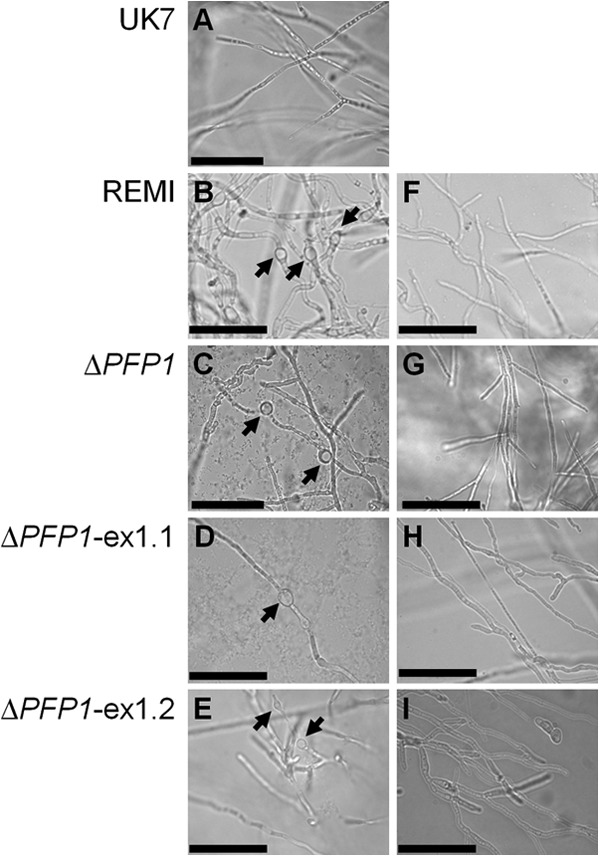
Morphological mutant phenotype. Chlamydospore-like structures/hyphal swellings were found only in the REMI (B) and the PFP1 and PFP1-exon 1 deletion mutants (C to E, arrows) and not in wild-type strains UK7 (A) or the corresponding complementation mutants (F to I; G, mutant 132). For microscopy of fungal mycelia in liquid medium, 5 × 103 conidia were added to 2 ml of sucrose-supplemented Fries medium no. 3 using a Lab-Tek chamber slide system (Nalge Nunc International). After 24 or 48 h, the liquid medium and the chambers were removed and a coverslip was added. Bars = 50 μm.
Complementation of PFP1 deletion.
To verify the causal relationship between the loss of PFP1 and the observed loss of pathogenicity or very low virulence in planta as well as the slightly aberrant fungal development ex planta, the deletion mutants were retransformed with the complete original 5,977-bp PFP1 genomic fragment. For each of the deletion mutants, three independent complementation mutants were selected, grown on lima bean agar plates, and inoculated onto and reisolated from primary leaves of barley cultivar ‘Ingrid' for further analysis.
When the ex planta development of the three UK7ΔPFP1 complementation mutants (mutants 55, 132, and 209) was compared to that of wild-type strain UK7 and that of mutant UK7ΔPFP1, no significant morphological differences occurred during growth in liquid culture. The biomass of the mutants tended to be somewhat higher than that of the wild type (Fig. 8). However, the chlamydospore-like structures produced by the deletion mutants could no longer be observed in the complementation mutants (Fig. 9). The PFP1 mRNA abundance in the mutants growing ex planta was similar to that of wild-type strain UK7 (Fig. 4). Again, the PFP1 protein could not be detected by LC/MS analysis.
When inoculated onto barley leaves, all complementation mutants showed pathogenicity. However, symptom development and, as shown for the three UK7ΔPFP1 complementation mutants, fungal biomass in planta did not reach wild-type levels. Both varied consistently, with mutant 132 producing both the most expanded lesions and the highest biomass (Fig. 5 and 7). In contrast, the level of PFP1 expression in the three complementation mutants was the same as that in the wild type (Fig. 4). These results suggest that retransformation of the deletion mutant with the PFP1 gene partially reestablished the wild-type growth phenotype in planta. Furthermore, these data confirm the results from the preliminary REMI complementation. The mutants generated by transforming the REMI mutant with the PFP1 sequence also regained pathogenicity without expressing full virulence (see Fig. S3 in the supplemental material). Fungal ex planta development was not substantially affected by the PFP1 gene, except for the occurrence of the chlamydospore-like structures/hyphal swellings in the absence of the gene (Fig. 9).
DISCUSSION
Insertion mutagenesis of R. commune led to the identification of the PFP1 gene, which is highly expressed during the early development of R. commune on its host plant, barley. Fungal pathogenesis is characterized by 4 phases: (i) germination and penetration (phase A), (ii) leaf colonization by the formation and spread of thin hyphae (phase B), (iii) rapid growth and multiplication of fungal biomass through thick hyphae (phase C), and (iv) formation of a dense stroma and sporulation (phase D) (cf. Fig. 3). It can take one to several weeks postinoculation until disease symptoms start to occur during phase C. Due to this long period of symptomless development, the fungus is now classified as a hemibiotroph. Most likely, all developmental phases are characterized by the expression of sets of specific genes. For instance, the abundance of mRNA of effector genes, such as NIP1, NIP2, and NIP3, runs through a maximum during phase B, thereby reaching values substantially higher than the value for PFP1 mRNA (13). In contrast, the PFP1 transcript level is already high at 0 dpi (i.e., it is probably already in the spores prior to inoculation) and drops drastically to a constant level until 6 dpi. Effectors are mainly required in the stage when the fungus makes itself at home in the host leaves. In contrast, genes expressed very early are probably needed for penetration and the first steps inside the host tissue. In this context, PFP1 can be expected, on the one hand, to control the genes required during early pathogenesis. On the other hand, the low but constant mRNA level during later stages suggests an additional function during late fungal growth as well. This is supported by the observation that the PFP1 deletion mutant not only shows a reduced penetration frequency (biotrophic stage) compared to that of the wild type but also its mycelia subsequently show slower growth than wild-type mycelia.
The most prominent structural feature of the PFP1 gene product is an Epc-N domain, which is named after the N terminus of the protein encoded by the enhancer of polycomb E (Pc) gene in Drosophila (53). This domain, consisting of a PHD motif, a Zn-binding domain, and a PHD-like domain, is described as a protein-protein interaction module found in subunits of large protein complexes. It is conserved across eukaryotes and is predicted to form a right-handed orthogonal four-helix bundle with β-strands at both termini (50). On the basis of the presence of additional structural features, two major Epc-N families, each consisting of two subfamilies, were identified. Several Epc-N-containing proteins have been shown to be involved in epigenetic events as components of histone acetyltransferase (HAT) complexes. PFP1 belongs to Epc-N subfamily IV, which also contains yeast NTO1 and human JADE-1/2/3 proteins (50).
The PHD motif has been identified to regulate chromatin structure and dynamics in a wide variety of eukaryotic proteins. Also in yeast and filamentous fungi, dozens of PHD-containing proteins with similarity to PFP1 are present. A function has not yet been demonstrated for most of these proteins. However, a few were presumed to be subunits of HAT complexes. In addition, several HAT complex components were found among the better-characterized Epc-N proteins. Particularly well studied are the yeast protein NTO1 and the human proteins JADE-1/2/3 belonging to Epc-N subfamily IV (50). These proteins are scaffold subunits. Together with a catalytic subunit and one or more associated proteins, such as members of the PHD-containing ING (in yeast, YNG) protein family, which bind to lysine 4-methylated histone (54, 55), they form the core of HAT complexes. NTO1 has this function in the yeast NuA3 histone H3 complex, whereas the JADE-1/2/3 proteins are the human NTO1 homologs in the HBO1 complex acetylating histone H4 (TIP60 H4/H2A HAT complex) (56). The human histone H3-acetylating HAT complex, MOZ/MORF, contains the proteins BRPF1/2/3 (57). These proteins belong to Epc-N subfamily III, which is absent from the Ascomycota. It is characterized by two additional structural features, a bromodomain recognizing acetylated lysine and a domain consisting of the amino acid sequence PWWP that recognizes the methylated lysine of histones (50, 58). In contrast, EPL1, the scaffold subunit of the yeast histone H4 HAT complex, Piccolo NuA4, belongs to Epc-N subfamily II, which lacks the PZPM domain.
Notwithstanding their low overall similarities (see Table S2A in the supplemental material), the amino acid sequences of PFP1, BRPF1/2/3, NTO1, and JADE-1/2/3 are highly homologous in their PHD regions (Table 1) and Zn-knuckle/PHD-like regions (see Tables S2B and C in the supplemental material). Furthermore, PFP1 contains the conserved domains I, IIa, and IIb (Fig. 2), which were described for NTO1 and EPL1 as well as for the JADE and BRPF proteins (56). Domain I covers the predicted N-terminal β-sheets and helices 1 and 2 of the Epc-N domain, while domain IIa comprises helix 4 and the C-terminal sheet. Domain IIb lies immediately outside the Epc-N domain. For JADE-1 it was shown that it coordinates HAT complex assembly by interacting through domain I with the catalytic subdomain HBO1 and through domain IIb with the ING protein. The respective interactions were confirmed with domains I and II of EPL1 from the HAT complex Piccolo NuA4 (56). The high homology of the PFP1 domains with the domains of NTO1, BRPF1, and JADE-1 provides strong evidence for a HAT function of PFP1. The protein may be required as the platform for assembly of a histone H3 HAT complex, analogous to the role of NTO1 and BRPF proteins in the histone H3-targeting complexes NuA3 (yeast) and MOZ/MORF (human) (57) and of EPL1 and JADE-1 in the histone H4-targeting complexes Piccolo NuA4 (yeast) and HBO1 (human), respectively. The high level of consensus found with the NTO1 and the BRPF proteins suggests a scaffold role of PFP1 in the formation of an active HAT complex targeting histone H3 in R. commune.
TABLE 1.
PHD amino acid sequence comparison
| Protein domainc | Identity (%) with: |
|||||||
|---|---|---|---|---|---|---|---|---|
| PFP1 | NTO1 | BRPF1a | BRPF2b | BRPF3 | JADE-1 | JADE-2 | JADE-3 | |
| PFP1IV | 66 | 70 | 70 | 66 | 50 | 46 | 52 | |
| NTO1IV | 78 | 56 | 54 | 58 | 54 | 48 | 56 | |
| BRPF1III | 90 | 74 | 96 | 86 | 50 | 50 | 50 | |
| BRPF2III | 86 | 74 | 96 | 86 | 64 | 50 | 50 | |
| BRPF3III | 82 | 74 | 90 | 92 | 50 | 50 | 52 | |
| JADE-1IV | 66 | 66 | 68 | 68 | 64 | 92 | 82 | |
| JADE-2IV | 64 | 64 | 66 | 66 | 62 | 98 | 82 | |
| JADE-3IV | 64 | 68 | 68 | 68 | 64 | 96 | 94 | |
Alternative names, BR140 and peregrin.
Alternative names, BRD1 and BRL.
Roman numerals indicate Epc-N classes. Proteins belonging to mainly histone 3-acetylating HAT complexes are indicated in boldface. GenBank accession numbers are as follows: NTO1, NP_015356; BRPF1, NP_001003694; BRPF2, XP_005261529; BRPF3, AAI17388; JADE-1, NP_955352; JADE-2, XP_005272000; and JADE-3, NP_055550. Sequences were aligned pairwise using the matrix BLOSUM62 of the European Molecular Biology Open Source Software Suite (http://www.ebi.ac.uk/tools/psa/emboss_needle/).
The Epc-N domain-containing proteins play important roles in epigenetic processes. Nevertheless, members of Epc-N subfamily IV appear to have nonessential, rather specialized functions (50). Furthermore, loss of histone H3 acetylation in most cases does not cause severe phenotypes. In yeast, for instance, a knockout of EPL1 (Epc-N subclass II, histone H4 HAT complex) is homozygous lethal, whereas yeasts with an NTO1 knockout (Epc-N subclass IV, histone H3 HAT complex) are viable.
In R. commune, the PFP1 deletion mutant is viable. Ex planta, i.e., on agar and in liquid medium, mycelial development is delayed during the first few days, but it does not deviate from the development of wild-type mycelia at later stages. Hyphal morphology is wild type-like, except for the occurrence of chlamydospore-like structures/hyphal swellings. Similar swellings were described previously for fungal mycelia grown in the presence of gramine and were interpreted as a stress response (11). In contrast to the only minor growth deviation observed ex planta, the deletion mutant differs drastically from the wild type in planta. Few small fungal mycelia that caused only minor disease symptoms were detected in inoculated host leaves.
Modulation of the chromatin structure by posttranscriptional modification of histones is essential for establishing and maintaining distinct states of gene transcription during development and differentiation of an organism. In this context, HAT-mediated histone acetylation is generally associated with increased DNA accessibility, active transcription, and histone deacetylation by HDACs with suppression of gene expression (59). The opposing activities of HATs and HDACs need to be tightly regulated because inactivation of either of them will cause a strong shift in histone acetylation, thereby distorting proper gene regulation. A few previous studies on different fungi suggest that this global mechanism can be utilized in regulating pathogenesis.
The HDC1 gene of the maize pathogen Cochliobolus carbonum is related to the yeast HDAC gene HOS2 (30). Due to reduced penetration efficiency, disruption mutants were strongly reduced in virulence, whereas growth ex planta was normal on glucose, slightly reduced on sucrose, and strongly affected on other different media. The FTL1 gene of the wheat and barley pathogen Fusarium graminearum is homologous to the yeast gene SIF2, which encodes a transducin-β-like component of the HDAC complex Set3 (28). A deletion mutant showed a slightly reduced growth rate ex planta but strongly reduced virulence in planta by failing to normally colonize the host and to cause typical disease symptoms. In addition, conidiation was strongly reduced. The FTL1 homolog TIG1 of the rice pathogen Magnaporthe oryzae was shown to be a component of an HDAC complex that also comprises Set3, Hos2, and Snt1 (29). A deletion mutant showed defective plant infection, reduced conidiation, and abnormal conidium morphology but only slightly reduced growth ex planta. Disruption of the other three genes in M. oryzae as well as of their homologs in F. graminearum resulted in similarly nonpathogenic mutants. This suggests that the HDAC complex is crucial for allowing invasive growth of the respective wild-type fungi, supposedly by histone deacetylation-mediated simultaneous repression of a large number of genes.
If the PFP1 gene of R. commune is part of the opposing beam of the histone acetylation balance, disruption of the PFP1-associated HAT complex may cause strongly reduced virulence of the mutant through increased histone deacetylation. This suggests that in the fungal wild type the protein complex that includes PFP1 may be required for maintaining the expression of a group of fungal genes essential for pathogenesis. The HAT/HDAC system may therefore form a switch which controls fungal differentiation toward invasion and pathogenicity. This differentiation switch needs to be activated early during fungal development on the plant, which is in agreement with the very early expression of the PFP1 gene during pathogenesis, with the highest mRNA abundance already being detected immediately after inoculation. Switching precision may be further augmented by a rapid turnover of the PFP1 protein, as suggested by the presence of three putative PEST domains near the C terminus. Interestingly, this type of motif is also found in the proteins JADE-1/2/3 (60) and in BRPF1 and BRPF2 (not shown). However, PFP1 turnover rates remain to be shown in future studies.
Many HATs/HDACs have a wide range of nonhistone protein substrates, such as transcription factors (61). Therefore, the possibility that instead of a general histone acetylation effect more specific nonhistone acetylation may alternatively be responsible for controlling fungal pathogenicity cannot be excluded. Furthermore, transformation of the PFP1 deletion mutant with the wild-type PFP1 fragment reestablished pathogenicity and abolished the formation of chlamydospore-like structures. Unexpectedly, however, the complementation mutants failed to exert full virulence, although PFP1 transcription reached the wild-type level during growth ex planta as well as in planta. This observation suggests an additional regulatory principle that is involved in controlling PFP1 activity during fungal growth in planta. Due to the nonhomologous integration of the PFP1 wild-type gene at different genomic sites, some kind of position-affected long-range control of gene expression may go awry in the complementation mutants. This has been observed for a number of regulator genes which control defined developmental stages in higher organisms (62). Future experiments need to show whether enhancer or repressor elements outside the PFP1 transcription unit are involved in controlling correct gene expression during R. commune differentiation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Leibniz Association.
Susanne Kirsten and Annette Böttcher are gratefully acknowledged for excellent technical assistance, and Petra Majovski and Ines Lassowskat are acknowledged for carrying out and evaluating the LC/MS analyses.
Footnotes
Published ahead of print 6 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00043-14.
REFERENCES
- 1.Caldwell RM. 1937. Rhynchosporium scald of barley, rye, and other grasses. J. Agric. Res. 55:175–198 [Google Scholar]
- 2.Shipton WA, Boyd WJR, Ali SM. 1974. Scald of barley. Rev. Plant Pathol. 53:839–861 [Google Scholar]
- 3.Avrova A, Knogge W. 2012. Rhynchosporium commune: a persistent threat to barley cultivation. Mol. Plant Pathol. 13:986–997. 10.1111/j.1364-3703.2012.00811.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbott DC, Lagudah ES, Brown AHD. 1995. Identification of RFLPs flanking a scald resistance gene on barley chromosome 6. J. Hered. 86:152–154 [DOI] [PubMed] [Google Scholar]
- 5.Goodwin SB. 2002. The barley scald pathogen Rhynchosporium secalis is closely related to the discomycetes Tapesia and Pyrenopeziza. Mycol. Res. 106:645–654. 10.1017/S0953756202006007 [DOI] [Google Scholar]
- 6.Perfect SE, Green JR. 2001. Infection structures of biotrophic and hemibiotrophic fungal plant pathogens. Mol. Plant Pathol. 2:101–108. 10.1046/j.1364-3703.2001.00055.x [DOI] [PubMed] [Google Scholar]
- 7.Jones P, Ayres PG. 1974. Rhynchosporium leaf blotch of barley studied during the subcuticular phase by electron microscopy. Physiol. Plant Pathol. 4:229–233. 10.1016/0048-4059(74)90011-3 [DOI] [Google Scholar]
- 8.Ayesu-Offei EN, Clare BG. 1970. Processes in the infection of barley leaves by Rhynchosporium secalis. Aust. J. Biol. Sci. 23:299–307 [DOI] [PubMed] [Google Scholar]
- 9.Lehnackers H, Knogge W. 1990. Cytological studies on the infection of barley cultivars with known resistance genotypes by Rhynchosporium secalis. Can. J. Bot. 68:1953–1961 [Google Scholar]
- 10.Horbach R, Navarro-Quesada AR, Knogge W, Deising HB. 2011. When and how to kill a plant cell: infection strategies of plant pathogenic fungi. J. Plant Physiol. 168:51–62. 10.1016/j.jplph.2010.06.014 [DOI] [PubMed] [Google Scholar]
- 11.Kirsten S, Siersleben S, Knogge W. 2011. A GFP-based assay to quantify the impact of effectors on the ex planta development of the slowly growing barley pathogen Rhynchosporium commune. Mycologia 103:1019–1027. 10.3852/10-306 [DOI] [PubMed] [Google Scholar]
- 12.Zhan J, Fitt BDL, Pinnschmidt HO, Oxley SJP, Newton AC. 2008. Resistance, epidemiology and sustainable management of Rhynchosporium secalis populations on barley. Plant Pathol. 57:1–14. 10.1111/j.1365-3059.2007.01691.x [DOI] [Google Scholar]
- 13.Kirsten S, Navarro-Quezada A, Penselin D, Wenzel C, Matern A, Leitner A, Baum T, Seiffert U, Knogge W. 2012. Necrosis-inducing proteins of Rhynchosporium commune, effectors in quantitative disease resistance. Mol. Plant Microbe Interact. 25:1314–1325. 10.1094/MPMI-03-12-0065-R [DOI] [PubMed] [Google Scholar]
- 14.Auriol P, Strobel G, Pio Beltran J, Gray G. 1978. Rhynchosporoside, a host-selective toxin produced by Rhynchosporium secalis, the causal agent of scald disease of barley. Proc. Natl. Acad. Sci. U. S. A. 75:4339–4343. 10.1073/pnas.75.9.4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wevelsiep L, Kogel K-H, Knogge W. 1991. Purification and characterization of peptides from Rhynchosporium secalis inducing necrosis in barley. Physiol. Mol. Plant Pathol. 39:471–482. 10.1016/0885-5765(91)90013-8 [DOI] [Google Scholar]
- 16.Wevelsiep L, Rüpping E, Knogge W. 1993. Stimulation of barley plasmalemma H+-ATPase by phytotoxic peptides from the fungal pathogen Rhynchosporium secalis. Plant Physiol. 101:297–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van't Slot KAE, Knogge W. 2002. A dual role of microbial pathogen-derived proteins in plant disease and resistance. Crit. Rev. Plant Sci. 21:229–271. 10.1080/0735-260291044223 [DOI] [Google Scholar]
- 18.Hahn M, Jüngling S, Knogge W. 1993. Cultivar-specific elicitation of barley defense reactions by the phytotoxic peptide NIP1 from Rhynchosporium secalis. Mol. Plant Microbe Interact. 6:745–754. 10.1094/MPMI-6-745 [DOI] [PubMed] [Google Scholar]
- 19.Rohe M, Gierlich A, Hermann H, Hahn M, Schmidt B, Rosahl S, Knogge W. 1995. The race-specific elicitor, NIP1, from the barley pathogen, Rhynchosporium secalis, determines avirulence on host plants of the Rrs1 resistance genotype. EMBO J. 14:4168–4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van't Slot KAE, van den Burg HA, Kloks CPAM, Hilbers CW, Knogge W, Papavoine CHM. 2003. Solution structure of the plant disease resistance-triggering protein NIP1 from the fungus Rhynchosporium secalis shows a novel β-sheet fold. J. Biol. Chem. 278:45730–45736. 10.1074/jbc.M308304200 [DOI] [PubMed] [Google Scholar]
- 21.van't Slot KAE, Gierlich A, Knogge W. 2007. A single binding site mediates resistance- and disease-associated activities of the effector protein NIP1 from the barley pathogen Rhynchosporium secalis. Plant Physiol. 144:1654–1666. 10.1104/pp.106.094912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Idnurm A, Howlett BJ. 2001. Pathogenicity genes of phytopathogenic fungi. Mol. Plant Pathol. 2:241–255. 10.1046/j.1464-6722.2001.00070.x [DOI] [PubMed] [Google Scholar]
- 23.Tudzynski B, Sharon A. 2003. Fungal pathogenicity genes, p 187–212 In Arora DK, Khachatourians GG. (ed), Applied mycology and biotechnology, vol 3 Fungal genomics; Elsevier, Berlin, Germany [Google Scholar]
- 24.van de Wouw AP, Howlett BJ. 2011. Fungal pathogenicity genes in the age of ‘omics.'. Mol. Plant Pathol. 12:507–514. 10.1111/j.1364-3703.2010.00680.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shim WB, Woloshuk CP. 2001. Regulation of fumonisin B-1 biosynthesis and conidiation in Fusarium verticillioides by a cyclin-like (C-type) gene, FCC1. Appl. Environ. Microbiol. 67:1607–1612. 10.1128/AEM.67.4.1607-1612.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou XY, Heyer C, Choi YE, Mehrabi R, Xu JR. 2010. The CID1 cyclin C-like gene is important for plant infection in Fusarium graminearum. Fungal Genet. Biol. 47:143–151. 10.1016/j.fgb.2009.11.001 [DOI] [PubMed] [Google Scholar]
- 27.Choi YE, Goodwin SB. 2011. Gene encoding a c-type cyclin in Mycosphaerella graminicola is involved in aerial mycelium formation, filamentous growth, hyphal swelling, melanin biosynthesis, stress response, and pathogenicity. Mol. Plant Microbe Interact. 24:469–477. 10.1094/MPMI-04-10-0090 [DOI] [PubMed] [Google Scholar]
- 28.Ding SL, Mehrabi R, Koten C, Kang ZS, Wei YD, Seong KY, Kistler HC, Xu JR. 2009. Transducin β-like gene FTL1 is essential for pathogenesis in Fusarium graminearum. Eukaryot. Cell 8:867–876. 10.1128/EC.00048-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding SL, Liu WD, Iliuk A, Ribot C, Vallet J, Tao A, Wang Y, Lebrun MH, Xu JR. 2010. The Tig1 histone deacetylase complex regulates infectious growth in the rice blast fungus Magnaporthe oryzae. Plant Cell 22:2495–2508. 10.1105/tpc.110.074302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baidyaroy D, Brosch G, Ahn JH, Graessle S, Wegener S, Tonukari NJ, Caballero O, Loidl P, Walton JD. 2001. A gene related to yeast HOS2 histone deacetylase affects extracellular depolymerase expression and virulence in a plant pathogenic fungus. Plant Cell 13:1609–1624. 10.2307/3871389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhadauria V, Banniza S, Wei YD, Peng YL. 2009. Reverse genetics for functional genomics of phytopathogenic fungi and oomycetes. Comp. Funct. Genomics 2009:380719. 10.1155/2009/380719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riggle PJ, Kumamoto CA. 1998. Genetic analysis in fungi using restriction-enzyme-mediated integration. Curr. Opin. Microbiol. 1:395–399. 10.1016/S1369-5274(98)80055-6 [DOI] [PubMed] [Google Scholar]
- 33.Kahmann R, Basse C. 1999. REMI (restriction enzyme mediated integration) and its impact on the isolation of pathogenicity genes in fungi attacking plants. Eur. J. Plant Pathol. 105:221–229. 10.1023/A:1008757414036 [DOI] [Google Scholar]
- 34.Kuspa A. 2006. Restriction enzyme-mediated integration (REMI) mutagenesis. Methods Mol. Biol. 346:201–209. 10.1385/1-59745-144-4:201 [DOI] [PubMed] [Google Scholar]
- 35.Manivasakam P, Schiestl RH. 1998. Nonhomologous end joining during restriction enzyme-mediated DNA integration in Saccharomyces cerevisiae. Mol. Cell. Biol. 18:1736–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohe M, Searle J, Newton AC, Knogge W. 1996. Transformation of the plant pathogenic fungus, Rhynchosporium secalis. Curr. Genet. 29:587–590. 10.1007/BF02426964 [DOI] [PubMed] [Google Scholar]
- 37.Punt PJ, Oliver RP, Dingemanse MA, Pouwels MA, van den Hondel CAMJJ. 1987. Transformation of Aspergillus based on the hygromycin B resistance marker from E. coli. Gene 56:117–124. 10.1016/0378-1119(87)90164-8 [DOI] [PubMed] [Google Scholar]
- 38.Mattern IE, Punt PJ, van den Hondel CAMJJ. 1988. A vector of Aspergillus transformation conferring phleomycin resistance. Fungal Genet. Newsl. 35:25 [Google Scholar]
- 39.Drocourt D, Calmels T, Reynes JP, Baron M, Tiraby G. 1990. Cassettes of the Streptoalloteichus hindustanus ble gene for transformation of lower and higher eukaryotes to phleomycin resistance. Nucleic Acids Res. 18:4009. 10.1093/nar/18.13.4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuwayama H, Obara S, Morio T, Katoh M, Urushihara H, Tanaka Y. 2002. PCR-mediated generation of a gene disruption construct without the use of DNA ligase and plasmid vectors. Nucleic Acids Res. 30:e2. 10.1093/nar/30.2.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sweigard JA, Carroll AM, Farrall L, Chumley FG, Valent B. 1998. Magnaporthe grisea pathogenicity genes obtained through insertional mutagenesis. Mol. Plant Microbe Interact. 11:404–412. 10.1094/MPMI.1998.11.5.404 [DOI] [PubMed] [Google Scholar]
- 42.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devic M, Albert S, Delseny M, Roscoe TJ. 1997. Efficient PCR walking on plant genomic DNA. Plant Physiol. Biochem. 35:331–339 [Google Scholar]
- 44.Liu YG, Whittier RF. 1995. Thermal asymmetric interlaced PCR—automatable amplification and sequencing of insert end fragments from P1 and Yac clones for chromosome walking. Genomics 25:674–681. 10.1016/0888-7543(95)80010-J [DOI] [PubMed] [Google Scholar]
- 45.Scotto-Lavino E, Du GW, Frohman MA. 2006. 5′ end cDNA amplification using classic RACE. Nat. Protoc. 1:2555–2562. 10.1038/nprot.2006.480 [DOI] [PubMed] [Google Scholar]
- 46.Scotto-Lavino E, Du GW, Frohman MA. 2006. 3′ end cDNA amplification using classic RACE. Nat. Protoc. 1:2742–2745. 10.1038/nprot.2006.481 [DOI] [PubMed] [Google Scholar]
- 47.Lassowskat I, Naumann K, Lee J, Scheel D. 2013. PAPE (prefractionation-assisted phosphoprotein enrichment): a novel approach for phosphoproteomic analysis of green tissues from plants. Proteomes 1:254–274. 10.3390/proteomes1030254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Köcher T, Swart R, Mechtler K. 2011. Ultra-high-pressure RPLC hyphenated to an LTQ-Orbitrap Velos reveals a linear relation between peak capacity and number of identified peptides. Anal. Chem. 83:2699–2704. 10.1021/ac103243t [DOI] [PubMed] [Google Scholar]
- 49.Motulsky HJ. 2003. Prism 4 statistics guide—statistical analyses for laboratory and clinical researchers. GraphPad Prism Software Inc., San Diego, CA [Google Scholar]
- 50.Perry J. 2006. The Epc-N domain: a predicted protein-protein interaction domain found in select chromatin associated proteins. BMC Genomics 7:6. 10.1186/1471-2164-7-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aravind L, Landsman D. 1998. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 26:4413–4421. 10.1093/nar/26.19.4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rechsteiner M, Rogers SW. 1996. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 21:267–271. 10.1016/0968-0004(96)10031-1 [DOI] [PubMed] [Google Scholar]
- 53.Stankunas K, Berger J, Ruse C, Sinclair DAR, Randazzo F, Brock HW. 1998. The enhancer of polycomb gene of Drosophila encodes a chromatin protein conserved in yeast and mammals. Development 125:4055–4066 [DOI] [PubMed] [Google Scholar]
- 54.Martin DGE, Baetz K, Shi XB, Walter KL, MacDonald VE, Wlodarski MJ, Gozani O, Hieter P, Howe L. 2006. The Yng1p plant homeodomain finger is a methyl-histone binding module that recognizes lysine 4-methylated histone H3. Mol. Cell. Biol. 26:7871–7879. 10.1128/MCB.00573-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taverna SD, Ilin S, Rogers RS, Tanny JC, Lavender H, Li HT, Baker L, Boyle J, Blair LP, Chait BT, Patel DJ, Aitchison JD, Tackett AJ, Allis CD. 2006. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell 24:785–796. 10.1016/j.molcel.2006.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Avvakumov N, Lalonde ME, Saksouk N, Paquet E, Glass KC, Landry AJ, Doyon Y, Cayrou C, Robitaille GA, Richard DE, Yang XJ, Kutateladze TG, Cote J. 2012. Conserved molecular interactions within the HBO1 acetyltransferase complexes regulate cell proliferation. Mol. Cell. Biol. 32:689–703. 10.1128/MCB.06455-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ullah M, Pelletier N, Xiao L, Zhao SP, Wang K, Degerny C, Tahmasebi S, Cayrou C, Doyon Y, Goh SL, Champagne N, Cote J, Yang XJ. 2008. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell. Biol. 28:6828–6843. 10.1128/MCB.01297-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu H, Zeng H, Lam R, Tempel W, Amaya MF, Xu C, Dombrovski L, Qiu W, Wang YM, Min JR. 2011. Structural and histone binding ability characterizations of human PWWP domains. PLoS One 6:e18919. 10.1371/journal.pone.0018919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brosch G, Loidl P, Graessle S. 2008. Histone modifications and chromatin dynamics: a focus on filamentous fungi. FEMS Microbiol. Rev. 32:409–439. 10.1111/j.1574-6976.2007.00100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou MI, Wang HM, Foy RL, Ross JJ, Cohen HT. 2004. Tumor suppressor von Hippel-Lindau (VHL) stabilization of Jade-1 protein occurs through plant homeodomains and is VHL mutation dependent. Cancer Res. 64:1278–1286. 10.1158/0008-5472.CAN-03-0884 [DOI] [PubMed] [Google Scholar]
- 61.Glozak MA, Sengupta N, Zhang XH, Seto E. 2005. Acetylation and deacetylation of non-histone proteins. Gene 363:15–23. 10.1016/j.gene.2005.09.010 [DOI] [PubMed] [Google Scholar]
- 62.Kleinjan DA, van Heyningen V. 2005. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 76:8–32. 10.1086/426833 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.