

Abstract
Brilacidin (PMX30063) has shown potent bactericidal activity against drug-resistant and -susceptible strains of multiple Gram-negative and Gram-positive pathogens. In this study, we demonstrate that brilacidin causes membrane depolarization in the Gram-positive bacterium Staphylococcus aureus, to an extent comparable to that caused by the lipopeptidic drug daptomycin. Transcriptional profiling of Staphylococcus aureus by deep sequencing shows that the global response to brilacidin treatment is well correlated to those of treatment with daptomycin and the cationic antimicrobial peptide LL37 and mostly indicates abrogation of cell wall and membrane functions. Furthermore, the upregulation of various chaperones and proteases by brilacidin and daptomycin indicates that cytoplasmic protein misfolding stress may be a contributor to the mechanism of action of these drugs. These stress responses were orchestrated mainly by three two-component systems, GraSR, VraSR, and NsaSR, which have been implicated in virulence and drug resistance against other clinically available antibiotics.
INTRODUCTION
The recent rise of multidrug-resistant pathogenic bacteria is an alarming health care crisis that has outpaced the discovery of effective and novel therapeutics (1, 2). Antimicrobial peptides (AMPs), which are evolutionarily conserved, first-line host defense mechanisms, offer an attractive platform for the development of new antibiotics (3–5). Most AMPs are believed to interact with bacterial membranes and cause cell death by dysregulating the properties of the phospholipid bilayer or by causing membrane leakage, although some have been identified to have downstream cytoplasmic targets as well (6). Despite the variety of sequences and secondary and tertiary structures, most AMPs share an amphiphilic topology, with a charged, mostly positive face that allows for interaction with the negatively charged bacterial membrane and a hydrophobic face that allows for insertion into the membrane and interaction with the apolar acyl chains of the bilayer (4, 7–10). Several mechanisms have been suggested for the nature of this interaction with the membrane, including carpet, toroidal pore, and barrel stave mechanisms (6). Development of resistance to these peptides is limited (11), presumably due to the membrane being the primary target (12). As such, several strategies have been employed to mimic the activity of AMPs in order to improve efficacy, selectivity for bacteria, and bioavailability while circumventing issues associated with peptidic drugs, such as proteolytic degradation and difficulties with large-scale synthesis. These include the use of scaffolds such as D-L peptides, β-amino acid helices, and antimicrobial polymers (13–15).
In previous work, we developed a series of small-molecule arylamide mimics of AMPs that showed potent activity against a broad range of drug-susceptible and multidrug-resistant Gram-negative and Gram-positive bacteria (15–19). These compounds feature a small arylamide backbone that is stabilized by intramolecular hydrogen bonding and decorated with cationic and hydrophobic substitutions, resulting in potent and selective amphiphilic molecules with molecular masses of ∼1,000 Da. The optimization of these compounds for activity against Staphylococcus aureus resulted in a lead compound, brilacidin (PMX30063) (Fig. 1), which features a planar, conformationally restrained scaffold with four positive guanadinyl and pyridinyl substitutions and two trifluoromethane hydrophobic substitutions. Brilacidin has shown great efficacy in phase II clinical trials against acute Staphylococcus aureus skin and skin structure infections, comparable to that of the lipopeptidic drug daptomycin, which is currently used clinically to treat drug-resistant staph infections (20). Brilacidin also has potent broad-spectrum activity in vitro against several other Gram-positive and Gram-negative pathogenic bacteria, including several multidrug-resistant strains (16, 21).
FIG 1.
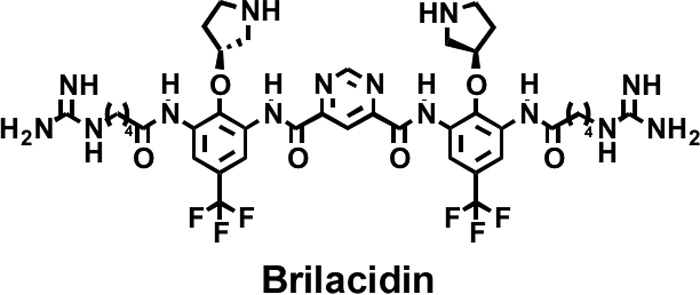
Structure of brilacidin (PMX30063).
Earlier precursors of brilacidin were shown to have bactericidal activity against the Gram-negative bacterium Escherichia coli resulting from their effects on bacterial membrane properties (22). While these precursors showed permeabilization of the outer membrane to small polar substrates comparable to that of the lipopeptide polymyxin B, they showed little change in the permeability of the inner membrane to these substrates. However, protein translocation across the inner membrane was compromised by arylamide treatment, suggesting that the proton motive force (PMF) and/or physiochemical properties of the inner membrane are affected. This is further corroborated by the transcriptional induction of the Kdp operon, which is responsive to K+ homeostasis and turgor pressure (23, 24), and transmission electron microscopy (TEM) imaging studies, which showed wide-scale destabilization of the outer membrane but relatively intact cell morphology with increased uptake of uranyl acetate stain into the cytoplasm (22). Most of the genes upregulated by arylamide treatment were found to be under the control of two-component systems (TCSs) that primarily respond to membrane stress (Rcs and Kdp) (25, 26) and periplasmic misfolding stress (Bae and Cpx) (27, 28).
In this study, we compare brilacidin to the lipopeptidic antibiotic daptomycin (20), a Ca2+-dependent anionic lipopeptide with activity against Gram-positive bacteria, and the antimicrobial peptide LL16 (a 16-residue truncation of human cathelicidin LL37) (29). LL37 is an alpha-helical antimicrobial peptide known to exert antibacterial activity via membrane permeabilization. In Escherichia coli, the peptide has been shown to cause permeabilization of both the outer and inner membranes with kinetics that vary according to the external salt concentration and can cooperatively bind to lipopolysaccharide (29). Previous studies have also shown that LL37 causes an immediate depolarization of the Staphylococcus aureus membrane (30). The mechanism of daptomycin, however, is less well understood. Previous studies have shown that daptomycin treatment causes membrane depolarization (31–33) and the induction of genes associated with cell wall stress (34, 35), with some reports of inhibition of lipoteichoic acid synthesis (36, 37), although the latter has been shown not to be a universal feature of the bactericidal activity of daptomycin (38).
We found that brilacidin treatment causes levels of membrane depolarization in Staphylococcus aureus similar to those of daptomycin, suggesting that membrane activity is the primary initial target. Transcriptional profiling shows that brilacidin, daptomycin, and LL16 treatments cause a significant induction of the NsaSR (nisin sensitivity-associated) two-component system (39) as well as the cell wall stress-responsive VraSR (40, 41) and WalKR (42, 43) two-component system regulons. While these regulons were upregulated by all three treatments, brilacidin caused the greatest magnitude of changes in these regulons, suggesting that it may cause more potent dysregulation of the bacterial cell wall. Interestingly, brilacidin treatment also caused a modest induction of the antimicrobial peptide-sensing two-component system GraSR (41) and the lysine biosynthesis operon (Dap) (44), which, along with MprF (a member of the GraSR regulon), is involved in the lysinylation of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) lipids in the cytoplasmic membrane (45). These pathways were upregulated to a comparable degree by treatment with LL16, as expected from a cationic AMP with potent activity against Staphylococcus aureus. We also report the upregulation of proteases and chaperones involved in the cell stress response by brilacidin and daptomycin treatments. However, LL16 treatment did not cause a significant upregulation of these proteins. We hypothesize that brilacidin acts primarily on the membrane, as supported by depolarization of the membrane potential upon treatment and the induction of NsaSR, GraSR, and Dap regulons. In addition, brilacidin causes a large upregulation of the VraSR and WalKR regulons and cytoplasmic proteases and chaperones, indicating that it causes significant cell wall stress as well as stress due to the accumulation of misfolded proteins.
MATERIALS AND METHODS
Antibiotics.
Daptomycin (Cubicin) was used without further purification. Brilacidin was purified as an HCl salt by reverse-phase high-performance liquid chromatography (HPLC). LL16 was prepared by 9-fluorenylmethoxy carbonyl (Fmoc) solid-phase peptide synthesis and purified as a trifluoroacetic acid (TFA) salt by reverse-phase HPLC.
Bacterial strains.
All bacterial studies were conducted on Staphylococcus aureus Newman (46), which was a gift from Jeffrey Cox (University of California, San Francisco [UCSF]).
Membrane depolarization assay.
A culture of strain Newman was grown to an optical density at 600 nm (OD600) of 0.5 at 37°C, pelleted, washed twice in a solution containing 5 mM HEPES (pH 7.2) and 5 mM glucose, and resuspended in a solution containing 5 mM HEPES (pH 7.2), 5 mM glucose, 100 mM KCl, and 1 mM CaCl2 to a final OD600 of 0.02. The suspension was then incubated with drugs at room temperature (RT) for 30 min, 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)] was added to a final concentration of 30 μM, and the mixture was incubated for 15 min. Cells were then analyzed by using flow cytometry with 488-nm excitation, and green (530-nm/30-nm) and red (585-nm/42-nm) emission channels were monitored for 30,000 cells. Red/green ratios were then calculated for each cell by using Flo-Jo software.
Transcriptional profiling.
A culture of strain Newman was grown to an OD600 of 0.5 and split 2-fold into prewarmed LB medium with drug, and aliquots were collected by centrifugation every 20 min for 2 h. Pellets were flash-frozen in liquid N2 to halt transcription. Total RNA was purified from pellets by using TRIzol reagent according to the manufacturer's instructions. mRNA was enriched by rRNA removal using a Microb-express bacterial mRNA purification kit (Ambion).
Illumina sequencing library generation.
Barcoded Illumina sequencing libraries were constructed by using a modified protocol obtained from the DeRisi laboratory at UCSF (47). Briefly, random-hexamer primers with a 5′ adapter sequence (primer 1 [3Sol_N]) were used to synthesize first-strand cDNA from enriched mRNA by using a cDNA synthesis kit (Invitrogen). The second strand was synthesized by using the same primer and Thermo Sequenase DNA polymerase (GE Health Care). The cDNA library was PCR amplified by using Kapa polymerase (10 cycles) with primer 2 (3Sol). After PCR cleanup, concentrations were determined by using a Nanodrop system, and samples were normalized to a 2-ng/μl concentration. This library was barcoded by using custom-built 7-bp-barcode Illumina sequencing primers (primer 3 [SolM2] and primer 4 [5SolM2]; DeRisi laboratory) with Kapa Hi-fi polymerase (Kapa Biosystems) for 2 amplification cycles, and the barcoded library was amplified in the same reaction by using hot-start primers (primer 5 [5SolM2_18] and primer 6 [5SolM2_19]) for an additional 6 cycles. Hot-start primers were activated by heating the reaction mixture at 94°C for 10 min. The resulting barcoded libraries were multiplexed (3 samples for 15 conditions and 1 sample for 16 conditions), size selected by using a labChip XT DNA 750 assay kit (300 to 400 bp), and quantified by using high-sensitivity DNA chips (Agilent). These 4 samples were then single-read sequenced (50-bp reads) by using a HiSeq Illumina sequencer with 7-bp-barcode sequencing, according to the manufacturer's instructions, at the Center for Advanced Technology at UCSF.
Deep-sequencing data analysis.
Sequencing data were first checked for sequencing quality and then demultiplexed by using perfect matches to the 7-bp barcodes using an in-house script. Demultiplexed data were then mapped onto the Newman strain genome by using BowTie (Galaxy project) (48–50). Mapped reads were then binned into gene open reading frames (ORFs) and analyzed for differential expression. Out of a total of ∼551 million raw sequences, 472 million sequences were successfully demultiplexed and indexed. About 51% of these reads were mapped to mRNA coding regions (49% were rRNA and tRNA coding regions), which roughly corresponds to a 25-fold enrichment of mRNA reads. Fold enrichment was calculated by using an expectation-weighted normalization to minimize overestimation of fold changes in genes with low read counts, as follows:
| (1) |
where
| (2) |
In order to determine the statistical significance of differential expression of each gene, the various time points were treated as replicates, and a standard t test was used to measure the difference between the nontreated and treated samples. Treating the various time points as replicates biases the test toward the null hypothesis, which increases the stringency of the significance test. Maximal upregulation was also assessed for significance by comparison to a normal distribution fitted to the nontreated samples. P values of <0.05 were considered significant based on these tests.
RESULTS
Mechanism of action against Staphylococcus aureus.
Earlier generations of arylamides have been shown to cause depolarization of the Staphylococcus aureus membrane, which could contribute to the lethality of these compounds in Gram-positive bacteria (16). In order to examine the mechanism of brilacidin more fully, we compared its effects to those of daptomycin and, where applicable, LL16 (residues 17 to 32 of LL37) (51). Here, we report the extent of membrane depolarization caused by brilacidin and conduct detailed profiling of the transcriptional response of Staphylococcus aureus to brilacidin, comparing these responses to those of daptomycin and LL16 treatments.
Brilacidin treatment causes membrane depolarization in Staphylococcus aureus.
We determined the extent of membrane depolarization caused by these agents using the membrane potential-sensitive dye DiOC2(3), which is thought to accumulate in the cytoplasm of cells with hyperpolarized membranes (52). The high concentration of DiOC2(3) in polarized cells causes dye-stacking interactions that result in a red shift in its emission. Therefore, the red/green ratio of its emission when excited with blue light is indicative of the extent of polarization of cellular membranes. Newman cultures were grown to mid-log phase (OD600 = 0.5), washed, resuspended in HEPES buffer, and treated with different concentrations of brilacidin and daptomycin for 30 min. DiOC2(3) was then added to cultures, the cultures were incubated for 15 min, and cells were analyzed by flow cytometry. Brilacidin was found to cause dose-dependent depolarization of the membrane (Fig. 2). The extent of depolarization was comparable to that caused by daptomycin and not as severe as that caused by the protonophore carbonyl cyanide m-chlorophenylhydrazone (CCCP). Untreated cells had a red/green emission ratio of ∼0.65, while cells treated with brilacidin and daptomycin had a red/green ratio of ∼0.4 at the highest doses. Since cell death brought about by both of these antibacterials requires >30 min, membrane depolarization happens prior to the loss of cell viability rather than concomitantly. Therefore, it is important to examine what other effects brilacidin treatment has on cells and what the downstream consequences of membrane depolarization may be.
FIG 2.

Membrane depolarization of Staphylococcus aureus. Newman cultures suspended in buffer supplemented with Ca2+ were treated with 1, 2, and 4 μg/ml of brilacidin and daptomycin for 30 min and stained with DiOC2(3) for 15 min. Red/green fluorescence emission ratios were determined for 30,000 cells by flow cytometry, with a decrease in the red/green ratio indicating depolarization of the membrane. The protonophore CCCP was used as a positive control for membrane depolarization.
Transcriptional profiling of Staphylococcus aureus treated with brilacidin, daptomycin, and LL16.
The transcriptional response of bacteria to sublethal concentrations of antimicrobials is an invaluable tool for understanding how bacteria perceive the physiological insult caused by drug treatments and consequently serves to inform their possible mechanism of action. We employed next-generation sequencing technology (Illumina deep sequencing) (53, 54) to examine the transcriptional response of strain Newman to brilacidin treatment over an extended period of time, using daptomycin and LL16 as comparators. Three doses of each treatment, which caused very slight to half-maximal inhibition of culture growth (see Fig. S1 in the supplemental material), were used, along with a no-treatment control, and samples were analyzed every 20 min for 2 h (61 total drug/concentration/time combinations). These concentrations corresponded to 1- to 4-fold the MIC of brilacidin, 2- to 6-fold the MIC for daptomycin (the reported MIC against strain Newman is 1 μg/ml), and 0.16- to 0.48-fold the MIC for LL16 (the measured MIC is 12.5 μg/ml, which is likely due to inactivation of LL16 by proteolytic degradation during extended incubation). Using a modified protocol developed by the DeRisi laboratory (see Methods and Materials), we constructed barcoded Illumina libraries from isolated and enriched mRNAs, which were then single-read sequenced in 4 multiplexed runs. These reads were then demultiplexed and mapped to the Newman genome, and fold changes in transcription were computed as discussed in Materials and Methods.
Due to differences in growth rate and stationary-phase transition between drug-treated and untreated samples, we used a binomial distribution-weighted 5-time-point sliding window to normalize drug-treated samples against nontreated controls [gene counts for treated samples at time tn, where n is the sequential number of time points, were normalized with a 1:4:6:4:1 weighted average count of the five time points centered around tn for the nontreated control, i.e., (1tn-2:4tn-1:6tn:4tn+1:1tn+2)/16]. This normalization was effective in removing any artifacts resulting from a delayed transition to stationary phase caused by antibiotic treatment.
Transcriptional regulation by brilacidin treatment is globally well correlated with daptomycin and LL16 treatments.
All three treatments caused the upregulation of a large set of genes, with increasing concentrations of antibiotics generally causing the upregulation of a larger number of genes and to a greater magnitude. At the highest concentration of treatment, brilacidin caused the upregulation of 698 genes at a >3-fold change threshold (at any time point) (P < 0.05). A total of 365 genes were upregulated by the highest concentration of daptomycin, and 359 genes were upregulated by the highest concentration of LL16. The numbers of differentially regulated genes at each concentration of these 3 treatments are listed in Table 1. Pairwise Pearson correlations show that the different concentrations and time points are well correlated within a given drug treatment, with correlation coefficients being as high as 0.85 (Fig. 3). Moreover, there is significant correlation between the transcriptional profiles of brilacidin and daptomycin as well as those of brilacidin and LL16, particularly at the highest concentrations of treatment used. However, the profiles of daptomycin and LL16 treatments are less correlated. This suggests that the insult perceived as a result of brilacidin treatment is intermediate between those of daptomycin and LL16. Notably, there is a high correlation between brilacidin and LL16 at corresponding treatment times, indicating a similar progression of gene induction, whereas the earlier time points of brilacidin treatment correlate well with the later time points of daptomycin treatment. This is also mirrored in the time of maximal gene induction and follows the growth profiles of Newman cultures in the presence of these antibiotics (see Fig. S1 in the supplemental material). Brilacidin causes maximal induction after ∼40 min of treatment, whereas daptomycin causes maximal induction 1 to 2 h after treatment. LL16 treatment caused an immediate maximal induction of genes, with several genes showing maximal induction at 20 min. Similarly, brilacidin and LL16 have an immediate effect on the growth rate of treated cultures, whereas daptomycin shows an appreciable effect only after ∼1 h of treatment.
TABLE 1.
Number of genes upregulated >3-fold by drug treatment
| Treatment condition | Concn (μg/ml) | No. of genes upregulated >3-folda |
|---|---|---|
| Control | 0 | |
| Brilacidin | 0.39 | 171 |
| 0.78 | 415 | |
| 1.17 | 698 | |
| Daptomycin | 2.0 | 258 |
| 4.0 | 293 | |
| 6.0 | 365 | |
| LL16 | 2.0 | 276 |
| 4.0 | 436 | |
| 6.0 | 359 |
P < 0.05.
FIG 3.

Global pairwise correlations of transcript levels. Pearson correlation coefficients were calculated for all possible 61-by-61 combinations of conditions evaluated by RNA-seq. Correlations range from −0.79 to 0.85, with significant positive correlation within drug treatments.
The most highly upregulated genes belong to stress-responsive TCSs.
A closer examination of the most upregulated genes, as defined by the maximal upregulation at any time point for a given concentration of treatment, revealed that most of the annotated genes belonged to three stress-responsive two-component systems: GraSR, VraSR, and NsaSR. There was also a large number of genes that are thought to be members of the WalKR (YycGF) TCS, a large regulon that is involved in cell wall and membrane synthesis, degradation, and maintenance (55). Several genes regulated by GraSR, VraSR, and NsaSR also belong to this regulon. Figure 4 shows the maximal fold changes of genes in these four regulons at the highest treatment concentrations. Although the direct regulation of several genes by these TCSs has been confirmed, it is important to note that most of these TCSs have been identified by using differential expression between wild-type strains and strains in which these TCSs were deleted. As such, the full extent of their regulation is unknown.
FIG 4.

Upregulation of Staphylococcus aureus two-component systems. Shown are fold changes in expression levels of genes belonging to the GraSR (A), NsaSR (B), VraSR (C), and WalKR (D) two-component systems. The y axis is log2 transformed for clarity.
The VraSR TCS was the pathway most upregulated by brilacidin treatment (Fig. 4C and Table 2). This regulon is also upregulated by daptomycin and LL16 treatments, although the induction by brilacidin was generally greater in magnitude and more sustained. Most of the genes were maximally upregulated 40 min after treatment. VraSR (vancomycin resistance associated) primarily responds to cell wall stress and upregulates genes involved in lipoteichoic acid and peptidoglycan synthesis (56). Thirty-two of the 42 genes thought to belong to the VraSR regulon were upregulated >3-fold, with 18 of these genes being upregulated >10-fold. In contrast, 22 of these genes were upregulated >3-fold by daptomycin, but only 2 genes were upregulated >10-fold. Similarly, 27 genes were upregulated >3-fold by LL16 treatment, with only 4 being upregulated >10-fold. We also noted the upregulation of the WalKR regulon, which is thought to be involved in cell wall and membrane turnover and maintenance. Several of these genes were also identified as members of the VraSR, NsaSR, and GraSR TCSs. The full list of WalKR-regulated genes and their differential expression levels due to treatments are listed in Table S3 in the supplemental material.
TABLE 2.
Differential expression of VraSR-regulated genesa
| Gene | Gene description | Fold change in expression |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Control |
PMX30063 at concn (μg/ml) of: |
Daptomycin at concn (μg/ml) of: |
LL16 at concn (μg/ml) of: |
||||||||
| 0.39 | 0.78 | 1.17 | 2.0 | 4.0 | 6.0 | 2.0 | 4.0 | 6.0 | |||
| VraSR regulated | |||||||||||
| drp35 | Lactonase drp35 | 1.48*+ | 1.66*+ | 33.12* | 46.55* | 2.35* | 3.25*+ | 4.41*+ | 2.04*+ | 13.59* | 16.65* |
| vraS | Sensor histidine kinase VraS | 1.22*+ | 5.66* | 14.94* | 28.01*+ | 2.36*+ | 5.90*+ | 5.93*+ | 5.18* | 6.14*+ | 10.31* |
| NWMN_2332 | Hypothetical protein | 1.18*+ | 2.47*+ | 12.2*+ | 22.85*+ | 2.2*+ | 4.52*+ | 4.99*+ | 4.48* | 5.61*+ | 7.80*+ |
| mgt | Monofunctional glycosyltransferase | 1.18*+ | 4.55*+ | 13.74* | 21.74*+ | 2.49*+ | 3.82*+ | 5.53*+ | 5.07* | 7.39* | 9.06*+ |
| vraR | DNA-binding response regulator VraR | 1.13*+ | 4.36*+ | 12.30* | 19.61*+ | 2.01*+ | 4.74*+ | 5.48*+ | 4.25* | 6.27*+ | 9.10* |
| NWMN_2405 | Hypothetical protein | 1.23*+ | 2.95*+ | 10.13* | 19.50*+ | 1.69*+ | 2.38*+ | 3.27*+ | 2.16*+ | 5.53*+ | 7.46* |
| NWMN_1824 | Hypothetical protein | 1.22*+ | 2.88*+ | 12.38* | 18.73*+ | 2.73*+ | 4.29*+ | 4.47*+ | 3.39* | 6.01*+ | 6.99*+ |
| proP | Similar to glycine betaine-proline ABC transporter permease | 1.10*+ | 2.98*+ | 12.39* | 15.50*+ | 2.54*+ | 4.52*+ | 5.47*+ | 4.55*+ | 5.46*+ | 10.83*+ |
| NWMN_2211 | Cell envelope-related transcriptional attenuator | 1.23*+ | 2.60*+ | 7.09*+ | 14.66*+ | 2.14*+ | 3.96*+ | 5.44*+ | 2.91*+ | 5.40*+ | 4.82*+ |
| NWMN_1835 | RNA methyltransferase TrmA family protein | 1.18*+ | 2.21*+ | 8.99*+ | 14.44*+ | 1.00*+ | 1.55*+ | 2.01*+ | 1.80*+ | 2.63*+ | 3.50*+ |
| NWMN_1621 | Similar to serine proteinase | 1.24*+ | 2.19*+ | 8.41* | 14.26*+ | 2.03*+ | 3.44*+ | 3.31*+ | 2.34*+ | 4.21*+ | 7.53* |
| NWMN_2331 | Glycerate kinase | 1.07*+ | 2.50*+ | 9.36*+ | 13.55*+ | 1.75*+ | 3.64*+ | 5.09*+ | 2.74*+ | 3.49*+ | 5.36*+ |
| NWMN_1732 | Hypothetical protein | 1.59* | 1.08*+ | 12.37* | 3.66*+ | 3.73*+ | 2.21*+ | 3.29*+ | 2.36* | 6.35* | 2.53*+ |
| NWMN_1825 | Hypothetical protein | 1.36*+ | 2.03* | 4.28*+ | 11.97*+ | 1.96*+ | 2.40*+ | 3.77*+ | 2.11*+ | 2.36*+ | 3.33*+ |
| NWMN_2404 | Transcriptional regulator MerR family protein | 1.28*+ | 1.25*+ | 4.97*+ | 9.07*+ | 1.68*+ | 2.13*+ | 2.90*+ | 1.94*+ | 2.27*+ | 2.91*+ |
| tcaA | Teicoplanin resistance-associated protein A | 1.32*+ | 2.46* | 7.82* | 7.91*+ | 1.61*+ | 2.65*+ | 2.78*+ | 2.56* | 2.62*+ | 4.07*+ |
| ctpA | Carboxyl-terminal protease | 1.17*+ | 2.14*+ | 4.73* | 6.93*+ | 1.73*+ | 2.86*+ | 2.11*+ | 1.52*+ | 2.33*+ | 3.12*+ |
| murZ | UDP-N-acetylglucosamine 1-carboxyvinyltransferase 2 | 1.09*+ | 2.56*+ | 5.39*+ | 6.65*+ | 1.69*+ | 2.72*+ | 2.76*+ | 2.76*+ | 3.10*+ | 3.75*+ |
| NWMN_0931 | Similar to chitinase | 1.33*+ | 1.35* | 3.60*+ | 6.43*+ | 2.41*+ | 3.04*+ | 1.97*+ | 2.40*+ | 2.32*+ | 2.91*+ |
| plsC | 1-Acyl-sn-glycerol-3-phosphate acyltransferase | 1.26*+ | 1.55* | 5.29* | 2.59* | 1.88*+ | 2.52* | 1.99* | 1.44* | 3.36*+ | 4.12* |
| NWMN_0977 | Hypothetical protein | 1.41*+ | 1.23*+ | 1.92*+ | 2.97*+ | 2.74*+ | 3.29* | 2.19*+ | 2.52*+ | 5.19* | 2.40*+ |
| NWMN_2518 | Sulfite reductase flavoprotein | 1.22*+ | 2.01*+ | 2.58*+ | 5.18*+ | 1.6*+ | 2.40*+ | 2.46*+ | 1.89*+ | 2.17*+ | 1.92*+ |
| NWMN_0946 | Hypothetical protein | 1.21*+ | 1.66*+ | 2.24*+ | 5.13*+ | 1.66*+ | 1.83*+ | 2.51*+ | 1.87*+ | 2.39*+ | 2.01*+ |
| NWMN_2406 | Hypothetical protein | 1.40*+ | 1.07*+ | 2.42*+ | 2.82*+ | 1.84*+ | 2.14*+ | 3.21*+ | 1.85*+ | 4.30*+ | 2.65*+ |
| copA | Cation-transporting ATPase E1-E2 family protein | 1.32* | 1.54* | 3.01*+ | 3.85*+ | 1.16* | 2.06* | 1.68*+ | 1.42*+ | 1.90*+ | 2.54* |
| pbpB (pbp2) | Penicillin-binding protein 2 | 1.28*+ | 1.50* | 3.14*+ | 3.10*+ | 1.14*+ | 1.73*+ | 1.94*+ | 2.53* | 2.59*+ | 3.25*+ |
| NWMN_1730 | Hypothetical protein | 1.23*+ | 1.77* | 1.97* | 3.10*+ | 1.92* | 1.67* | 1.84* | 1.70*+ | 1.99*+ | 2.46* |
| NWMN_0622 | Hypothetical protein | 1.45* | 1.93* | 2.76*+ | 3.09*+ | 1.66* | 1.49*+ | 1.47*+ | 2.30* | 2.42*+ | 2.72* |
| recU | Recombination protein U | 1.30* | 1.33* | 2.04*+ | 2.65*+ | 1.38*+ | 1.64*+ | 1.57*+ | 2.58* | 2.03*+ | 2.75*+ |
| NWMN_0687 | Similar to sulfatase | 1.13*+ | 1.58*+ | 2.38*+ | 2.75*+ | 1.35*+ | 1.59*+ | 1.91*+ | 1.91*+ | 1.94*+ | 2.31*+ |
| spsA | Type I signal peptidase A component | 1.45* | 1.18* | 2.13* | 2.72*+ | 1.72* | 1.48* | 1.24* | 2.03*+ | 1.90*+ | 2.27* |
| NWMN_2333 | Drug resistance transporter | 1.44* | 1.47* | 1.94* | 2.16* | 1.32* | 1.48* | 1.42* | 1.91* | 1.77*+ | 1.76*+ |
| tagA | N-Acetylglucosaminyldiphospho-undecaprenol N-acetyl-beta-d-mannosaminyltransferase | 1.23* | 1.74* | 1.31*+ | 1.40*+ | 1.25* | 1.50* | 1.75* | 2.01*+ | 1.99*+ | 1.96*+ |
| opuD | Glycine betaine transporter 1 | 1.44* | 1.59* | 1.29*+ | 1.43* | 1.54* | 1.37* | 1.03 | 1.77*+ | 1.94*+ | 1.46*+ |
| tcaB | Teicoplanin resistance-associated protein B | 1.22* | 1.40* | 1.56* | 1.13 | 0.94*+ | 1.20+ | 1.04+ | 1.20 | 1.39* | 1.22* |
| VraSR and WalKR regulated | |||||||||||
| vraX | Hypothetical protein | 1.13*+ | 6.24* | 40.07*+ | 105.06*+ | 10.01*+ | 47.59* | 37.97*+ | 12.37* | 18.84*+ | 23.66*+ |
| NWMN_1552 | Hypothetical protein | 1.25*+ | 4.18* | 5.86*+ | 34.68* | 5.01*+ | 9.39*+ | 14.35*+ | 4.08*+ | 7.36*+ | 4.92*+ |
| NWMN_1834 | Hypothetical protein | 1.22*+ | 2.14*+ | 7.75*+ | 18.32*+ | 1.38*+ | 2.40*+ | 3.06*+ | 1.64*+ | 3.60*+ | 3.84*+ |
| prsA | Peptidyl-prolyl cis-trans-isomerase | 1.22*+ | 2.47*+ | 6.76*+ | 11.31*+ | 1.96*+ | 4.31*+ | 3.45*+ | 3.32*+ | 3.63*+ | 6.79*+ |
| NWMN_2456 | Hypothetical protein | 1.09*+ | 1.44*+ | 5.86* | 10.92*+ | 6.24*+ | 4.03*+ | 6.80*+ | 5.08* | 3.66*+ | 3.30*+ |
| fmtA | Autolysis and methicillin resistance-related protein FmtA | 1.37*+ | 3.39*+ | 6.05*+ | 7.16*+ | 1.67*+ | 3.49*+ | 2.70*+ | 4.05* | 3.73*+ | 5.60*+ |
| dinP | DNA polymerase IV | 1.15*+ | 1.43* | 4.88*+ | 6.52*+ | 1.05*+ | 1.31*+ | 1.35*+ | 1.73* | 2.25*+ | 2.66*+ |
The maximal upregulation at any time point under each given condition is reported. Genes with >3-fold maximal regulation are highlighted in boldface type and underlining. Fold changes with significance at a P value of <0.05 are marked with asterisks for the Z-factor test and crosses for the t test. Genes are arranged by decreasing maximal upregulation under any of the conditions.
The NsaSR (nisin sensitivity-associated) regulon was also upregulated by all 3 treatments, with brilacidin treatment being the more potent inducer (Fig. 4B). Again, the induction of this regulon mirrored the growth profiles of the treated cultures and the induction of the VraSR regulon, with most genes being maximally upregulated at 40 min for brilacidin, ∼80 min for daptomycin, and 20 to 40 min for LL16 treatment. Although this regulon was first identified due to the increased susceptibility to nisin, it has since been reported to be upregulated by several membrane- and cell wall-active antibiotics. A recent study identified ∼200 genes that may be regulated by NsaSR, including genes involved in cell wall synthesis, membrane transport, redox stress, DNA remodeling, and general cell metabolism (39). The full set of NsaSR-regulated genes and their regulation by various treatments are listed in Table S2 in the supplemental.
All three treatments also caused the upregulation of the GraSR two-component system, which was recently identified as a sensor of antimicrobial peptides in Staphylococcus aureus. GraSR upregulates the Dlt operon, which is involved in d-Ala modification of teichoic acids, as well as MprF (FmtC), which lysinylates POPG phospholipid in the membrane. These responses are aimed at reducing the overall negative charge of the bacterial cell surface, thereby reducing the efficacy of cationic AMPs. This is further aided by the upregulation of the lysine biosynthesis operon Dap, which feeds into MprF. GraSR also upregulates efflux ABC transporters in response to drug treatment and as such represents a concerted response against membrane-active drugs. While this regulon is upregulated by all 3 treatments, LL16 caused the highest and most sustained upregulation of the Dlt and Dap operons, while brilacidin caused a more transient induction of these operons (Fig. 5A and B). Daptomycin treatment did not seem to induce the GraSR TCS to as great an extent as brilacidin and LL16. Table S1 in the supplemental material lists the maximal fold changes of the GraSR regulon by all three treatments.
FIG 5.

Induction of the Dap operon, the Dlt operon, and proteases/chaperones. Heat maps of gene induction (log2 transformed) of the Dlt operon (A), the Dap operon (B), and select proteases and chaperones (C) are shown. Brilacidin and LL16 caused a strong induction of the Dap operon as well as a modest upregulation of the Dlt operon, while daptomycin and higher concentrations of brilacidin treatment caused upregulation of proteases and chaperones.
Another notable difference in responses to treatments was the induction of proteases and chaperones by brilacidin and daptomycin treatments, which was not caused by LL16 treatment (Fig. 5C). Daptomycin induces this stress response concomitant with its effect on culture growth and induction of the TCS-mediated response, even at the lowest concentration of treatment. A similar trend was observed for the higher concentrations of brilacidin, although daptomycin appears to be a more potent inducer at lower treatment concentrations.
DISCUSSION
Brilacidin is a clinical lead against Staphylococcal infections and has shown performance on par with that of daptomycin in phase II clinical trials against Staphylococcus aureus skin and skin structure infections. Our work shows that brilacidin causes a dose-dependent depolarization of the Staphylococcus aureus membrane comparable to that of daptomycin. Transcriptome sequencing shows the induction of major stress regulons, particularly those involved in cell wall and membrane stress. The kinetics of gene induction are well correlated with the effects of brilacidin treatment on culture growth. Global comparison with the transcriptional responses to daptomycin and LL16 shows that brilacidin has certain similarities to both treatments, specifically in its ability to induce the GraSR, NsaSR, VraSR, and WalKR two-component systems (TCSs), with an enhanced upregulation of the cell wall stress regulons. These TCSs are induced by cell membrane- and cell wall-damaging agents and are also involved in resistance to these antibiotics (57–59), which is consistent with the hypothesized mechanism of action of these drugs. Interestingly, recent reports on the mechanism of a novel vancomycin-derivative lipoglycopeptidic drug, telavancin, show curious similarities and key differences in the induction of these TCSs. Telavancin is reported to have a dual mechanism of action that involves inhibition of cell wall synthesis by direct binding to d-Ala–d-Ala moieties of lipid II peptidoglycan precursors as well as the disruption of the membrane potential due to interactions via the apolar decylaminoethyl side chain appended to the vancosamine sugar moiety (60). Transcriptional response studies have shown a strong induction of the VraSR regulon in response to telavancin treatment, as observed for brilacidin treatment and as expected from a cell wall synthesis inhibitor. Moreover, telavancin highly upregulated the BraDE and VraDE ABC transporters, which are regulated by the NsaSR TCS (61). While brilacidin, daptomycin, and LL16 treatments did cause a large upregulation of the NsaSR regulon, not all members of this regulon were equally affected, and vraD and vraE were significantly upregulated by LL16 treatment only (22- and 35-fold, respectively) (see Table S2 in the supplemental material). Thus, the regulation of VraDE ABC transporters appears to include components beyond NsaSR that allow differential responses to these agents (39). In contrast to LL16 treatment, however, no significant induction of the VraFG transporter or the d-alanylation pathway via the GraSR TCS was observed for telavancin treatment (Fig. 5B; see also Table S1 in the supplemental material).
Apart from the induction of these TCSs, we also noted an upregulation of pathways involved in altering cell surface charge as well as some cytoplasmic chaperones and proteases. However, only brilacidin and LL16 lead to a significant induction of the lysine biosynthesis pathway (Dap operon), while only brilacidin and daptomycin treatment upregulated cellular proteases and chaperones. The induction of the Dap operon as well as cytoplasmic chaperones/proteases in response to telavancin treatment was also observed (61) and is thought to be a hallmark of membrane depolarization and misfolded protein accumulation stress, respectively. Several of the responses observed have been implicated in the decreased susceptibility of Staphylococcus aureus to agents such as daptomycin (e.g., MprF and the WalKR TCS) (62) and AMPs (e.g., the Dlt operon) (63). However, there has been no observed increase in susceptibility to brilacidin thus far, despite the upregulation of these pathways.
Despite several reports on the importance of these TCSs in the sensing of and resistance to cell wall-active antibiotics, the mechanism by which GraS, VraS, and NsaS sense either the direct presence or the indirect consequence of these drugs is largely unknown. These histidine kinases are among the shortest sensor kinases in Gram-positive bacteria and feature only a short periplasmic loop (3 to 10 residues), unlike most other TCS histidine kinases, which have defined extracellular or cytoplasmic sensor domains. It was recently shown that signaling via the GraS kinase involves 3 additional proteins, the accessory protein GraX and the ABC transporter proteins VraF and VraG. The VraFG transporter was found not to be involved in detoxification but rather in AMP sensing and signaling via the GraXSR complex (64). The extracellular loop of GraS is also enriched with acidic residues (conserved in homologs in related species) and may be involved in the direct sensing of cationic AMPs and mimetics as well. VraS and NsaS do not have any apparent sensory domains; however, they both have ORFs coding for small unidentified proteins immediately upstream of the response regulator (VraX in VraSR and an unannotated 66-residue peptide, NWMN_2525, in NsaSR), and the NsaSR cassette also contains two ABC transporters immediately downstream of the response regulator (BraDE and VraDE), which may similarly be involved in sensing and signaling as well as detoxification. Further studies on how these histidine kinases sense the presence of membrane-disrupting agents may yield more detailed insight into the mechanism of action of these drugs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jessica Lund at the UCSF Center for Advanced Technologies for assistance with Illumina HiSeq experiments. We also thank Gabriel Gonzalez and Brett Hannigan for their help in RNA-seq data analysis. We are grateful to Susan Gottesman (National Cancer Institute), Ken Keiler (Penn State University), and Jeffrey Cox (UCSF) for the gift of several strains used in this publication.
This work was supported by NIH grant AI074866.
Footnotes
Published ahead of print 16 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02955-14.
REFERENCES
- 1.Opar A. 2007. Bad bugs need more drugs. Nat. Rev. Drug Discov. 6:943–944. 10.1038/nrd2477 [DOI] [Google Scholar]
- 2.Weinstein RA. 2001. Controlling antimicrobial resistance in hospitals: infection control and use of antibiotics. Emerg. Infect. Dis. 7:188–192. 10.3201/eid0702.010206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boman HG. 2000. Innate immunity and the normal microflora. Immunol. Rev. 173:5–16. 10.1034/j.1600-065X.2000.917301.x [DOI] [PubMed] [Google Scholar]
- 4.Tossi A, Sandri L, Giangaspero A. 2000. Amphipathic, α-helical antimicrobial peptides. Biopolymers 55:4–30. [DOI] [PubMed] [Google Scholar]
- 5.Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395. 10.1038/415389a [DOI] [PubMed] [Google Scholar]
- 6.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250. 10.1038/nrmicro1098 [DOI] [PubMed] [Google Scholar]
- 7.Glukhov E, Burrows LL, Deber CM. 2008. Membrane interactions of designed cationic antimicrobial peptides: the two thresholds. Biopolymers 89:360–371. 10.1002/bip.20917 [DOI] [PubMed] [Google Scholar]
- 8.Hancock RE, Diamond G. 2000. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 8:402–410. 10.1016/S0966-842X(00)01823-0 [DOI] [PubMed] [Google Scholar]
- 9.Hancock RE, Rozek A. 2002. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol. Lett. 206:143–149. 10.1111/j.1574-6968.2002.tb11000.x [DOI] [PubMed] [Google Scholar]
- 10.Powers JP, Hancock RE. 2003. The relationship between peptide structure and antibacterial activity. Peptides 24:1681–1691. 10.1016/j.peptides.2003.08.023 [DOI] [PubMed] [Google Scholar]
- 11.Perron GG, Zasloff M, Bell G. 2006. Experimental evolution of resistance to an antimicrobial peptide. Proc. Biol. Sci. 273:251–256. 10.1098/rspb.2005.3301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nizet V. 2006. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr. Issues Mol. Biol. 8:11–26 [PubMed] [Google Scholar]
- 13.Schmitt MA, Weisblum B, Gellman SH. 2007. Interplay among folding, sequence, and lipophilicity in the antibacterial and hemolytic activities of alpha/beta-peptides. J. Am. Chem. Soc. 129:417–428. 10.1021/ja0666553 [DOI] [PubMed] [Google Scholar]
- 14.Shandler SJ, Shapovalov MV, Dunbrack RL, Jr, DeGrado WF. 2010. Development of a rotamer library for use in beta-peptide foldamer computational design. J. Am. Chem. Soc. 132:7312–7320. 10.1021/ja906700x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tew GN, Scott RW, Klein ML, Degrado WF. 2010. De novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications. Acc. Chem. Res. 43:30–39. 10.1021/ar900036b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. 2009. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. U. S. A. 106:6968–6973. 10.1073/pnas.0811818106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu D, Choi S, Chen B, Doerksen RJ, Clements DJ, Winkler JD, Klein ML, DeGrado WF. 2004. Nontoxic membrane-active antimicrobial arylamide oligomers. Angew. Chem. Int. Ed. Engl. 43:1158–1162. 10.1002/anie.200352791 [DOI] [PubMed] [Google Scholar]
- 18.Su Y, DeGrado WF, Hong M. 2010. Orientation, dynamics, and lipid interaction of an antimicrobial arylamide investigated by 19F and 31P solid-state NMR spectroscopy. J. Am. Chem. Soc. 132:9197–9205. 10.1021/ja103658h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tew GN, Liu D, Chen B, Doerksen RJ, Kaplan J, Carroll PJ, Klein ML, DeGrado WF. 2002. De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. U. S. A. 99:5110–5114. 10.1073/pnas.082046199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vilhena C, Bettencourt A. 2012. Daptomycin: a review of properties, clinical use, drug delivery and resistance. Mini Rev. Med. Chem. 12:202–209. 10.2174/1389557511209030202 [DOI] [PubMed] [Google Scholar]
- 21.Tew GN, Scott RW, Klein ML, DeGrado WF. 2010. De novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications. Acc. Chem. Res. 43:30–39. 10.1021/ar900036b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mensa B, Kim YH, Choi S, Scott R, Caputo GA, DeGrado WF. 2011. Antibacterial mechanism of action of arylamide foldamers. Antimicrob. Agents Chemother. 55:5043–5053. 10.1128/AAC.05009-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malli R, Epstein W. 1998. Expression of the Kdp ATPase is consistent with regulation by turgor pressure. J. Bacteriol. 180:5102–5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asha H, Gowrishankar J. 1993. Regulation of kdp operon expression in Escherichia coli: evidence against turgor as signal for transcriptional control. J. Bacteriol. 175:4528–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laubacher ME, Ades SE. 2008. The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J. Bacteriol. 190:2065–2074. 10.1128/JB.01740-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Callewaert L, Vanoirbeek KG, Lurquin I, Michiels CW, Aertsen A. 2009. The Rcs two-component system regulates expression of lysozyme inhibitors and is induced by exposure to lysozyme. J. Bacteriol. 191:1979–1981. 10.1128/JB.01549-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danese PN, Snyder WB, Cosma CL, Davis LJ, Silhavy TJ. 1995. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev. 9:387–398. 10.1101/gad.9.4.387 [DOI] [PubMed] [Google Scholar]
- 28.Leblanc SK, Oates CW, Raivio TL. 2011. Characterization of the induction and cellular role of the BaeSR two-component envelope stress response of Escherichia coli. J. Bacteriol. 193:3367–3375. 10.1128/JB.01534-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. 1998. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 42:2206–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Senyurek I, Paulmann M, Sinnberg T, Kalbacher H, Deeg M, Gutsmann T, Hermes M, Kohler T, Gotz F, Wolz C, Peschel A, Schittek B. 2009. Dermcidin-derived peptides show a different mode of action than the cathelicidin LL-37 against Staphylococcus aureus. Antimicrob. Agents Chemother. 53:2499–2509. 10.1128/AAC.01679-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Straus SK, Hancock RE. 2006. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta 1758:1215–1223. 10.1016/j.bbamem.2006.02.009 [DOI] [PubMed] [Google Scholar]
- 32.Alborn WE, Jr, Allen NE, Preston DA. 1991. Daptomycin disrupts membrane potential in growing Staphylococcus aureus. Antimicrob. Agents Chemother. 35:2282–2287. 10.1128/AAC.35.11.2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen NE, Alborn WE, Jr, Hobbs JN., Jr 1991. Inhibition of membrane potential-dependent amino acid transport by daptomycin. Antimicrob. Agents Chemother. 35:2639–2642. 10.1128/AAC.35.12.2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muthaiyan A, Silverman JA, Jayaswal RK, Wilkinson BJ. 2008. Transcriptional profiling reveals that daptomycin induces the Staphylococcus aureus cell wall stress stimulon and genes responsive to membrane depolarization. Antimicrob. Agents Chemother. 52:980–990. 10.1128/AAC.01121-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peleg AY, Miyakis S, Ward DV, Earl AM, Rubio A, Cameron DR, Pillai S, Moellering RC, Jr, Eliopoulos GM. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One 7:e28316. 10.1371/journal.pone.0028316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boaretti M, Canepari P, Lleo MM, Satta G. 1993. The activity of daptomycin on Enterococcus faecium protoplasts: indirect evidence supporting a novel mode of action on lipoteichoic acid synthesis. J. Antimicrob. Chemother. 31:227–235. 10.1093/jac/31.2.227 [DOI] [PubMed] [Google Scholar]
- 37.Canepari P, Boaretti M, Lleo MM, Satta G. 1990. Lipoteichoic acid as a new target for activity of antibiotics: mode of action of daptomycin (LY146032). Antimicrob. Agents Chemother. 34:1220–1226. 10.1128/AAC.34.6.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laganas V, Alder J, Silverman JA. 2003. In vitro bactericidal activities of daptomycin against Staphylococcus aureus and Enterococcus faecalis are not mediated by inhibition of lipoteichoic acid biosynthesis. Antimicrob. Agents Chemother. 47:2682–2684. 10.1128/AAC.47.8.2682-2684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolar SL, Nagarajan V, Oszmiana A, Rivera FE, Miller HK, Davenport JE, Riordan JT, Potempa J, Barber DS, Koziel J, Elasri MO, Shaw LN. 2011. NsaRS is a cell-envelope-stress-sensing two-component system of Staphylococcus aureus. Microbiology 157:2206–2219. 10.1099/mic.0.049692-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belcheva A, Golemi-Kotra D. 2008. A close-up view of the VraSR two-component system. A mediator of Staphylococcus aureus response to cell wall damage. J. Biol. Chem. 283:12354–12364. 10.1074/jbc.M710010200 [DOI] [PubMed] [Google Scholar]
- 41.Li M, Lai Y, Villaruz AE, Cha DJ, Sturdevant DE, Otto M. 2007. Gram-positive three-component antimicrobial peptide-sensing system. Proc. Natl. Acad. Sci. U. S. A. 104:9469–9474. 10.1073/pnas.0702159104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bisicchia P, Lioliou E, Noone D, Salzberg LI, Botella E, Hubner S, Devine KM. 2010. Peptidoglycan metabolism is controlled by the WalRK (YycFG) and PhoPR two-component systems in phosphate-limited Bacillus subtilis cells. Mol. Microbiol. 75:972–989. 10.1111/j.1365-2958.2009.07036.x [DOI] [PubMed] [Google Scholar]
- 43.Bisicchia P, Noone D, Lioliou E, Howell A, Quigley S, Jensen T, Jarmer H, Devine KM. 2007. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Mol. Microbiol. 65:180–200. 10.1111/j.1365-2958.2007.05782.x [DOI] [PubMed] [Google Scholar]
- 44.Wiltshire MD, Foster SJ. 2001. Identification and analysis of Staphylococcus aureus components expressed by a model system of growth in serum. Infect. Immun. 69:5198–5202. 10.1128/IAI.69.8.5198-5202.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Staubitz P, Neumann H, Schneider T, Wiedemann I, Peschel A. 2004. MprF-mediated biosynthesis of lysylphosphatidylglycerol, an important determinant in staphylococcal defensin resistance. FEMS Microbiol. Lett. 231:67–71. 10.1016/S0378-1097(03)00921-2 [DOI] [PubMed] [Google Scholar]
- 46.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190:300–310. 10.1128/JB.01000-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stenglein MD, Sanders C, Kistler AL, Ruby JG, Franco JY, Reavill DR, Dunker F, Derisi JL. 2012. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. mBio 3(4):e00180-12. 10.1128/mBio.00180-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goecks J, Nekrutenko A, Taylor J. 2010. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. 2010. Galaxy: a Web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. Chapter 19:Unit 19.10.1-21. 10.1002/0471142727.mb1910s89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A. 2005. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15:1451–1455. 10.1101/gr.4086505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Li Y, Han H, Miller DW, Wang G. 2006. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 128:5776–5785. 10.1021/ja0584875 [DOI] [PubMed] [Google Scholar]
- 52.Shapiro HM, Natale PJ, Kamentsky LA. 1979. Estimation of membrane potentials of individual lymphocytes by flow cytometry. Proc. Natl. Acad. Sci. U. S. A. 76:5728–5730. 10.1073/pnas.76.11.5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett S. 2004. Solexa Ltd. Pharmacogenomics 5:433–438. 10.1517/14622416.5.4.433 [DOI] [PubMed] [Google Scholar]
- 54.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Cheetham RK, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53–59. 10.1038/nature07517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delaune A, Dubrac S, Blanchet C, Poupel O, Mader U, Hiron A, Leduc A, Fitting C, Nicolas P, Cavaillon JM, Adib-Conquy M, Msadek T. 2012. The WalKR system controls major staphylococcal virulence genes and is involved in triggering the host inflammatory response. Infect. Immun. 80:3438–3453. 10.1128/IAI.00195-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuroda M, Kuroda H, Oshima T, Takeuchi F, Mori H, Hiramatsu K. 2003. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49:807–821. 10.1046/j.1365-2958.2003.03599.x [DOI] [PubMed] [Google Scholar]
- 57.Kawada-Matsuo M, Yoshida Y, Zendo T, Nagao J, Oogai Y, Nakamura Y, Sonomoto K, Nakamura N, Komatsuzawa H. 2013. Three distinct two-component systems are involved in resistance to the class I bacteriocins, nukacin ISK-1 and nisin A, in Staphylococcus aureus. PLoS One 8:e69455. 10.1371/journal.pone.0069455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoo JI, Kim JW, Kang GS, Kim HS, Yoo JS, Lee YS. 2013. Prevalence of amino acid changes in the yvqF, vraSR, graSR, and tcaRAB genes from vancomycin intermediate resistant Staphylococcus aureus. J. Microbiol. 51:160–165. 10.1007/s12275-013-3088-7 [DOI] [PubMed] [Google Scholar]
- 59.Kato Y, Suzuki T, Ida T, Maebashi K. 2010. Genetic changes associated with glycopeptide resistance in Staphylococcus aureus: predominance of amino acid substitutions in YvqF/VraSR. J. Antimicrob. Chemother. 65:37–45. 10.1093/jac/dkp394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lunde CS, Hartouni SR, Janc JW, Mammen M, Humphrey PP, Benton BM. 2009. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob. Agents Chemother. 53:3375–3383. 10.1128/AAC.01710-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song Y, Lunde CS, Benton BM, Wilkinson BJ. 2012. Further insights into the mode of action of the lipoglycopeptide telavancin through global gene expression studies. Antimicrob. Agents Chemother. 56:3157–3164. 10.1128/AAC.05403-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 1277:139–158. 10.1111/j.1749-6632.2012.06819.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 274:8405–8410. 10.1074/jbc.274.13.8405 [DOI] [PubMed] [Google Scholar]
- 64.Falord M, Karimova G, Hiron A, Msadek T. 2012. GraXSR proteins interact with the VraFG ABC transporter to form a five-component system required for cationic antimicrobial peptide sensing and resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 56:1047–1058. 10.1128/AAC.05054-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.