

Abstract
Botulinum neurotoxin A is a category A bioterrorism agent. Current antitoxin therapies are scarce and produce adverse reactions. XOMA 3AB consists of 3 IgG1 monoclonal antibodies (MAbs), each with a distinct human or humanized variable region, which bind to distinct epitopes on botulinum neurotoxin serotype A. This first-in-human study evaluated the safety and pharmacokinetics (PK) of escalating doses of XOMA 3AB administered intravenously (i.v.) to healthy adults. In this double-blind placebo-controlled dose escalation study, 3 cohorts of 8 healthy subjects received a single intravenous dose of XOMA 3AB or placebo at a 3:1 ratio. Follow-up examinations included physical examinations, hematology and chemistry blood tests, electrocardiograms, and pharmacokinetics. Pharmacokinetic parameters were estimated using noncompartmental methods. There were no infusion discontinuations or hypersensitivity reactions. Two or more subjects experienced headache, hyperglycemia, or anemia; none was dose related. All adverse events (AEs) were mild to moderate except for an episode of exercise-induced elevation of a subject's creatine phosphokinase (CPK) level, unrelated to XOMA 3AB. Concentration-time plots demonstrated a peak in MAb concentrations 1 to 2 h after completion of the infusion, after which the levels declined in a biexponential decay pattern for all analytes. For each MAb, the maximum concentration of drug in serum (Cmax) and the area under the concentration-time curve from 0 to infinity (AUCinf) increased as the dose increased. Clearance of the humanized mouse MAb was more rapid than that of the two fully human MAbs, particularly at the lowest dose. None of the MAbs was immunogenic. At the doses administered, XOMA 3AB was well tolerated. These safety findings support further investigation of XOMA 3AB as a potential agent for botulism treatment and postexposure prophylaxis. (This study has been registered at ClinicalTrials.gov under registration no. NCT01357213.)
INTRODUCTION
Botulism is a class A bioterror threat caused by the obligately anaerobic Gram-positive spore-forming bacterium Clostridium botulinum. This organism expresses eight botulism neurotoxin (BoNT) serotypes, A, B, C, D, E, F, G, and H (1, 2). Serotypes A, B, E, F, and H cause naturally occurring human disease, of which there are three types, food borne, wound, and intestinal. Sporadic intestinal cases in adults are usually associated with ingestion of the toxin from home-prepared and improperly processed foods (3); intoxication in infants is associated with the ingestion of C. botulinum spores and bacterial colonization of the intestines. The diagnosis of botulism is made clinically and is confirmed by either direct identification and/or serotyping of the toxin or isolation of the pathogen. Common presenting symptoms of all forms of the disease include diplopia, dysarthria, and dry mouth, followed by progressive symmetric descending weakness or paralysis. Left untreated, death can occur within 2 weeks (4).
BoNTs are classified as category A biothreats; aerosolized BoNT leads to inhalational botulism, a potential bioterrorism weapon (3, 5). The BoNT/A serotype family, containing BoNT subtypes A1, A2, A3, A4, and A5, is the most potent of all serotypes and the one that most commonly intoxicates humans (6, 7). BoNT/A may be the most likely to be used as a biothreat due to its potency, ease of production, and long duration of action. There have already been several attempts to use botulism as a bioweapon; members of the Japanese cult Aum Shinrikyo dispersed aerosols at a number of sites in downtown Tokyo in 1990 and 1995, and the Iraqi government loaded 10,000 liters of concentrated toxin into military weapons after the 1991 Persian Gulf War (5). Large-scale toxin exposure could cause significant mortality and morbidity. Epidemiologic modeling suggests that an aerosol release over a metropolitan area with exposure to 100,000 individuals would lead to 50,000 botulism cases, 30,000 fatalities, and $8.6 billion in estimated costs (8). Treatment of an exposed population would require rapid mobilization and administration of therapy that is effective, nontoxic, and easily administered.
The current primary treatment for botulism is antitoxin (9). Minute quantities of human botulism immunoglobulin, produced by plasmapheresis of laboratory workers who were immunized with an investigational toxoid vaccine, are available to treat infant botulism; however, large-scale manufacture of this product is impossible (10). Equine BONT/A and BONT/B antitoxins [F(ab′)2 fragments] can be used to treat adult botulism, but they have short half-lives and an approximately 10% chance of causing severe acute allergic reactions and late-onset serum sickness, making them inappropriate for prophylactic use (11). Additionally, antibodies to the equine F(ab′)2 fragments quickly form and limit treatment to a single use.
Traditional antitoxins are not easily produced, as they require immunization of animals or humans, plasmapheresis or bleeding, and processing of serum for each lot. Furthermore, each lot differs in its antibody composition, potency, and, possibly, safety profile. The development of monoclonal antibodies (MAbs) that can be produced on a large scale and at high quality has revolutionized therapeutics development. Human and humanized MAbs can provide an essentially unlimited supply of botulinum antitoxin free of any infectious risk. Previous work found that no single MAb neutralizes BoNT/A with a potency of >1,000 mouse 50% lethal doses (LD50s)/mg of antibody (12, 13). However, combining three MAbs that each bind nonoverlapping epitopes results in highly potent BoNT neutralization due to multiple mechanisms, including an increase in the functional binding affinity of the Ab mixture for toxin (12), blockade of multiple epitopes on the toxin-binding domain surface that bind to cellular receptors (12), and first-pass hepatic clearance of the immune complexes (12). XOMA 3AB was developed as a potential therapeutic for the treatment of BoNT/A disease. XOMA 3AB is an equimolar mixture of three IgG1 MAbs, referred to as Aa, Ab, and Ac (Table 1), that target different regions of BoNT/A. Each MAb has been engineered to have distinct human or humanized variable regions that bind BoNT/A subtypes A1, A2, A3, and A4. The MAbs have a common human light and heavy chain constant region and are individually expressed by separate stably transfected Chinese hamster ovary cell lines.
TABLE 1.
Characteristics of the monoclonal antibodies that comprise XOMA 3AB
| Antibody | Origin | No. of amino acids | Molecular mass (Da) |
|---|---|---|---|
| Aa | Human | 1,336 | 146,568 |
| Ac | Human | 1,342 | 146,122 |
| Ab | Mouse/human | 1,332 | 146,480 |
Aa and Ac were derived from a human phage library and are composed of 1,336 amino acid residues (molecular mass, 146 kDa) and 1,342 amino acid residues (molecular mass, 146 kDa), respectively. Ab is a humanized mouse MAb composed of 1,332 amino acid residues with a molecular mass of 146 kDa. The combination of the three antibodies was a potent neutralizer of BoNT/A in the mouse neutralization assay (MNA) and demonstrated 100% protection against 40,000 mouse 50% lethal dose (LD50) units of BoNT/A1 and BoNT/A2. In vivo studies in mice found that the highest level of potency was dependent on the presence of all three antibodies (12, 14). Preclinical toxicology studies with Sprague-Dawley rats and cynomolgus monkeys showed no significant changes in clinical findings, serum chemistry, hematology, urinalysis, or histopathology (rats only) at a dose of 50 mg/kg of body weight intravenously (i.v.). This product therefore shows promise for human use as treatment and postexposure prophylaxis.
MATERIALS AND METHODS
We performed an inpatient first-in-human double-blinded single-center placebo-controlled dose escalation study to evaluate the safety, pharmacokinetic characteristics, and immunogenicity of XOMA 3AB at doses of 0.033 (cohort A), 0.165 (cohort B), and 0.33 mg/kg (cohort C). The starting dose was calculated based on the no-observable-adverse-effect-level (NOAEL) values from rat toxicity studies and on the projected effective human dose (14). The placebo was normal saline.
Subjects.
The study enrolled 24 volunteers. Eligible research participants were healthy adults who met the NIH healthy volunteer policy criteria (15). Briefly, healthy research participants were 18 to 45 years old, had a body mass index of <35, had a negative illicit drug screen, and, if sexually active, were using adequate contraception. Exclusion criteria included history of a chronic medical condition, severe allergic reaction to any medication, pregnancy, and active drug or alcohol dependence. Research participants who had received any monoclonal antibody in the past, any antibody or blood products in the 6 months before study day 0, any live vaccine in the previous 3 months, or any killed vaccine or prescription medication other than oral contraceptives in the previous 30 days were also excluded. Additional exclusion criteria were abnormal findings on standard chemistry, hematology, and urinalysis profiles; positive serology for hepatitis B, hepatitis C, or HIV; prolongation of the QTc interval on electrocardiography; or previous exposure to botulinum toxin.
Study design.
We randomized research participants in three sequential dose escalation cohorts of eight participants each. Within each dosing cohort, research participants were randomized to receive either active drug or placebo at an overall 3:1 ratio. This study was conducted between 22 June 2011 and 24 May 2012 in the Clinical Research Unit of Johns Hopkins University. The study was approved by the Johns Hopkins University Institutional Review Board, and written informed consent was obtained from all participants prior to study screening.
Research participants were asked to refrain from using nonstudy medications or herbal supplements during the study and were instructed not to drink caffeine-containing products for 24 h prior to and during the inpatient visit and to avoid alcohol from 24 h prior to study product receipt through day 7.
The study drug was manufactured by the XOMA Corporation and filled by Althea Technologies (San Diego, CA). Assays developed for the characterization and release of a single MAb drug substance have been qualified, including two biological tests, a plate-based enzyme-linked immunosorbent assay (ELISA) binding assay and an in vitro single-MAb protection assay (time-to-death assay [MTTDA]). The ELISAs were developed and qualified at XOMA with the use of nontoxic antibody-specific botulinum protein domains (16), whereas the MTTDAs were developed and qualified in collaboration with SRI International (Menlo Park, CA). XOMA produced XOMA 3AB as an i.v. formulation for testing in human clinical trials. Quality control testing and release were performed at XOMA and SRI International. Drug product assays also have been developed and qualified, including two biological tests, a plate-based ELISA binding assay for the individual MAbs in the mixture (16) and an in vivo triple-antibody protection assay. The ELISAs were developed and qualified at XOMA; the protection assays were developed and qualified in collaboration with SRI International. The investigational product was formulated in a pH 6.0 buffered vehicle to achieve a strength of 5 mg/ml with an equal molar ratio of each antibody component (∼1.7 mg/ml of each MAb) and was supplied as a sterile liquid in 2-ml single-use vials.
Active drug and placebo infusions were prepared in similar packaging by the Johns Hopkins Investigational Drug Service Pharmacy by a nonblinded pharmacist. For each dosing cohort, the first two research participants were randomized in a 1:1 fashion to ensure that one of the first two participants received active treatment and the other received the placebo. The product assignments for the remaining six research participants in each cohort were a simple random sample to ensure the 3:1 ratio for the dosing cohort.
On the day before dosing, research participants were admitted to the Johns Hopkins Inpatient Clinical Research Unit. A complete medical history and physical examination were performed, and samples were collected for complete blood count, chemistries, urinalysis, and pregnancy testing. The following morning, they received a single intravenous infusion of XOMA 3AB or placebo administered over 1 h. Infusions were observed by a study physician, and vital signs were closely monitored. Electrocardiograms were obtained following infusion. Research participants were discharged the next day and followed with outpatient visits for 90 days (cohort A) or 120 days (cohorts B and C). The safety monitoring committee reviewed data through day 7 before escalation to the next dosing cohort was permitted.
Safety analyses.
Adverse events (AEs) were assessed by interviews for symptoms, physical examinations, and analyses of clinical analytes from the time of study product administration through day 90 for cohort A or through day 120 for cohorts B and C. AEs were graded, using a standard table (see Table S1 in the supplemental material), as mild (grade 1, transient or mild discomfort, no medical intervention required), moderate (grade 2, mild to moderate limitation in activity, minimal medical intervention required), severe (grade 3, marked limitation in activity, medical intervention required), or life-threatening (grade 4, extreme limitation in activity, hospitalization likely). The relationship of AEs to the study drug was categorized as possibly associated or not associated based on the temporal relationship of the event to administration of the study product and on the identification of an alternative etiology and biological plausibility.
Laboratory analyses were conducted at the Johns Hopkins Hospital clinical laboratory. Safety laboratory tests included a complete blood count with differential (hemoglobin, hematocrit, total leukocyte count, leukocyte differential, and platelets), a comprehensive metabolic panel (sodium, potassium, chloride, carbon dioxide, blood urea nitrogen, creatinine, calcium, total bilirubin, alanine aminotransferase [ALT], aspartate aminotransferase [AST], and nonfasting glucose), and a urinalysis (protein, blood, pH, and specific gravity). Laboratory test results found to be outside the normal limits were reported as AEs.
Pharmacokinetic and immunogenicity analysis.
Blood for pharmacokinetic analysis was collected at baseline, at the end of the infusion (hour 1), 2, 4, 8, 24, 48, and 72 h following the infusion, and at days 7, 14, 28, 42, 56, and 90 for all three cohorts and at day 120 for cohorts B and C. The shorter period of sampling for cohort A was specified in the protocol and based on the low dose given to members of that cohort.
Three immunoassays were developed to measure the concentrations of Aa, Ab, and Ac in the serum samples. A bridging electrochemiluminescence (ECL) assay was used (16). These immunoassays used the bivalent binding capabilities of the antibodies to form immune complexes with their nonoverlapping BoNT/A domains for detection. A mixture of biotinylated and ruthenylated (Sulfo-Tag) bacterial expressed domains, specific to each of the three MAbs, Aa, Ab, or Ac (heavy-chain C terminus [HCC], heavy-chain N terminus [HCN], or light-chain heavy-chain N terminus [LCHN], respectively), was used in each assay. ECL signals generated from captured immune complexes formed when one arm of an MAb bound to a biotinylated domain and the other arm to a ruthenium-conjugated domain were detected with use of a Meso-Scale Discovery (MSD) SECTOR Imager 6000 and reported in ECL units. Calibration standards and quality control samples were prepared by spiking known amounts of each MAb into human serum. The concentration of each MAb in the quality control and study samples was calculated using calibration curves. With the minimum required dilution of 1:10, the lower limit of quantification (LLOQ) for the Aa assay was 10 ng/ml, with a quantifiable range of 10 to 2,000 ng/ml. The LLOQ for Ab and that for Ac were 80 and 20 ng/ml, respectively, with ranges of 80 to 1,100 and 20 to 500 ng/ml, respectively. MAb Ab was of mouse origin and of lower affinity, which accounted for its higher LLOQ than the two fully human MAbs (12).
Blood samples for immunogenicity analysis were drawn at study day 0, 28, 56, 90, and 120. Immunoassays were developed and validated to detect anti-Aa, anti-Ab, and anti-Ac antibodies in human serum. Detection of anti-drug antibodies (ADA) was done with a tiered process consisting of assays for screening, confirmation, and titer. Bridging ECL assays use labeled MAbs for the capture and detection of ADA in serum. A mixture of biotinylated and ruthenylated Aa, Ab, and Ac MAbs was incubated in each assay. ECL signals were generated when one arm of an ADA bound to a biotinylated MAb while the other arm bound a ruthenium-conjugated MAb. Samples that generated a signal above the screening cut point (SCP) were considered to be potentially positive. Positive samples were confirmed with the use of a competitive assay in which serum samples were incubated with and without nonlabeled MAbs. Samples with ECL signals above the confirmation cut point were considered to have ADA and were analyzed further for titers by determining the dilution that brought the ECL signal to the SCP.
Statistical analyses.
Categorical variables were summarized as numbers and percentages of subjects and continuous data with standard deviations, means, and medians. Pharmacokinetic (PK) parameters included area under the concentration-time curve (AUC), the maximum observed concentration (Cmax), the time to maximum concentration (Tmax), the elimination rate constant (kel), unweighted parameter estimation after exploration of 1/y and 1/y2 (found not to improve the goodness of fit over no weighting), the elimination half-life (t1/2), clearance, and volume of distribution. All parameters were estimated by noncompartmental methods using WinNonLin (version 6.3; Pharsight Inc, Cary, NC). All PK analyses were done for each MAb separately for each dosing cohort.
RESULTS
Demographic characteristics.
Twenty four research participants were enrolled, randomized, and administered the study product or the placebo. Two participants discontinued the study. One research participant from cohort A moved out of the state, and one from cohort B was lost to follow up due to incarceration. These two research participants were determined later to have received the active drug product. Eighteen research participants, six per cohort, received XOMA 3AB (0.033 mg/kg, 0.165 mg/kg, or 0.33 mg/kg); two research participants per cohort, six in total, received placebo.
The research participants' ages ranged from 20 to 43 years. The majority of the research participants were white (15 [62.5%]), of non-Hispanic or Latino ethnicity (23 [95.8%]), and male (14 [58.3%]). The demographic characteristics were similar across the treatment groups, except for cohort C, in which all participants who received the active study product were male. The demographic characteristics are summarized in Table 2.
TABLE 2.
Demographics of the study population
| Characteristic | Dataa for: |
||||
|---|---|---|---|---|---|
| Subjects receiving XOMA 3AB at a dosage (mg/kg) of: |
Subjects receiving placebo (combined) (n = 6) | All subjects (n = 24) | |||
| 0.033 (n = 6) | 0.165 (n = 6) | 0.330 (n = 6) | |||
| Age (yr) | |||||
| Mean (SD) | 33.5 (8.7) | 30.2 (6.4) | 29.2 (3.2) | 32.2 (6.6) | 31.3 (6.3) |
| Median | 36.0 | 28.0 | 30.0 | 34.0 | 30.0 |
| Min, Maxb | 20, 42 | 26, 43 | 24, 32 | 23, 39 | 20, 43 |
| Gender | |||||
| Male | 4 (66.7) | 1 (16.7) | 6 (100) | 4 (66.7) | 15 (62.5) |
| Female | 2 (33.3) | 5 (83.3) | 0 (0) | 2 (33.3) | 9 (37.5) |
| Race | |||||
| White | 2 (33.3) | 4 (66.7) | 5 (83.3) | 3 (50.0) | 14 (58.3) |
| Black or African American | 3 (50.0) | 1 (16.7) | 1 (16.7) | 3 (50.0) | 8 (33.3) |
| American Indian/Alaskan Native | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Native Hawaiian or other Pacific Islander | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Asian | 1 (16.7) | 1 (16.7) | 0 (0) | 0 (0) | 2 (8.3) |
| Other | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Ethnicity | |||||
| Hispanic or Latino | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 1 (4.2) |
| Non-Hispanic or Latino | 6 (100) | 6 (100) | 6 (100) | 5 (83.3) | 23 (95.8) |
Data are no. (%) unless stated otherwise.
Min, minimum; Max, maximum.
Safety profile.
Treatment emergent adverse events (AEs) were reported by 23 of the research participants (95.8%), 17 who received the active drug and all 6 who received the placebo. A summary of the AEs is presented in Table 3. There were no serious AEs or deaths, and no AEs occurred during infusion of the study drug. AEs between the three cohorts of treatment groups and placebo recipients were similar in frequency and severity. The highest number of AEs was seen in cohort A. The AEs most frequently reported were increased blood glucose (9 [37.5%]), headache (8 [33.3%]), nasopharyngitis (6 [25%]), and increased ALT level (5 [20.8%]). The majority of the AEs were classified as grade 1 in severity (121 [87.1%]), 17 (12.2%) were grade 2, and one (0.7%) was classified as grade 4. The grade 4 AE, an increased AST level from day 7 to day 14, occurred in the 0.33-mg/kg treatment group. After this result was reported, the research participant was contacted and brought in for evaluation. He reported having started an intensive physical exercise regimen 3 days into his study participation, including several hours per day of aerobic exercise and weight lifting, and at his day 7 study visit, he reported mild myalgias. At the reevaluation visit, his creatine phosphokinase and aldolase levels were elevated to 31,060 IU and 770 U, respectively. His gamma glutamyl transpeptidase level was normal, and hepatitis serologies were negative. With rest, oral fluids, and no other intervention by the study team, his myalgia symptoms resolved, and his AST level returned to normal within 7 days. Given the temporal relation to the exercise regimen and normalization without significant intervention, we concluded that this adverse event was not related to the study product and that the clinical diagnosis was exercise-induced myolysis (17, 18).
TABLE 3.
Adverse events with an overall rate of ≥10%
| Adverse event | Data (no. [%]) for: |
|||||
|---|---|---|---|---|---|---|
| Subjects receiving XOMA 3AB at a dosage (mg/kg) of: |
Subjects receiving placebo (combined) (n = 6) | All subjects (n = 24) | ||||
| 0.033 (n = 6) | 0.165 (n = 6) | 0.330 (n = 6) | Combined (n = 18) | |||
| Blood glucose increased | 1 (16.7) | 3 (50.0) | 1 (16.7) | 5 (27.8) | 4 (66.7) | 9 (37.5) |
| Headache | 3 (50.0) | 3 (50.0) | 1 (16.7) | 7 (38.9) | 1 (16.7) | 8 (33.3) |
| Nasopharyngitis | 1 (16.7) | 4 (66.7) | 0 (0) | 5 (27.8) | 1 (16.7) | 6 (25.0) |
| ALT increased | 1 (16.7) | 0 (0) | 2 (33.3) | 3 (16.7) | 2 (33.3) | 5 (20.8) |
| AST increased | 1 (16.7) | 0 (0) | 1 (16.7) | 2 (11.1) | 2 (33.3) | 4 (16.7) |
| Hemoglobin decreased | 1 (16.7) | 1 (16.7) | 1 (16.7) | 3 (16.7) | 1 (16.7) | 4 (16.7) |
| Upper respiratory tract infection | 0 (0) | 1 (16.7) | 1 (16.7) | 2 (11.1) | 2 (33.3) | 4 (16.7) |
| CO2 decreased | 1 (16.7) | 1 (16.7) | 1 (16.7) | 3 (16.7) | 0 (0) | 3 (12.5) |
| Muscle strain | 0 (0) | 0 (0) | 2 (33.3) | 2 (11.1) | 1 (16.7) | 3 (12.5) |
| Myalgia | 0 (0) | 0 (0) | 2 (33.3) | 2 (11.1) | 1 (16.7) | 3 (12.5) |
Most of the AEs were considered not associated with the study drug (total, 123 [88.5%]). Sixteen AEs (11.5%), the majority of which occurred in cohort A, were considered possibly associated with the study drug. Increased AST (2 [8.3%]) and ALT (2 [8.3%]) levels were the most commonly reported events that were possibly associated with the study drug.
Pharmacokinetic analysis.
A summary of the pharmacokinetic data is presented in Table 4. Peak concentrations generally occurred 1 to 2 h after the end of the 1-h infusion in most of the research participants. In one participant, concentrations increased over the 8 h after the end of the infusion. After the peak, the concentrations of all three antibodies declined biexponentially (Fig. 1). Ac and Aa were detected in plasma for >28 days at all doses and for >45 days at the highest doses. Ab also was detected >28 days after infusion of the higher two doses but for <7 days after infusion of the lowest dose.
TABLE 4.
Summary of PK dataa
| MAb | Cohort | Tmax (day) | Cmax (μg/ml) | AUCinf (μg/day/ml) | t1/2 (days) | Vzb (ml/kg) | CLc (ml/day/kg) |
|---|---|---|---|---|---|---|---|
| Aa | A | 0.05 (0.046, 0.079) | 0.23 (0.23, 0.24) | 2.08 (2.04, 2.1) | 8.9 (8.8, 10.1) | 69 (67, 77) | 5.3 (5.3, 5.5) |
| B | 0.09 (0.084, 0.148) | 1.2 (1.1, 1.23) | 11.63 (9.91, 16.3) | 12.4 (10, 14.1) | 92 (72, 97) | 4.7 (3.4, 5.6) | |
| C | 0.085 (0.083, 0.094) | 2.35 (2.27, 2.88) | 22.09 (20.4, 30.06) | 12.3 (11.3, 13.7) | 88 (82, 93) | 4.9 (3.7, 5.4) | |
| Ab | A | 0.046 (0.046, 0.077) | 0.2 (0.19, 0.21) | 0.66 (0.62, 0.79) | 2.5 (2.3, 2.8) | 59 (56, 65) | 16.6 (14.1, 17.6) |
| B | 0.129 (0.057, 0.17) | 1.16 (1.13, 1.24) | 9.42 (6.86, 12.97) | 12.8 (7.8, 14) | 85 (81, 105) | 5.9 (4.3, 8.2) | |
| C | 0.085 (0.083, 0.094) | 2.62 (2.07, 2.93) | 18.64 (14.43, 21.44) | 10.3 (8.6, 11.3) | 95 (88, 99) | 6 (5, 7.7) | |
| Ac | A | 0.11 (0.048, 0.167) | 0.23 (0.22, 0.24) | 2.9 (2.48, 3.25) | 15.5 (12.1, 21.2) | 87 (79, 112) | 3.8 (3.4, 4.4) |
| B | 0.085 (0.055, 0.091) | 1.19 (1.07, 1.3) | 18.64 (16.01, 23.13) | 26.9 (23.7, 32.1) | 110 (83, 128) | 3 (2.4, 3.5) | |
| C | 0.085 (0.083, 0.144) | 2.4 (2.32, 2.84) | 35.08 (33.33, 41.4) | 24.4 (23.2, 25.5) | 105 (96, 110) | 3.1 (2.8, 3.3) |
All data are median (interquartile range).
Vz, apparent volume of distribution during terminal phase.
CL, clearance.
FIG 1.
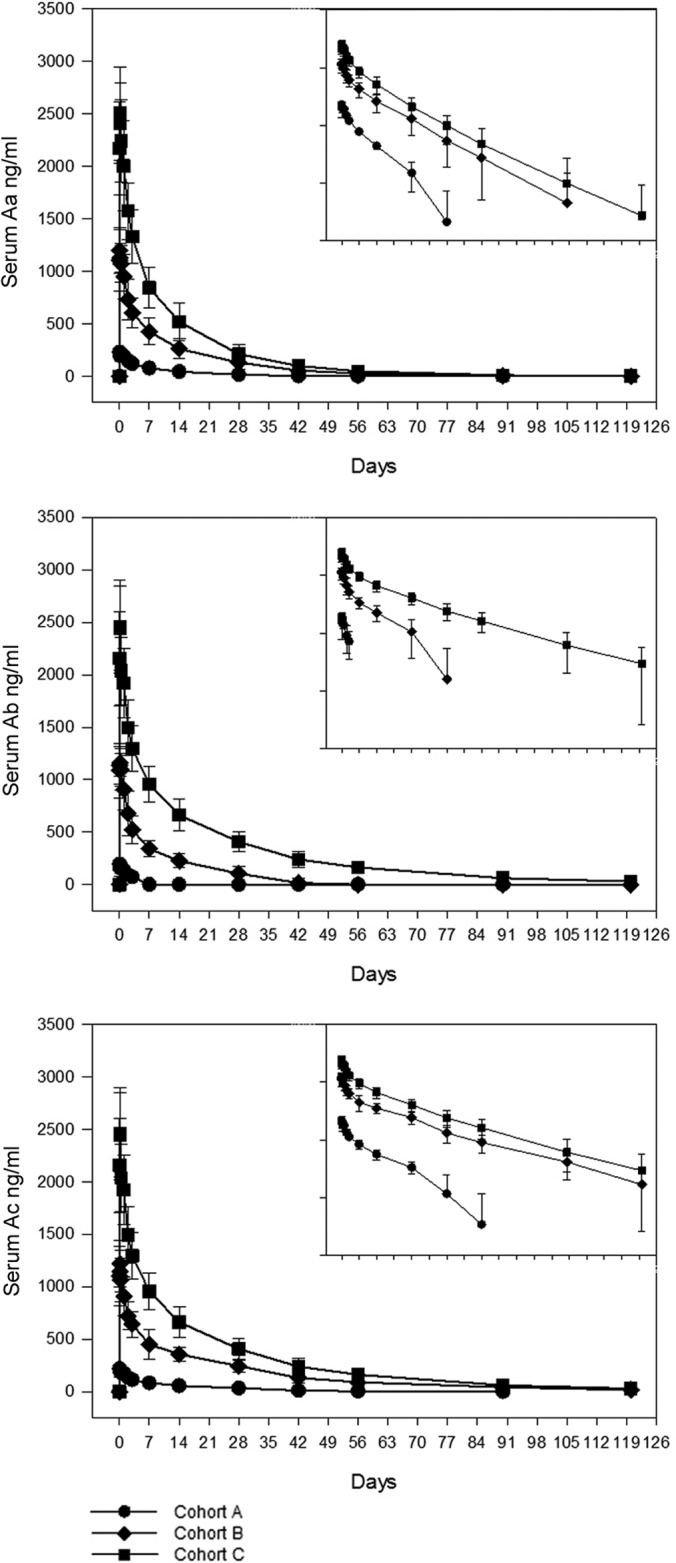
Plasma concentrations over time of Aa, Ab, and Ac monoclonal antibodies after a single i.v. infusion.
The dose-adjusted Cmax and the area under the concentration-time curve from 0 to infinity (AUCinf) for Aa and Ac were not different across all three dose cohorts, which indicated dose proportionality over the 10-fold dose range. The one exception was the AUCinf for Ab. Because many of the Ab concentrations in cohort A fell below the 80-ng/ml LLOQ (a higher LLOQ compared to that of the other monoclonal antibodies) prior to the sample times associated with terminal decay in all other cohorts and monoclonal antibodies, the dose-adjusted AUCinf and terminal half-life were poorly estimated for cohort A, and dose proportionality was not demonstrated for the AUCinf.
The pharmacokinetics of the three individual antibodies were roughly similar to each other except that the AUCinf and half-life of Ac were substantially greater than those of Aa, which were slightly greater than those of Ab. This order was reversed for clearance. There were no consistent trends in the other PK parameters among the three monoclonal antibodies.
Immunogenicity analysis.
No ADA developed against the MAbs in any of the research participants during the 3 months postinfusion. One research participant in cohort A and one in cohort B with very low-titer Aa antibodies before dosing no longer had antibodies at day 56 and 28, respectively. A second participant in cohort A and one in cohort C, found to have low titers before dosing, maintained stable low titers of Aa antibodies over the 3 months. Another cohort C participant had a modest titer of antibodies (1:350) against Ab before dosing that was not found at day 28 or afterward. This was the only research participant throughout the study with antibodies to any MAb other than Aa. One cohort C participant developed a very low titer of antibodies (1:24) against Aa at day 120, although the appearance of antibodies that long after infusion and at that low of a level likely does not represent a response to the MAb but, rather, the acquisition of the background antibodies found in preinfusion samples from other participants. A summary of immunogenicity data is in Table 5.
TABLE 5.
Titers of antibodies against XOMA 3AB MAbs
| Cohort | Subject no. | MAb | Titera on day: |
||||
|---|---|---|---|---|---|---|---|
| 0 | 28 | 56 | 84 | 120 | |||
| A | 2 | Aa | 22 | 19 | NA | ||
| A | 7 | Aa | 69 | 74 | 72 | 63 | NA |
| B | 11 | Aa | 22 | ||||
| C | 19 | Aa | 37 | 31 | 27 | 31 | |
| Ab | 350 | ||||||
| C | 21 | Aa | 24 | ||||
Titer is the reciprocal of the dilution of the sample that brings the signal to the limit of detection (cut point). NA, not available (serum not drawn on day 120).
DISCUSSION
Clostridium botulinum strains produce eight serologically distinct toxins, of which serotype A most commonly intoxicates humans (1). These category A biothreats are some of the most toxic substances in existence (19). Botulism causes significant morbidity and mortality; current antitoxins produce severe side effects and are in short supply. There is no current logistically feasible prophylactic agent. The relatively new drug class of monoclonal antibodies can potently neutralize BoNT/A. XOMA 3AB is a mixture of three human or humanized MAbs engineered to neutralize BONT/A subclasses. This agent solves two issues that plague current therapeutic strategies, side effects from anti-toxin derived from nonhomologous species and the risk of infectious disease transmission from human anti-toxin antibodies produced from immunized volunteers. XOMA 3AB also can be mass produced, unlike human anti-toxin antibodies, and it provided postexposure protection for up to 23 h in preclinical studies in mice (XOMA, unpublished data).
This study was the first-in-human assessment of XOMA 3AB in human subjects. We found that single escalating doses of XOMA 3AB administered intravenously to healthy research participants were safe and well tolerated and had an acceptable immunogenicity profile over the investigated dose range. AEs between the three cohorts of treatment groups and placebo recipients were similar in frequency and severity. All but one of the AEs were mild to moderate. The single grade 4 adverse event resolved without intervention and was not thought to be study product related.
Because animal studies suggested that the effectiveness of XOMA 3AB in neutralizing botulinum neurotoxin A is dependent on the presence of all three antibodies, the duration of the effectiveness of the combination may be determined by the monoclonal antibody that drops below effective levels most rapidly. At the lowest dose used, the humanized mouse MAb, Ab, was cleared more rapidly than the two fully human MAbs, but at the highest dose, all the monoclonal antibodies were detected for a minimum of 4 weeks after infusion. The protective level of the MAbs in humans is not known, and in mice it varies depending on the BoNT/A subtype (11, 14). However, XOMA 3AB is more than 400- to 600-fold more potent than the licensed equine antitoxin in mouse protection studies with BoNT/A1 and ∼160-fold more potent against challenge with BoNT/A2. Whether the concentrations present in human serum 4 weeks after infusion would prevent BoNT/A intoxication is speculative, but the increased potency of XOMA 3AB, compared with that of the equine antitoxin, and its prolonged half-life suggest that some level of effective neutralization of botulinum neurotoxin A would be provided for that length of time.
Most of the antibodies that were found to bind any one of the MAbs in this study were present before drug administration and should therefore be considered preexisting background antibodies. Only one research participant developed extremely low-titer antibodies against one of the three antibodies in XOMA 3AB at 120 days after infusion. Even though it is not uncommon for subjects who receive biologics to develop antibodies against the biologic agent, such a low antibody level that long after infusion makes it extremely unlikely to be related to the study product.
This study was the first assessment of XOMA 3AB in humans and only provides an early evaluation of the monoclonal antibody mixture's safety profile. XOMA 3AB reduced mortality in mice during preclinical studies when administered prior to or concurrently with the BONT/A challenge and also when administered up to 23 h after the toxin challenge. This finding, along with the increased potency and prolonged half-life of XOMA 3AB, compared with those of the currently available equine antitoxin, supports the conclusion that XOMA 3AB can clinically treat BONT/A intoxication and can have utility in disease prevention after exposure.
Supplementary Material
ACKNOWLEDGMENTS
This project has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract no. HHSN272200800026C (to J.M.G., principal investigator). This publication was made possible by the Johns Hopkins Institute for Clinical and Translational Research (ICTR), which is funded in part by grant UL1 TR 000424-06 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the Johns Hopkins ICTR, NCATS, or NIH.
Jeffrey L. Blumer provided invaluable help in the preparation of the manuscript.
Footnotes
Published ahead of print 9 June 2014
Supplemental for this article can be found at http://dx.doi.org/10.1128/AAC.02830-14.
REFERENCES
- 1.Lacy DB, Stevens RC. 1999. Sequence homology and structural analysis of the clostridial neurotoxins. J. Mol. Biol. 291:1091–1104. 10.1006/jmbi.1999.2945 [DOI] [PubMed] [Google Scholar]
- 2.Barash JR, Arnon SS. 2014. A novel strain of Clostridium botulinum that produces type B and type H botulinum toxins. J. Infect. Dis. 209:183–191. 10.1093/infdis/jit449 [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. 1998. Botulism in the United States 1899–1996. In Handbook for epidemiologists, clinicians and laboratory workers. Centers for Disease Control and Prevention, Atlanta, GA [Google Scholar]
- 4.Dembek ZF, Smith LA, Rusnak JM. 2007. Botulism: cause, effects, diagnosis, clinical and laboratory identification and treatment modalities. Disaster Med. Public Health Prep. 1:122–134. 10.1097/DMP.0b013e318158c5fd [DOI] [PubMed] [Google Scholar]
- 5.Arnon SS, Schecter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K, Working Group on Civilian Biodefense 2001. Botulinum toxin as a biological weapon: medical and public health management. JAMA 285:1059–1070. 10.1001/jama.285.8.1059 [DOI] [PubMed] [Google Scholar]
- 6.Arndt JW, Jacobson MJ, Abola EE, Forsyth CM, Tepp WH, Marks JD, Johnson EA, Stevens RC. 2006. A structural perspective of the sequence variability within botulinum neurotoxin subtypes A1-A4. J. Mol. Biol. 362:733–742. 10.1016/j.jmb.2006.07.040 [DOI] [PubMed] [Google Scholar]
- 7.Hill KK, Smith TJ, Helma CH, Ticknor LO, Foley BT, Svennsson RT, Brown JL, Johnson EA, Smith LA, Okinaka RT, Jackson PJ, Marks JD. 2007. Genetic diversity among botulinum neurotoxin producing clostridial strains. J. Bacteriol. 189:818–832. 10.1128/JB.01180-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.St John R, Finlay B, Blair C. 2001. Bioterrorism in Canada: an economic assessment of prevention and postattack response. Can. J. Infect. Dis. 12:275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franz DR, Pitt LM, Clayton MA, Hanes MA, Rose KJ. 1993. Efficacy of prophylactic and therapeutic administration of antitoxin for inhalational botulism, p 473–476 In DasGupta BR. (ed), Botulinum and tetanus neurotoxins: neurotransmission and biochemical aspects. Plenum, New York, NY [Google Scholar]
- 10.Arnon SS, Schecter R, Maslanka SE, Jewell NP, Hatheway CL. 2006. Human botulism immune globulin for the treatment of infant botulism. N. Engl. J. Med. 354:462–471. 10.1056/NEJMoa051926 [DOI] [PubMed] [Google Scholar]
- 11.Black RE, Gunn RA. 1980. Hypersensitivity reactions associated with botulinal antitoxin. Am. J. Med. 69:567–570. 10.1016/0002-9343(80)90469-6 [DOI] [PubMed] [Google Scholar]
- 12.Nowakowski A, Wang C, Powers DB, Amersdorfer P, Smith TJ, Montgomery VA, Sheridan R, Blake R, Smith LA, Marks JD. 2002. Potent neutralization of botulinum neurotoxin by recombinant oligoclonal antibody. Proc. Natl. Acad. Sci. U. S. A. 99:11346–11350. 10.1073/pnas.172229899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pless DD, Torres ER, Reinke EK, Bavari S. 2001. High-affinity, protective antibodies to the binding domain of botulinum neurotoxin type A. Infect. Immun. 69:570–574. 10.1128/IAI.69.1.570-574.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.XOMA Corporation. 2012. XOMA 3AB: investigator's brochure, version 3.0. XOMA, Berkeley, CA [Google Scholar]
- 15.National Institute of Allergy and Infectious Diseases Division of Microbiology and Infectious Diseases Office of Clinical Research Affairs. 2011. Establishing criteria for volunteer participation in DMID clinical research. National Institute of Allergy and Infectious Diseases, Rockville, MD [Google Scholar]
- 16.Meng Q, Garcia-Rodriquez C, Manzanarez G, Silberg MA, Conrad F, Bettencourt J, Pan X, Breece T, To R, Li M, Lee D, Thorner L, Tomic MT, Marks JD. 2012. Engineered domain-based assays to identify individual antibodies in oligoclonal combinations targeting the same protein. Anal. Biochem. 430:141–150. 10.1016/j.ab.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarkson PM, Kearns AK, Rouzier P, Rubin R, Thompson PD. 2006. Serum creatinine kinase levels and renal function measures in exertional muscle damage. Med. Sci. Sports Exerc. 38:623–627. 10.1249/01.mss.0000210192.49210.fc [DOI] [PubMed] [Google Scholar]
- 18.Pettersson J, Hindorf U, Persson P, Bengtsson T, Malmqvist U, Werkstrom V, Ekelund M. 2008. Muscular exercise can cause highly pathological liver function tests in healthy men. Br. J. Clin. Pharmacol. 65:253–259. 10.1111/j.1365-2125.2007.03001.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broussard LA. 2001. Biological agents: weapons of warfare and bioterrorism. Mol. Diagn. 6:323–333 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.