

Abstract
Chloroquine (CQ) has been under clinical use for several decades, and yet little is known about CQ sensing and signaling mechanisms or about their impact on various biological pathways. We employed the budding yeast Saccharomyces cerevisiae as a model organism to study the pathways targeted by CQ. Our screening with yeast mutants revealed that it targets histone proteins and histone deacetylases (HDACs). Here, we also describe the novel role of mitogen-activated protein kinases Hog1 and Slt2, which aid in survival in the presence of CQ. Cells deficient in Hog1 or Slt2 are found to be CQ hypersensitive, and both proteins were phosphorylated in response to CQ exposure. CQ-activated Hog1p is translocated to the nucleus and facilitates the expression of GPD1 (glycerol-3-phosphate dehydrogenase), which is required for the synthesis of glycerol (one of the major osmolytes). Moreover, cells treated with CQ exhibited an increase in intracellular reactive oxygen species (ROS) levels and the effects were rescued by addition of reduced glutathione to the medium. The deletion of SOD1, the superoxide dismutase in yeast, resulted in hypersensitivity to CQ. We have also observed P38 as well as P42/44 phosphorylation in HEK293T human cells upon exposure to CQ, indicating that the kinds of responses generated in yeast and human cells are similar. In summary, our findings define the multiple biological pathways targeted by CQ that might be useful for understanding the toxicity modulated by this pharmacologically important molecule.
INTRODUCTION
Chloroquine (CQ) has been used extensively for decades, but the molecular targets of CQ are still not completely known. There is evidence that CQ may affect multiple cellular processes, including activation of apoptosis, by inhibiting autophagic protein degradation (1–3) and cellular stress response pathways (4), antigen presentation (5), and oxidative stress responses (6). Apart from its antimalarial activity, CQ has emerged as a potential anticancer agent (2) and antifungal agent (7–10), and it is also known to possess antiviral activity (11, 12). The cytotoxic effects of CQ have been demonstrated for tumor cells derived from different types of human cancers (2, 13, 14). Recently, CQ has been shown to improve dengue-related symptoms in infected patients (15). CQ also alters cell cycle-related protein expression and downregulates mitochondrial transmembrane potential in Bcap-37 cells (16). Evidence suggests that CQ can also target the genome of the host cells by directly intercalating into double-stranded DNA without causing physical damage to the DNA (17). Due to these diverse biological effects, CQ is also effective in the treatment of rheumatoid arthritis, systemic lupus erythematosus, and many other rheumatic and skin diseases (18). In several cases, the fundamental molecular mechanisms of the therapeutic and hazardous effects of CQ are not well understood.
Elucidation of the underlying mechanisms by which CQ shows its effects will immensely facilitate the rational designing of advanced drug analogs. The model organism yeast Saccharomyces cerevisiae is an excellent system for discovering conserved targets of bioactive compounds (19, 20). Recently, there have been reports of CQ activity against fungal pathogens (8, 9). CQ has also been shown to inhibit thiamine transport in yeast as well as human cells (21). The relationship between CQ toxicity and iron acquisition is also being studied in yeast cells (22). Another report indicated a potential role for the yeast pleiotropic drug resistance (PDR) ABC transporter in mediating CQ sensitivity (23). Hence, the objective of this study was to apply the yeast tool to gain new insights into CQ action.
Under stress conditions, yeast cells have developed a variety of mechanisms to give a specific and adaptive response. The cellular response to stress usually involves mitogen-activated protein kinase (MAPK) cascades, which are common and well-conserved signaling components present in both higher and lower eukaryotic cells (24). The high-osmolarity glycerol (HOG) pathway, which is one of the conserved pathway pathways, operates mainly during osmotic stress. It is composed of membrane-associated osmosensors, an intracellular signaling pathway whose core is the Hog1 MAPK cascade, and cytoplasmic and nuclear effector members (25). However, it has been demonstrated that CQ markedly stimulates p38 MAPK (the human homologue of yeast Hog1) activity in C6 glioma cells (26) but its effect on the HOG pathway has not been not established. The cell wall integrity (CWI) pathway is another conserved pathway which plays a central role in ensuring cell survival under various stress conditions, including cell wall damage (27). Therefore, the present study was designed to elucidate the stress response generated by the budding yeast S. cerevisiae upon CQ exposure.
Here, we show that yeast cells exhibit an Hog1-mediated osmoresponse in the presence of CQ by inducing its phosphorylation. We also found activation of Slt2, which is a central kinase of the CWI pathway, in response to CQ exposure. Our results indicated that phosphorylated Hog1 migrates to the nucleus and upregulates GPD1 (glycerol-3-phosphate dehydrogenase). Taking the results together, we have identified Hog1 and Slt2 as important mediators of the cellular response to CQ exposure.
MATERIALS AND METHODS
Strains, chemicals, and growth media.
All the chemicals, unless otherwise stated, were purchased from Sigma-Aldrich. The S. cerevisiae strains used in this study are listed in Table 1. For making synthetic complete (SC) media, all amino acids, YNB (yeast nitrogen base), and ammonium sulfate were mixed together following a standard protocol (28). Yeast cells were grown at 30°C in SC media containing 2% dextrose (SCD). For making solid-agar plates, 2% agar was added to SCD.
TABLE 1.
List of strains used in present study
| Strain no. | S. cerevisiae strain name | Description | Mutation(s) | Source |
|---|---|---|---|---|
| 1 | WT 15884C | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | WT | Toshi Tsukiyama |
| 2 | H2A (1–20) | MATa his3-1 leu2-0 met15-0 ura3-0 hht1-hhf1::KAN hhf-2hht2::NAT hta1-htb1::HPH hta2-htb2::NAT p[CEN LEU2 hta1Δ(1-20)-HTB1-HHT2-HHF2] | H2A Δ(1-20) | C. D. Allis |
| 3 | H3 (1–30) | MATa his3-1 leu2-0 met15-0 ura3-0 hht1-hhf1::KAN hhf-2hht2::NAT hta1-htb1::HPH hta2-htb2::NAT p[CEN LEU2 HTA1-HTB1-hht2Δ(1-30)-HHT2-HHF2] | H3 Δ(1-30) | C. D. Allis |
| 4 | H4 (1–16) | MATa his3-1 leu2-0 met15-0 ura3-0 hht1-hhf1::KAN hhf-2hht2::NAT hta1-htb1::HPH hta2-htb2::NAT p[CEN LEU2 HTA1-HTB1-HHT2-hhf2 Δ(1-16)] | H4 Δ(1-16) | C. D. Allis |
| 5 | H2A Δ(1–20), H3 Δ(1–30) | MATa his3-1 leu2-0 met15-0 ura3-0 hht1-hhf1::KAN hhf-2hht2::NAT hta1-htb1::HPH hta2-htb2::NAT p[CEN LEU2 hta1Δ(1-20)-HTB1- hht2 Δ(1-30)-HHF2] | H2A Δ(1-20), H3 Δ(1-30) | C. D. Allis laboratory |
| 6 | H3 WT | MATa his3Δ200 leu2Δ0 lys2Δ0 trp1Δ63 ura3Δ0met15Δ0 can1::MFA1pr-HIS3 hht1- hhf1::NatMX4 hht2-hhf2::[HHTS-HHFS]-URA3 | WT | R. Nicholas Laribee |
| 7 | H3K56A | H3 WT [H3K56A]-URA3 | H3K56A | R. Nicholas Laribee |
| 8 | WZY 42 WT | MATa ura3-52 leu2D1 trp1D63 his3D200 lys2-801 ade2-101 hht1-hhf1::pWZ 405-F2F9-LEU2 hht2- hf2::pWZ403-F4F10-HIS3 YCp50-copyII (HHT2-HHF2) | WT | Eun-Jung Cho |
| 9 | H3 PKR16–18A | WZY 42/pWZ414-F13::H3 PKR16-18A | H3 PKR16-18A | Eun-Jung Cho |
| 10 | H3 QL19,20A | WZY 42/pWZ414-F13::H3 QL19,20A | H3 QL19,20A | Eun-Jung Cho |
| 11 | H3 SK22,23A | WZY 42/pWZ414-F13::H3 SK22,23A | H3 SK22,23A | Eun-Jung Cho |
| 12 | H3 RK26,27A | WZY 42/pWZ414-F13::RK26,27A | H3 RK26,27A | Eun-Jung Cho |
| 13 | H3 SP28,30A | WZY 42/pWZ414-F13::SP28,30A | H3 SP28,30A | Eun-Jung Cho |
| 14 | H3 STG31–33A | WZY 42/pWZ414-F13::STG31-33A | H3 STG31-33A | Eun-Jung Cho |
| 15 | NSY 430 | MATα ura3-52 leu2-3,112 trp1-289 his3Δ1 Δ(hhf1-hht1) (hhf2-hht2) pNS329[CEN TRP1 HHF1-HHT1] rpd3::LEU2 | rpd3Δ | Morse laboratory |
| 16 | CLY 460 | MATα ura3-52 leu2-3,112 trp1-289 his3Δ1 Δ(hhf1-hht1) (hhf2-hht2) pCL460 [CEN TRP1 HHF1 hht1-3 H3 (K4,9,14,18,23,27Q)] | H3 (K4,9,14,18,23,27Q) | Morse laboratory |
| 17 | RMY 491 | MATα ura3-52 leu2-3,112 trp1-289 his3Δ1 Δ(hhf1-hht1) (hhf2-hht2) pNS491 [CEN TRP1 hhf1-10 H4 (K5,8,12,16Q) HHT1] | H4 (K5,8,12,16Q) | Morse laboratory |
| 18 | RMY 492 | MATα ura3-52 leu2-3,112 trp1-289 his3Δ1 Δ(hhf1-hht1) (hhf2-hht2) pNS491[CEN TRP1hhf1-10H4(K5,8,12,16Q)HHT1] rpd3::LEU2 | H4 (K5,8,12,16Q), rpd3Δ | Morse laboratory |
| 19 | EYO 690 | w303 MATa | WT | Erin K. O'Shea laboratory |
| 20 | Hog1 GFP | EYO 690 Hog1-GFP(His) Nhp6a-RFP(KanMX6) MATa | Hog1 GFP | Erin K. O'Shea laboratory |
| 21 | hog1Δ | EYO 690 hog1::Ura MATa | hog1Δ | Erin K. O'Shea laboratory |
| 22 | hot1Δ | EYO 690 sko1::Leu2 MATa | hot1Δ | Erin K. O'Shea laboratory |
| 23 | msn2Δ msn4Δ | EYO 690 msn2::Leu2 msn4::His3 MATa | msn2Δ, msn4Δ | Erin K. O'Shea laboratory |
| 24 | hog1Δ msn2Δ msn4Δ | EYO 690 hog1::Ura msn2::Leu2 msn4::His3 MATa | hog1Δ, msn2Δ, msn4Δ | Erin K. O'Shea laboratory |
| 25 | sko1Δ hot1Δ hog1Δ msn2Δ msn4Δ | EYO 690 sko1::Ura hot1::G418 hog1::Ura msn2::Leu2 msn4::His3 MATa | sko1Δ, hot1Δ, hog1Δ, msn2Δ, msn4Δ | Erin K. O'Shea laboratory |
| 26 | WT 4743 | MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 met15Δ0/MET15 ura3Δ0/ura3Δ0 | WT | Yeast deletion collection (O.B.)a |
| 27 | hos3Δ hos3Δ | Isogenic to BY4743 hos2Δ::KANMX4 | hos3Δ | Yeast deletion collection (O.B.) |
| 27 | hos2Δ hos2Δ | Isogenic to BY4743 hos2Δ::KANMX4 | hos2Δ | Yeast deletion collection(O.B.) |
| 29 | hda1Δ hda1Δ | Isogenic to BY4743 hda1Δ::KANMX4 | hda1Δ | Yeast deletion collection(O.B.) |
| 30 | hda3Δ hda3Δ | Isogenic to BY4743; hda3Δ::KANMX4 | hda3Δ | Yeast deletion collection (O.B.) |
| 31 | sap30Δ sap30Δ | Isogenic to BY4743; sap30Δ::KANMX4 | sap30Δ | Yeast deletion collection (O.B.) |
| 32 | hst3Δ hst3Δ | Isogenic to BY4743; hst3Δ::KANMX4 | hst3Δ | Yeast deletion collection(O.B.) |
| 33 | hst4Δ hst4Δ | Isogenic to BY4743; hst4Δ::KANMX4 | hst4Δ | Yeast deletion collection (O.B.) |
| 34 | gcn5Δ gcn5Δ | Isogenic to BY4743; gcn5Δ::KANMX4 | gcn5Δ | Yeast deletion collection (O.B.) |
| 35 | hat1Δ hat1Δ | Isogenic to BY4743; hat1Δ::KANMX4 | hat1Δ | Yeast deletion collection (O.B.) |
| 36 | rtt109Δ rtt109Δ | Isogenic to BY4743; rtt109Δ::KANMX4 | rtt109Δ | Yeast deletion collection (O.B.) |
| 37 | atf2Δ atf2Δ | Isogenic to BY4743; atf2Δ::KANMX4 | atf2Δ | Yeast deletion collection (O.B.) |
| 38 | hpa2Δ hpa2Δ | Isogenic to BY4743; hpa2Δ::KANMX4 | hpa2Δ | Yeast deletion collection (O.B.) |
| 39 | sas2Δ sas2Δ | Isogenic to BY4743; sas2Δ::KANMX4 | sas2Δ | Yeast deletion collection(O.B.) |
| 40 | nut1Δ nut1Δ | Isogenic to BY4743; nut1Δ::KANMX4 | nut1Δ | Yeast deletion collection (O.B.) |
| 41 | wsc2Δ wsc2Δ | Isogenic to BY4743; wsc2Δ::KANMX4 | wsc2Δ | Yeast deletion collection (O.B.) |
| 42 | wsc3Δ wsc3Δ | Isogenic to BY4743; wsc3Δ::KANMX4 | wsc3Δ | Yeast deletion collection (O.B.) |
| 43 | rom2Δ rom2Δ | Isogenic to BY4743; rom2Δ::KANMX4 | rom2Δ | Yeast deletion collection (O.B.) |
| 44 | skn7Δ skn7Δ | Isogenic to BY4743; skn7Δ::KANMX4 | skn7Δ | Yeast deletion collection (O.B.) |
| 45 | fks2Δ fks2Δ | Isogenic to BY4743; fks2Δ::KANMX4 | fks2Δ | Yeast deletion collection (O.B.) |
| 46 | bni1Δ bni1Δ | Isogenic to BY4743; bni1Δ::KANMX4 | bni1Δ | Yeast deletion collection (O.B.) |
| 47 | mkk1Δ mkk1Δ | Isogenic to BY4743; mkk1Δ::KANMX4 | mkk1Δ | Yeast deletion collection (O.B.) |
| 48 | mkk2Δ mkk2Δ | Isogenic to BY4743; mkk2Δ::KANMX4 | mkk2Δ | Yeast deletion collection (O.B.) |
| 49 | slt2Δ slt2Δ | Isogenic to BY4743; slt2Δ::KANMX4 | slt2Δ | Yeast deletion collection (O.B.) |
| 50 | bck1Δ bck1Δ | Isogenic to BY4743; bck1Δ::KANMX4 | bck1Δ | Yeast deletion collection (O.B.) |
| 51 | chr1Δ chr1Δ | Isogenic to BY4743; chr1Δ::KANMX4 | chr1Δ | Yeast deletion collection (O.B.) |
| 52 | mid2Δ mid2Δ | Isogenic to BY4743; mid2Δ::KANMX4 | mid2Δ | Yeast deletion collection (O.B.) |
| 53 | wsc1Δ wsc1Δ | Isogenic to BY4743; wsc1Δ::KANMX4 | wsc1Δ | Yeast deletion collection (O.B.) |
| 54 | swi4Δ swi4Δ | Isogenic to BY4743; swi4Δ::KANMX4 | swi4Δ | Yeast deletion collection (O.B.) |
| 55 | rlm1Δ rlm1Δ | Isogenic to BY4743; rlm1Δ::KANMX4 | rlm1Δ | Yeast deletion collection (O.B.) |
| 56 | cln1Δ cln1Δ | Isogenic to BY4743; cln1Δ::KANMX4 | cln1Δ | Yeast deletion collection (O.B.) |
| 57 | cln2Δ cln2Δ | Isogenic to BY4743; cln2Δ::KANMX4 | cln2Δ | Yeast deletion collection (O.B.) |
| 58 | pcl1Δ pcl1Δ | Isogenic to BY4743; pcl1Δ::KANMX4 | pcl1Δ | Yeast deletion collection (O.B.) |
| 59 | pcl2Δ pcl2Δ | Isogenic to BY4743; pcl2Δ::KANMX4 | pcl2Δ | Yeast deletion collection (O.B.) |
O.B., Open Biosystems.
Growth sensitivity, growth curve, and clonogenic cell survival assays.
The growth sensitivity to CQ of yeast cells was analyzed by performing spot tests as described earlier (29). Briefly, different wild-type (WT) yeast cells (WT 1588-4C, BY4741 and W3031b) were grown overnight to saturation in SCD medium at 30°C and 10-fold serial dilutions (10−1, 10−2, 10−3, and 10−4) were made in sterile distilled water. A 3-μl volume of each of these dilutions was then spotted on SCD plates without or with CQ (50, 100, and 150 mM). For analyzing the effect of antioxidants such as reduced glutathione (GSH) or N-acetyl-l-cysteine (NAC) on CQ sensitivity, 10 mM GSH or 10 mM NAC was added in synthetic complete agar (SCA) plates along with CQ. All plates were incubated at 30°C, and growth of the yeast strains was recorded at 48 h by scanning the plates using an HP scanner. Growth curve analysis was performed as described earlier (30, 31). Briefly, exponentially growing wild-type yeast cells (WT 1588-4C) were treated with 150 mM CQ in SCD medium and growth was monitored spectrophotometrically for 24 h by measuring the optical density at 600 nm. A clonogenic cell survival assay was performed as explained previously (28). Briefly, equal numbers of cells from untreated or CQ (150 mM)-treated cultures (3, 6, and 9 h) were spread on solid-agar plates. The plates were incubated at 30°C, and survival was evaluated after 48 h of incubation by calculating the number of colonies per plate. Experiments were done in triplicate, and cell survival was calculated with respect to the results seen with untreated cells.
FACS analysis of yeast cells.
Fluorescence-activated cell sorter (FACS) analysis was performed as described earlier (32). Briefly, exponentially growing yeast cells were treated for 3 h with 100 and 150 mM CQ. A 1-ml volume of cells was harvested and washed with ice-cold water. Ethanol was added to cell pellets, and a vigorous vortex procedure was followed by one wash with 50 mM sodium citrate buffer (pH 7.0). RNase A was added to the samples, and the reaction mixture was incubated at 37°C for 1 h. RNase A-treated samples were transferred to BD FACS flow fluid (Becton-Dickinson) containing 20 mg/ml propidium iodide (Sigma). Cellular DNA was detected using a BD FACS Aria III cell sorter with CellQuest software (Becton Dickinson). For cell cycle synchronization experiments, exponentially growing yeast cells were synchronized by alpha factor treatment for 2 h. After synchronization, G1-arrested cells were released into untreated media or media containing 150 mM CQ. Cells were harvested at regular intervals until 6 h and processed for FACS analysis as described above.
Detection of intracellular ROS levels.
To measure reactive oxygen species (ROS) production, we used DCFH-DA (2,7-dichlorodihydrofluorescein diacetate) (catalog no. D6883; Sigma) as described previously (33). DCFH-DA is membrane permeative and is trapped intracellularly following deacetylation. The resulting compound, DCFH, reacts with ROS (primarily with H2O2 and hydroxyl radicals) to produce the oxidized fluorescent form 2,7-dichlorofluorescein (DCF). ROS analysis using DCFH-DA was performed as follows. Yeast cells were treated with 10 μM DCFH-DA in culture media for 1 h prior to harvesting. Cells were washed twice in ice-cold phosphate-buffered saline (PBS), resuspended in the same buffer, and immediately subjected to FACS analysis.
Western blot analysis.
Whole-cell extracts from untreated and CQ-treated samples were prepared using the trichloroacetic acid (TCA) extraction method. Protein extracts were resolved using SDS-PAGE, and Western blotting was conducted as described earlier (34). The following primary antibodies were used: green fluorescent protein (GFP) (catalog no. G1544; Sigma), p38/phospho-Hog1 (catalog no. 92115; Cell Signaling), and anti-Slt2p-P/phospho-p42/44 MAPK (catalog no. 4370; Cell Signaling).
Isolation of total RNA and real-time PCR.
Exponentially growing wild-type yeast cells (WT 1588-4C) were treated with CQ (150 mM), and cells were harvested at regular intervals (0, 30, 60, and 180 min). Total RNA was extracted by a heat/freeze phenol method as described earlier (34). A 1-μg volume of total RNA was reverse transcribed to synthesize cDNA using a High Capacity RNA-to-cDNA kit (Bio-Rad) according to the manufacturer's instructions. Real-time PCR experiments were performed by using SYBR green mix (Roche Diagnostics) in an ABI real-time PCR instrument. Melting curve analysis was performed for each primer pair, and relative changes in mRNA levels between control and treated groups were calculated by using the 2-ΔΔCT method (35). The GPD1 relative mRNA level was detected by using forward primer 5′-CATTGCCACCGAAGTCGCTC-3′ and reverse primer 5′-AACCACAACCTAAGGCAACAACG-3′. ACT1 (forward primer 5′-CCTTCTGTTTTGGGTTTGGA-3′ and reverse primer 5′-CGGTGATTTCCTTTTGCATT-3′) was used as the reference control gene.
Cell culture and Western blot analysis.
HEK293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco Corporation, Carlsbad, CA) supplemented with 10% newborn bovine serum (Gibco Corporation) at 37°C and 5% CO2. Equal numbers of cells was treated with 50, 100, 200, and 500 μM CQ for 30 min. Cells treated with 300 mM NaCl were taken as a positive control for P38 phosphorylation. Cells were harvested, lysate was prepared in radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% sodium deoxycholate, 1% NP-40, 1 mM EDTA, 0.1% SDS), and Western blot analysis was done.
RESULTS
CQ inhibits growth of S. cerevisiae in a dose-dependent manner.
As an initial step toward examining the consequences of CQ treatment for S. cerevisiae cell growth, we performed a spot test assay with wild-type cells on synthetic complete agar (SCA) plates incorporated with increasing concentrations (50, 100, and 150 mM) of CQ. We have chosen concentrations of CQ similar to those which has been used on yeast cells in several published reports (22, 23, 36–38). We used three wild-type yeast strains, namely, WT 1588-4C, BY4741, and W3031b, for performing spot tests to ensure that the effect of CQ is not dependent on a particular strain background. CQ exhibited similar growth-inhibitory effects on all WT strains (Fig. 1A). The results obtained in spot tests were further validated by the growth curve experiment, which showed the reduction in the growth rate of yeast cells upon exposure to CQ (Fig. 1B). To further check the viability of cells in the presence of CQ, a clonogenic assay was performed. After drug treatment, the cells were washed with drug-free medium, diluted, and then plated onto SCA plates to determine the frequency of viable cells. Our results illustrate that there was a significant decrease in the number of colonies upon treatment with CQ (150 mM) compared to the results seen with untreated samples (Fig. 1C and D).
FIG 1.

CQ has a growth-inhibitory effect on yeast cells. (A) The wild types of strains of different backgrounds (WT 1588-4C, W3031b, and BY4741) were inoculated into 5 ml of yeast extract-peptone-dextrose (YPD) and grown overnight. Yeast saturated cultures were serially diluted (10−1, 10−2, 10−3, and 10−4) in 1.0 ml of sterile double-distilled water, and a 3-μl volume of each dilution was spotted on SCA control plates (untreated) or CQ plates (50, 100, and 150 mM). All plates were incubated at 30°C, and growth of the yeast strains was recorded at 48 h by scanning the plates using a HP scanner. (B) Wild-type yeast cells (WT 1588-4C) were grown in SC medium until the exponential phase was reached and then treated with 100 or 150 mM CQ for 24 h. Growth was monitored by recording the optical density at 600 nm (OD600) of untreated (Un) and treated cultures at regular intervals. Error bars represent the standard deviations (SD) of the results of three independent repeats. (C) A clonogenic assay was performed by taking equal numbers of cells from untreated or CQ (150 mM)-treated cultures (3, 6, and 9 h) and spreading the cells on solid-agar plates. The plates were incubated at 30°C, and survival was evaluated after 48 h of incubation. The representative images of plates are shown. (D) The numbers of colonies that appeared on plates were counted and are represented in the form of a bar diagram showing cell survival upon 150 mM CQ treatment for 3, 6, or 9 h. Error bars represent the SD of the results of three independent repeats. (E) Fluorescence-activated cell sorting (FACS) analysis, showing the effect of the CQ on yeast cell cycle. Wild-type cells were cultured in SC medium to the exponential phase followed by treatment with CQ (100 or 150 mM) for 3 h. As a control, untreated cells were also grown for same duration of time. The cultured cells were sampled, and cellular DNA content was analyzed by FACS. (F) Wild-type cells were cultured in SC medium to the exponential phase and treated with alpha factor to synchronize all cells in G1 phase. After synchronization, cells were released in either fresh (control) or 150 mM CQ-containing media. The culture was sampled at indicated time points, and cellular DNA content was analyzed by FACS.
Based on the above-mentioned experiments, we observed the growth-inhibitory effect of CQ, which indicated that the drug could possibly affect cell cycle progression. To validate this hypothesis, we performed FACS analysis and observed the cell cycle progression in the presence or absence of CQ. Exponentially growing yeast cells were treated with CQ (100 or 150 mM) for 3 h. Cells were processed for FACS analysis. We observed that CQ exposure led to accumulation of yeast cells in the G2/M phase (Fig. 1E). Furthermore, we performed FACS analysis after synchronizing cells in G1 phase by the use of alpha factor. After synchronization, G1-arrested cells were released in fresh media (control) or media containing 150 mM CQ. In the control experiment, cells progressed to the G2 phase within 60 min of release from alpha factor arrest (Fig. 1F), while CQ treatment delayed the movement of cells from G1 to G2/M (Fig. 1F). Once cells entered the G2/M phase, they remained in same phase even after 360 min of release, suggesting that CQ treatment causes a delay in the cell cycle. Our results are consistent with an earlier report where it is demonstrated that CQ causes cell cycle arrest in the G2/M phase in breast cancer cell lines (16), suggesting that the effects of CQ are the same in yeast and higher eukaryotes. Taken together, these results reveal that CQ is inhibitory to S. cerevisiae cell growth.
CQ targets histones and chromatin-modifying enzymes.
It was previously reported that several antimalarial drugs, including CQ, regulate acetylation of histones in breast cancer cell lines (39), suggesting that CQ might target chromatin-associated processes/epigenetics. To understand the underlying mechanism through which CQ shows its effect on host epigenetics, we employed yeast strains harboring mutations in the histone tails, including strains with amino acid deletions in the N-terminal tail of H2A, H3, and H4. Removal of the N-terminal tail of H4 (positions 1 to 16) led to hypersensitivity to CQ, while not much growth inhibition was observed with deletion of the N-terminal tails of H2A (positions 1 to 20 [1-20]), H3 (1-30), and H3 (1-5) (Fig. 2A), suggesting that histone H4 is crucial for providing resistance to this drug. We also observed mild sensitivity of H3 point mutants (H3 PKR16-18A, H3 QL19-20A, H3 SK22-23A, H3 RK26-27A, H3 SP28-30A, and H3 STG31-33A) to CQ (Fig. 2A). Interestingly, the H3K-Q mutant, in which the lysines at positions 4, 9, 14, 18, 23, and 27 were substituted by glutamine, was resistant to CQ whereas a more-severe growth defect was seen with the H4K-Q mutant (K5, 8, 12, 16Q). These results suggest that either deletion of the H4 N-terminal tail or mutation of those lysine residues leads to enhanced growth-inhibitory effects of CQ. Histone proteins are posttranslationally modified by various chromatin-modifying enzymes. Therefore, we next examined the effect of CQ on yeast mutants lacking some of the chromatin-modifying enzymes such as histone acetyl transferases (HATs) (gcn5Δ, hat1Δ, rtt109Δ, nlp3Δ, atf2Δ, hpa2Δ, sas2Δ, and nut1Δ mutants) or histone deacetylases (HDACs) (hos2Δ, hst3Δ, hst4Δ, hda1Δ, hda3Δ, sap30Δ, and sos3Δ mutants). In particular, we observed that rtt109Δ, gcn5Δ, and rpd3Δ mutants were sensitive to CQ (Fig. 2B). RPD3 acts as a histone deacetylase for H4 N-terminal lysine residues; the hypersensitivity to CQ exhibited by H4 K-Q, H4 (1-16), and rpd3Δ mutants indicates that this drug was specifically activating those signaling pathways and operating through the combined action of these molecules.
FIG 2.

CQ functions through targeting histone proteins and its modifying enzymes. For a growth assay, the wild-type and different mutant yeast strains were grown until saturation. A 3-μl volume of each undiluted and 10-fold serially diluted culture was spotted onto control (untreated) plates or SCA plates containing 150 mM CQ. All plates were incubated at 30°C for 48 h and photographed.
Hog1 protein is phosphorylated upon exposure to CQ.
On the basis of the results of a sensitivity assay (Fig. 2), we propose that CQ might induce osmotic stress. One of the hallmarks of the osmotic stress response is the phosphorylation of Hog1; hence, we examined the effect of CQ on Hog1 phosphorylation in yeast cells. To determine whether the exposure of yeast cells to CQ results in the activation of Hog1 protein, we measured its active form, the dually phosphorylated form, by Western blot analysis using a selective antibody appropriate for the phosphorylated form of Hog1 protein. We observed increased phosphorylation of Hog1 protein with increasing concentrations of CQ (Fig. 3A). Also, it was possible to detect phosphorylated Hog1 within 15 min of incubation with CQ, with maximum phosphorylation occurring after 60 min, after which the level of phosphorylation began to decrease (Fig. 3B). The CQ-induced phosphorylation of Hog1 persisted for a prolonged time (at least 9 h) as revealed by Western blotting (Fig. 3B). As a positive control, samples treated with 0.8 M NaCl were analyzed for Hog1 phosphorylation and, per previous reports (25, 40, 41), phosphorylation of Hog1 started within 15 min and remained detectable for 60 min, after which it started to decrease.
FIG 3.

CQ induces hog1p phosphorylation. (A) A yeast strain harboring a Hog1-GFP tag was grown until the exponential phase. Protein extracts were made from cells incubated for 1 h with 0.8 M NaCl and from cells incubated with increasing concentrations (2, 5, 10, 25, 50, 100, and 150 mM) of CQ. The phosphorylated form of Hog1 was detected using an anti-phospho-p38 antibody (phospho-hog1). The Western blot membranes were probed for total Hog1, using a polyclonal anti-GFP antibody, and served as a loading control. (B) Protein extracts were made from either cells incubated with 0.8 M NaCl or cells incubated with 150 mM CQ at the time points indicated in the figure. The Western blot membranes were probed for anti-phospho-p38 (phosphor-hog1) or total Hog1 (anti-GFP). The Western signal of anti-GFP antibody served as a loading control. The values written above the GFP lanes correspond to the relative intensities of the phosphorylated Hog1p. (C) Growth assay. Wild-type and different mutant yeast strains were grown until saturation. Serial dilution was made in double-distilled water. A 3-μl volume of each undiluted and 10-fold serially diluted culture was spotted onto control (untreated) plates or SCA plates containing 150 mM CQ. All plates were incubated at 30°C for 48 h and photographed. (D) Wild-type and HOG pathway mutant yeast strains were grown in SC media until the exponential phase. Cells were treated with either 0.8 M NaCl (30 min) or 150 mM CQ (60 min), and protein extracts were made. The Western blot membranes were probed for anti-phospho-p38 (phosphor-hog1). The Ponceau S stain of the Western membrane served as a loading control.
Hog1 is a member of the MAPK family, and there are several upstream kinases which are involved in mediating the osmostress signal for hog1 through phosphorylation. We investigated the growth sensitivity of the nonessential gene deletions within the entire HOG pathway and performed spot test assays to determine its sensitivity to CQ. Yeast knockouts for sho1 and msb2 (regulatory sensors), ssk1 (upstream control system), ste50 (upstream kinase), ssk2 (MAPK kinase kinase [MAPKKK]), pbs2 (MAPK kinase [MAPKK]), hog1 (MAPK), and ptc2 (protein phosphatases) showed patterns of sensitivity to CQ similar to those seen with NaCl (Fig. 3C). In particular, we observed that the MAPKK of Hog1 protein (Pbs2), which is activated under conditions of severe osmotic stress, was sensitive to CQ (150 mM). Interestingly, mutants with deletions of phosphatases such as Ptc1, which dephosphorylates Pbs2p, and Ptp2, which dephosphorylates Hog1p, were found to be hypersensitive to CQ but not to NaCl (Fig. 3C), suggesting that Ptc1 and Ptp2 dephosphatase activity is indispensable to overcome CQ-induced osmotic stress. Next, we sought to evaluate the phosphorylation of Hog1 protein in HOG pathway mutants to investigate the role of these mutants in mediating Hog1 phosphorylation in response to CQ exposure. Western blotting was conducted to analyze the phosphorylation of Hog1 upon treatment with CQ or NaCl. We did not observe significant phosphorylation of Hog1 in the ssk1Δ, ssk2Δ, and pbs2Δ strains (Fig. 3D) upon CQ treatment, suggesting that Ssk1, Ssk2, and Pbs2 are important upstream components of the HOG pathway that are required for optimal activation of Hog1 in response to CQ-induced stress. Other mutants induced Hog1 phosphorylation in a manner somewhat similar to that exhibited in response to NaCl treatment. The results indicated that the mode of action of CQ is similar to but not exactly the same as that of NaCl in generating an osmoresponse.
A hog1Δ mutant is sensitive to CQ.
To examine the role of Hog1 protein in yeast in the presence of CQ, we examined the hog1Δ mutant through spot tests, where serially diluted cultures were spotted on SCA plates with and without CQ. We observed that the hog1Δ mutant was hypersensitive to CQ compared to the wild-type cells (Fig. 4A). Apart from Hog1, there are several additional transcription factors such as Msn2, Msn4, and Hot1 which play an important role under conditions of osmotic stress (42, 43). To explore the possible role of these different transcription factors in CQ-induced stress, we performed sensitivity assays. Interestingly, neither of the other mutants (hot1Δ or msn2Δ/msn4Δ) showed hypersensitivity to CQ similar to that exhibited by the hog1Δ mutant (Fig. 4A). The growth-inhibitory effect of CQ was enhanced when the hog1Δ mutation was combined with either msn2Δ/msn4Δ or sko1Δ/hot1Δ/msn2Δ/msn4Δ (Fig. 4A), suggesting that the Hog1 protein plays a central role in providing resistance to CQ. Furthermore, we also performed growth curve analyses with wild-type and hog1Δ yeast strains. We observed that the growth-inhibitory effect of CQ was more prominent in the absence of hog1p (Fig. 4B). Taken together, these results indicate the requirement of Hog1 for mediating downstream signaling in response to CQ-induced stress for survival in yeast.
FIG 4.

Deletion of Hog1 potentiates CQ sensitivity. (A) Growth assay. Wild-type and different mutant yeast strains were grown until saturation. Serial dilution was made in double-distilled water. A 3-μl volume of each undiluted and 10-fold serially diluted culture was spotted onto control (untreated) plates or SCA plates containing 150 mM CQ. All plates were incubated at 30°C for 48 h and photographed. (B) The hog1Δ strain and its wild type were grown in SC medium until the exponential phase and then treated with 100 or 150 mM CQ for 24 h. Growth was monitored by recording the optical density at 600 nm (OD600) of untreated and treated cultures at regular intervals. Error bars represent the SD of the results of three independent repeats.
Upon CQ exposure, phosphorylated Hog1 translocates to the nucleus, resulting in transient activation of GPD1.
In response to osmotic stress, Hog1 is phosphorylated and translocated into the nucleus, where it regulates the expression of many osmoresponsive genes at the transcriptional level (44, 45). Therefore, we desired to determine whether phosphorylated Hog1 was being translocated inside the nucleus after CQ exposure. We used a Hog1-GFP-tagged strain to perform confocal microscopy to examine untreated and CQ-treated yeast cells (30 or 60 min) and monitored the localization of Hog1. In untreated cells, Hog1 was distributed throughout the cells, while upon exposure to CQ or NaCl, the bright foci of hog1 were evident inside yeast nuclei as it colocalized with DAPI (4′,6-diamidino-2-phenylindole) (Fig. 5A). Microscopy results indicated that upon phosphorylation, hog1 gets translocated to the nucleus. Once translocated into the nucleus, hog1 regulates expression of various osmoresponse genes (43, 46, 47). Therefore, we confirmed the expression of one such gene, GPD1 (glycerol-3-phosphate dehydrogenase), involved in glycerol biosynthesis during osmotic stress (48). For that purpose, wild-type yeast cells were treated with CQ (150 mM) and cells were harvested at regular intervals (0, 30, 60, and 180 min). Total RNA was extracted and reverse transcribed to cDNA. The expression of GPD1 was analyzed through real-time PCR normalized to the reference gene ACT1. We observed a 9-fold increase in the expression of GPD1 within 30 min of CQ exposure which decreased significantly at later time points (Fig. 5B). This strongly indicates that upon CQ exposure, Hog1 not only is phosphorylated and translocated to the nucleus but is also in an enzymatically active state causing upregulation of GPD1 mRNA levels.
FIG 5.

CQ-induced phosphorylated Hog1 gets translocated to the nucleus, leading to transient upregulation of GPD1 mRNA. (A) A HOG1-GFP-tagged yeast strain was treated with 150 mM CQ (30 or 60 min), and the same strain was treated with 0.8 M NaCl (control) for 5 min, prior to visualization by confocal microscopy. DAPI was used to stain nuclei. (B) An exponentially growing wild-type yeast strain was exposed to 150 mM CQ for 3 h. Samples were taken at 0, 30, 60, and 180 min of incubation, and total mRNA was extracted and reverse transcribed to cDNA. The expression of GPD1 mRNA levels was examined by SYBR green real-time PCR. The fold change of GPD1 mRNA levels was normalized to the ACT1 reference gene levels. The error bars represent the SD of the results of three independent repeats.
CQ exposure causes activation of Slt2 MAPK.
Yeast MAPK pathways respond to diverse signals leading to specific physiological outcomes, but evidence of cross talk between such pathways is being reported; e.g., the HOG pathway is known to be involved in cell wall integrity (CWI) (49). The Hog1 kinase has been recently described to operate with Slt2 in adaptation to zymolyase-mediated cell wall stress (50). This led us to propose that CQ might also induce cell wall stress along with the osmoresponse. To validate our hypothesis, we examined the sensitivity to CQ of different members of the CWI pathway by performing spot test assays using SCA plates impregnated with 150 mM CQ or left untreated. The central kinase of the CWI pathway, Slt2, was hypersensitive to CQ (Fig. 6A), suggesting that CQ was inducing cell wall damage. We also observed that bni1Δ, wsc1Δ, swi4Δ, and rlm1Δ mutants exhibited enhanced growth inhibition in response to CQ exposure compared to wild-type cells. Moreover, the effect of cell wall damage caused by CQ can be rescued by supplementation with 1 M sorbitol (Fig. 6B). Interestingly, as shown in Fig. 6B, the growth-inhibitory effect of CQ on the slt2Δ mutant was rescued by addition of 1 M sorbitol only at lower doses (2 or 10 mM CQ).
FIG 6.

CQ activates Slt2 by inducing its phosphorylation. (A) Growth assay. Wild-type and different mutant yeast strains related to the CWI pathway were grown until saturation. Serial dilutions were made in double-distilled water. A 3-μl volume of each undiluted and 10-fold serially diluted culture was spotted onto control (untreated) plates or SCA plates containing 150 mM CQ. All plates were incubated at 30°C for 48 h and photographed. (B) Exponential-phase cultures of S. cerevisiae BY4743 (wild type) and slt2Δ slt2Δ strains were serially diluted and spotted onto agar supplemented with increasing concentrations (2, 10, and 50 mM) of CQ in the absence or presence of 1 M sorbitol. (C) Cells were grown at 24°C to the mid-exponential phase, and cell extracts were fractionated and analyzed by Western blotting using phospho-p44/p42 MAPK antibodies to detect dually phosphorylated Slt2p. An asterisk (*) indicates that the nonspecific band appeared with phospho-p44/p42, used as a loading control, in the Western blot analysis. Calcofluor white (CFW)-treated yeast whole-cell extract was taken as a positive control.
Current information supports the idea that the cell wall damage-induced transcriptional responses are mainly governed by MAPK Slt2. The cellular response to cell wall stress is governed by the cell wall integrity pathway. To investigate this, activation of Slt2 by CQ was analyzed in the wild-type yeast cells. We observed that treatment with CQ caused a rapid enhancement of the levels of phosphorylated Slt2 (Fig. 6C), as revealed by Western blotting using an antibody against the dually phosphorylated form of Slt2 MAPK. The time point kinetics demonstrated a consistent increase of Slt2 phosphorylation until 1 to 9 h of treatment with CQ. Taken together, our results describe the activation of Hog1 as well as Slt2 upon CQ exposure.
Exposure to CQ leads to generation of reactive oxygen species.
Because CQ treatment causes significant growth inhibition in slt2Δ or hog1Δ mutants, we sought to investigate whether the sensitivity of cells to CQ is the result of the generation of reactive oxygen species (ROS). The level of ROS in yeast cells was detected by DCFH-DA (2,7-dichlorodihydrofluorescein diacetate), which is oxidized by ROS to the fluorescent chromophore DCF (2,7-dichlorofluorescein). As indicated by FACS analysis (Fig. 7A), after treatment with CQ, the DCF fluorescence was significantly increased in the wild-type cells. To further confirm the elevated levels of ROS upon CQ exposure, we used sod1Δ cells, which are known to be sensitive to ROS-inducing agents (51). SOD1 is one of the main antioxidative enzymes that are generated under different stress conditions in response to ROS. We compared the growth of sod1Δ cells to the growth of wild-type cells in the presence of 150 mM CQ, and, as shown in Fig. 7B, the sod1Δ mutant was hypersensitive to CQ. Next, we analyzed the effect of antioxidants, such as reduced glutathione (GSH) or N-acetyl-l-cysteine (NAC), on the growth phenotype of the sod1Δ mutant. As shown in Fig. 7B, addition of 10 mM GSH or 10 mM NAC restored the growth of the sod1Δ mutant in the presence of CQ, suggesting the role of ROS in the toxicity exhibited by CQ.
FIG 7.

CQ treatment increases reactive oxygen species production in S. cerevisiae cells. (A) A yeast wild-type strain was grown in SC media until the exponential phase. Cells were treated with a 150 mM concentration of CQ for 3 h. The cells were then stained with DCF-DA and examined by FACS analysis as described in Materials and Methods. (B) Growth assay. Wild-type and sod1Δ strains were grown to the log phase. A 3-μl volume of each undiluted and 10-fold serially diluted culture was spotted onto control SCA plates, SCA plates containing 150 mM CQ, and SCA plates impregnated with a combination of 150 mM CQ and 10 mM-GSH or 10 mM NAC. All plates were incubated at 30°C for 72 h and photographed. (C and D) Spot test of hog1Δ (C) or slt2Δ (D) mutants on SC agar plates supplemented with either NAC (10 mM) or GSH (10 mM) or plates containing the indicated concentration of CQ with either NAC or GSH. Yeast saturated cultures were serially diluted (10−1, 10−2, 10−3, and 10−4) in 1.0 ml of sterile double-distilled water and spotted onto the plates. Cells were cultured at 30°C for 48 h, and pictures were taken using an HP scanner.
We also analyzed the effect of the antioxidants on the growth of the slt2Δ or hog1Δ mutant in the presence of CQ. As shown in Fig. 7C and D, the addition of 10 mM GSH or 10 mM NAC restored the growth of both of the mutants in the presence of CQ. These results suggested that the growth inhibition observed in the slt2Δ or hog1Δ mutant in response to CQ is primarily due to generation of ROS.
CQ treatment causes phosphorylation of both P38 and P42/44 in the HEK293T human cell line.
To further consolidate our results obtained with yeast cells, we analyzed the effect of CQ on the HEK293T human cell line. HEK293T cells were grown as described in Materials and Methods followed by treatment with increasing concentrations (50, 100, 200, and 500 μM) of CQ for 30 min. Our Western blotting results showed a dose-dependent increase in p38 phosphorylation (Fig. 8A) upon CQ exposure. Similarly, CQ also induces P42/44 phosphorylation (Fig. 8B) in HEK293T cells. These results suggest that CQ activates similar kinds of responses in yeast and human cell lines.
FIG 8.
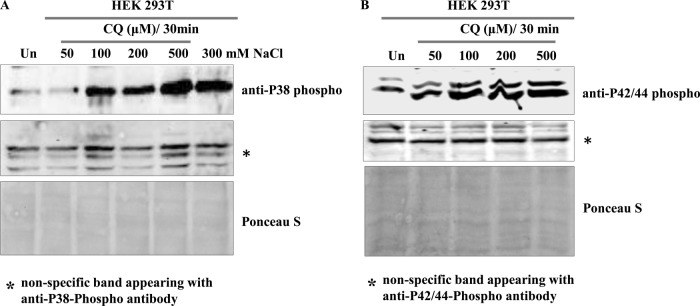
Effect of CQ on HEK293T cell line. HEK293T cells were grown as described in Materials and Methods. Equal numbers of cells were treated with increasing concentrations of CQ for 30 min. Protein extracts were made, and Western blotting was performed with anti-P38 phosphoantibody (A) or anti-P42/44 (B) phosphoantibody. NaCl treated cells were used as a positive control for P38 phosphorylation. A nonspecific band appearing with P38 or P42/44 antibody and a Ponceau S-stained image of representative Western blots were used as loading controls.
DISCUSSION
In this study, we used yeast as a model organism as they exhibit protective responses as a consequence of various kinds of environmental stresses. We tried to understand the response of yeast to CQ and elucidated the mechanism of action of this drug. To understand the biological pathways that might be the target of CQ, we employed several yeast mutants for screening and found that the members involved in the osmoresponse and cell wall integrity are the major targets of CQ. In this report, we have shown for the first time that Hog1 and the Slt2 MAP kinase play central roles in responses to the antimalarial drug CQ.
Mammalian p38 and the yeast Hog1 protein are stress-activated kinases and were long thought to be activated exclusively by osmotic stress. But recent studies have indicated that this MAPK has in a role in mediating tolerance of a variety of stress conditions such as osmotic stress (52), oxidative stress (53, 54), heat stress (55), and citric acid stress (56). Exposure of yeast cells to high osmolarity results in rapid activation of the MAPK Hog1 protein, which coordinates the transcriptional program necessary for cell survival of osmostress (25, 57). The primary sequences of p38 and its yeast homologue Hog1 show similarity at phosphorylation sites (52), and that distinguishes Hog1 from most other known members of the MAPK family. Both Hog1 and p38 are tyrosine phosphorylated after extracellular changes in osmolarity (57). Our results are consistent with earlier observations which showed that CQ markedly stimulates p38 MAPK activity in C6 glioma cells (26), although direct phosphorylation of p38 was not shown previously. We have demonstrated that Hog1 and Slt2 play a key role in the yeast adaptive response to CQ challenge since the growth of hog1Δ and slt2Δ mutants was inhibited by exposure to CQ compared to the growth of the wild type.
To understand the molecular targets of CQ, we extended our preliminary observations by examining factors which function together with Hog1. Rpd3 is a member of a family of five related histone deacetylases in yeast that also comprise Hda1, Hos1, Hos2, and Hos3. It has been established that deletion of the gene encoding the histone deacetylase Rpd3 rendered cells osmosensitive (58), while deleting the genes encoding the other related deacetylases did not affect cellular osmosensitivity in the presence of NaCl (58). Rpd3 affects the regulation of a substantial number of genes in S. cerevisiae (59). Similarly to these observations, our results show that CQ specifically targeted Rpd3 whereas other deacetylases were not affected (Fig. 2A), suggesting that genes regulated specifically by Rpd3 are required to provide tolerance of CQ-induced stress. These results also indicate that CQ-induced stress closely resembles osmostress generated due to NaCl. Rpd3 mainly deacetylates acetyl groups in histones H3 and H4 (60). Previously, Rahim and Strobl (39) demonstrated that CQ stimulates histone hyperacetylation. Similarly, our study also revealed that histone H4 (K-Q) mutant growth was completely abolished in the presence of CQ (Fig. 2A), suggesting that Rpd3 deacetylates H4 N-terminal lysine residues to regulate the downstream target genes which are required to confer tolerance of stress generated by CQ treatment. Our results are consistent with an earlier observation where a histone H4 (K-Q) and rpd3Δ mutant strains were found to be osmosensitive (58), suggesting that CQ was inducing osmotic stress similar to that induced by NaCl. The osmotic stress-induced Hog1 phosphorylation declined after 60 min of NaCl exposure, while the CQ-induced Hog1 phosphorylation peaked at 60 to 120 min and persisted for a prolonged duration of 9 h (Fig. 3B). Based on earlier reported results, it has been well established that different osmotically active substances exhibit different kinetics of hog1 phosphorylation, subcellular translocation, and gene expression profiles (43, 61–63). On the other hand, apart from this kinetic difference, the stress response to CQ exposure and to high osmolarity (NaCl) in yeast appears to follow a comparable Hog1-dependent pathway. In contrast, other types of stress conditions (exposure to weak organic acids, ethanol, metalloid salts, or heat) do not lead to importation of phosphorylated Hog1 into the nucleus (44, 61, 64), while we observed that CQ efficiently brings about the translocation of phosphorylated Hog1 protein into the nucleus.
As a result of phosphorylation, Hog1 gets translocated into the nucleus (44, 61), where it targets various transcription factors, leading to a change of gene expression (43, 46, 47). Notably, glycerol-3-phosphate dehydrogenase encoded by GPD1, required for the synthesis of a major osmolyte glycerol, is overexpressed (65). Elevated glycerol production is a prerequisite for the adaptation of S. cerevisiae to hyperosmotic stress (66–68). Most importantly, for the first time, we report that exposure of S. cerevisiae to CQ leads to Hog1 phosphorylation, followed by its translocation to the nucleus, leading to the activation of GPD1 gene expression (Fig. 5). Although we have observed the induction of an increase in the GPD1 mRNA level upon CQ exposure, whether there is a subsequent increase in intracellular glycerol levels needs to be tested. Our results are consistent with previous reports in which osmotic swelling of acidic organelles was found upon treatment with CQ in rat hepatocytes (69).
It was previously reported that osmotic stress and cell wall damage are closely related (49). Consistent with that, we have demonstrated that CQ treatment induced the activation of Slt2 along with Hog1, suggesting that both of the pathways contribute to the growth-inhibitory effect of CQ. Our results are consistent with a recently reported study by Islahudin et al. (70), in which they demonstrated that CQ causes cell wall perturbation. Here, we have provided additional insight by demonstrating that Slt2, which is a central member of cell wall integrity MAPK pathway, is phosphorylated upon CQ treatment. In yeast, Slt2 is known to be activated under conditions that stress the cell surface, such as hypo-osmotic shock, growth at high temperatures, actin perturbation, polarized growth, and the presence of compounds or mutations that interfere with cell wall biosynthesis (71). Furthermore, we have also identified an increase in the level of reactive oxygen species (ROS) as one of the reasons behind the growth inhibition of yeast cells caused by CQ. Previously, it was reported that CQ increases the intracellular level of ROS in human astroglial cells (6) as well as in a host-parasite system (72, 73), and those reports are consistent with our study on yeast cells.
To further substantiate our study results, we analyzed the effects of CQ on the HEK293T human cell line. P38 (human orthologue of yeast Hog1) and P42/44 (human orthologue of yeast Slt2) were phosphorylated in response to CQ exposure (Fig. 8). These results suggest that similar kinds of responses to CQ exposure are generated in yeast and human cells. Our work is primarily based on a model system, S. cerevisiae, and the CQ concentrations used in this system are higher than those required to kill malarial parasites. However, CQ is accumulated in the parasite food vacuole to millimolar concentrations similar to those used in these experiments, and thus, this work may have some biological relevance to the in vivo parasite system (74, 75). We have not tested our hypothesis on malarial parasites; hence, the effect of CQ on Hog1 and Slt2 homologues of malarial parasites should be analyzed in future studies. In conclusion, our present findings can be summarized in a model depicting the proposed molecular mode of action of CQ in S. cerevisiae (Fig. 9). Here, we have shown that CQ targets histone proteins and the modifying enzymes, including HATs and HDACs. Our study also revealed that CQ causes activation of the osmotic stress response as well as cell wall damage. Based on our results, we can conclude that CQ causes ROS generation leading to cell wall damage which eventually gives rise to an imbalance in the osmotic potential, causing activation of the osmotic response. Moreover, Hog1 and Slt2 are phosphorylated in response to CQ exposure. Activated Hog1 gets translocated to the nucleus, where it regulates expression of genes, leading to cell survival. Taken together, our data indicate that the HOG and CWI pathways are essential for providing resistance to CQ-induced stress. Further analysis of global and gene-specific changes in response to CQ exposure will give more insights into the mechanisms of toxicity exhibited by them. We anticipate that these facts would assist development of better approaches for assessment of toxicity caused by the pharmacologically important drug CQ.
FIG 9.

Diagrammatic representation of the model, showing the molecular mode of action of CQ on budding S. cerevisiae yeast. CQ exposure to yeast cells leads to generation of reactive oxygen species. ROS causes damage to yeast cell wall, leading to osmotic stress. Along with oxidative stress, CQ also targets histone proteins and chromatin-modifying enzymes (HATs and HDACs). An imbalance in osmotic stress as well as cell wall perturbation eventually causes activation of both Slt2 and Hog1 MAPK. Finally, these two central kinases induce downstream effectors to mediate the stress response which is required to provide tolerance of CQ-induced stress.
ACKNOWLEDGMENTS
We thank Toshi Tsukiyama, Randall H. Morse, C. D. Allis, Eun-Jung Cho, Erin K. O'Shea, Peter Samson, and Zhiguo Zhang for providing yeast strains.
This work was financially supported by the Department of Biotechnology (DBT) and Department of Science & Technology (DST), government of India, to R.S.T. DBT is acknowledged for fellowship support of S.B. Members of the Laboratory of Chromatin Biology are acknowledged for helpful discussions throughout this work.
Footnotes
Published ahead of print 14 July 2014
REFERENCES
- 1.Boya P, Gonzalez-Polo RA, Poncet D, Andreau K, Vieira HL, Roumier T, Perfettini JL, Kroemer G. 2003. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene 22:3927–3936. 10.1038/sj.onc.1206622 [DOI] [PubMed] [Google Scholar]
- 2.Fan C, Wang W, Zhao B, Zhang S, Miao J. 2006. Chloroquine inhibits cell growth and induces cell death in A549 lung cancer cells. Bioorg. Med. Chem. 14:3218–3222. 10.1016/j.bmc.2005.12.035 [DOI] [PubMed] [Google Scholar]
- 3.Choi JH, Yoon JS, Won YW, Park BB, Lee YY. 2012. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. APMIS 120:597–604. 10.1111/j.1600-0463.2012.02876.x [DOI] [PubMed] [Google Scholar]
- 4.Wellems TE. 1992. Malaria. How chloroquine works. Nature 355:108–109 [DOI] [PubMed] [Google Scholar]
- 5.Fox R. 1996. Anti-malarial drugs: possible mechanisms of action in autoimmune disease and prospects for drug development. Lupus 5(Suppl 1):S4–S10. 10.1177/096120339600500103 [DOI] [PubMed] [Google Scholar]
- 6.Park J, Choi K, Jeong E, Kwon D, Benveniste EN, Choi C. 2004. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia 47:9–20. 10.1002/glia.20017 [DOI] [PubMed] [Google Scholar]
- 7.Dias-Melicio LA, Calvi SA, Bordon AP, Golim MA, Peracoli MT, Soares AM. 2007. Chloroquine is therapeutic in murine experimental model of paracoccidioidomycosis. FEMS Immunol. Med. Microbiol. 50:133–143. 10.1111/j.1574-695X.2007.00243.x [DOI] [PubMed] [Google Scholar]
- 8.Jahn B, Langfelder K, Schneider U, Schindel C, Brakhage AA. 2002. PKSP-dependent reduction of phagolysosome fusion and intracellular kill of Aspergillus fumigatus conidia by human monocyte-derived macrophages. Cell. Microbiol. 4:793–803. 10.1046/j.1462-5822.2002.00228.x [DOI] [PubMed] [Google Scholar]
- 9.Levitz SM, Harrison TS, Tabuni A, Liu X. 1997. Chloroquine induces human mononuclear phagocytes to inhibit and kill Cryptococcus neoformans by a mechanism independent of iron deprivation. J. Clin. Invest. 100:1640–1646. 10.1172/JCI119688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rolain JM, Colson P, Raoult D. 2007. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int. J. Antimicrob. Agents 30:297–308. 10.1016/j.ijantimicag.2007.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keyaerts E, Li S, Vijgen L, Rysman E, Verbeeck J, Van Ranst M, Maes P. 2009. Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice. Antimicrob. Agents Chemother. 53:3416–3421. 10.1128/AAC.01509-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savarino A, Di Trani L, Donatelli I, Cauda R, Cassone A. 2006. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 6:67–69. 10.1016/S1473-3099(06)70361-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim EL, Wüstenberg R, Rübsam A, Schmitz-Salue C, Warnecke G, Bücker EM, Pettkus N, Speidel D, Rohde V, Schulz-Schaeffer W, Deppert W, Giese A. 2010. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro-Oncol. 12:389–400. 10.1093/neuonc/nop046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, Khodyakova AV, Ganapathi R, Ganapathi M, Tararova ND, Bosykh D, Lvovskiy D, Webb TR, Stark GR, Gudkov AV. 2005. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc. Natl. Acad. Sci. U. S. A. 102:17448–17453. 10.1073/pnas.0508888102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borges MC, Castro LA, Fonseca BA. 2013. Chloroquine use improves dengue-related symptoms. Mem. Inst. Oswaldo Cruz 108:596–599. 10.1590/S0074-02762013000500010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang PD, Zhao YL, Shi W, Deng XQ, Xie G, Mao YQ, Li ZG, Zheng YZ, Yang SY, Wei YQ. 2008. Cell growth inhibition, G2/M cell cycle arrest, and apoptosis induced by chloroquine in human breast cancer cell line Bcap-37. Cell. Physiol. Biochem. 22:431–440. 10.1159/000185488 [DOI] [PubMed] [Google Scholar]
- 17.Mitscher LA. 2005. Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev. 105:559–592. 10.1021/cr030101q [DOI] [PubMed] [Google Scholar]
- 18.Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. 2012. Hydroxychloroquine: from malaria to autoimmunity. Clin. Rev. Allergy Immunol. 42:145–153. 10.1007/s12016-010-8243-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melese T, Hieter P. 2002. From genetics and genomics to drug discovery: yeast rises to the challenge. Trends Pharmacol. Sci. 23:544–547. 10.1016/S0165-6147(02)02097-7 [DOI] [PubMed] [Google Scholar]
- 20.Hughes TR. 2002. Yeast and drug discovery. Funct. Integr. Genomics 2:199–211. 10.1007/s10142-002-0059-1 [DOI] [PubMed] [Google Scholar]
- 21.Huang ZW, Srinivasan S, Zhang JH, Chen KF, Li YX, Li W, Quiocho FA, Pan XW. 2012. Discovering thiamine transporters as targets of chloroquine using a novel functional genomics strategy. PloS Genet. 8:e1003083. 10.1371/journal.pgen.1003083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emerson LR, Nau ME, Martin RK, Kyle DE, Vahey M, Wirth DF. 2002. Relationship between chloroquine toxicity and iron acquisition in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 46:787–796. 10.1128/AAC.46.3.787-796.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emerson LR, Skillman BC, Wolfger H, Kuchler K, Wirth DF. 2004. The sensitivities of yeast strains deficient in PDR ABC transporters, to quinoline-ring antimalarial drugs. Ann. Trop. Med. Parasit. 98:643–649. 10.1179/000349804225021523 [DOI] [PubMed] [Google Scholar]
- 24.Garrington TP, Johnson GL. 1999. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 11:211–218. 10.1016/S0955-0674(99)80028-3 [DOI] [PubMed] [Google Scholar]
- 25.Hohmann S. 2002. Osmotic stress signaling and osmoadaptation in yeasts. Microbiol. Mol. Biol. Rev. 66:300–372. 10.1128/MMBR.66.2.300-372.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen TH, Chang PC, Chang MC, Lin YF, Lee HM. 2005. Chloroquine induces the expression of inducible nitric oxide synthase in C6 glioma cells. Pharmacol. Res. 51:329–336. 10.1016/j.phrs.2004.10.004 [DOI] [PubMed] [Google Scholar]
- 27.Bermejo C, Rodriguez E, Garcia R, Rodriguez-Pena JM, Rodriguez de la Concepcion ML, Rivas C, Arias P, Nombela C, Posas F, Arroyo J. 2008. The sequential activation of the yeast HOG and SLT2 pathways is required for cell survival to cell wall stress. Mol. Biol. Cell 19:1113–1124. 10.1091/mbc.E07-08-0742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azad GK, Singh V, Golla U, Tomar RS. 2013. Depletion of cellular iron by curcumin leads to alteration in histone acetylation and degradation of Sml1p in Saccharomyces cerevisiae. PLoS One 8:e59003. 10.1371/journal.pone.0059003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azad GK, Balkrishna SJ, Sathish N, Kumar S, Tomar RS. 2012. Multifunctional Ebselen drug functions through the activation of DNA damage response and alterations in nuclear proteins. Biochem. Pharmacol. 83:296–303. 10.1016/j.bcp.2011.10.011 [DOI] [PubMed] [Google Scholar]
- 30.Shah JB, Kumar S, Azad GK, Bhakuni BS, Panini P, Ahalawat N, Tomar RS, Detty M, Kumar S. 8 January 2014. An ebselen like catalyst with enhanced GPx activity via a selenol intermediate. Org. Biomol. Chem. 10.1039/c4ob00027g [DOI] [PubMed] [Google Scholar]
- 31.Singh V, Azad GK, Reddy M A, Baranwal S, Tomar RS. 2 May 2014. Anti-cancer drug KP1019 induces Hog1 phosphorylation and protein ubiquitylation in Saccharomyces cerevisiae. Eur. J. Pharmacol. 10.1016/j.ejphar.2014.04.032 [DOI] [PubMed] [Google Scholar]
- 32.Singh V, Azad GK, Mandal P, Reddy MA, Tomar RS. 20 February 2014. Anti-cancer drug KP1019 modulates epigenetics and induces DNA damage response in Saccharomyces cerevisiae. FEBS Lett. 10.1016/j.febslet.2014.02.017 [DOI] [PubMed] [Google Scholar]
- 33.Azad GK, Singh V, Mandal P, Singh P, Golla U, Baranwal S, Chauhan S, Tomar RS. 6 January 2014. Ebselen induces reactive oxygen species (ROS)-mediated cytotoxicity in Saccharomyces cerevisiae with inhibition of glutamate dehydrogenase being a target. FEBS Open Bio 10.1016/j.fob.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Golla U, Singh V, Azad GK, Singh P, Verma N, Mandal P, Chauhan S, Tomar RS. 2013. Sen1p contributes to genomic integrity by regulating expression of ribonucleotide reductase 1 (RNR1) in Saccharomyces cerevisiae. PLoS One 8:e64798. 10.1371/journal.pone.0064798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 36.Baro NK, Pooput C, Roepe PD. 2011. Analysis of chloroquine resistance transporter (CRT) isoforms and orthologues in S. cerevisiae yeast. Biochemistry 50:6701–6710. 10.1021/bi200922g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Islahudin F, Khozoie C, Bates S, Ting KN, Pleass RJ, Avery SV. 2013. Cell wall perturbation sensitizes fungi to the antimalarial drug chloroquine. Antimicrob. Agents Chemother. 57:3889–3896. 10.1128/AAC.00478-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baro NK, Callaghan PS, Roepe PD. 2013. Function of resistance conferring Plasmodium falciparum Chloroquine resistance transporter isoforms. Biochemistry 52:4242–4249. 10.1021/bi400557x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rahim R, Strobl JS. 2009. Hydroxychloroquine, chloroquine, and all-trans retinoic acid regulate growth, survival, and histone acetylation in breast cancer cells. Anti-Cancer Drugs 20:736–745. 10.1097/CAD.0b013e32832f4e50 [DOI] [PubMed] [Google Scholar]
- 40.Maeda T, Wurgler-Murphy SM, Saito H. 1994. A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature 369:242–245. 10.1038/369242a0 [DOI] [PubMed] [Google Scholar]
- 41.Tamás MJ, Rep M, Thevelein JM, Hohmann S. 2000. Stimulation of the yeast high osmolarity glycerol (HOG) pathway: evidence for a signal generated by a change in turgor rather than by water stress. FEBS Lett. 472:159–165. 10.1016/S0014-5793(00)01445-9 [DOI] [PubMed] [Google Scholar]
- 42.Rep M, Krantz M, Thevelein JM, Hohmann S. 2000. The transcriptional response of Saccharomyces cerevisiae to osmotic shock. Hot1p and Msn2p/Msn4p are required for the induction of subsets of high osmolarity glycerol pathway-dependent genes. J. Biol. Chem. 275:8290–8300. 10.1074/jbc.275.12.8290 [DOI] [PubMed] [Google Scholar]
- 43.Rep M, Reiser V, Gartner U, Thevelein JM, Hohmann S, Ammerer G, Ruis H. 1999. Osmotic stress-induced gene expression in Saccharomyces cerevisiae requires Msn1p and the novel nuclear factor Hot1p. Mol. Cell. Biol. 19:5474–5485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrigno P, Posas F, Koepp D, Saito H, Silver PA. 1998. Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1. EMBO J. 17:5606–5614. 10.1093/emboj/17.19.5606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Proft M, Mas G, de Nadal E, Vendrell A, Noriega N, Struhl K, Posas F. 2006. The stress-activated Hog1 kinase is a selective transcriptional elongation factor for genes responding to osmotic stress. Mol. Cell 23:241–250. 10.1016/j.molcel.2006.05.031 [DOI] [PubMed] [Google Scholar]
- 46.Proft M, Serrano R. 1999. Repressors and upstream repressing sequences of the stress-regulated ENA1 gene in Saccharomyces cerevisiae: bZIP protein Sko1p confers HOG-dependent osmotic regulation. Mol. Cell. Biol. 19:537–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Proft M, Pascual-Ahuir A, de Nadal E, Arino J, Serrano R, Posas F. 2001. Regulation of the Sko1 transcriptional repressor by the Hog1 MAP kinase in response to osmotic stress. EMBO J. 20:1123–1133. 10.1093/emboj/20.5.1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albertyn J, Hohmann S, Thevelein JM, Prior BA. 1994. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in Saccharomyces cerevisiae, and its expression is regulated by the high-osmolarity glycerol response pathway. Mol. Cell. Biol. 14:4135–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bermejo C, Rodriguez E, Garcia R, Rodriguez-Pena JM, de la Concepcion MLR, Rivas C, Arias P, Nombela C, Posas F, Arroyo J. 2008. The sequential activation of the yeast HOG and SLT2 pathways is required for cell survival to cell wall stress. Mol. Biol. Cell 19:1113–1124. 10.1091/mbc.E07-08-0742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.García R, Rodríguez-Peña JM, Bermejo C, Nombela C, Arroyo J. 2009. The high osmotic response and cell wall integrity pathways cooperate to regulate transcriptional responses to zymolyase-induced cell wall stress in Saccharomyces cerevisiae. J. Biol. Chem. 284:10901–10911. 10.1074/jbc.M808693200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azad GK, Singh V, Tomar RS. 2014. Assessment of the biological pathways targeted by isocyanate using N-succinimidyl N-methylcarbamate in budding yeast Saccharomyces cerevisiae. PLoS One 9:e92993. 10.1371/journal.pone.0092993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. 1993. An osmosensing signal transduction pathway in yeast. Science 259:1760–1763. 10.1126/science.7681220 [DOI] [PubMed] [Google Scholar]
- 53.Bilsland E, Molin C, Swaminathan S, Ramne A, Sunnerhagen P. 2004. Rck1 and Rck2 MAPKAP kinases and the HOG pathway are required for oxidative stress resistance. Mol. Microbiol. 53:1743–1756. 10.1111/j.1365-2958.2004.04238.x [DOI] [PubMed] [Google Scholar]
- 54.Alonso-Monge R, Navarro-Garcia F, Roman E, Negredo AI, Eisman B, Nombela C, Pla J. 2003. The Hog1 mitogen-activated protein kinase is essential in the oxidative stress response and chlamydospore formation in Candida albicans. Eukaryot. Cell 2:351–361. 10.1128/EC.2.2.351-361.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winkler A, Arkind C, Mattison CP, Burkholder A, Knoche K, Ota I. 2002. Heat stress activates the yeast high-osmolarity glycerol mitogen-activated protein kinase pathway, and protein tyrosine phosphatases are essential under heat stress. Eukaryot. Cell 1:163–173. 10.1128/EC.1.2.163-173.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawrence CL, Botting CH, Antrobus R, Coote PJ. 2004. Evidence of a new role for the high-osmolarity glycerol mitogen-activated protein kinase pathway in yeast: regulating adaptation to citric acid stress. Mol. Cell. Biol. 24:3307–3323. 10.1128/MCB.24.8.3307-3323.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han J, Lee JD, Bibbs L, Ulevitch RJ. 1994. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265:808–811. 10.1126/science.7914033 [DOI] [PubMed] [Google Scholar]
- 58.De Nadal E, Zapater M, Alepuz PM, Sumoy L, Mas G, Posas F. 2004. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature 427:370–374. 10.1038/nature02258 [DOI] [PubMed] [Google Scholar]
- 59.Bernstein BE, Tong JK, Schreiber SL. 2000. Genomewide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. U. S. A. 97:13708–13713. 10.1073/pnas.250477697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurdistani SK, Grunstein M. 2003. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 4:276–284. 10.1038/nrm1075 [DOI] [PubMed] [Google Scholar]
- 61.Reiser V, Ruis H, Ammerer G. 1999. Kinase activity-dependent nuclear export opposes stress-induced nuclear accumulation and retention of Hog1 mitogen-activated protein kinase in the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell 10:1147–1161. 10.1091/mbc.10.4.1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O'Rourke SM, Herskowitz I. 2004. Unique and redundant roles for HOG MAPK pathway components as revealed by whole-genome expression analysis. Mol. Biol. Cell 15:532–542. 10.1091/mbc.E03-07-0521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nahas N, Molski TF, Fernandez GA, Sha'afi RI. 1996. Tyrosine phosphorylation and activation of a new mitogen-activated protein (MAP)-kinase cascade in human neutrophils stimulated with various agonists. Biochem. J. 318(Pt 1):247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thorsen M, Di Y, Tangemo C, Morillas M, Ahmadpour D, Van der Does C, Wagner A, Johansson E, Boman J, Posas F, Wysocki R, Tamas MJ. 2006. The MAPK Hog1p modulates Fps1p-dependent arsenite uptake and tolerance in yeast. Mol. Biol. Cell 17:4400–4410. 10.1091/mbc.E06-04-0315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Albertyn J, Hohmann S, Prior BA. 1994. Characterization of the osmotic-stress response in Saccharomyces cerevisiae: osmotic stress and glucose repression regulate glycerol-3-phosphate dehydrogenase independently. Curr. Genet. 25:12–18. 10.1007/BF00712960 [DOI] [PubMed] [Google Scholar]
- 66.Nevoigt E, Stahl U. 1997. Osmoregulation and glycerol metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 21:231–241. 10.1111/j.1574-6976.1997.tb00352.x [DOI] [PubMed] [Google Scholar]
- 67.André L, Hemming A, Adler L. 1991. Osmoregulation in Saccharomyces cerevisiae. Studies on the osmotic induction of glycerol production and glycerol-3-phosphate dehydrogenase (NAD+). FEBS Lett. 286:13–17 [DOI] [PubMed] [Google Scholar]
- 68.Blomberg A, Adler L. 1989. Roles of glycerol and glycerol-3-phosphate dehydrogenase (NAD+) in acquired osmotolerance of Saccharomyces cerevisiae. J. Bacteriol. 171:1087–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Michihara A, Toda K, Kubo T, Fujiwara Y, Akasaki K, Tsuji H. 2005. Disruptive effect of chloroquine on lysosomes in cultured rat hepatocytes. Biol. Pharm. Bull. 28:947–951. 10.1248/bpb.28.947 [DOI] [PubMed] [Google Scholar]
- 70.Islahudin F, Khozoie C, Bates S, Ting KN, Pleass RJ, Avery SV. 2013. Cell wall perturbation sensitizes fungi to the antimalarial drug chloroquine. Antimicrob. Agents Chemother. 57:3889–3896. 10.1128/AAC.00478-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harrison JC, Bardes EG, Zyla TR, Lew DJ. 2002. Stress-specific activation mechanisms for the “cell integrity” MAPK pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 13:294a–294a. 10.1074/jbc.M306110200 [DOI] [PubMed] [Google Scholar]
- 72.Becker K, Tilley L, Vennerstrom JL, Roberts D, Rogerson S, Ginsburg H. 2004. Oxidative stress in malaria parasite-infected erythrocytes: host-parasite interactions. Int. J. Parasitol. 34:163–189. 10.1016/j.ijpara.2003.09.011 [DOI] [PubMed] [Google Scholar]
- 73.Loria P, Miller S, Foley M, Tilley L. 1999. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem. J. 339:363–370. 10.1042/0264-6021:3390363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aikawa M. 1972. High-resolution autoradiography of malarial parasites treated with 3 H-chloroquine. Am. J. Pathol. 67:277–284 [PMC free article] [PubMed] [Google Scholar]
- 75.Yayon A, Cabantchik ZI, Ginsburg H. 1984. Identification of the acidic compartment of Plasmodium falciparum-infected human erythrocytes as the target of the antimalarial drug chloroquine. EMBO J. 3:2695–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]