

Abstract
Increasing evidence indicates that the Trypanosoma brucei flagellum (synonymous with cilium) plays important roles in host-parasite interactions. Several studies have identified virulence factors and signaling proteins in the flagellar membrane of bloodstream-stage T. brucei, but less is known about flagellar membrane proteins in procyclic, insect-stage parasites. Here we report on the identification of several receptor-type flagellar adenylate cyclases (ACs) that are specifically upregulated in procyclic T. brucei parasites. Identification of insect stage-specific ACs is novel, as previously studied ACs were constitutively expressed or confined to bloodstream-stage parasites. We show that procyclic stage-specific ACs are glycosylated, surface-exposed proteins that dimerize and possess catalytic activity. We used gene-specific tags to examine the distribution of individual AC isoforms. All ACs examined localized to the flagellum. Notably, however, while some ACs were distributed along the length of the flagellum, others specifically localized to the flagellum tip. These are the first transmembrane domain proteins to be localized specifically at the flagellum tip in T. brucei, emphasizing that the flagellum membrane is organized into specific subdomains. Deletion analysis reveals that C-terminal sequences are critical for targeting ACs to the flagellum, and sequence comparisons suggest that differential subflagellar localization might be specified by isoform-specific C termini. Our combined results suggest insect stage-specific roles for a subset of flagellar adenylate cyclases and support a microdomain model for flagellar cyclic AMP (cAMP) signaling in T. brucei. In this model, cAMP production is compartmentalized through differential localization of individual ACs, thereby allowing diverse cellular responses to be controlled by a common signaling molecule.
INTRODUCTION
African trypanosomes, including Trypanosoma brucei and related species, are the causative agents of African trypanosomiasis, also known as sleeping sickness in humans and nagana in animals. Sleeping sickness is recognized to be one of the world's most neglected diseases and poses a threat to 60 million people living in sub-Saharan Africa (1). The disease is fatal if left untreated, and therapeutic treatments are antiquated, difficult to administer, and increasingly ineffective (2, 3). Due to its ability to infect livestock, T. brucei also hinders economic growth and agricultural development and as such represents a significant contributor to poverty in some of the most impoverished regions of the world (4).
T. brucei is heteroxenous, requiring a tsetse fly vector and a mammalian host in order to complete its life cycle. In both hosts, the parasite must sense and respond to extracellular signals, but very little is known about how trypanosomes accomplish this. In other eukaryotes, the flagellum (synonymous with cilium) harbors membrane proteins and signal transduction pathways that mediate cellular responses to changing extracellular signals (5). In mammals, for example, ciliary receptor-guanylate cyclases, ion channels, and G-protein-coupled receptors (GPCRs) control development in response to external signals (5–7). The T. brucei flagellar membrane is a direct interface with the host, and accumulating evidence indicates that flagellar proteins of these parasites play important roles in mediating the interaction with the host environment (8–15). For example, proteomic analysis of the flagellum in bloodstream-form (BSF) T. brucei parasites identified receptor and transporter proteins predicted to function in signaling, as well as corresponding effector proteins (9). In addition, recent forward genetic screens for downstream effectors in quorum sensing and cyclic AMP (cAMP) signaling pathways in bloodstream-stage T. brucei parasites identified putative flagellar proteins (16, 17).
Perhaps the best-characterized flagellar protein involved in host-parasite interaction is expression site-associated gene 4 (ESAG4), a bloodstream-form-specific adenylate cyclase (AC) that is localized along the length of the flagellar membrane (18). ESAG4 contributes to virulence in mice and upon encountering host cells is postulated to be activated to drive cAMP production, which in turn inhibits host tumor necrosis factor alpha production, thereby resisting the host's early innate immunity attack (15). Several other virulence factors are also localized to the T. brucei flagellum, including glycosylphosphatidylinositol-phospholipase C (11), calflagin (13), and metacaspase 4 (14). The precise role of these proteins in host interaction is not known, but each is required for full virulence, as mice infected with corresponding knockout or knockdown parasites show prolonged survival compared to mice infected with control parasites.
The flagellum is also important for parasite interaction within the tsetse fly vector. For example, flagellum-dependent motility is required for transmission through the tsetse fly (19), and parasite attachment to the fly salivary gland epithelium is mediated by outgrowths of the flagellar membrane (10). Flagellum attachment is a critical step in the transmission cycle, as it enables the parasite to establish a permanent infection in the salivary gland and marks the onset of differentiation into forms infectious for mammals (20, 21). Little is known about flagellar membrane and matrix proteins in insect-stage T. brucei (22), but one interesting family of proteins is a set of adenylate cyclases encoded by genes related to ESAG4 (GRESAG4) (23). T. brucei encodes approximately 65 GRESAG4 proteins (15), some of which cross-react with anti-ESAG4 antibodies and are localized along the flagellum in both bloodstream and procyclic (fly midgut-stage) cells (18). Trypanosomal ACs (ESAG4 and GRESAG4s) have a domain structure that differs from the canonical architecture of mammalian adenylate cyclases. Canonical ACs are multi-transmembrane-pass proteins that have two catalytic domains on a single polypeptide and lack direct receptor activity, relying instead on upstream GPCR signaling pathways. Trypanosomal ACs, on the other hand, resemble mammalian receptor-guanylate cyclases, having an intracellular catalytic domain connected by a single transmembrane segment to a large, extracellular, putative ligand binding domain (23, 24). The trypanosome AC extracellular domain exhibits homology to bacterial periplasm binding proteins, which bind small ligands to direct chemotaxis and other cellular responses in bacteria (25–27). Trypanosomes have no known GPCRs, and it has been suggested that trypanosomal ACs function directly as receptors, similar to the function of the mammalian receptor-guanylate cyclases that they resemble (24, 28).
In vitro differentiation of bloodstream-form cells into procyclic cells and subsequent proliferation are associated with bursts of cAMP production, suggesting that procyclic stage-specific cAMP-dependent processes are important for parasite differentiation (29–31). Trypanosomal adenylate cyclases exhibit sequence diversity in their extracellular domains, which suggests a mechanism for ligand-specific regulation of AC activity (24, 32), making these proteins well-suited for directing cAMP signaling in response to host-specific signals. To date, however, all GRESAG4 genes studied have been found to be expressed in both bloodstream-stage and insect-stage cells (18, 23, 29, 33, 34), raising questions about whether they are responsible for procyclic stage-specific regulation of cAMP production. Here we report on the identification of a group of T. brucei adenylate cyclases whose expression is upregulated in procyclic cells. We show that procyclic stage-specific ACs are glycosylated, assemble into multimeric complexes, exhibit catalytic activity, and are localized to the flagellum, where they are surface exposed. Interestingly, individual ACs are located in distinct subdomains of the flagellum, indicating specialized functions and trafficking mechanisms. Our studies provide the first analysis of individual trypanosome adenylate cyclases within insect-stage cells and support a model for the microdomain organization of cAMP signaling in the T. brucei flagellum.
MATERIALS AND METHODS
Cell culture and RNAi knockdown.
Procyclic-form cells were used for all experiments and cultured in Cunningham's SM medium as previously described (35). Transfections and selection of clonal lines by limiting dilution were done as described previously (35). The Fla1-knockdown cell line was generated by transfection of 2913 cells (36) with the p2T7-Fla1 plasmid, as described previously (37). For knockdown of AC protein 1 (ACP1), the RNA interference (RNAi) target region, corresponding to 311 bp of the 3′ untranslated region (UTR), was PCR amplified using the following forward (F) and reverse (R) primers (restriction sites are in italics): ACP1-RNAi-F (ATAAGCTTTCCTTCTGGCTTCGTCACTT) and ACP1-RNAi-R (ATTCTAGATTCATCCCGGAACAAAACTC). The resulting DNA was ligated into the p2T7-Ti-B RNAi vector (37). Insertion was verified by sequencing by Genewiz, Inc. The p2T7-ACP1-RNAi vector was linearized with NotI and transfected into 2913 cells, and stable transfectants were selected with 10 μg/ml phleomycin. Transfected cells were maintained in selective medium, and clonal lines were generated by limiting dilution.
Proteomic identification of flagellar adenylate cyclases.
Procyclic T. brucei Fla1-knockdown cells (37) were induced for 25 h with 1 μg/ml tetracycline. After addition of 0.2 M sucrose to cells at a density of 4 × 106 cells/ml, cells were sonicated for 6 min and spun at 2,000 × g for 5 min to pellet cell bodies (this is referred to as the P1 fraction), leaving flagella in the supernatant (this is referred to as the S1 fraction). The S1 fraction was spun again at 2,000 × g for 5 min to remove debris and then subjected to a high-speed centrifugation at 20,000 × g for 35 min. The pellet (the P2 fraction) was resuspended in phosphate-buffered saline (PBS), layered on top of a 13-ml step gradient of sucrose (10, 20, 30, 40, 55, and 68%), and then centrifuged at 245,000 × g for 4 h at 4°C in a Beckman Optima L-90K ultracentrifuge using an SW41 rotor. Fourteen fractions of approximately 1 ml each were collected from the top of the gradient and spun at 14,000 × g for 1 h at 4°C to concentrate the samples for examination by phase-contrast microscopy. Flagella were primarily found in fractions 8 and 9, corresponding to the interface between the 40% and 55% sucrose layers. To solubilize the membranes, this flagellum fraction was incubated for 10 min at room temperature with 0.1% NP-40 in PBS and then centrifuged at 10,000 × g for 10 min to separate the axoneme-containing pellet (P3 fraction) from the supernatant (S3 fraction) harboring flagellar membranes and matrix proteins. Since NP-40 can degrade the quality of the spectra obtained by mass spectrometry, proteins were precipitated from the flagellum (S3) fraction by trichloroacetic acid (TCA), followed by two washes with acetone. Cell bodies (P1 fraction) were disrupted by hypotonic lysis (38), sonicated for 2 min, and incubated for 10 min at room temperature with 0.1% NP-40 in PBS. Solubilized proteins were precipitated by TCA and washed with acetone. TCA-precipitated proteins from solubilized flagellum and cell body fractions were digested by the sequential addition of Lys-C and trypsin proteases (39, 40) and subjected to analysis by use of the multidimensional protein identification technology as described previously (9). Proteins were considered present in the analysis if they were identified by two or more peptides using a 5% peptide-level false discovery rate (41–43).
The majority of proteins were uniquely identified by specific peptides (unique, “U”). Proteins identified only by peptides shared with other proteins were assigned to groups. The set of proteins identified in the cell body fraction was subtracted from the combined flagellum fractions, yielding a subtracted data set of 175 proteins arranged in 157 groups (see Table S2 in the supplemental material). In situ tagging and immunofluorescence localization analyses (44) were done for four proteins of the subtracted data set. All of these localized to the cytoplasm or cell body surface, but not the flagellum (not shown), indicating that the subtracted data set contained substantial cell body contamination and precluding analysis of the data set as a stand-alone flagellar proteome. Interestingly, however, the subtracted data set included a group of six receptor-type adenylate cyclases that were not found in previous analysis of bloodstream-form flagellar membranes (9) (see Tables S1 and S2 in the supplemental material). These proteins were selected for further study. Sequence comparisons were done by pairwise alignments using VectorNTI's AlignX module (Invitrogen) with sequences obtained from the TriTryp database (91).
Quantitative real-time PCR.
Cells were harvested at a density of approximately 5 × 106 cells/ml (procyclic culture form [PCF]) and 1 × 106 cells/ml (BSF). For ACP1 knockdowns, cells were grown with or without tetracycline at 1 μg/ml for 72 h prior to harvesting. Total RNA was extracted using a Qiagen RNeasy kit, and quantitative reverse transcriptase, real-time PCR (qRT-PCR) was performed as described previously (45). Gene-specific primer sets were designed using the Trypanofan RNAit algorithm (46) and the NCBI Primer-BLAST program (47). The primers used were ACP1-F (CGTTGACTTCACGGCTTACA), ACP1-R (ACATTTCGTTCTCCCACTGC), ACP2-F (GCCATGTCGTTGATTTCACA), ACP2-R (CCAACCAGACCACAGACCTT), ACP4-F (AGCTTACGAGGGCTGTGAAA), ACP4-R (AAATACACTGCCCCTTGTCG), ACP5-F (TCTGCTTATGCAGGACGATG), ACP5-R (CCTCAAAAGTCTCGAGGTGC), FS33-F (GCGCTAGCATAAGACGTGGT), FS33-R (GAACCGTTCTCACACCAACA), ISG65-F (CATGACAGAGGAGTGGCAGA), and ISG65-R (CATGCTCGGTTGAAGCACTA). qRT-PCR was conducted on a DNA Engine Opticon 2 real-time cycler (MJ Research, Bio-Rad) using iQ SYBR green Supermix (Bio-Rad) according to the manufacturer's instructions. All analyses were performed in technical duplicate on at least two independent RNA preparations, and values were normalized against those for two stage-independent control genes, TERT and PFR2 (48), using the 2−ΔΔCT method (45, 49).
In situ epitope tagging.
In situ tagging was done by amplifying short (300- to 600-bp) fragments of DNA homologous to the target gene's open reading frame (ORF) or 3′ UTR and cloning these upstream of the 3× hemagglutinin (HA) tag or downstream of the puromycin resistance gene of the pMOTag2H vector or the 3× Myc tag and phleomycin resistance gene of pMOTag53M (44). The primers used, with restriction sites italicized, were ACP1orfF (ATGGTACCACATGGCTGCCCGCACAGAG), ACP1orfR (ATCTCGAGGTTTTCCTCCTTTGGGGTTGA), ACP1utrF (ATGGATCCGGGTCGTAGGCATGCGCCAT), ACP1utrR (ATTCTAGAGGGGGAGCAGGCGCCTCAAT), ACP2orfF (ATGGTACCTTGTGAATTGGGTCAGTCGCA), ACP2orfR (ATCTCGAGTTCTCGTTCGCTGCTTGT), ACP2utrF (ATGGATCCCATCAAGAAGGAAACCGAGTA), ACP2utrR (ATTCTAGATGAACTAAATTGCAGTCTCCCA), ACP4orfF (GATGGTACCTGCGCGGACGGAAAATGTGACGAAC), ACP4orfR (GATCTCGAGAAACTTATCAAAATCCGTGGTCCGATTGGGGA), ACP4utrF (GATGGATCCGGGTTTTGGGGGGTTAATGGCACAA), ACP4utrR (GATTCTAGAATTTCACCGCCGGAGACGTTGTTGA), ACP5orfF (ATGGTACCATGGCTGCGGGACGGAGAG), ACP5orfR (ATCTCGAGCCGCTGCGCTTCGGGGTTTC), ACP5utrF (ATGGATCCAAACCACTCCACGAACTAATGAC), and ACP5utrR (ATTCTAGAGAAGGAGTGTTCCCTGCGATAA). For ACP1 and ACP2 truncations, the following primers for amplification of the ORF were designed to eliminate the final 45 codons: ACP1ΔC45F (ATATGGTACCAGGGTTACGACTACTATGGTCA), ACP1ΔC45R (ATATCTCGAAACGACGTGTCCCACTTTTG), ACP2ΔC46F (ATGGTACCCCGTCAATGAGCTTCAGAGACCCTAGCGAAGGAAAACTC), and ACP2ΔC46R (ATCTCGAGCATATGAACGACATGGCCCACTTTTGTTGCTATACGCCGCATAAC). All sequences were verified by direct sequencing at the UCLA Sequencing and Genotyping Core Center. Tagging cassettes were excised by restriction digestion, purified, and transfected into 2913 cells. Transfected cells were maintained in selective medium, and clonal lines were generated by limiting dilution.
Southern blotting.
Genomic DNA was isolated using a PureLink genomic DNA kit (Invitrogen) according to the manufacturer's instructions. Restriction enzyme digestions (New England BioLabs) were done for 8 h, and samples were then separated on a 0.8% agarose gel. Gels were treated for 15 min each in depurination buffer (0.25 M HCl), denaturation buffer (0.5 M NaOH, 1.0 M NaCl), and neutralization buffer (1 M Tris-HCl [pH 7.5], 3 M NaCl) and rinsed in distilled water. DNA was transferred to a Hybond-XL membrane (GE Healthcare) using capillary transfer overnight in SSC buffer (3 M NaCl, 300 mM Na3C6H5O7). The membrane was cross-linked using 1,500 J of heat on a UV Stratalinker cross linker (Stratagene). Digoxigenin-labeled probes were PCR generated using the following primers: PuroProbe_F (CACCGAGCTGCAAGAACT), PuroProbe_R (CTCGTAGAAGGGGAGGTTG), ACP1_probeF (ATTCTAGAGGGGGAGCAGGCGCCTCAAT), and ACP1_probeR (ATGGATCCGGTCGTAGGCATGCGCCAT). Hybridization, washes, and detection were done according to the manufacturer's instructions using DIG EasyHyb buffer, CDP-Star substrate, and a PCR DIG-probe synthesis kit (Roche, Inc.).
Cell fractionation and immunoblotting.
To assay for an association with detergent-resistant membranes (see Fig. 2A), cells were washed once in PBS and lysed at either 4°C or 37°C in PBS with 1% Triton X-100 and protease inhibitors (SigmaFAST cocktail; Sigma-Aldrich) as described previously (50). Lysates were centrifuged for 20 min at 15,000 × g at either 4°C or room temperature to separate solubilized proteins (S) from the insoluble pellet (P) fraction. Immunoblotting was done as previously described (45).
FIG 2.

Trypanosomal adenylate cyclases are glycosylated surface proteins that dimerize. (A) Western blot analysis of the proteins in the soluble (S) and insoluble pellet (P) fraction from cells extracted with Triton X-100 at the indicated temperature (4° or 37°C). Protein samples from cells expressing the indicated HA-tagged ACP (ACP1, ACP2, ACP4, or ACP5) were probed with antibodies against HA, calflagin, and tubulin. (B) Cells expressing HA-tagged ACP4 or ACP5 were surface biotinylated, and protein extracts harvested as indicated in the schematic were subjected to Western blot analysis with antibodies against HA or BiP. (C) Whole-cell lysates were prepared from 2913 control cells or cells expressing HA-tagged ACP1, ACP2, ACP4, or ACP5. Samples were untreated (−) or treated (+) with PNGase F and then subjected to Western blot analysis with antibodies against HA or EIF4AI. (D) Whole-cell lysates prepared from 2913 control cells or cells expressing HA-tagged ACP1, ACP2, ACP4, or ACP5 were separated by Blue Native gel electrophoresis and then transferred to a PVDF membrane and probed with anti-HA antibodies. (E) Cells with two tagged alleles of ACP1 (ACP1-Myc/HA) or two tagged alleles of ACP2 (ACP2-Myc/HA) were used for coimmunoprecipitation to test for homodimerization. In test samples, one allele of the indicated AC gene contains an HA tag and the other allele has a Myc tag. Samples were immunoprecipitated with anti-HA antibody, and input (I), unbound (U), and bound (B) fractions were probed in Western blots with anti-HA or anti-Myc antibody. As a negative control, ACP1-Myc lacks any HA tag and is not precipitated by anti-HA antibody (see Fig. S2 in the supplemental material). The numbers below each lane indicate the relative number of cell equivalents analyzed. (F) Cells expressing ACP1-HA were mixed with cells expressing ACP1-Myc, and the cells in the mixture were lysed and immunoprecipitated using anti-HA antibody. Input, unbound, and bound fractions were probed in Western blots with anti-HA or anti-Myc antibody.
Blue Native gel analysis and deglycosylation.
Cells were harvested and washed in PBS and then resuspended into PEME buffer (100 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 1 mM MgSO4, 0.1 mM EDTA, 2 mM EGTA, pH 6.9) with 1% NP-40 and protease inhibitors (SigmaFAST cocktail; Sigma-Aldrich). Following a 10-min incubation at room temperature, lysates were spun for 15 min at 13,000 rpm and 4°C. Supernatants were transferred to new tubes and spun again for 10 min to clear the debris, and the resultant soluble fractions were used. For Blue Native gel analysis, NativePAGE sample buffer and 5% G-250 sample additive (Invitrogen) were added, and samples were run on a NativeMARK 4 to 16% bis-Tris native gel and transferred to a polyvinylidene difluoride (PVDF) membrane per the manufacturer's suggested protocols (Invitrogen), followed by immunoblotting. For deglycosylation, the soluble fraction was denatured and treated with peptide N-glycosidase F (PNGase F; New England BioLabs) for 1 h at 37°C using the manufacturer's suggested protocols, followed by SDS-PAGE and immunoblotting.
Immunoprecipitation.
Cells were harvested, washed in PBS, and then lysed in IP buffer (150 mM NaCl, 50 mM HEPES, 5 mM EDTA, 5 mM EGTA, 1% NP-40, 10% glycerol, 1× SigmaFAST protease inhibitors). After 10 min on ice, lysates were centrifuged at 4°C for 30 min to remove insoluble material. A fraction of the soluble fraction was retained, the rest was added to EZview red anti-HA affinity matrix (Sigma-Aldrich), and the mixture was incubated for approximately 3.5 h at 4°C on a nutator mixer. Beads were collected by centrifugation and washed several times in IP buffer. Input, unbound, and bead fractions were boiled in sample buffer and analyzed by SDS-PAGE and immunoblotting.
Surface biotinylation and streptavidin purification.
Surface biotinylation was done as described previously (9). Cells were harvested and washed in ice-cold PBS and then resuspended in 3 ml of cold 0.5 mg/ml biotin (catalog no. 21331; Pierce) solution and incubated for 10 min on ice. Tris (2 M, pH 6.8) was added to a final concentration of 100 mM to block unreacted biotin, and the mixture was incubated on ice for 10 min. Cells were pelleted, washed with cold PBS–100 mM Tris, and extracted with 1% NP-40 in PBS with 100 mM Tris and SigmaFast protease inhibitors. After a 10-min incubation on ice, samples were centrifuged at 14,000 rpm for 10 min at 4°C to pellet insoluble material. A fraction of the supernatant (the input) was retained, and the remainder was transferred to a new tube with 50 μl of GE Healthcare streptavidin Sepharose high-performance beads and incubated for 1 h at 4°C on a nutator mixer to allow biotin-streptavidin binding. The beads were pelleted and washed as described previously (9) and then resuspended and boiled in native sample buffer (bound samples) for analysis by immunoblotting.
Yeast complementation.
For yeast complementation, adenylate cyclase open reading frames were subcloned into a modified version of the Saccharomyces cerevisiae pRS315 expression vector (51), pRS315-GPDp-CYCt, which contains a glyceraldehyde 3-phosphate dehydrogenase (GPD) promoter to allow constitutive high expression, with a cytochrome c isoform 1 (CYC1) terminator. The pRS315-GPDp-CYCt plasmid was kindly provided by Giancarlo Costaguta and Gregory Payne (UCLA). The S. cerevisiae wild-type adenylate cyclase (CYR1) coding sequence was amplified from the YCp50-CYR1 plasmid (52), and full-length T. brucei adenylate cyclase-coding sequences were amplified from genomic DNA using the following forward (F) and reverse (R) primers (restriction sites are italicized): CYR1F (ATATGGATCCATGTCATCAAAACCTGATACTGGTTCG), CYR1R (ATATGTCGACTCAAGTTGATAAATCCTTTGCGTTC), ACP1F (ATATACTAGTACTGCGATGAATATGCTTCACTTG), ACP1R (ATATCTCGAGGTCGACACGACCCTCAGTTTTCCTCC), ACP2F (ATATAAGCTTACTAGTATGAATATGCTTCACTTGGACGAC), ACP2R (ATATCTCGAGGTCGACTTATTCTCGTTCGCTGCTTGTG), ACP4F (ATATACTAGTATGAAAGCACCAGCCTTGC), ACP4R (ATATCTCGAGTTAAAACTTATCAAAATCCGTGGTC), ACP5F (ATATACTAGTATGACCACCACGAAGGCTTCGTGTC), and ACP5R (ATATAAGCTTTCACCGCTGCGCTTCGGGGTTTC). Amplified coding sequences were cloned into the pRS315-GPDp-CYCt vector using the corresponding restriction sites (italicized in the primer sequences), and the resulting plasmids were transformed into the S. cerevisiae cyr1-2 mutant (53) using standard methods (54). Transformants were selected and maintained on SD-Leu selective medium as described previously (54). Clonal strains were resuspended in PBS at an optical density at 600 nm of 1.0, and 5-fold serial dilutions were spotted onto yeast extract-peptone-dextrose-rich medium agar plates, which were incubated at permissive (22°C) or restrictive (35°C) temperatures.
Antibody production.
Anti-ACP1 antibody against a synthetic peptide corresponding to the C-terminal 15 amino acids of ACP1 (CAVGERNVSTPKEEN), which is unique and distinguishes ACP1 from ACP2, was generated by Pacific Immunology, Inc. Antibodies were raised in New Zealand White rabbits, and ACP1-specific antibodies were affinity purified by the vendor using peptide coupled to agarose beads. Antibody specificity was determined by immunoblotting with total protein extracts from ACP1 RNAi-knockdown cells grown with or without tetracycline. For PFR antibodies, a fragment encoding the N-terminal 328 amino acids of PFR2 (NCBI GenBank accession number XP_847327.1) was amplified from genomic DNA and expressed using a pETDuet-1 system (Novagen). Recombinant protein was purified using the manufacturer's suggested protocols and used to raise polyclonal antibodies in rabbits by Pacific Immunology, Inc.
Immunofluorescence microscopy.
Immunofluorescence on whole cells was done as described previously (9). Monoclonal anti-HA antibody HA.11 (Covance) was used at a 1:250 dilution and detected using donkey antimouse secondary antibody coupled to Alexa Fluor 488 (Molecular Probes) at 1:750. Polyclonal PFR antiserum was used at a 1:1,250 dilution, polyclonal affinity-purified anti-ACP1 antibodies were used at a 1:500 dilution, and both were detected using donkey antirabbit antibody coupled to either Alexa Fluor 488 or 594 (Molecular Probes) at a 1:750 dilution. Coverslips were mounted in VectaShield mounting medium with DAPI (4′,6-diamidino-2-phenylindole). Samples were imaged on a Zeiss Axioskop II microscope (Zeiss, Inc.).
RESULTS
Identification of procyclic stage-specific adenylate cyclases.
We previously reported on the isolation of intact flagella and proteomic analysis of flagellar membrane and matrix proteins from bloodstream-form (BSF) T. brucei parasites (9). We attempted a similar analysis using procyclic culture-form (PCF) T. brucei parasites, corresponding to insect midgut-stage parasites, but our analyses indicated a high level of contamination by cellular proteins, likely owing to the use of sonication to remove flagella from cell bodies (see Materials and Methods). Among the proteins identified, however, was a group of receptor-type adenylate cyclases that were not detected in BSF flagellum preparations (see Table S1 in the supplemental material). These adenylate cyclases are part of a previously described family of genes related to expression site-associated gene 4 (GRESAG4), on the basis of their sequence similarity to ESAG4 (23). ESAG4 is expressed only in BSF parasites, while previously examined GRESAG4 genes were found to be constitutively expressed in both BSF and PCF parasites (18, 23, 29, 33, 34). We were therefore surprised to uncover a group of GRESAG4 genes that were detected only in procyclic flagella, and we investigated these further. We termed these proteins ACP1 through ACP6, to reflect the fact that they were found in procyclic culture-form, but not bloodstream-form, proteomic analyses.
The T. brucei genome includes approximately 65 GRESAG4 genes, with the protein encoded by each gene having a large extracellular domain at the N terminus, followed by a single transmembrane region and a cytoplasmic catalytic domain (15). The catalytic domain is followed by a short C-terminal region of approximately 150 to 175 amino acids. Sequence relationships among trypanosomal ACs, including ACP1 to ACP6, have been described previously (15, 55). The amino acid sequence diversity among ACP1 to ACP6 was found to be the highest within the N-terminal and C-terminal regions (Fig. 1B). Pairwise alignments revealed that ACP1 and ACP2 are approximately 90% identical in amino acid sequence throughout their length, with differences lying primarily within the C terminus. The other ACs identified exhibited considerable sequence differences between one another (Fig. 1B).
FIG 1.

Procyclic stage-specific receptor adenylate cyclases. (A) The schematic shows the general architecture of T. brucei adenylate cyclases, including a signal peptide (SP), a single-pass transmembrane domain (TM), and a cyclase catalytic domain (CYC), followed by a short intracellular C-terminal region. Within the N-terminal region are one to two domains homologous to periplasmic binding proteins (PBPs) of bacteria. The average sequence identity among ACP1 to ACP6 is shown above each section. (B) Pairwise amino acid sequence identities within the N-terminal, catalytic, and C-terminal domains of ACP1 to ACP6 (P1 to P6). (C) The chart shows the relative mRNA abundance in procyclic culture-form (PCF) and bloodstream-form (BSF) cells for the genes for adenylate cyclases ACP1 to ACP6 and FS33, as determined by qRT-PCR. ISG65 is a bloodstream-form-enriched gene (90) used as a control. The expression levels for each gene are normalized to the levels for the life cycle stage expressing the genes at higher levels.
Peptides specific to ACP1 to ACP6 were not detected in a proteomic analysis of flagella from bloodstream-form cells (9), suggesting that these proteins are expressed only in the procyclic life cycle stage. However, sequence similarities among the AC protein family make it difficult to unambiguously identify specific isoforms using proteomics alone. For example, while one or more peptides uniquely mapped to each of ACP1 and ACP3 to ACP6 in the current study (see Table S1 in the supplemental material), the five peptides that mapped to ACP2 also mapped to ACP1. We therefore used quantitative reverse transcriptase, real-time PCR (qRT-PCR) with gene-specific primers to directly determine the developmental expression profile for ACP1 through ACP6. We found that ACP1 and ACP3 to ACP6 were each expressed primarily in procyclic-form parasites (Fig. 1C), while ACP2 expression was similar in both life cycle stages. Notably, qRT-PCR also demonstrated that expression of FS33, an AC identified in BSF flagella (9) but not PCF flagella, was indeed upregulated in BSF cells (Fig. 1C). Thus, ACP1 and ACP3 to ACP6 show developmentally regulated expression distinct from that reported for all other GRESAG4 genes studied to date.
In situ epitope tagging enables analysis of individual ACs.
The large size of the AC gene family, together with the extensive sequence homology among individual genes, has complicated efforts to analyze any single AC gene or protein. Our proteomic analyses identified a small subset of ACs as being expressed in procyclic cells, thereby allowing prioritization of individual genes for direct analysis. We focused our studies here on ACP1, ACP2, ACP4, and ACP5, while ACP3 and ACP6 are the focus of separate work. To study each protein individually, in situ tagging (44) was used to incorporate an HA epitope tag at the 3′ end of each gene. Western blot analysis of cell lysates demonstrated that a single HA-tagged protein of the expected size was expressed in each tagged cell line, and Southern blotting demonstrated integration of the HA epitope tag at the expected locus in each case (see Fig. S1 in the supplemental material). Having established gene-specific tags for each AC, we set out to characterize the individual proteins.
Biochemical analysis reveals that T. brucei ACs are surface-exposed glycoprotein multimers.
The cellular distribution of adenylate cyclases in procyclic T. brucei has previously been examined only in conglomerate (18). The availability of gene-specific epitope tags provided a unique opportunity to monitor the fractionation and distribution of individual AC proteins. The ACs studied here were identified in detergent-solubilized cell fractions. To determine whether this represented the entire cellular pool for each protein, we performed Western blot analysis of detergent-soluble and detergent-insoluble fractions. Each AC fractionated exclusively in detergent-soluble supernatants, consistent with what would be anticipated for membrane-associated proteins (Fig. 2A). Some flagellar membrane proteins are associated with detergent-resistant membranes (56). To determine whether this is the case for ACs, we asked whether solubilization with Triton X-100 at 4°C shifts ACs to the pellet fraction, as seen for proteins in detergent-resistant membranes, such as calflagin (56). The AC fractionation pattern was unchanged at 4°C and 37°C, indicating that ACs are not associated with detergent-resistant membranes. T. brucei ACs are predicted to be surface exposed, and this has been demonstrated for ESAG4 and FS33 in BSF parasites, as well as for a group of AC proteins in conglomerate in procyclic cells (9, 18). We asked whether ACP4 and ACP5 were surface exposed using surface biotinylation, followed by affinity purification with streptavidin. Western blot analysis of bound and unbound fractions demonstrated that each AC eluted with the bound, i.e., surface-biotinylated, fraction, while the intracellular marker BiP eluted in the unbound fraction (Fig. 2B). Therefore, ACP4 and ACP5 are exposed on the cell surface of procyclic-form T. brucei parasites.
The calculated molecular masses of ACP1 and ACP2 are nearly identical, 137.5 and 137.9 kDa, respectively (see Table S1 in the supplemental material). However, Western blots revealed significant size differences between these proteins (see Fig. S1 in the supplemental material), suggesting differential posttranslational modifications. Trypanosomal ACs have a receptor-type structure and are predicted to function in recognition of extracellular ligands. Glycosylation is a common feature of surface proteins and can be critically important for receptor-ligand interactions (57). Moreover, there are several putative glycosylation sites present in both proteins, and all are predicted to be within the extracellular N-terminal domains (data not shown). We therefore asked whether glycosylation accounted for the size difference between ACP1 and ACP2. To test for glycosylation, we used digestion with peptide N-glycosidase F (PNGase F), which cleaves N-linked carbohydrate groups. PNGase F treatment caused a significant reduction in size for each AC, as seen by SDS-PAGE (Fig. 2C). As a control, the cytoplasmic protein EIF4AI (58) showed no change in size with PNGase F treatment. Therefore, individual ACs are differentially glycosylated, although some difference in size remains, suggesting that additional modifications may be present.
All characterized nucleotide cyclase catalytic domains operate as dimers, with catalysis occurring at the dimer interface (59, 60). Unlike conventional adenylate cyclases, which have two cyclase domains on a single polypeptide, trypanosomal cyclases have only a single catalytic domain per protein (Fig. 1A). In vitro studies with recombinant catalytic domains previously demonstrated that T. brucei AC catalytic domains require dimerization for catalytic activity (34, 59), but whether the full-length proteins form dimers in vivo is not known. We therefore asked whether native T. brucei ACs formed multimeric complexes by examining cell lysates using Blue Native gels under nonreducing conditions. Western blots of Blue Native gels showed that each of the ACs examined migrated as multiple species ranging in size from approximately 160 kDa to 500 kDa, with the major species in each case being the one with the larger molecular mass (>300 kDa) (Fig. 2D). The size of the smaller species in each case agrees with the predicted size of the monomeric protein, ∼150 kDa. Therefore, AC proteins form multimeric complexes under native conditions. The multimeric complexes observed on Blue Native gels suggested dimerization, but they could be due to interaction with proteins other than ACs themselves. To test for dimerization directly, we generated doubly tagged lines, in which one ACP1 allele was HA tagged and the other was Myc tagged. We also did this for ACP2. Immunoprecipitation with anti-HA in each case demonstrated that the Myc-tagged protein coprecipitated with the HA-tagged protein (Fig. 2E), while singly tagged ACP1-Myc was not precipitated with anti-HA antibody (Fig. S2 in the supplemental material). Notably, coimmunoprecipitation was observed only if the HA-tagged and Myc-tagged proteins were expressed in the same cells. Using a mixture of cells in which one half expressed HA-tagged protein and the other half expressed Myc-tagged protein, coimmunoprecipitation was not observed (Fig. 2F). Although dimerization was anticipated, this is the first direct evidence that ACs dimerize in vivo.
To test whether the ACs identified here are catalytically active, we tested their ability to rescue the growth of Saccharomyces cerevisiae mutants that lack a functional adenylate cyclase. The yeast cyr1-2 strain harbors a temperature-sensitive mutation that disrupts function of the endogenous adenylate cyclase, CYR1, rendering cyr1-2 yeast nonviable at the restrictive temperature due to a cAMP deficiency (53). Expression of T. brucei ACP1, ACP2, ACP4, or ACP5 restored the viability of the cyr1-2 mutant at restrictive temperatures (Fig. 3). Thus, each of these proteins individually possesses adenylate cyclase catalytic activity in vivo.
FIG 3.
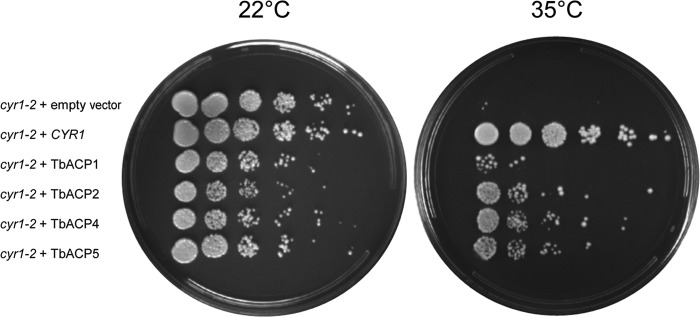
Trypanosomal adenylate cyclases are catalytically active. A temperature-sensitive Saccharomyces cerevisiae adenylate cyclase mutant (cyr1-2) was transformed with a yeast expression vector containing either the S. cerevisiae wild-type adenylate cyclase (CYR1) or the gene for T. brucei ACP1, ACP2, ACP4, or ACP5 (TbACP1, TbACP2, TbACP4, and TbACP5, respectively). An empty vector was transformed as a control, and yeast viability was assessed at the permissive (22°C) and restrictive (35°C) temperatures.
Individual adenylate cyclases are localized to different domains of the flagellar membrane.
Subcellular distribution has not been determined for most adenylate cyclases in T. brucei. Two individual ACs, ESAG4 (18) and FS33 (9), have been shown to localize along the length of the flagellum in bloodstream-form parasites. Immunofluorescence using pan-specific antibodies showed localization along the length of the flagellum for a group of GRESAG4 proteins in procyclic-form parasites, but individual proteins were not examined in this life cycle stage and it is not known to which specific GRESAG4 genes the labeling corresponds (18). The availability of clonal lines, each having a single, uniquely epitope-tagged AC protein, made it possible to examine the location of each AC individually by immunofluorescence microscopy. Using immunofluorescence with anti-HA antibody, we found ACP1, ACP2, ACP4, and ACP5 to be localized exclusively to the flagellum (Fig. 4; see also Fig. S3 in the supplemental material). Notably, the specific distribution within the flagellum was different for individual ACs. ACP1 and ACP4 were localized primarily to the distal tip of the flagellum, whereas ACP2 was evenly distributed along the entire length of the flagellum. ACP5 was concentrated at the flagellum tip, with a weaker signal seen along the flagellum. The tip-specific localization of ACP1 and ACP4 distinguishes them from trypanosomal ACs studied previously and is, to our knowledge, a novel finding for a transmembrane protein in T. brucei.
FIG 4.

Trypanosomal adenylate cyclases localize to flagellum subdomains. (A) Trypanosomes expressing the indicated HA-tagged adenylate cyclase were subjected to immunofluorescence. Cells were stained with anti-HA antibodies (green), and nuclear and kinetoplast DNA were visualized with DAPI (blue). (B) Immunofluorescence analysis as described for panel A showing dividing cells. Cells have a single nucleus and two kinetoplasts, indicating that they have not completed mitosis, and possess a mature flagellum (filled arrowhead) as well as a newly forming daughter flagellum (unfilled arrowhead).
Localization of individual trypanosomal ACs to distinct regions indicates that different structural features must be present to distinguish the tip of the flagellum from the length of the flagellum in order to enable tip-specific targeting. To assess when such features are established, we examined AC protein localization as a function of the cell cycle. Trypanosome cultures grow asynchronously, and morphogenetic markers are available to easily define the cell cycle stage for any given cell in the population (61). Cells that have completed kinetoplast division but not mitosis contain two kinetoplasts and a single nucleus (2K1N). These cells possess one fully formed flagellum and one newly forming flagellum, whose tip connects to the side of the old flagellum (61). Anti-HA immunofluorescence showed that 2K1N cells expressing ACP1-HA or ACP4-HA have two spots of fluorescence, one corresponding to the tip of the old flagellum and one corresponding to the tip of the newly forming flagellum (Fig. 5B). Therefore, any cellular features required for flagellum tip-specific localization are established prior to the completion of mitosis, while the nascent flagellum is still growing.
FIG 5.

Endogenous ACP1 localizes to the flagellum tip. (A) mRNA levels for ACP1 and ACP2 were determined by qRT-PCR in ACP1-knockdown cells grown in the absence (−) or presence (+) of tetracycline (Tet). (B) Total protein extracts were prepared from BSF cells and from procyclic ACP1-knockdown cells (ACP1-KD) grown without (−) or with (+) tetracycline and then subjected to Western blot analysis using affinity-purified anti-ACP1 antibodies (top). (Bottom) Total protein in the same samples visualized by Coomassie staining of SDS-polyacrylamide gels. (C) Immunofluorescence of ACP1-knockdown cells grown without or with tetracycline and probed with affinity-purified anti-ACP1 antibodies. Nuclear and kinetoplast DNA are stained with DAPI (blue).
To assess whether the HA epitope tag influenced protein localization, we raised ACP1-specific antibodies and used these antibodies to determine the location of the endogenous protein. To test the specificity of the anti-ACP1 antibody, we generated a gene-specific RNAi knockdown of ACP1. qRT-PCR demonstrated that specific and efficient knockdown of ACP1 did not affect the expression of ACP2 (Fig. 5A), which is the protein most closely related to ACP1. Knockdown of ACP1 did not affect parasite growth or motility (not shown). Western blotting with anti-ACP1 antibodies detected a single band of the expected size that was lost following the induction of RNAi (Fig. 5B). Anti-ACP1 antibody failed to detect any signal in lysates from bloodstream-form parasites, corroborating the qRT-PCR results demonstrating that ACP1 is a procyclic stage-specific protein. These results further demonstrate that the antibody distinguishes ACP1 from ACP2, as ACP2 expression is unaffected by ACP1 knockdown. Immunofluorescence with ACP1-specific antibody showed that endogenous ACP1 is located at the distal tip of the flagellum (Fig. 5C), as seen for the HA-tagged protein, and the tip signal is lost upon RNAi induction against ACP1. Therefore, the HA-tagged protein (Fig. 4) correctly reports the localization of the endogenous protein.
Protein targeting to specific subcellular locations requires cis-acting targeting sequences within the protein. ACP1 and ACP2 are almost identical in sequence, except for their C termini, suggesting that this region is important for specifying flagellar and/or subflagellar localization. We therefore generated epitope-tagged deletion mutants lacking the C-terminal 45 or 46 amino acids of ACP1 (ACP1ΔC45) and ACP2 (ACP2ΔC46), respectively, to assess the influence of these amino acids on flagellar localization. In both cases, expression was reduced and the deletion mutants exhibited a punctate distribution throughout the cell but were completely absent from the flagellum (Fig. 6; see also Fig. S3 in the supplemental material), demonstrating that these residues are critical for targeting to the flagellum.
FIG 6.

C-terminal sequences are required for targeting to the flagellum. (A) Western blot analysis of whole-cell lysates from cells expressing the indicated HA-tagged protein. Blots were probed with anti-HA or anti-tubulin antibodies. (B) Trypanosomes expressing the indicated HA-tagged deletion mutants were subjected to immunofluorescence. Cells were stained with anti-HA antibodies (green) and anti-PFR antibodies (red). Nuclear and kinetoplast DNA were visualized with DAPI (blue). (C) (Top) Schematic diagram illustrating amino acid sequence identities between ACP1 and ACP2, which are nearly identical except for a short region at the C terminus. (Bottom) Alignment of the C termini of ACP1 to ACP6 (P1 to P6). ACP1 residues that differ from ACP2 are boxed, and among these, those residues that are conserved between ACP1 and ACP4 are highlighted in gray.
DISCUSSION
Insect stage-specific adenylate cyclases.
Our studies reveal a new paradigm for trypanosomal ACs through identification of a group of AC genes upregulated in the procyclic life cycle stage, indicating a specific role within the tsetse fly. There are approximately 65 chromosome-internal adenylate cyclase genes in the T. brucei genome (55), but only a few of these have been studied directly. Among the ACs identified here, ACP4 corresponds to previously studied GRESAG4.2/4.3 (33, 55). ACP1 and ACP2 correspond to two ESAG4-like genes identified as being upregulated in BSF parasites following knockout of ESAG4 (55), though they were not examined in PCF parasites. ACP3, ACP5, and ACP6 do not correspond to previously studied GRESAG4 genes. Procyclic stage-specific expression is a novel finding, as all ACs examined previously have been found to be either BSF specific, ESAG4 (23, 29) and FS33 (Fig. 1) (9), or constitutively expressed, GRESAG4.1, GRESAG4.2/4.3, and GRESAG4.4 (18, 23, 33, 34). Prior analysis of GRESAG4.2/4.3, corresponding to ACP4, reported equal expression in BSF and PCF parasites by Northern blotting (23), while our qRT-PCR analysis shows 6-fold upregulation of ACP4 in PCF versus BSF parasites. We suspect that the discrepancy may lie in the greater capacity for gene-specific analysis in qRT-PCR. Additionally, earlier studies may have underestimated the potential for cross-reactivity in Northern blots, because the size and extent of sequence similarity among the members of the AC/GRESAG gene family were not known.
ESAG4 functions in manipulation of host immune responses to trypanosome infection (15) and potentially in cytokinesis in bloodstream-form parasites (55). However, the functions of chromosome-internal GRESAG4 ACs and the reasons for expansion of the AC gene family remain unknown. ACP1 and ACP2 are upregulated in bloodstream cells following the knockout of ESAG4, compensating for the loss of ESAG4 (55). Thus, some ACs may substitute for others under selective pressure. Nonetheless, our data argue against a strictly redundant role for T. brucei ACs, because we see developmentally regulated expression profiles that differ from the expression profiles described for ESAG4 and all other GRESAG4 genes studied to date. Distinct functions for individual ACs are also supported by sequence diversity among different isoforms (28) and our finding here that individual isoforms show distinct distributions along the flagellum.
Nonredundant functions for individual ACs in parasite-tsetse fly interactions are also consistent with the observation that expansion of the AC gene family varies among different tsetse fly-transmitted trypanosomes. T. vivax, for example, which develops only in the fly mouthparts and foregut (62), has 14 AC genes (15). In comparison, development of T. congolense and T. brucei occurs not only in the mouthparts and foregut but also the midgut, with T. brucei additionally advancing through the salivary glands (62). Correspondingly, T. congolense and T. brucei have a larger cohort of AC genes, with approximately 45 and 65 chromosome-internal AC genes, respectively (15). Therefore, the size of the AC gene family in different African trypanosomes directly correlates with the complexity of the parasite's developmental cycle and tissue distribution in the tsetse fly, consistent with the idea that ACs function in tsetse fly-parasite interactions. The in vitro differentiation of bloodstream-form parasites into procyclic forms and subsequent proliferation are accompanied by spikes in cellular cAMP levels (18, 29), and procyclic stage-specific ACs provide a potential source for this cAMP.
Functional consequences of tip-localized adenylate cyclases.
Previous studies showed flagellum localization for ACs in procyclic parasites, but the antibodies could not distinguish between isoforms (18). By using gene-specific epitope tagging, we were able to determine the unique distribution of individual AC proteins. Surprisingly, we discovered distinct patterns of localization for different isoforms. Most notably, ACP1 and ACP4 were localized to the flagellum tip, which has not been reported for any previously studied T. brucei AC. Hundreds of T. brucei flagellar proteins have been described (9, 22, 63). However, aside from the ACs reported here, only three flagellar tip proteins have been identified. These are two axonemal proteins, kinesin KIF13.2 (64) and FLAM8 (22), and one membrane protein, calpain1.3, which associates with the membrane via acylation (65). KIF13.2 functions in flagellum length control, while the functions of calpain1.3 and FLAM8 are not known. Flagellum tip proteins in other organisms play important functions in cell signaling. Examples include vertebrate polycystins and Gli proteins that function in Ca2+ and Hedgehog signaling, respectively (66–68). Examples in other protists include flagellar tip agglutinins in Chlamydomonas that mediate cell adhesion events associated with cAMP signaling and mating (69, 70). Thus, identification of tip-localized ACs that could modulate cAMP signaling in T. brucei is of great interest.
Unlike mammalian ACs, trypanosomal ACs are postulated to function as receptors modulating signaling, with specificity conferred by divergent N-terminal ligand binding domains (24, 28). Consistent with this idea, the AC N-terminal domain shares homology with bacterial periplasmic binding proteins (PBPs) that function in chemotaxis and signaling through recognition of diverse ligands (Fig. 1) (25, 71). A receptor function for trypanosome ACs remains speculative at present, but demonstration that procyclic stage-specific ACs dimerize and are catalytically active supports their function in cAMP production. Differential localization would provide a mechanism to spatially restrict the cAMP output from different AC isoforms, allowing them to interface with distinct effector proteins. Thus, segregating individual isoforms to distinct flagellum subcompartments would allow two different ACs to initiate specific responses, despite using a common output. Such an arrangement provides support for a microdomain model for cAMP signaling postulated for T. brucei and observed in other eukaryotic cells, where the close proximity of ACs, phosphodiesterases, and effectors confines cAMP signaling to distinct foci (72–74). Importantly, the T. brucei cAMP-specific phosphodiesterase PDEB1 is distributed along the length of the flagellum (75) and is thus positioned to act as a diffusion barrier, limiting cAMP to the site of production by differentially localized adenylate cyclases (74).
Protein localization to the flagellum tip is also interesting in the context of parasite development because the tip initiates attachment to the fly salivary gland prior to differentiation into metacyclic parasites infectious for mammals (20). The reorganization of cytoskeletal filaments and the flagellum membrane at the site of attachment (76), as well as subsequent cellular and molecular changes that accompany differentiation into metacyclic parasites (20, 21), presumably involves signaling events triggered by flagellum contact. Consistent with this idea, metacyclogenesis in T. congolense can be triggered in vitro via flagellum contact with plastic surfaces (77). There is also a precedent for flagellum tip attachment triggering cellular differentiation in the protist Chlamydomonas reinhardtii, whereby flagellum tip adhesion between two gametes triggers a cAMP signaling cascade resulting in gamete fusion (69, 70, 78). Recent ex vivo reconstruction of the T. brucei mating cycle identified gamete-like cells that interact via their flagella prior to fusion (79), raising the intriguing possibility that flagellum-dependent interactions may be part of the T. brucei mating cycle. The flagellum functions in mechanosensation in other protists (80, 81) and may therefore function in contact-mediated signaling in T. brucei. Surface-exposed adenylate cyclases at the tip of the flagellar membrane are ideally positioned to perceive and transduce responses to flagellum attachment. Tests of this hypothesis will require tsetse fly infection experiments using trypanosomes lacking specific ACs. The insect stage-specific ACs identified here present excellent candidates for testing this idea, and the lack of any growth defect following RNAi knockdown of ACP1 demonstrates that such experiments are now feasible. Apart from host-parasite interaction, the flagellum tip is also distinguished structurally from the rest of the flagellum and is the site of flagellar structures important to cell division (82, 83). Thus, tip-specific ACs are located in a unique region of the cell that plays several important functions in parasite biology.
Flagellum tip-targeting signals.
The C-terminal 45 and 46 amino acids are required for flagellar targeting of ACP1 and ACP2, respectively. Previous work showed that a 21-amino-acid fragment near the C terminus of calpain1.3 is required for flagellar targeting (65). We did not observe any obvious sequence similarities between the C-terminal sequences of ACs and calpain1.3, nor did we identify similarities to published targeting sequences in flagellar membrane proteins of other organisms (84). Protein localization within specific flagellum subdomains is emerging as an important aspect of flagellum biology (22, 85–87), but the sequences responsible for directing subflagellar targeting are unknown (88, 89). In this regard, ACP1 and ACP2 in T. brucei offer potential insights, because they are ∼90% identical in amino acid sequence and differences are primarily restricted to the C-terminal region that is required for flagellum localization. This region is expected to be intracellular and thus accessible to targeting machinery. Alignment of the C-terminal 42 amino acids of ACP1 and ACP2 revealed differences at 31 positions (Fig. 6). Of these 31 positions, only 5 are conserved between ACP1 and the other tip-localized AC, ACP4. Therefore, these 5 residues are likely to be important for specifying subcompartment localization within the flagellum. Notably, 3 of these 5 residues are conserved in ACP5, consistent with the intermediate localization observed for ACP5; i.e., it is enriched at the tip plus along the length. The localization of ACP3 and ACP6 was not determined, but on the basis of the sequence conservation within the putative tip-targeting domain (Fig. 6), these proteins are likely to be tip localized or enriched at the tip.
The flagellum and cAMP signaling in trypanosomes.
Cyclic AMP is important in T. brucei development and pathogenesis (15, 29, 32). ACs are the source of cAMP production and are therefore critical for cAMP signaling. All of the adenylate cyclases so far studied in T. brucei are concentrated in the flagellum, which also contains cAMP-specific phosphodiesterases (75) and cAMP effectors (16). Together, this indicates an important role for the flagellum in T. brucei cAMP signaling. Future studies aimed at understanding AC function as well as mechanisms of targeting to the flagellum and specific flagellum subcompartments offer opportunities for understanding key aspects of trypanosome biology and host-parasite interaction.
Supplementary Material
ACKNOWLEDGMENTS
We thank Carl Mann (CEA/Saclay, Gif-sur-Yvette, France) for the cyr1-2 yeast strain and YCp50-CYR1 plasmid and Giancarlo Costaguta and Gregory Payne (UCLA) for the pRS315-GPDp-CYCt plasmid used for yeast complementation experiments. We thank Eden Freire and Osvaldo Pompílio de Melo Neto (Universidade de Brasilia, Brasilia, Brazil) for the anti-EIF4AI antibody and David Engman for the calflagin antiserum. We thank Walter Hardesty and Stephanie De Marco for assistance in generating plasmid constructs for the yeast complementation assays and ACP1ΔC45, respectively, and thank members of the K. L. Hill laboratory for many helpful discussions on the work.
Funding and support for the work were provided by grants from the National Institutes of Health (NIH) (grant AI052348 to K.L.H.), the Burroughs Wellcome Fund (to K.L.H.), NIGMS (grant GM089778 to J.A.W.), and the Jonsson Cancer Center at UCLA (to J.A.W.), as well as fellowships from NIH-USPHS-NRSA (GM07104 to E.A.S.) and NIH-NRSA (F31AI085961 to M.L.), NIH Ruth L. Kirschstein NRSA grant AI094750 (to M.M.S.), a UCLA Warsaw Fellowship (to G.L.), and an HHMI Gilliam Fellowship (to A.R.).
Footnotes
Published ahead of print 30 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00019-14.
REFERENCES
- 1.Yamey G, Torreele E. 2002. The world's most neglected diseases. BMJ 325:176–177. 10.1136/bmj.325.7357.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamey G. 2002. Public sector must develop drugs for neglected diseases. BMJ 324:698. 10.1136/bmj.324.7339.698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baral TN. 2010. Immunobiology of African trypanosomes: need of alternative interventions. J. Biomed. Biotechnol. 2010:389153. 10.1155/2010/389153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nussbaum K, Honek J, Cadmus CM, Efferth T. 2010. Trypanosomatid parasites causing neglected diseases. Curr. Med. Chem. 17:1594–1617. 10.2174/092986710790979953 [DOI] [PubMed] [Google Scholar]
- 5.Christensen ST, Pedersen LB, Schneider L, Satir P. 2007. Sensory cilia and integration of signal transduction in human health and disease. Traffic 8:97–109. 10.1111/j.1600-0854.2006.00516.x [DOI] [PubMed] [Google Scholar]
- 6.Berberi N, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. 2008. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc. Natl. Acad. Sci. U.S.A. 105:4242–4246. 10.1073/pnas.0711027105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaupp UB, Kashikar ND, Weyand I. 2008. Mechanisms of sperm chemotaxis. Annu. Rev. Physiol. 70:93–117. 10.1146/annurev.physiol.70.113006.100654 [DOI] [PubMed] [Google Scholar]
- 8.Maric D, Epting CL, Engman DM. 2010. Composition and sensory function of the trypanosome flagellar membrane. Curr. Opin. Microbiol. 13:466–472. 10.1016/j.mib.2010.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberholzer M, Langousis G, Nguyen HT, Saada EA, Shimogawa MM, Jonsson ZO, Nguyen SM, Wohlschlegel JA, Hill KL. 2011. Independent analysis of the flagellum surface and matrix proteomes provides insight into flagellum signaling in mammalian-infectious Trypanosoma brucei. Mol. Cell. Proteomics 10:M111.010538. 10.1074/mcp.M111.010538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tetley L, Vickerman K. 1985. Differentiation in Trypanosoma brucei: host-parasite cell junctions and their persistence during acquisition of the variable antigen coat. J. Cell Sci. 74:1–19 [DOI] [PubMed] [Google Scholar]
- 11.Webb H, Carnall N, Vanhamme L, Rolin S, Van Den Abbeele J, Welburn S, Pays E, Carrington M. 1997. The GPI-phospholipase C of Trypanosoma brucei is nonessential but influences parasitemia in mice. J. Cell Biol. 139:103–114. 10.1083/jcb.139.1.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rotureau B, Morales MA, Bastin P, Spath GF. 2009. The flagellum-mitogen-activated protein kinase connection in trypanosomatids: a key sensory role in parasite signaling and development? Cell. Microbiol. 11:710–718. 10.1111/j.1462-5822.2009.01295.x [DOI] [PubMed] [Google Scholar]
- 13.Emmer BT, Daniels MD, Taylor JM, Epting CL, Engman DM. 2010. Calflagin inhibition prolongs host survival and suppresses parasitemia in Trypanosoma brucei infection. Eukaryot. Cell 9:934–942. 10.1128/EC.00086-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Proto WR, Castanys-Munoz E, Black A, Tetley L, Moss CX, Juliano L, Coombs GH, Mottram JC. 2011. Trypanosoma brucei metacaspase 4 is a pseudopeptidase and a virulence factor. J. Biol. Chem. 286:39914–39925. 10.1074/jbc.M111.292334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salmon D, Vanwalleghem G, Morias Y, Denoeud J, Krumbholz C, Lhomme F, Bachmaier S, Kador M, Gossmann J, Dias FB, De Muylder G, Uzureau P, Magez S, Moser M, De Baetselier P, Van Den Abbeele J, Beschin A, Boshart M, Pays E. 2012. Adenylate cyclases of Trypanosoma brucei inhibit the innate immune response of the host. Science 337:463–466. 10.1126/science.1222753 [DOI] [PubMed] [Google Scholar]
- 16.Gould MK, Bachmaier S, Ali JA, Alsford S, Tagoe DN, Munday JC, Schnaufer AC, Horn D, Boshart M, de Koning HP. 2013. Cyclic AMP effectors in African trypanosomes revealed by genome-scale RNA interference library screening for resistance to the phosphodiesterase inhibitor CpdA. Antimicrob. Agents Chemother. 57:4882–4893. 10.1128/AAC.00508-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mony BM, Macgregor P, Ivens A, Rojas F, Cowton A, Young J, Horn D, Matthews K. 2013. Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature 505:681–685. 10.1038/nature12864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paindavoine P, Rolin S, Van Assel S, Geuskens M, Jauniaux JC, Dinsart C, Huet G, Pays E. 1992. A gene from the variant surface glycoprotein expression site encodes one of several transmembrane adenylate cyclases located on the flagellum of Trypanosoma brucei. Mol. Cell. Biol. 12:1218–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rotureau B, Ooi CP, Huet D, Perrot S, Bastin P. 2014. Forward motility is essential for trypanosome infection in the tsetse fly. Cell. Microbiol. 16:425–433. 10.1111/cmi.12230 [DOI] [PubMed] [Google Scholar]
- 20.Vickerman K, Tetley L, Hendry KA, Turner CM. 1988. Biology of African trypanosomes in the tsetse fly. Biol. Cell 64:109–119. 10.1016/0248-4900(88)90070-6 [DOI] [PubMed] [Google Scholar]
- 21.Rotureau B, Subota I, Buisson J, Bastin P. 2012. A new asymmetric division contributes to the continuous production of infective trypanosomes in the tsetse fly. Development 139:1842–1850. 10.1242/dev.072611 [DOI] [PubMed] [Google Scholar]
- 22.Subota I, Julkowska D, Vincensini L, Reeg N, Buisson J, Blisnick T, Huet D, Perrot S, Santi-Rocca J, Duchateau M, Hourdel V, Rousselle JC, Cayet N, Namane A, Chamot-Rooke J, Bastin P. 16 April 2014. Proteomic analysis of intact flagella of procyclic Trypanosoma brucei cells identifies novel flagellar proteins with unique sub-localisation and dynamics. Mol. Cell. Proteomics 10.1074/mcp.M113.033357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandre S, Paindavoine P, Tebabi P, Pays A, Halleux S, Steinert M, Pays E. 1990. Differential expression of a family of putative adenylate/guanylate cyclase genes in Trypanosoma brucei. Mol. Biochem. Parasitol. 43:279–288. 10.1016/0166-6851(90)90152-C [DOI] [PubMed] [Google Scholar]
- 24.Seebeck T, Gong K, Kunz S, Schaub R, Shalaby T, Zoraghi R. 2001. cAMP signalling in Trypanosoma brucei. Int. J. Parasitol. 31:491–498. 10.1016/S0020-7519(01)00164-3 [DOI] [PubMed] [Google Scholar]
- 25.Felder CB, Graul RC, Lee AY, Merkle HP, Sadee W. 1999. The Venus flytrap of periplasmic binding proteins: an ancient protein module present in multiple drug receptors. AAPS PharmSci. 1:E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emes RD, Yang Z. 2008. Duplicated paralogous genes subject to positive selection in the genome of Trypanosoma brucei. PLoS One 3:e2295. 10.1371/journal.pone.0002295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anantharaman V, Iyer LM, Aravind L. 2007. Comparative genomics of protists: new insights into the evolution of eukaryotic signal transduction and gene regulation. Annu. Rev. Microbiol. 61:453–475. 10.1146/annurev.micro.61.080706.093309 [DOI] [PubMed] [Google Scholar]
- 28.Pays E, Nolan DP. 1998. Expression and function of surface proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 91:3–36. 10.1016/S0166-6851(97)00183-7 [DOI] [PubMed] [Google Scholar]
- 29.Rolin S, Paindavoine P, Hanocq-Quertier J, Hanocq F, Claes Y, Le Ray D, Overath P, Pays E. 1993. Transient adenylate cyclase activation accompanies differentiation of Trypanosoma brucei from bloodstream to procyclic forms. Mol. Biochem. Parasitol. 61:115–125. 10.1016/0166-6851(93)90164-S [DOI] [PubMed] [Google Scholar]
- 30.Rolin S, Hanocq-Quertier J, Paturiaux-Hanocq F, Nolan D, Salmon D, Webb H, Carrington M, Voorheis P, Pays E. 1996. Simultaneous but independent activation of adenylate cyclase and glycosylphosphatidylinositol-phospholipase C under stress conditions in Trypanosoma brucei. J. Biol. Chem. 271:10844–10852. 10.1074/jbc.271.18.10844 [DOI] [PubMed] [Google Scholar]
- 31.Vassella E, Reuner B, Yutzy B, Boshart M. 1997. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell Sci. 110(Pt 21):2661–2671 [DOI] [PubMed] [Google Scholar]
- 32.Gould MK, de Koning HP. 2011. Cyclic-nucleotide signalling in protozoa. FEMS Microbiol. Rev. 35:515–541. 10.1111/j.1574-6976.2010.00262.x [DOI] [PubMed] [Google Scholar]
- 33.Alexandre S, Paindavoine P, Hanocq-Quertier J, Paturiaux-Hanocq F, Tebabi P, Pays E. 1996. Families of adenylate cyclase genes in Trypanosoma brucei. Mol. Biochem. Parasitol. 77:173–182. 10.1016/0166-6851(96)02591-1 [DOI] [PubMed] [Google Scholar]
- 34.Naula C, Schaub R, Leech V, Melville S, Seebeck T. 2001. Spontaneous dimerization and leucine-zipper induced activation of the recombinant catalytic domain of a new adenylyl cyclase of Trypanosoma brucei, GRESAG4.4B. Mol. Biochem. Parasitol. 112:19–28. 10.1016/S0166-6851(00)00338-8 [DOI] [PubMed] [Google Scholar]
- 35.Oberholzer M, Lopez MA, Ralston KS, Hill KL. 2009. Approaches for functional analysis of flagellar proteins in African trypanosomes. Methods Cell Biol. 93:21–57. 10.1016/S0091-679X(08)93002-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wirtz E, Leal S, Ochatt C, Cross GA. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99:89–101. 10.1016/S0166-6851(99)00002-X [DOI] [PubMed] [Google Scholar]
- 37.LaCount DJ, Barrett B, Donelson JE. 2002. Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J. Biol. Chem. 277:17580–17588. 10.1074/jbc.M200873200 [DOI] [PubMed] [Google Scholar]
- 38.Kubata BK, Duszenko M, Kabututu Z, Rawer M, Szallies A, Fujimori K, Inui T, Nozaki T, Yamashita K, Horii T, Urade Y, Hayaishi O. 2000. Identification of a novel prostaglandin f(2alpha) synthase in Trypanosoma brucei. J. Exp. Med. 192:1327–1338. 10.1084/jem.192.9.1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tagwerker C, Flick K, Cui M, Guerrero C, Dou Y, Auer B, Baldi P, Huang L, Kaiser P. 2006. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivo cross-linking. Mol. Cell. Proteomics 5:737–748. 10.1074/mcp.M500368-MCP200 [DOI] [PubMed] [Google Scholar]
- 40.Wohlschlegel JA. 2009. Identification of SUMO-conjugated proteins and their SUMO attachment sites using proteomic mass spectrometry. Methods Mol. Biol. 497:33–49. 10.1007/978-1-59745-566-4_3 [DOI] [PubMed] [Google Scholar]
- 41.Tabb DL, McDonald WH, Yates JR., III 2002. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1:21–26. 10.1021/pr015504q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yates JR, III, Eng JK, McCormack AL, Schieltz D. 1995. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67:1426–1436. 10.1021/ac00104a020 [DOI] [PubMed] [Google Scholar]
- 43.Cociorva D, Tabb DL, Yates JR. 2007. Validation of tandem mass spectrometry database search results using DTASelect. Curr. Protoc. Bioinformatics Chapter 13:Unit 13.4. 10.1002/0471250953.bi1304s16 [DOI] [PubMed] [Google Scholar]
- 44.Oberholzer M, Morand S, Kunz S, Seebeck T. 2006. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol. Biochem. Parasitol. 145:117–120. 10.1016/j.molbiopara.2005.09.002 [DOI] [PubMed] [Google Scholar]
- 45.Kabututu ZP, Thayer M, Melehani JH, Hill KL. 2010. CMF70 is a subunit of the dynein regulatory complex. J. Cell Sci. 123:3587–3595. 10.1242/jcs.073817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Redmond S, Vadivelu J, Field MC. 2003. RNAit: an automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol. Biochem. Parasitol. 128:115–118. 10.1016/S0166-6851(03)00045-8 [DOI] [PubMed] [Google Scholar]
- 47.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134. 10.1186/1471-2105-13-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brenndorfer M, Boshart M. 2010. Selection of reference genes for mRNA quantification in Trypanosoma brucei. Mol. Biochem. Parasitol. 172:52–55. 10.1016/j.molbiopara.2010.03.007 [DOI] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 50.Tyler KM, Fridberg A, Toriello KM, Olson CL, Cieslak JA, Hazlett TL, Engman DM. 2009. Flagellar membrane localization via association with lipid rafts. J. Cell Sci. 122:859–866. 10.1242/jcs.037721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dubacq C, Guerois R, Courbeyrette R, Kitagawa K, Mann C. 2002. Sgt1p contributes to cyclic AMP pathway activity and physically interacts with the adenylyl cyclase Cyr1p/Cdc35p in budding yeast. Eukaryot. Cell 1:568–582. 10.1128/EC.1.4.568-582.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morishita T, Matsuura A, Uno I. 1993. Characterization of the cyr1-2 UGA mutation in Saccharomyces cerevisiae. Mol. Gen. Genet. 237:463–466 [DOI] [PubMed] [Google Scholar]
- 54.Gietz D, St Jean A, Woods RA, Schiestl RH. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20:1425. 10.1093/nar/20.6.1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salmon D, Bachmaier S, Krumbholz C, Kador M, Gossmann JA, Uzureau P, Pays E, Boshart M. 2012. Cytokinesis of Trypanosoma brucei bloodstream forms depends on expression of adenylyl cyclases of the ESAG4 or ESAG4-like subfamily. Mol. Microbiol. 84:225–242. 10.1111/j.1365-2958.2012.08013.x [DOI] [PubMed] [Google Scholar]
- 56.Emmer BT, Maric D, Engman DM. 2010. Molecular mechanisms of protein and lipid targeting to ciliary membranes. J. Cell Sci. 123:529–536. 10.1242/jcs.062968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walsh CT. 2006. Posttranslational modification of proteins: expanding nature's inventory. Roberts and Co. Publishers, Englewood, CO [Google Scholar]
- 58.Dhalia R, Reis CR, Freire ER, Rocha PO, Katz R, Muniz JR, Standart N, de Melo Neto OP. 2005. Translation initiation in Leishmania major: characterisation of multiple eIF4F subunit homologues. Mol. Biochem. Parasitol. 140:23–41. 10.1016/j.molbiopara.2004.12.001 [DOI] [PubMed] [Google Scholar]
- 59.Bieger B, Essen LO. 2001. Structural analysis of adenylate cyclases from Trypanosoma brucei in their monomeric state. EMBO J. 20:433–445. 10.1093/emboj/20.3.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Linder JU. 2006. Class III adenylyl cyclases: molecular mechanisms of catalysis and regulation. Cell. Mol. Life Sci. 63:1736–1751. 10.1007/s00018-006-6072-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vaughan S. 2010. Assembly of the flagellum and its role in cell morphogenesis in Trypanosoma brucei. Curr. Opin. Microbiol. 13:453–458. 10.1016/j.mib.2010.05.006 [DOI] [PubMed] [Google Scholar]
- 62.Rotureau B, Van Den Abbeele J. 2013. Through the dark continent: African trypanosome development in the tsetse fly. Front. Cell. Infect. Microbiol. 3:53. 10.3389/fcimb.2013.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Broadhead R, Dawe HR, Farr H, Griffiths S, Hart SR, Portman N, Shaw MK, Ginger ML, Gaskell SJ, McKean PG, Gull K. 2006. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 440:224–227. 10.1038/nature04541 [DOI] [PubMed] [Google Scholar]
- 64.Chan KY, Matthews KR, Ersfeld K. 2010. Functional characterisation and drug target validation of a mitotic kinesin-13 in Trypanosoma brucei. PLoS Pathog. 6:e1001050. 10.1371/journal.ppat.1001050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu W, Apagyi K, McLeavy L, Ersfeld K. 2010. Expression and cellular localisation of calpain-like proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 169:20–26. 10.1016/j.molbiopara.2009.09.004 [DOI] [PubMed] [Google Scholar]
- 66.Bloodgood RA. 2012. The future of ciliary and flagellar membrane research. Mol. Biol. Cell 23:2407–2411. 10.1091/mbc.E12-01-0073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kottgen M, Walz G. 2005. Subcellular localization and trafficking of polycystins. Pflugers Arch. 451:286–293. 10.1007/s00424-005-1417-3 [DOI] [PubMed] [Google Scholar]
- 68.Kim J, Kato M, Beachy PA. 2009. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. U. S. A. 106:21666–21671. 10.1073/pnas.0912180106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goodenough UW, Adair WS, Caligor E, Forest CL, Hoffman JL, Mesland DA, Spath S. 1980. Membrane-membrane and membrane-ligand interactions in Chlamydomonas mating. Soc. Gen. Physiol. Ser. 34:131–152 [PubMed] [Google Scholar]
- 70.Pan J, Snell WJ. 2000. Signal transduction during fertilization in the unicellular green alga, Chlamydomonas. Curr. Opin. Microbiol. 3:596–602. 10.1016/S1369-5274(00)00146-6 [DOI] [PubMed] [Google Scholar]
- 71.Dwyer MA, Hellinga HW. 2004. Periplasmic binding proteins: a versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 14:495–504. 10.1016/j.sbi.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 72.Buxton IL, Brunton LL. 1983. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 258:10233–10239 [PubMed] [Google Scholar]
- 73.Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, Karpen JW. 2000. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 116:147–161. 10.1085/jgp.116.2.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oberholzer M, Bregy P, Marti G, Minca M, Peier M, Seebeck T. 2007. Trypanosomes and mammalian sperm: one of a kind? Trends Parasitol. 23:71–77. 10.1016/j.pt.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 75.Oberholzer M, Marti G, Baresic M, Kunz S, Hemphill A, Seebeck T. 2007. The Trypanosoma brucei cAMP phosphodiesterases TbrPDEB1 and TbrPDEB2: flagellar enzymes that are essential for parasite virulence. FASEB J. 21:720–731. 10.1096/fj.06-6818com [DOI] [PubMed] [Google Scholar]
- 76.Fenn K, Matthews KR. 2007. The cell biology of Trypanosoma brucei differentiation. Curr. Opin. Microbiol. 10:539–546. 10.1016/j.mib.2007.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gray MA, Cunningham I, Gardiner PR, Taylor AM, Luckins AG. 1981. Cultivation of infective forms of Trypanosoma congolense from trypanosomes in the proboscis of Glossina morsitans. Parasitology 82:81–95. 10.1017/S0031182000041883 [DOI] [PubMed] [Google Scholar]
- 78.Mesland DA, Hoffman JL, Caligor E, Goodenough UW. 1980. Flagellar tip activation stimulated by membrane adhesions in Chlamydomonas gametes. J. Cell Biol. 84:599–617. 10.1083/jcb.84.3.599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peacock L, Bailey M, Carrington M, Gibson W. 2014. Meiosis and haploid gametes in the pathogen Trypanosoma brucei. Curr. Biol. 24:181–186. 10.1016/j.cub.2013.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bloodgood RA. 2010. Sensory reception is an attribute of both primary cilia and motile cilia. J. Cell Sci. 123:505–509. 10.1242/jcs.066308 [DOI] [PubMed] [Google Scholar]
- 81.de Miguel N, Riestra A, Johnson PJ. 2012. Reversible association of tetraspanin with Trichomonas vaginalis flagella upon adherence to host cells. Cell. Microbiol. 14:1797–1807. 10.1111/cmi.12003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moreira-Leite FF, Sherwin T, Kohl L, Gull K. 2001. A trypanosome structure involved in transmitting cytoplasmic information during cell division. Science 294:610–621. 10.1126/science.1063775 [DOI] [PubMed] [Google Scholar]
- 83.Hughes L, Towers K, Starborg T, Gull K, Vaughan S. 2013. A cell-body groove housing the new flagellum tip suggests an adaptation of cellular morphogenesis for parasitism in the bloodstream form of Trypanosoma brucei. J. Cell Sci. 126:5748–5757. 10.1242/jcs.139139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pazour G, Bloodgood R. 2008. Targeting proteins to the ciliary membrane. Curr. Top. Dev. Biol. 85:115–149. 10.1016/S0070-2153(08)00805-3 [DOI] [PubMed] [Google Scholar]
- 85.Dorn KV, Hughes CE, Rohatgi R. 2012. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 23:823–835. 10.1016/j.devcel.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blacque OE, Sanders AA. 2014. Compartments within a compartment: what C. elegans can tell us about ciliary subdomain composition, biogenesis, function, and disease. Organogenesis 10:126–137. 10.4161/org.28830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cevik S, Sanders AA, Van Wijk E, Boldt K, Clarke L, van Reeuwijk J, Hori Y, Horn N, Hetterschijt L, Wdowicz A, Mullins A, Kida K, Kaplan OI, van Beersum SE, Man Wu K, Letteboer SJ, Mans DA, Katada T, Kontani K, Ueffing M, Roepman R, Kremer H, Blacque OE. 2013. Active transport and diffusion barriers restrict Joubert syndrome-associated ARL13B/ARL-13 to an Inv-like ciliary membrane subdomain. PLoS Genet. 9:e1003977. 10.1371/journal.pgen.1003977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pazour GJ, Bloodgood RA. 2008. Targeting proteins to the ciliary membrane. Curr. Top. Dev. Biol. 85:115–149. 10.1016/S0070-2153(08)00805-3 [DOI] [PubMed] [Google Scholar]
- 89.Nachury MV, Seeley ES, Jin H. 2010. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu. Rev. Cell Dev. Biol. 26:59–87. 10.1146/annurev.cellbio.042308.113337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chung WL, Carrington M, Field MC. 2004. Cytoplasmic targeting signals in transmembrane invariant surface glycoproteins of trypanosomes. J. Biol. Chem. 279:54887–54895. 10.1074/jbc.M409311200 [DOI] [PubMed] [Google Scholar]
- 91.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, Gardner MJ, Gingle A, Grant G, Harb OS, Heiges M, Hertz-Fowler C, Houston R, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Logan FJ, Miller JA, Mitra S, Myler PJ, Nayak V, Pennington C, Phan I, Pinney DF, Ramasamy G, Rogers MB, Roos DS, Ross C, Sivam D, Smith DF, Srinivasamoorthy G, Stoeckert CJ, Jr, Subramanian S, Thibodeau R, Tivey A, Treatman C, Velarde G, Wang H. 2010. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 38:D457–D462. 10.1093/nar/gkp851 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.