

Abstract
Berberine is a quaternary ammonium salt from the protoberberine group of isoquinoline alkaloids. Some reports show that berberine exhibits anti-inflammatory, antitumor, and antiviral properties by modulating multiple cellular signaling pathways, including p53, nuclear factor κB (NF-κB), and mitogen-activated protein kinase. In the present study, we investigated the antiviral effect of berberine against herpes simplex virus (HSV) infection. Current antiherpes medicines such as acyclovir can lessen the recurring activation when used early at infection but are unable to prevent or cure infections where treatment has selected for resistant mutants. In searching for new antiviral agents against herpesvirus infection, we found that berberine reduced viral RNA transcription, protein synthesis, and virus titers in a dose-dependent manner. To elucidate the mechanism of its antiviral activity, the effect of berberine on the individual steps of viral replication cycle of HSV was investigated via time-of-drug addition assay. We found that berberine acted at the early stage of HSV replication cycle, between viral attachment/entry and genomic DNA replication, probably at the immediate-early gene expression stage. We further demonstrated that berberine significantly reduced HSV-induced NF-κB activation, as well as IκB-α degradation and p65 nuclear translocation. Moreover, we found that berberine also depressed HSV-induced c-Jun N-terminal kinase (JNK) phosphorylation but had little effect on p38 phosphorylation. Our results suggest that the berberine inhibition of HSV infection may be mediated through modulating cellular JNK and NF-κB pathways.
INTRODUCTION
Herpes simplex virus (HSV) infection causes several distinct medical disorders. Common infection of the skin or mucosa may affect the face and mouth (orofacial herpes), genitalia (genital herpes), or hands (herpetic whitlow) (1, 2). More serious disorders occur when the virus infects and damages the eye (herpes keratitis) or invades the central nervous system, damaging the brain (herpes encephalitis). HSV infection is highly prevalent worldwide and is shown to facilitate human immunodeficiency virus type 1 (HIV-1) infection and transmission (3–6). HSV-1 and HSV-2, both large DNA viruses, are the etiological agents for orofacial and genital infections, respectively, although HSV-1 infection of genitalia was also reported (1, 7).
HSV establishes lifelong infection, and the virus cannot yet be eradicated once the virus establishes latency. Unfortunately, there are no cures or approved vaccines to prevent HSV infection and transmission. Antiviral medication is most effective if it is taken when patients have the prodromal symptoms of a recurrent genital herpes outbreak. Acyclovir, famcyclovir, valacyclovir, and penciclovir are the currently available medications which act in similar mechanisms by inhibiting viral DNA polymerase and causing premature chain termination when they compete with guanine triphosphate for incorporation into newly synthesized viral DNA (8–10). Acyclovir is thus an effective inhibitor of HSV and causes only mild side effects. However, drug resistance occurs especially in immunocompromised individuals (11–13). Due to the limited effectiveness of the current medication and resistant viruses, it is necessary to continue the search for new potential antiviral agents that act in distinct antiviral mechanisms.
Previous studies have shown that berberine (Fig. 1A) possesses in vitro activities in suppressing the growth of colonic carcinoma (14), neuroblastoma (15), and a number of other tumor cells (16). Although the mechanisms of these antipathogenic and antitumor activities have not been well illustrated, its inhibitory effects on several intracellular signaling pathways, including p53 nuclear factor κB (NF-κB) (17) and mitogen-activated protein kinase (MAPK) (18), have been investigated.
FIG 1.

Berberine inhibited HSV viral replication. (A) Molecular structure of berberine. (B and C) Berberine inhibited the formation of intracellular HSV infectious particles. Confluent HEC-1-A cells were mixed with serial concentrations of berberine prior to infection with HSV-1(HF) or HSV-2(G) (MOI = 1) for 24 h. The infectious viral particles were released by three cycles of freezing and thawing the infected cells, and viral infectivity was titrated by measuring the PFU as described in the text. The titration results of HSV-1(HF) or HSV-2(G) are means ± the standard deviations (SD) from three separate experiments.
We demonstrate here the antiviral activity of berberine against the infection of both HSV-1 and HSV-2 and provide evidence showing that its inhibitory activity is mediated by modulating host cell NF-κB and MAPK pathway activation. The treatment of berberine could result in the inhibition of virus-induced IκB-α degradation and p65 nuclear translocation. We also provide evidence that treatment with berberine suppressed c-Jun N-terminal kinase (JNK) activation in HSV-infected cells. These findings demonstrate that berberine may possess a unique mechanism of antiviral action and may serve as a potential antiherpesvirus agent.
MATERIALS AND METHODS
Reagents, cell lines, plasmids, and viruses.
Berberine and acyclovir were obtained from National Institutes for Food and Drug Control in China (Beijing, China). SB203580, SP600125, BAY11-7082, MG132, and PMA (phorbol-12-myristate-13-acetate) were purchased from Beyotime (Haimen, Jiangsu, China). Alexa Fluor 488-conjugated goat anti-mouse IgG(H+L) and DAPI (4′,6′-diamidino-2-phenylindole) were obtained from Life Technologies (Carlsbad, CA). IRDye 680-conjugated goat anti-rabbit and IRDye 800-conjugated goat anti-mouse antibodies were obtained from LI-COR (Lincoln, NE). Antibodies specific for gD-1/2, ICP4-1, ICP-8, ICP27-1, JNK2, p38, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and β-catenin, and radioimmunoprecipitation assay (RIPA) lysis buffer were purchased from Santa Cruz (Santa Cruz, CA). Antibodies specific for p65, p-p38, p-c-Jun, p-JNK1/2, p-ATF-2, and IκB-α were obtained from Cell Signaling Technology (Beverly, MA). Anti-ICP5-1/2 was obtained from Abcam (Cambridge, United Kingdom). Recombinant human tumor necrosis factor alpha (TNF-α) was obtained from PeproTech (Rocky Hill, NJ).
HEK293T, Vero, and HEC-1-A cells were obtained from the American Type Culture Collection (Manassas, VA). NF-κB-luc and AP-1-luc reporter plasmids were purchased from Clontech (Palo Alto, CA). pGL4/TNF-α promoter was constructed by inserting the TNF-α promoter (ca. bp −100 to +900) into pGL4.17 (Promega, Madison, WI). HSV-1(HF), HSV-1/blue, and HSV-2(G) were propagated and titrated on Vero cells as described previously (19).
In vitro antiviral assay.
The in vitro antiviral activity of berberine was determined by titrating the infectious virions in berberine-treated cells as described previously (20). HEC-1-A cells were seeded into 96-well plates at a density of 2 × 104 per well and cultured for 24 h and then pretreated with serial concentrations of berberine and infected with HSV-1 or HSV-2 (multiplicity of infection [MOI] = 1). At 24 h postinfection (p.i.), the culture medium was discarded, and then 200 μl of fresh medium was dispensed into each well. HSV-1/2-infected cells were frozen and thawed with three cycles to release the virions. The virion-containing medium was then diluted and dispensed on confluent Vero cell monolayers. Viral titration was performed by counting the numbers of plaques.
In vitro cytotoxicity assay.
The in vitro cytotoxicity of berberine was measured by using a commercial CCK-8 kit (Dojindo, Kumamoto, Japan) via the colorimetric method according to the manufacturer's instructions. Briefly, 2 × 104 cells per well were seeded into 96-well plates and cultured for 24 h before serial concentrations of berberine were added in triplicate. After 24 h, 10 μl of CCK-8 reagent was dispensed into each well, and the plates were incubated at 37°C for 3 h. The absorbance at 450 nm was measured using a TECAN Infinite M200 microplate reader (Männedorf, Switzerland). Cell viability was plotted as the percent viable cells of the mock-treated control cells.
Western blotting and in-cell Western assay.
The cells were lysed using RIPA lysis buffer on ice for 30 min and then centrifuged at 12,000 × g for 10 min at 4°C. Total protein concentrations in the supernatants were determined using BCA protein assay kit (Pierce, Rockford, IL). After they were separated using SDS-PAGE, the proteins were transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). The membranes were blocked using Odyssey blocking buffer (LI-COR) and then incubated with primary antibodies for 2 h at room temperature. After five washes with PBS-T buffer (phosphate-buffered saline, 0.1% Tween 20), the membranes were incubated in IRDye IgG (1:10,000) for 1 h at room temperature and visualized using a LI-COR Odyssey infrared imager.
An in-cell Western assay was performed in a 96-well plate. The cells cultured in a 96-well plate were fixed with 4% paraformaldehyde for 20 min at room temperature and permeabilized by five washes in PBS–0.1% Triton X-100 with 5 min for each wash. The cell monolayers were blocked for 90 min in blocking buffer (4% nonfat dry milk) and then incubated with primary antibodies diluted in blocking buffer (1:200) for 2 h at room temperature. After washing with PBS-T buffer, the cell layers were stained with IRDye IgG (1:1500) for 1 h, rinsed and scanned in an Odyssey infrared imager. The relative protein expression level was normalized against β-catenin.
Synergy analysis.
Anti-HSV-2 activity of berberine and acyclovir were tested individually in serial concentrations in HEC-1-A cells through an in-cell Western assay, and the 50% maximal effective concentrations (EC50s) of the single drugs were calculated. The two drug combinations were tested at a fixed molar concentration ratio, which was optimized to give the greatest synergism over a range of serial dilutions. The EC50s of single drugs and the combination index (CI) of the two drugs were calculated using CalcuSyn software (21) according to the method of Chou and Talalay (22). The synergy was estimated from the CI values (22) and scored as follows: CI < 0.1, very strong synergism; CI = 0.1 to 0.3, strong synergism; CI = 0.3 to 0.7, synergism; CI = 0.7 to 0.85, moderate synergism; CI = 0.85 to 0.90, slight synergism; CI = 0.9 to 1.1, nearly additive synergism; and CI = > 1.1, antagonism.
Immunofluorescence staining and microscopic imaging.
HEC-1-A cells grown on 10-mm glass coverslips in a 24-well plate were rinsed with PBS and then fixed with 4% paraformaldehyde for 15 min at room temperature. The cells were then permeabilized with PBS–0.2% Triton X-100 for 15 min, followed by two washes with PBS. The coverslips were blocked with 1% bovine serum albumin in PBS for 30 min at room temperature. Target proteins were immunolabeled using respective primary antibodies, followed by Alexa Fluor 488-conjugated goat anti-mouse 488 IgG and Alexa Fluor-conjugated goat anti-rabbit 594 IgG (Life Technologies). Nuclei were visualized by staining with DAPI. Images were acquired using an Olympus FluoView FV10i confocal microscope (Tokyo, Japan).
Cell transfection and luciferase assay.
HEC-1-A cells cultured in a 96-well plate were transiently transfected with luciferase reporter plasmid (100 ng/well) using Lipofectamine 2000 transfection reagent (Life Technologies). The cells were further cultured for 24 h and treated as described above. The relative luciferase activity was determined using a Bright-Glo luciferase assay system (Promega).
RNA extraction and real-time PCR.
Cellular total RNA was extracted using TRIzol reagent (Life Technologies) according to the manufacturer's protocol. cDNA was reverse transcribed using a ReverTra Ace qPCR-RT kit (Toyobo, Osaka, Japan). Real-time PCR was performed in triplicate on ABI Prism 7300 sequence detection system using the SYBR green PCR master mix (Life Technologies). The sequences of primer pairs are listed in Table 1. mRNA transcription levels were standardized against housekeeping gene GAPDH.
TABLE 1.
Primer pairs used in this study
| Gene | Primer sequence (5′–3′) |
|
|---|---|---|
| Forward | Reverse | |
| HSV-1 gD | AGCAGGGGTTAGGGAGTTG | CCATCTTGAGAGAGGCATC |
| HSV-2 gD | CCAAATACGCCTTAGCAGACC | CACAGTGATCGGGATGCTGG |
| IL-8 | ATTGAGAGTGGACCACACTG | ACTACTGTAATCCTAACACCTG |
| TNF-α | CCTGCCCCAATCCCTTTATT | CCCTAAGCCCCCAATTCTCT |
| GAPDH | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG |
HSV-1/blue assay.
Confluent HEC-1-A cells in a 96-well plate were preincubated with drugs for 30 min and then infected with HSV-1/blue (MOI = 1). The cells were lysed with 1% NP-40 in PBS at 12 h p.i. The cell lysates were then transferred into a new Costar 96-well flat plate and mixed with CPRG (chlorophenol red-β-d-galactopyranoside; Boehringer, Ingelheim, Germany), and the β-galactosidase (β-Gal) activities were measured in a TECAN Infinity M200 microplate reader at 570 nm after 1 h.
Statistics.
Statistical analysis was performed using a two-tailed Student t test. Statistical significance is indicated in the figures by asterisks (*, P < 0.05; **, P < 0.01).
RESULTS
Berberine inhibited HSV viral replication.
To investigate the antiviral effect of berberine on HSV-1 and HSV-2 replication, HEC-1-A cells were pretreated with serial concentrations of berberine and then infected with HSV-1(HF) and HSV-2(G), respectively. Berberine effectively inhibited the formation of an HSV-induced cytopathogenic effect (data not shown). Furthermore, the infectious viral particles were released by three cycles of freezing and thawing the infected cells, and viral infectivity was titrated by measuring the PFU. As shown in Fig. 1B and C, berberine inhibited the replication of both HSV-1 (Fig. 1B) and HSV-2 (Fig. 1C) in a dose-dependent manner. The EC50s for HSV-1 and HSV-2 were 6.77 ± 1.13 μM and 5.04 ± 1.07 μM, respectively.
We initially investigated its anti-HSV activity by monitoring the reduction of gD expression, an HSV late gene product. As shown in Fig. 2A and B, berberine was effective in inhibiting the expression of both HSV-1 (Fig. 2A) and HSV-2 gD (Fig. 2B). To confirm these results, we investigated the effect of berberine on HSV-1 or HSV-2 gD expression level by measuring the copy numbers of gD mRNA transcript via real-time PCR. Similarly, the inhibitory effect was in parallel to that of the protein expression (Fig. 2C and D). The expression of another late gene product, VP5, the major capsid protein, was also determined, and the results were consistent with the gD expression (Fig. 2E and F). Western blot analysis of gD expression in another HSV permissive cell line, HEK293T cells, further substantiated the inhibitory effect of berberine and ruled out a cell type-specific effect of the drug, as shown in Fig. 2G. We conclude from these findings that berberine inhibits the replication of HSV-1 and HSV-2 and viral late gene expression. The viral inhibitory activity was not due to the effect of cytotoxicity of the drug to the host cells, since we showed that berberine had low cytotoxicities to both HEK293T and HEC-1-A cell lines. As shown in Fig. 2H, the 50% cytotoxicity concentration (CC50) of berberine to HEC-1-A cells was >400 μM, which was significantly higher than the viral inhibitory dosage. The CC50 of berberine to HEK293T cells was 165.7 μM, which was still significantly higher than that used in its antiviral assay. These results suggest that the viral inhibitory activity of berberine is not due to its cytotoxicity.
FIG 2.

Berberine inhibited HSV late gene expression. (A to D) Berberine inhibited gD expression. HEC-1-A cells were treated with various concentrations of berberine and then infected with HSV-1 or HSV-2 (MOI = 1). The gD-1/2 protein expression level was determined via in-cell Western assay and normalized based on the β-catenin level at 24 h p.i. (A and B). The mRNA transcript level of gD was quantified via real-time PCR analysis (C and D). (E and F) Berberine interfered with viral ICP5 expression. HEC-1-A cells treated with serial concentrations of berberine were infected with HSV-1 or HSV-2, and ICP5 expression was determined via an in-cell Western assay at 24 h p.i. (G) Berberine inhibited HSV-1 and HSV-2 gD expression in HEK293T cells. HEK293T cells were treated with various concentrations of berberine (Berb) prior to infection with HSV-1 or HSV-2 (MOI = 1). The gD expression level was determined via Western blotting at 24 h p.i. (H) Cytotoxic effect of berberine on HEC-1-A and HEK293T cells. The cells were treated with serial concentrations of berberine. The cell viability was determined by CCK-8 colorimetric assay after 24 h. All experiments were performed three times. Representative results are shown. The data are means ± the SD from triplicate determinations.
Berberine inhibited HSV infection at a postentry step.
A time-of-drug-addition assay was performed to determine the steps of berberine action during HSV-2 replication cycle. As shown in Fig. 3, HSV-2-infected HEC-1-A cells were treated with berberine, acyclovir, and dextran sulfate (DXS; a polyanion that inhibits HSV virion attachment and entry) at the indicated time points postinfection. The results showed that HSV-2 began to escape the inhibition by berberine at 4 to 8 h p.i., whereas it escaped from the inhibition by DXS at 0 to 2 h p.i. Acyclovir inhibited HSV-2 gD expression during the entire period from 0 to 8 h p.i. Based on these observations, we postulate that berberine may act at an early stage of HSV replication cycle, between viral entry and viral genomic DNA replication, probably during the immediate-early (IE) gene expression stage.
FIG 3.

Berberine inhibited HSV infection at a postentry step. HEC-1-A cells were infected with HSV-2 (MOI = 1) and exposed to berberine (50 μM), acyclovir (50 μg/ml), or DXS (100 μg/ml) at the indicated time points. The viral infection level is represented by gD-2 expression as determined by an in-cell Western assay at 24 h p.i. The data represent means ± the SD from triplicate determinations from three dependent experiments.
Berberine inhibited HSV IE and early genes expression.
We further explored the mechanisms of berberine inhibition of gD expression by investigating the IE genes that regulate the late genes. Due to the importance of HSV IE expression in viral replication, we first investigated the inhibitory effect of berberine on one of the IE genes, infected cell polypeptide 4 (ICP4), at 24 h p.i. We found, using an in-cell Western assay (Fig. 4A), that berberine inhibited HSV-1 ICP4 expression in a dose-dependent manner, a finding consistent with the reduction of late gene expression (see Fig. 2). We also used HSV-1/blue recombinant virus to determine the inhibitory effect of berberine on reporter gene lacZ expression. This virus contains an HSV-1 ICP4 promoter-driven lacZ gene inserted into HSV-1 thymidine kinase (TK) gene loci (23, 24). As shown in Fig. 4B, berberine inhibited ICP4 promoter-driven lacZ gene expression in a dose-dependent manner, further demonstrating that berberine might inhibit HSV replication by interfering with viral IE gene expression.
FIG 4.

Berberine inhibited HSV IE gene expression. (A) Berberine inhibited ICP4-1 expression in a dose-dependent manner. Confluent HEC-1-A cells were treated with the indicated concentrations of berberine prior to infection with HSV-1 (MOI = 1). ICP4-1 expression was determined via an in-cell Western assay and normalized based on the β-catenin level at 24 h p.i. (B) Berberine inhibited HSV-1/blue ICP4 promoter-driven lacZ gene expression in a dose-dependent manner. HEC-1-A cells were treated with serial concentrations of berberine or MG132 (5 μg/ml) prior to infection with HSV-1/blue (MOI = 1). The β-Gal activity was measured as described in the text at 12 h p.i. (C) HEC-1-A cells were either mock treated or treated with berberine (50 μM) and then infected with HSV-1 (MOI = 1). The cells were lysed at each time point. ICP4 was visualized by Western blotting. (D) HEC-1-A cells were either mock treated or treated with berberine (50 μM) and then infected with HSV-1 (MOI = 1). The cells were lysed at each time point. ICP27 was visualized by Western blotting. (E) HEC-1-A cells were either mock treated or treated with berberine (50 μM) and then infected with HSV-1 (MOI = 1). The cells were lysed at each time point. ICP8 was visualized by Western blotting. The data represent means ± the SD of triplicate determinations from three dependent experiments.
We also investigated the time course of the inhibitory effect of berberine on ICP4 gene expression during the early stages of HSV infection, and the result showed that berberine could inhibit ICP4 expression at 8 and 12 h p.i. and completely inhibited gD expression (Fig. 4C). We also investigated the time course of the inhibitory effect of berberine on ICP27 gene expression during the early stages of HSV infection, and the result showed that berberine could also have an inhibitory effect on ICP27 expression at 8 and 12 h p.i. (Fig. 4D).
Whether berberine inhibited HSV-1 early gene expression was also determined. As shown in Fig. 4E, berberine could block ICP8 expression completely. ICP8 is a single-strand DNA-binding protein that is required for HSV genomic DNA replication (25). The inhibitory effect of berberine on ICP8 was consistent with the results of the late gene expression (Fig. 2).
Berberine inhibited HSV-2-induced NF-κB activation.
HSV infection induces a persistent NF-κB activation, which is necessary for viral replication and host cell survival during the early stage of viral infection (26–29). To better understand whether berberine could inhibit HSV-2-induced NF-κB activation, we used NF-κB luciferase reporter plasmid to evaluate its effect. As shown in Fig. 5A, berberine downregulated HSV-2-induced NF-κB activation in a dose-dependent manner. MG132, a specific proteasome and NF-κB inhibitor that served as a positive control, completely inhibited NF-κB activation. We also examined the level of IκB-α, an endogenous NF-κB inhibitor, and found that HSV-2 infection induced the degradation of IκB-α, whereas berberine could block the IκB-α degradation at 12 and 24 h p.i. (Fig. 5B). We also evaluated the effect of berberine on TNF-α-induced NF-κB activation, and the results showed that berberine at a concentration of 50 μM could suppress TNF-α-induced NF-κB and that treatment with berberine alone did not induce NF-κB activation (Fig. 5C). p65 is a subunit of NF-κB transcription complex, which plays a crucial role in inflammatory and immune responses. The inhibitory effect of IκB-α upon NF-κB in the cytoplasm is exerted primarily through the interaction with p65, and p65 nuclear translocation is often taken as an indication of NF-κB activation. We also demonstrated that berberine could inhibit virus-mediated p65 nuclear translocation and HSV-2 gD expression simultaneously (Fig. 5D). Together, the data suggest that berberine may inhibit HSV-2-induced NF-κB activation, which may result in an inhibition of HSV replication.
FIG 5.

Berberine inhibited HSV-2-induced NF-κB activation. (A) HEC-1-A cells were transfected with NF-κB-luc reporter plasmid. The cells were mock treated or treated with the indicated concentrations of berberine or MG132 (5 μg/ml) prior to being mock infected or infected with HSV-2 (MOI = 1). The relative luciferase activity was determined after 24 h and is expressed as the fold change compared to that of mock-treated cells. (B) Berberine inhibited HSV-2-induced IκB-α degradation. HEC-1-A cells were mock infected or infected with HSV-2 (MOI = 1) in the absence or presence of berberine (12.5 and 50 μM) or MG132 (5 μg/ml). The IκB-α, gD, and GAPDH levels were determined at 12 or 24 h p.i. by Western blotting. The band intensity was determined by using Odyssey v3.0 software. (C) Berberine suppressed TNF-α-induced NF-κB activation. HEC-1-A cells transfected with NF-κB-luc reporter plasmid were mock treated or treated with berberine (50 μM) or MG132 (5 μg/ml) prior to exposure to TNF-α (100 ng/ml). The relative luciferase activity was determined after 12 h and is expressed as the fold change versus that of mock-treated cells. (D) Berberine interfered with HSV-2-induced p65 nuclear translocation. HEC-1-A cells were mock infected or infected with HSV-2 (MOI = 1) in the presence or absence of berberine (50 μM). p65 translocation was determined via immunofluorescence assay at 24 h p.i. The data represent means ± the SD of triplicate determinations from three dependent experiments.
Berberine inhibited HSV-2-induced JNK activation but showed less effect on p38 MAPK activation.
The JNK and p38 MAPK pathways are stimulated by HSV-1 infection, and their activations play a central role on HSV-1 replication (30, 31). We investigated whether HSV-2 infection would activate both MAPK cascades in HEC-1-A cells and whether berberine would inhibit HSV-2-induced AP-1 activation, which was the main transcription factor downstream of JNK/p38 MAPK. We found that berberine inhibited HSV-2-induced AP-1 binding site-driven luciferase expression in a dose-dependent manner, implying that it could attenuate virus-mediated MAPK activation through modulating certain cascade(s) (Fig. 6A). We also found that HSV-2 infection resulted in the persistent activation of the JNK and p38 MAPK pathways in HEC-1-A cells (Fig. 6B) and that their specific inhibitors (SB203580 and SP600125, respectively) were able to reduce HSV-2 replication (Fig. 6C). Further evidence showed that berberine could disrupt the phosphorylation of the upstream activator JNK but not p38 MAPK. As shown in Fig. 6D, HSV-2 infection caused JNK phosphorylation, which could be suppressed by berberine. The phosphorylation of its substrate, c-Jun was also attenuated by the berberine treatment. In contrast, berberine would increase the phosphorylation level of p38 MAPK in mock-infected cells. Although the low concentrations of berberine (12.5 μM) partially inhibited HSV-2-induced p38 activation, high concentrations activated the p38 MAPK cascade instead. ATF-2, the substrate of both JNK and p38 MAPK, was also activated in berberine-treated uninfected cells but suppressed in infected ones which might be due to the JNK pathway. These results indicate that berberine inhibits HSV-2-induced JNK activation but only has a marginal inhibitory effect on p38 MAPK activation. The effect of berberine on PMA-induced MAPK activation was also investigated. As shown in Fig. 6E, berberine could inhibit PMA-induced phosphorylation of JNK and its specific substrate c-Jun but showed less effect on p38 MAPK and ATF-2. Together, the data suggest that berberine may inhibit HSV-2-induced JNK activation, which may result in an inhibition of HSV replication but not of the p38 MAPK cascade.
FIG 6.

Berberine inhibited HSV-2-induced JNK activation but had less effect on p38 MAPK activation. (A) Berberine inhibited HSV-2-induced AP-1 activation. HEC-1-A cells were transfected with AP-1-luc reporter plasmid. The cells were mock treated or treated with serial concentrations of berberine, SB203580 (20 μM), or SP600125 (20 μM) prior to being mock infected or infected with HSV-2 (MOI = 1). SB203580 and SP600125, p38 MAPK and JNK inhibitors, respectively, were used as controls. The relative luciferase activity was determined after 24 h and is expressed as the fold change versus that of the mock-treated cells. (B) HSV-2 infection induced the activation of JNK and p38 MAPK pathways in HEC-1-A cells. HEC-1-A cells were infected with HSV-2 (MOI = 1). The cells were lysed at each time point. The phosphorylation levels of p38 MAPK, JNK, and their substrates were determined via Western blotting. (C) SB203580 and SP600125 had an inhibitory effect on HSV-2 replication in HEC-1-A cells. HEC-1-A cells seeded in a 96-well plate were treated with serial concentrations of SB203580 and SP600125 prior to infection with HSV-2 (MOI = 1). The gD protein expression level was determined via an in-cell Western assay and normalized based on the β-catenin level at 24 h p.i. (D) Berberine inhibited virus-induced JNK phosphorylation but had less effect on p38 MAPK phosphorylation. HEC-1-A cells were mock infected or infected with HSV-2 (MOI = 1) in the presence or absence of berberine. The levels of JNK, p38 MAPK, and their phosphorylated forms and the downstream p-c-Jun, p-ATF-2, were determined at 12 h p.i. (E) Berberine inhibited PMA-induced JNK activation. HEC-1-A cells were mock treated or treated with PMA (4 μg/ml) in the presence or absence of berberine. The levels of JNK, p38 MAPK, and their phosphorylated forms and substrates were determined after 2 h. All experiments were performed three times, and representative results are shown. The data represent means ± the SD of triplicate determinations.
Berberine downregulated HSV-2-induced interleukin-8 (IL-8) and TNF-α expression.
Previous studies report that HSV infection could upregulate the expression of certain cytokines/chemokines, which is associated with cellular NF-κB and MAPK activation (32, 33). Therefore, whether berberine would inhibit the virus-induced upregulation of cytokines/chemokines was investigated via real-time PCR. As shown in Fig. 7A and B, berberine effectively inhibited the expression of virus-induced IL-8 and TNF-α, two hallmarks of chemotactic and proinflammatory factors, respectively. Using a TNF-α reporter plasmid, we also confirmed the inhibitory effect of berberine on TNF-α promoter activity (Fig. 7C).
FIG 7.
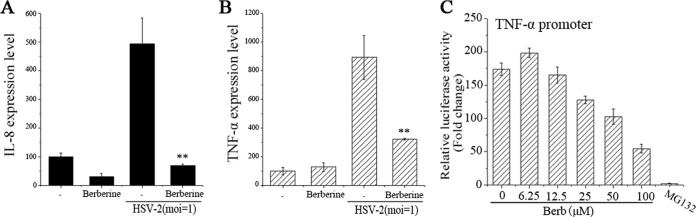
Berberine downregulated HSV-2-induced IL-8 and TNF-α expression. HEC-1-A cells were mock infected or infected with HSV-2 (MOI = 1) in the presence or absence of berberine (50 μM). The IL-8 (A) and TNF-α (B) expression levels were quantified by real-time PCR at 24 h p.i. (C) Berberine inhibited HSV-2-induced TNF-α promoter activation. HEC-1-A cells were transfected with TNF-α promoter luciferase reporter plasmid and, after 24 h, the cells were infected with HSV-2 (MOI = 1) in the presence of serial concentrations of berberine or MG132 (5 μg/ml). The relative luciferase activity was measured at 24 h p.i. The data represent the means ± the SD of triplicate determinations from three dependent experiments.
The synergistic effect of berberine and acyclovir against HSV-2 infection.
Since berberine and acyclovir, an inhibitor of herpesvirus TK, likely acted with distinct mechanisms against HSV (as demonstrated above), the viral inhibitory activity of combining berberine and acyclovir was investigated. As shown in Fig. 8, the combination index (CI) of berberine and acyclovir was 0.814, which represented a moderate synergism against HSV-2 infection when two drugs were used in combination, suggesting potential beneficial effects of using the two drugs together.
FIG 8.

Berberine showed a moderate synergistic effect with acyclovir against HSV-2 infection. The effective concentrations for the inhibition of HSV-2 infection by a compound alone or in combination were plotted in two curves. The CI values were calculated using CalcuSyn. The findings were scored as follows: CI < 0.1, very strong synergism; CI = 0.1 to 0.3, strong synergism; CI = 0.3 to 0.7, synergism; CI = 0.7 to 0.85, moderate synergism; CI = 0.85 to 0.90, slight synergism; CI = 0.9 to 1.1, nearly additive; and CI > 1.1, antagonism. The data represent means ± the SD of triplicate determinations from three dependent experiments.
DISCUSSION
Berberine is a natural product found in many traditional Chinese herbs. It is found in plants such as Berberis, Hydrastis canadensis (goldenseal), Xanthorhiza simplicissima (yellow root), and Phellodendron amurense (Amur cork tree) and has been used traditionally to treat fungal, Candida albicans, parasite, and bacterial/viral infections (17, 34–36). Some reports suggest it as a potential antitumor and anti-inflammatory agent (37, 38).
Berberine exhibits inhibitory activity against some viruses, including influenza A virus (39) and human cytomegalovirus (40). Chin et al. first reported that berberine from Coptidis rhizoma showed an anti-HSV effect (41). Due to its low cytotoxicity and minimal side effects, berberine is considered a promising antiviral drug candidate for alternative treatment. In the present study, we demonstrated that berberine could effectively inhibit HSV-1 and HSV-2 replication at concentrations below the 50% cytotoxicity dosage (Fig. 1 and 2). We also studied the synergistic effect of berberine and acyclovir against HSV-2 infection in vitro and showed that these two drugs exhibited moderate synergism against viral replication (Fig. 8). Combination treatment of acyclovir with a drug that has the ability to inhibit HSV replication could increase the anti-HSV activity in vitro and in vivo (42–45). In the present study, we found that berberine not only attenuated HSV replication but also depressed the inflammatory response and associated pathway activation. Thus, berberine and acyclovir may be potentially used in combination for the treatment of HSV infection.
The time-of-drug-addition analysis indicated that berberine exerted its inhibitory effect after viral attachment and entry but before HSV genomic DNA replication (Fig. 3). Further mechanistic analysis showed that berberine inhibited expression of the viral IE genes ICP4 and ICP27. We postulate that berberine might act at the stage of HSV IE gene expression and thus inhibit the expression of early (ICP8) and late (ICP5 and gD) gene expression, leading to reduced HSV infectivity and inhibition of viral replication. HSV IE gene expression plays significant roles in viral replication and regulates early and late gene expression. ICP4 and ICP27 are two major transcriptional activators for the viral early and late genes and are essential for viral growth (46, 47). Our evidence suggests that the inhibitory effect of berberine is mediated by acting on ICP4 and ICP27 expression, leading to the downmodulation of downstream early and late gene expression.
HSV can utilize certain cellular signaling pathways to facilitate its replication. In the present study, we found that berberine depressed the degradation of endogenous NF-κB inhibitor IκB-α and p65 nuclear translocation induced by HSV-2 infection, leading to the inhibition of NF-κB activation. It has been reported that HSV infection undergoes a sustained host cell NF-κB activation, which is necessary to prevent host cell from apoptosis at 3 to 6 h p.i. and lasts until the viral lytic phase (48). NF-κB belongs to the foremost transcription factors that mediate immune, inflammatory, or antiapoptotic responses. NF-κB can be activated by the exposure of cells to lipopolysaccharide (LPS), inflammatory cytokines (TNF-α, IL-1β, etc.), phorbol ester, UV irradiation, or viral infection or by the expression of certain viral gene products (49). Since NF-κB plays a key role in promoting HSV infection, inhibition of NF-κB signaling might be a promising therapeutic approach in HSV-induced inflammatory responses (50). The effect of berberine on NF-κB activation during HSV replication indicates that it might be a potential inhibitor in HSV multiplication. It is worth mentioning that berberine showed significant antiviral activity at low concentrations; however, it could not inhibit HSV-2-induced NF-κB activation at the same concentration. Only high concentrations of berberine could inhibit virus-induced NF-κB activation. This suggested to us that berberine is a multifunctional molecule and may have other mechanisms of action for an antiherpesvirus effect.
Previous reports have shown that HSV infection resulted in activation of the JNK/MAPK pathway. Both JNK and p38 MAPK are stimulated after HSV infection. A subset of cellular genes transactivated by AP-1 may ensure efficient viral gene expression and DNA replication and facilitate viral replication (31, 51). JNK and p38 MAPK are two main members of the MAPK family, and these two stress-activated protein kinases are activators for sensing various stimuli, such as proinflammatory cytokines, genotoxic agents, osmotic shock, and bacterial LPS (52). Activated JNK and p38 MAPK can transmit upstream signals to downstream factors and thus mediate apoptosis, differentiation, growth, or immune responses. JNK and p38 MAPK are also reported to be stimulated by many viruses or virus-associated proteins, including vaccinia virus (53), rotavirus (54), varicella-zoster virus (55), HIV-1 (56), HSV-1 (30), coxsackievirus B3 (57), influenza virus (58), and hepatitis B/C virus (59). Our data showed that berberine had an inhibitory effect on the activation of transcription factor AP-1, the main downstream factor of JNK and p38 MAPK, and that HSV infection led to a robust JNK phosphorylation, which was mitigated by berberine treatment. The virus-induced c-Jun and ATF-2 phosphorylations were also suppressed by the drug in a dose-dependent manner. Although berberine at 12.5 μM exhibited a slight inhibitory effect on HSV-induced p38 phosphorylation, the higher concentration (50 μM) increased the level of phosphorylated p38 in both mock-infected and HSV-infected cells (Fig. 6B). Besides, berberine could attenuate PMA-induced JNK activation but showed marginal effect on p38 MAPK (Fig. 6E). Based on the observations that berberine inhibited HSV-induced JNK phosphorylation, we postulate that berberine may act at upstream of the JNK cascade, which requires further investigation. IL-8 and TNF-α are important hallmarks of chemotactic and proinflammatory responses, respectively, and these two inflammatory cytokines are upregulated during HSV-1 infection, which is associated with the MAPK and NF-κB pathways (32, 33). The IL-8 promoter region has binding sites for NF-κB and AP-1, and this proinflammatory factor can be regulated by NF-κB and MAPK activation (60, 61). The TNF-α promoter has four NF-κB binding sites and one AP-1 binding site (62). We showed that berberine inhibited HSV-2-induced upregulation of IL-8 and TNF-α expression (Fig. 7), a finding consistent with our evidence that berberine downregulated these downstream factors via modulating the MAPK and NF-κB pathways.
In conclusion, we investigated the inhibitory mechanism of berberine on HSV-1 and HSV-2 infection and found that berberine significantly inhibited HSV-induced JNK and NF-κB activation. As a result, we conclude that the inhibitory effect of berberine on host cell JNK and NF-κB activation may thus inhibit HSV replication. However, further study is needed to delineate the mechanisms of the IE genes and the NF-κB and JNK pathways in detail. In view of its low cytotoxicity and significant antiherpetic activity, berberine might be a valuable candidate for further study as a promising anti-HSV drug.
ACKNOWLEDGMENTS
We thank Tao Peng from Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, for the generous gift of HSV-1/blue strain. This study was supported by the Major Research and Development Project from the Ministry of Health of China (grants 2012ZX10001-007-009-001 and 2013ZX10001005-003) and the Innovative Project for Graduate Students of Jiangsu Province (grant CXLX13_039).
Footnotes
Published ahead of print 9 June 2014
REFERENCES
- 1.Lafferty WE, Downey L, Celum C, Wald A. 2000. Herpes simplex virus type 1 as a cause of genital herpes: impact on surveillance and prevention. J. Infect. Dis. 181:1454–1457. 10.1086/315395 [DOI] [PubMed] [Google Scholar]
- 2.Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518. 10.1016/S0140-6736(00)04638-9 [DOI] [PubMed] [Google Scholar]
- 3.Corey L, Wald A, Celum CL, Quinn TC. 2004. The effects of herpes simplex virus-2 on HIV-1 acquisition and transmission: a review of two overlapping epidemics. J. Acquir. Immune Defic. Syndr. 35:435–445. 10.1097/00126334-200404150-00001 [DOI] [PubMed] [Google Scholar]
- 4.Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. 2006. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 20:73–83. 10.1097/01.aids.0000198081.09337.a7 [DOI] [PubMed] [Google Scholar]
- 5.Mbopi-Kéou F-X, Grésenguet G, Mayaud P, Weiss HA, Gopal R, Matta M, Paul J-L, Brown DW, Hayes RJ, Mabey DC. 2000. Interactions between herpes simplex virus type 2 and human immunodeficiency virus type 1 infection in African women: opportunities for intervention. J. Infect. Dis. 182:1090–1096. 10.1086/315836 [DOI] [PubMed] [Google Scholar]
- 6.Wald A, Link K. 2002. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J. Infect. Dis. 185:45–52. 10.1086/338231 [DOI] [PubMed] [Google Scholar]
- 7.Roberts CM, Pfister JR, Spear SJ. 2003. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex. Transm. Dis. 30:797–800. 10.1097/01.OLQ.0000092387.58746.C7 [DOI] [PubMed] [Google Scholar]
- 8.De Clercq E, Field HJ. 2006. Antiviral prodrugs–the development of successful prodrug strategies for antiviral chemotherapy. Br. J. Pharmacol. 147:1–11. 10.1038/sj.bjp.0706446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Earnshaw D, Bacon T, Darlison S, Edmonds K, Perkins R, Hodge RV. 1992. Mode of antiviral action of penciclovir in MRC-5 cells infected with herpes simplex virus type 1 (HSV-1), HSV-2, and varicella-zoster virus. Antimicrob. Agents Chemother. 36:2747–2757. 10.1128/AAC.36.12.2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wald A, Zeh J, Barnum G, Davis L, Corey L. 1996. Suppression of subclinical shedding of herpes simplex virus type 2 with acyclovir. Ann. Intern. Med. 124:8–15. 10.7326/0003-4819-124-1_Part_1-199601010-00002 [DOI] [PubMed] [Google Scholar]
- 11.Reyes M, Shaik NS, Graber JM, Nisenbaum R, Wetherall NT, Fukuda K, Reeves WC. 2003. Acyclovir-resistant genital herpes among persons attending sexually transmitted disease and human immunodeficiency virus clinics. Arch. Intern. Med. 163:76–80. 10.1001/archinte.163.1.76 [DOI] [PubMed] [Google Scholar]
- 12.Shin YK, Weinberg A, Spruance S, Bernard M, Bacon TH, Boon RJ, Levin MJ. 2003. Susceptibility of herpes simplex virus isolates to nucleoside analogues and the proportion of nucleoside-resistant variants after repeated topical application of penciclovir to recurrent herpes labialis. J. Infect. Dis. 187:1241–1245. 10.1086/373993 [DOI] [PubMed] [Google Scholar]
- 13.Stránská R, Schuurman R, Nienhuis E, Goedegebuure IW, Polman M, Weel JF, Wertheim-Van Dillen PM, Berkhout RJ, van Loon AM. 2005. Survey of acyclovir-resistant herpes simplex virus in the Netherlands: prevalence and characterization. J. Clin. Virol. 32:7–18. 10.1016/j.jcv.2004.04.002 [DOI] [PubMed] [Google Scholar]
- 14.Hsu W-H, Hsieh Y-S, Kuo H-C, Teng C-Y, Huang H-I, Wang C-J, Yang S-F, Liou Y-S, Kuo W-H. 2007. Berberine induces apoptosis in SW620 human colonic carcinoma cells through generation of reactive oxygen species and activation of JNK/p38 MAPK and FasL. Arch. Toxicol. 81:719–728. 10.1007/s00204-006-0169-y [DOI] [PubMed] [Google Scholar]
- 15.Choi MS, Yuk DY, Oh JH, Jung HY, Han SB, Moon DC, Hong JT. 2008. Berberine inhibits human neuroblastoma cell growth through induction of p53-dependent apoptosis. Anticancer Res. 28:3777–3784 [PubMed] [Google Scholar]
- 16.Tang J, Feng YB, Tsao S, Wang N, Curtain R, Wang YW. 2009. Berberine and Coptidis Rhizoma as novel antineoplastic agents: a review of traditional use and biomedical investigations. J. Ethnopharmacol. 126:5–17. 10.1016/j.jep.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 17.Pandey MK, Sung B, Kunnumakkara AB, Sethi G, Chaturvedi MM, Aggarwal BB. 2008. Berberine modifies cysteine 179 of IκBα kinase, suppresses nuclear factor-κB-regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res. 68:5370–5379. 10.1158/0008-5472.CAN-08-0511 [DOI] [PubMed] [Google Scholar]
- 18.Cui G, Qin X, Zhang Y, Gong Z, Ge B, Zang YQ. 2009. Berberine differentially modulates the activities of ERK, p38 MAPK, and JNK to suppress Th17 and Th1 T cell differentiation in type 1 diabetic mice. J. Biol. Chem. 284:28420–28429. 10.1074/jbc.M109.012674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLean C, Erturk M, Jennings R, Challanain DN, Minson A, Duncan I, Boursnell M, Inglis S. 1994. Protective vaccination against primary and recurrent disease caused by herpes simplex virus (HSV) type 2 using a genetically disabled HSV-1. J. Infect. Dis. 170:1100–1109. 10.1093/infdis/170.5.1100 [DOI] [PubMed] [Google Scholar]
- 20.Qiu M, Chen Y, Chu Y, Song S, Yang N, Gao J, Wu Z. 2013. Zinc ionophores pyrithione inhibits herpes simplex virus replication through interfering with proteasome function and NF-κB activation. Antiviral Res. 100:44–53. 10.1016/j.antiviral.2013.07.001 [DOI] [PubMed] [Google Scholar]
- 21.Chou T, Hayball M. 1991. CalcuSyn: Windows software for dose effect analysis. CalcuSyn, Ferguson, MO [Google Scholar]
- 22.Chou T-C, Talalay P. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22:27–55. 10.1016/0065-2571(84)90007-4 [DOI] [PubMed] [Google Scholar]
- 23.Dicker IB, Seetharam S. 1995. Herpes simplex type 1: lacZ recombinant viruses. I. Characterization and application to defining the mechanisms of action of known antiherpes agents. Antiviral Res. 28:191–212 [DOI] [PubMed] [Google Scholar]
- 24.Zhu Q-C, Wang Y, Peng T. 2010. Herpes simplex virus (HSV) immediate-early (IE) promoter-directed reporter system for the screening of antiherpetics targeting the early stage of HSV infection. J. Biomol. Screening 15:1016–1020. 10.1177/1087057110372804 [DOI] [PubMed] [Google Scholar]
- 25.Boehmer PE, Lehman I. 1993. Herpes simplex virus type 1 ICP8: helix-destabilizing properties. J. Virol. 67:711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodkin ML, Ting AT, Blaho JA. 2003. NF-κB is required for apoptosis prevention during herpes simplex virus type 1 infection. J. Virol. 77:7261–7280. 10.1128/JVI.77.13.7261-7280.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gregory D, Hargett D, Holmes D, Money E, Bachenheimer S. 2004. Efficient replication by herpes simplex virus type 1 involves activation of the IκB kinase-IκB-p65 pathway. J. Virol. 78:13582–13590. 10.1128/JVI.78.24.13582-13590.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Li X, Wei Y, Tan Y, Liu X, Wu X. 2009. HSV-2 induces TLRs and NF-κB-dependent cytokines in cervical epithelial cells. Biochem. Biophys. Res. Commun. 379:686–690. 10.1016/j.bbrc.2008.12.150 [DOI] [PubMed] [Google Scholar]
- 29.Yedowitz JC, Blaho JA. 2005. Herpes simplex virus 2 modulates apoptosis and stimulates NF-κB nuclear translocation during infection in human epithelial HEp-2 cells. Virology 342:297–310. 10.1016/j.virol.2005.07.036 [DOI] [PubMed] [Google Scholar]
- 30.Gillis PA, Okagaki LH, Rice SA. 2009. Herpes simplex virus type 1 ICP27 induces p38 mitogen-activated protein kinase signaling and apoptosis in HeLa cells. J. Virol. 83:1767–1777. 10.1128/JVI.01944-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLean T, Bachenheimer S. 1999. Activation of c-Jun N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 73:8415–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Zhang J, Kumar A, Zheng M, Atherton SS, Yu FSX. 2006. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 117:167–176. 10.1111/j.1365-2567.2005.02275.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melchjorsen J, Sirén J, Julkunen I, Paludan SR, Matikainen S. 2006. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-κB and IRF-3. J. Gen. Virol. 87:1099–1108. 10.1099/vir.0.81541-0 [DOI] [PubMed] [Google Scholar]
- 34.Birdsall TC, Kelly GS. 1997. Berberine therapeutic potential of an alkaloid found in several medicinal plants. Alternative Med. Rev. 2:94–103 [Google Scholar]
- 35.Mahata S, Bharti AC, Shukla S, Tyagi A, Husain SA, Das BC. 2011. Berberine modulates AP-1 activity to suppress HPV transcription and downstream signaling to induce growth arrest and apoptosis in cervical cancer cells. Mol. Cancer 10:39. 10.1186/1476-4598-10-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Remppis A, Bea F, Greten HJ, Buttler A, Wang H, Zhou Q, Preusch MR, Enk R, Ehehalt R, Katus H. 2010. Rhizoma Coptidis inhibits LPS-induced MCP-1/CCL2 production in murine macrophages via an AP-1 and NF-κB-dependent pathway. Mediators Inflamm. 2010:194896. 10.1155/2010/194896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuo CL, Chi CW, Liu TY. 2004. The anti-inflammatory potential of berberine in vitro and in vivo. Cancer Lett. 203:127–137. 10.1016/j.canlet.2003.09.002 [DOI] [PubMed] [Google Scholar]
- 38.Lee C-H, Chen J-C, Hsiang C-Y, Wu S-L, Wu H-C, Ho T-Y. 2007. Berberine suppresses inflammatory agents-induced interleukin-1β and tumor necrosis factor-α productions via the inhibition of IκB degradation in human lung cells. Pharmacol. Res. 56:193–201. 10.1016/j.phrs.2007.06.003 [DOI] [PubMed] [Google Scholar]
- 39.Cecil CE, Davis JM, Cech NB, Laster SM. 2011. Inhibition of H1N1 influenza A virus growth and induction of inflammatory mediators by the isoquinoline alkaloid berberine and extracts of goldenseal (Hydrastis canadensis). Int. Immunopharmacol. 11:1706–1714. 10.1016/j.intimp.2011.06.002 [DOI] [PubMed] [Google Scholar]
- 40.Hayashi K, Minoda K, Nagaoka Y, Hayashi T, Uesato S. 2007. Antiviral activity of berberine and related compounds against human cytomegalovirus. Bioorg. Med. Chem. Lett. 17:1562–1564. 10.1016/j.bmcl.2006.12.085 [DOI] [PubMed] [Google Scholar]
- 41.Chin LW, Cheng Y-W, Lin S-S, Lai Y-Y, Lin L-Y, Chou M-Y, Chou M-C, Yang C-C. 2010. Anti-herpes simplex virus effects of berberine from Coptidis rhizoma, a major component of a Chinese herbal medicine, Ching-Wei-San. Arch. Virol. 155:1933–1941. 10.1007/s00705-010-0779-9 [DOI] [PubMed] [Google Scholar]
- 42.Andersen JH, Jenssen H, Gutteberg TJ. 2003. Lactoferrin and lactoferricin inhibit Herpes simplex 1 and 2 infection and exhibit synergy when combined with acyclovir. Antiviral Res. 58:209–215. 10.1016/S0166-3542(02)00214-0 [DOI] [PubMed] [Google Scholar]
- 43.Schinazi R, Peters J, Williams C, Chance D, Nahmias A. 1982. Effect of combinations of acyclovir with vidarabine or its 5′-monophosphate on herpes simplex viruses in cell culture and in mice. Antimicrob. Agents Chemother. 22:499–507. 10.1128/AAC.22.3.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanwick TL, Schinazi RF, Campbell DE, Nahmias AJ. 1981. Combined antiviral effect of interferon and acyclovir on herpes simplex virus types 1 and 2. Antimicrob. Agents Chemother. 19:672–674. 10.1128/AAC.19.4.672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoosook C, Bunyapraphatsara N, Boonyakiat Y, Kantasuk C. 2000. Anti-herpes simplex virus activities of crude water extracts of Thai medicinal plants. Phytomedicine 6:411–419. 10.1016/S0944-7113(00)80068-9 [DOI] [PubMed] [Google Scholar]
- 46.DeLuca NA, Schaffer PA. 1985. Activation of immediate-early, early, and late promoters by temperature-sensitive and wild-type forms of herpes simplex virus type 1 protein ICP4. Mol. Cell. Biol. 5:1997–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGregor F, Phelan A, Dunlop J, Clements J. 1996. Regulation of herpes simplex virus poly(A) site usage and the action of immediate-early protein IE63 in the early-late switch. J. Virol. 70:1931–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel A, Hanson J, McLean TI, Olgiate J, Hilton M, Miller WE, Bachenheimer SL. 1998. Herpes simplex virus type 1 induction of persistent NF-κB nuclear translocation increases the efficiency of virus replication. Virology 247:212–222. 10.1006/viro.1998.9243 [DOI] [PubMed] [Google Scholar]
- 49.Baldwin AS., Jr 1996. The NF-κB and IκB proteins: new discoveries and insights. Annu. Rev. Immunol. 14:649–681. 10.1146/annurev.immunol.14.1.649 [DOI] [PubMed] [Google Scholar]
- 50.Amici C, Belardo G, Rossi A, Santoro MG. 2001. Activation of IκB kinase by herpes simplex virus type 1 a novel target for anti-herpetic therapy. J. Biol. Chem. 276:28759–28766. 10.1074/jbc.M103408200 [DOI] [PubMed] [Google Scholar]
- 51.Zachos G, Clements B, Conner J. 1999. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kinase pathways and activates transcription factor AP-1. J. Biol. Chem. 274:5097–5103. 10.1074/jbc.274.8.5097 [DOI] [PubMed] [Google Scholar]
- 52.Kyriakis JM, Avruch J. 2001. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81:807–869 [DOI] [PubMed] [Google Scholar]
- 53.Andrade A, Silva P, Pereira A, De Sousa L, Ferreira P, Gazzinelli R, Kroon E, Ropert C, Bonjardim C. 2004. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 381:437–446. 10.1042/BJ20031375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holloway G, Coulson BS. 2006. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J. Virol. 80:10624–10633. 10.1128/JVI.00390-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rahaus M, Desloges N, Wolff MH. 2004. Replication of varicella-zoster virus is influenced by the levels of JNK/SAPK and p38/MAPK activation. J. Gen. Virol. 85:3529–3540. 10.1099/vir.0.80347-0 [DOI] [PubMed] [Google Scholar]
- 56.Jacqué JM, Mann A, Enslen H, Sharova N, Brichacek B, Davis RJ, Stevenson M. 1998. Modulation of HIV-1 infectivity by MAPK, a virion–associated kinase. EMBO J. 17:2607–2618. 10.1093/emboj/17.9.2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Si X, Luo H, Morgan A, Zhang J, Wong J, Yuan J, Esfandiarei M, Gao G, Cheung C, McManus BM. 2005. Stress-activated protein kinases are involved in coxsackievirus B3 viral progeny release. J. Virol. 79:13875–13881. 10.1128/JVI.79.22.13875-13881.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR, Ludwig S. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signaling cascade. Nat. Cell Biol. 3:301–305. 10.1038/35060098 [DOI] [PubMed] [Google Scholar]
- 59.Panteva M, Korkaya H, Jameel S. 2003. Hepatitis viruses and the MAPK pathway: is this a survival strategy? Virus Res. 92:131–140. 10.1016/S0168-1702(02)00356-8 [DOI] [PubMed] [Google Scholar]
- 60.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. 2002. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 72:847–855 [PubMed] [Google Scholar]
- 61.Kunsch C, Rosen CA. 1993. NF-κB subunit-specific regulation of the interleukin-8 promoter. Mol. Cell. Biol. 13:6137–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bazzoni F, Kruys V, Shakhov A, Jongeneel C, Beutler B. 1994. Analysis of tumor necrosis factor promoter responses to ultraviolet light. J. Clin. Invest. 93:56. 10.1172/JCI116984 [DOI] [PMC free article] [PubMed] [Google Scholar]