

Abstract
A serology-based tiered approach has, to date, provided the most effective means of laboratory confirmation of clinically suspected cases of Lyme disease, but it lacks sensitivity in the early stages of disease and is often dependent on subjectively scored immunoblots. We recently demonstrated the use of immuno-PCR (iPCR) for detecting Borrelia burgdorferi antibodies in patient serum samples that were positive for Lyme disease. To better understand the performance of the Lyme disease iPCR assay, the repeatability and variability of the background of the assay across samples from a healthy population (n = 36) were analyzed. Both of these parameters were found to have coefficients of variation of <3%. Using eight antigen-specific iPCR assays and positive call thresholds established for each assay, iPCR IgM and/or IgG diagnosis from Lyme disease patient serum samples (n = 12) demonstrated a strong correlation with that of 2-tier testing. Furthermore, a simplified iPCR approach using a single hybrid antigen and detecting only IgG antibodies confirmed the 2-tier diagnosis in the Lyme disease patient serum samples (n = 12). Validation of the hybrid antigen IgG iPCR assay using a blinded panel of Lyme disease and non-Lyme disease patient serum samples (n = 92) resulted in a sensitivity of 69% (95% confidence interval [CI], 50% to 84%), compared to that of the 2-tier analysis at 59% (95% CI, 41% to 76%), and a specificity of 98% (95% CI, 91% to 100%) compared to that of the 2-tier analysis at 97% (95% CI, 88% to 100%). A single-tier hybrid antigen iPCR assay has the potential to be an improved method for detecting host-generated antibodies against B. burgdorferi.
INTRODUCTION
Lyme disease is the most commonly reported tick-borne illness in the United States, with approximately 30,000 cases reported to the Centers for Disease Control and Prevention (CDC) each year (1). New preliminary estimates released by the CDC indicate that the number of Americans diagnosed with Lyme disease each year is closer to 300,000, which is roughly 10 times higher than the annual reported number (2). This new estimate supports studies published in the 1990s, which suggested that the number of cases may be between 3- and 12-fold higher than the number of reported cases (3, 4), making Lyme disease a significant health concern in the United States. Accurate diagnosis provides a considerable obstacle for the clinical management of the disease and is necessary in order to differentiate Lyme disease from other diseases with similar clinical presentation. Misdiagnosis is common due to difficulties in detecting Borrelia burgdorferi, the causative agent of Lyme disease (5). Although a wide range of laboratory diagnostic approaches have been explored, the currently accepted method utilizes the detection of serological responses to B. burgdorferi antigens (6).
The currently accepted method for diagnosing Lyme disease in a clinical setting entails a two-tiered approach using a first-tier enzyme-linked immunosorbent assay (ELISA), followed by a second-tier immunoblot assay for both IgM and IgG B. burgdorferi-specific antibodies using whole-cell B. burgdorferi lysates, recombinant antigens, or various combinations, depending on the commercial kit used (7). The ELISA provides an objective and sensitive first-tier screen but lacks the specificity and broad strain applicability (8) required for a standalone test. The second-tier immunoblot provides a higher level of specificity but currently requires somewhat subjective analysis due to its qualitative nature and general lack of automation (9). A tiered approach has to date provided the most effective means of diagnosing Lyme disease in a clinical setting (7).
Other approaches for diagnosing Lyme disease have been developed, including live culture, PCR, and additional molecular-based approaches, with no method surpassing the effectiveness of a serology-based approach. The detection of typical erythema migrans (EM) can be sufficient for a clinical diagnosis of early localized Lyme disease in the absence of laboratory tests (7). However, this manifestation is not present in all patients (7), further highlighting the need for improved methods for early objective diagnosis of Lyme disease. In our previous study, we demonstrated the use of immuno-PCR (iPCR) for detecting host-generated antibodies in a murine model, and we presented preliminary data using serum samples collected from Lyme disease patients and healthy controls (10). Our results indicated that iPCR using B. burgdorferi whole-cell sonicates and a limited number of B. burgdorferi recombinant antigens provided higher sensitivity for detecting B. burgdorferi antibodies in infected mice and an equivalent sensitivity for detecting B. burgdorferi antibodies in Lyme disease patient serum compared to both ELISA and the immunoblot (10).
It is well established that multiple antigens are required for an accurate overall diagnosis of the multiple stages and types of Lyme disease (7). Furthermore, it is critical that the antigens used for diagnosis are demonstrated to have low cross-reactivity for diseases other than Lyme disease. The goals of this study were to (i) determine the range of the levels of background detection of the Lyme disease iPCR assays across a healthy human population, (ii) explore a larger subset of antigens for assay sensitivity and specificity, and (iii) compare the performance of the optimized Lyme disease iPCR protocol with that of the current 2-tier method of Lyme disease diagnosis.
MATERIALS AND METHODS
Healthy human sera.
The current study was approved by the University of Central Florida's institutional review board (UCF IRB) (FWA00000351 and IRB00001138). All procedures and investigators involved in the sample collection process were approved by the UCF IRB with Collaborative Institutional Training Initiative (CITI) training. All donors provided written consent to participate in the study. Sample collection was undertaken at the University of Central Florida campus. UCF is a diverse community of nearly 60,000 students and approximately 8,000 faculty and staff members of various ages and ethnic and racial backgrounds. Individuals were included in the study if they had not been previously diagnosed with and/or treated for Lyme disease, received a Lyme disease vaccination, or lived within the past 10 years in a state with a high incidence of Lyme disease (Connecticut, Delaware, Maine, Maryland, Massachusetts, Minnesota, New Hampshire, New Jersey, New York, Pennsylvania, Vermont, Virginia, and Wisconsin). Approximately 10 ml of blood was sampled, according to the IRB-approved protocol, from 36 individuals into serum separator tubes, inverted five times to mix the clot activator with the blood, and allowed to clot for ≥30 min. Serum fractions were collected by centrifugation at 1,200 × g for 10 min. The serum was further clarified by centrifugation at 9,100 × g for 5 min to remove any insoluble material and stored at 4°C for short-term or −80°C for long-term storage.
Lyme disease human serum panel.
The CDC research panel I consisted of patient serum samples collected from 32 individuals, including patients with stage 1, 2, or 3 Lyme disease (n = 12), look-alike diseases, including fibromyalgia, rheumatoid arthritis, multiple sclerosis, mononucleosis, syphilis, and severe periodontitis (n = 12), as well as healthy individuals from areas of endemicity (n = 4) and nonendemicity (n = 4) for Lyme disease. All Lyme disease patients were diagnosed by a physician, stage 1 and 2 patients were confirmed by culture and/or PCR detection of B. burgdorferi, and stage 3 patients were positive by two-tiered testing. The CDC-recommended two-tiered testing algorithm (6) was performed using FDA-cleared assays for Lyme disease and consisted of a first-tier whole-cell sonicate enzyme immunoassay (VIDAS Lyme IgM and IgG polyvalent assay; bioMérieux, Inc., Durham, NC), followed by second-tier IgM and IgG immunoblots (IB) (MarDx Diagnostics, Inc., Carlsbad, CA). The blinded CDC research panel II consisted of serum samples collected from 92 individuals, including patients with stage 1, 2, or 3 Lyme disease (n = 32), look-alike diseases, including fibromyalgia, rheumatoid arthritis, multiple sclerosis, mononucleosis, syphilis, and severe periodontitis (n = 36), as well as healthy individuals from areas of endemicity (n = 12) and nonendemicity (n = 12) for Lyme disease. The laboratory support of Lyme disease diagnosis was the same as for CDC research panel I. Prior to analysis, all serum samples were clarified by centrifugation at 9,100 × g for 5 min to remove any insoluble material and put in the short-term storage at 4°C.
Cloning and expression of recombinant antigens lacking GST fusion tags.
Recombinant glutathione S-transferase (rGST)-BmpA and rGST-OspC were constructed as previously described (10). In-frame glutathione S-transferase (GST) fusion proteins for BBK19, OspA, DbpA, RevA, Crasp-2, and BBK50 were generated by PCR amplification of the corresponding coding regions, without the signal sequences from B. burgdorferi genomic DNA, using primer pairs 1147 and 1148 (BBK19), 1151 and 1152 (OspA), 1145 and 1146 (DbpA), 1143 and 1144 (RevA), 1149 and 1150 (Crasp-2), or 1043 and 1044 (BBK50) engineered with BamHI and SalI or XhoI restriction sites (Table 1) and Phusion polymerase (New England BioLabs, Ipswich, MA). The PCR products were purified (Qiagen, Valencia, CA), digested with the appropriate restriction enzymes (New England BioLabs), and cloned into BamHI- and SalI- or XhoI-digested pGEX-6P-1 (GE Healthcare, Piscataway, NJ) to generate translational fusions with GST at the N terminus. Subsequent clones were selected and the sequence confirmed by sequence analysis. pGEX-6P-1 plasmids carrying the bmpA, ospC, bbk19, ospA, dbpA, revA, crasp-2, and bbk50 genes were transformed into Escherichia coli strain BL21 (Novagen, Billerica, MA). Protein expression was induced by the growth of BL21 cells containing the expression construct for each B. burgdorferi antigen in 50 to 100 ml of MagicMedia E. coli expression medium, according to the manufacturer's protocol (Invitrogen, Carlsbad, CA) for 24 h at 37°C with aeration. Recombinant protein purification was performed according to the procedures outlined in the Bulk GST purification module (GE Healthcare). The purified proteins were dialyzed in Tris-buffered saline (50 mM Tris-HCl, 150 mM NaCl [pH 7.5]) overnight at 4°C using D-Tube dialyzers (EMD Millipore Chemicals, Philadelphia, PA) and two buffer exchanges to remove excess glutathione. The dialyzed proteins were subjected to protease cleavage of the GST tag overnight at 4°C, according to procedures outlined in the PreScission protease kit (GE Healthcare). Cleaved proteins were purified from GST and excess protease using two rounds of Bulk GST purification (GE Healthcare) and collection of the eluent. Purified proteins lacking a GST tag were concentrated using Amicon Ultra-2 centrifugal filter devices (EMD Millipore Chemicals) to a volume of approximately 80 μl and stored at 4°C. The total protein content was quantified by absorbance spectrophotometry at a wavelength of 280 nm. Recombinant protein purity and seroreactivity were determined by Coomassie gel staining and immunoblot using infected mouse serum. Briefly, 100 ng of each recombinant protein was separated by 12.5% polyacrylamide gel electrophoresis. For Coomassie staining, the gels were incubated in Imperial protein stain (Thermo Scientific, Rockford, IL) for 1 h and destained in deionized water for 1 h prior to imaging. For immunoblot analysis, the proteins were transferred to a nitrocellulose membrane, and the membrane was blocked in 5% skim milk and incubated for 1 h with mouse serum samples collected 3 weeks postinoculation with wild-type B. burgdorferi, as previously described (10), diluted 1:200 in Tris-buffered saline–0.05% Tween (TBST) (pH 7.6), washed twice with TBST, incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG/IgM (Chemicon International, Billerica, MA) for 1 h, washed twice with TBST, and the signal was detected using the SuperSignal West Pico chemiluminescent substrate kit (Thermo Scientific).
TABLE 1.
iPCR DNA oligonucleotide sequences used in this study
| Oligo no. | Oligo IDa | Sequence (5′ to 3′)b |
|---|---|---|
| T1 | Template 1 (IgG coupled) | Biotin-agcctcagaccaagccagacaactgcctcgtgacgttgctgcccctaccaacgtacccctacgagtcc |
| T1F | Template 1 forward | agcctcagaccaagccagac |
| T1R | Template 1 reverse | ggactcgtaggggtacgttgg |
| T1P | Template 1 probe | FAM-actgcctcgtgacgttgctgcccct-BHQ1 |
| T2 | Template 2 (IgM coupled) | Biotin-aggaggagggtcaagtcaccaacgctgctccaggccatcgtgctgatctggaccctggatcgagtga |
| T2F | Template 2 forward | aggaggagggtcaagtcacc |
| T2R | Template 2 reverse | tcactcgatccagggtccag |
| T2P | Template 2 probe | MAX-acgctgctccaggccatcgtgctga-BHQ1 |
| 1147 | BBK19 F | CGGGATCCttttcaaaagattctcgatcacg |
| 1148 | BBK19 R | ACGCCTCGAGtcaattgttaggtttttcttttcc |
| 1151 | OspA F | CGGGATCCaagcaaaatgttagcagcc |
| 1152 | OspA R | ACGCCTCGAGttattttaaagcgtttttaatttcatcaag |
| 1145 | DbpA F | CGGGATCCggactaacaggagcaacaa |
| 1146 | DbpA R | ACGCCTCGAGttagttatttttgcatttttcatcag |
| 1143 | RevA F | CGGGATCCaaagcatatgtagaagaaaagaaag |
| 1144 | RevA R | ACGCCTCGAGttaattagtgccctcttcg |
| 1149 | Crasp2 F | CGGGATCCgatgttagtagattaaatcagagaaatatt |
| 1150 | Crasp2 R | ACGCCTCGAGctataataaagtttgcttaatagctttataag |
| 1043 | BBK50 F | CGGGATCCatgtgtaaattatatgaaaagcttacaaataaatcgc |
| 1044 | BBK50R | CCGCTCGAGttatctagagtccatatcttgcaattt |
| 1084 | DbpA_PEPC10 R | AGGTTTTTTTGGACTTTCTGCCACAACAGGgttatttttgcatttttcatcagtaaaagt |
| 1085 | C6_PEPC10 F | CCTGTTGTGGCAGAAAGTCCAAAAAAACCTatgaagaaggatgatcagattgc |
| 1023 | C6 Bb R | ACGCGTCGACttacttcacagcaaactttccatc |
ID, identification.
Uppercase letters indicate nontemplate sequence used for the addition of terminal restriction sites, epitope tags, or synthetic assembly. FAM, 6-carboxyfluorescein; BHQ1, black hole quencher 1.
Cloning and expression of the recombinant DOC antigen.
An in-frame glutathione S-transferase (GST) fusion protein for the DOC hybrid protein was generated using two distinct PCR amplification steps. First, the corresponding coding regions for DbpA and the C6 peptide of VlsE (11) were amplified separately from B. burgdorferi strain B31 genomic DNA, and the PEPC10 sequence (12) was added to each amplicon using the primer pairs 1145 and 1084 (DbpA-PEPC10) and 1085 and 1023 (C6-PEPC10), respectively, engineered with BamHI/SalI restriction sites (Table 1). Both PCR products were diluted 100-fold, combined, and synthetically assembled into the DOC construct by overlapping PCR using the primer pairs 1145 and 1023. The final constructs were sequenced and verified, and the recombinant protein was generated and purified as described above for the other B. burgdorferi antigens.
iPCR reagents, assay, and signal amplification.
iPCR reagents were prepared and the assays conducted as previously described (10), with minor modifications. Briefly, iPCR assays were assembled in a two-sided (sandwich) manner, as detailed in Fig. 1A, with the capability to simultaneously capture and report both IgM and IgG host-generated antibodies (Fig. 1B). Recombinant antigens lacking fusion tags were used to coat magnetic beads for host antibody capture using 10 to 20 μg of antigen per mg of beads. The beads were resuspended in 500 μl TBST for secondary antibody incubation. Signal amplification by real-time quantitative PCR was accomplished as previously described (10), and the quantification cycle (Cq) for each reaction was determined using a manual baseline determination (cycle 10 to 20) and a manual threshold setting of 1.0.
FIG 1.
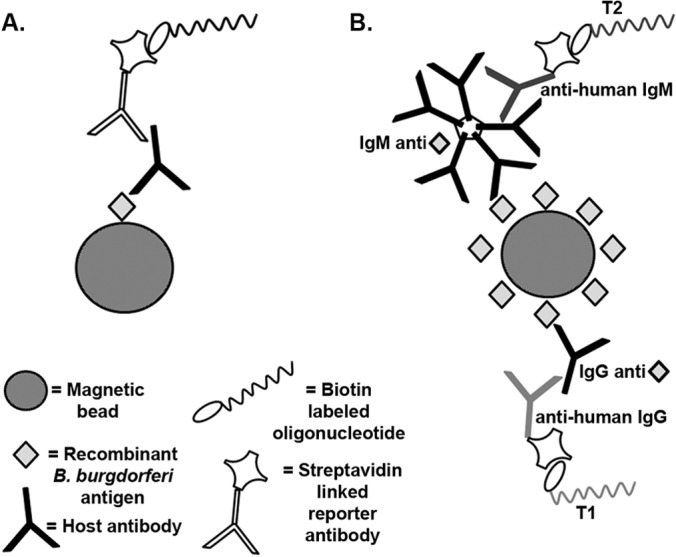
Schematic representation of the multiplex iPCR assay for detection of Lyme disease host antibodies using recombinant antigens. (A) A recombinant B. burgdorferi protein antigen coupled to magnetic beads was used to capture B. burgdorferi-specific host-generated antibodies. A biotinylated DNA oligonucleotide reporter molecule coupled to a streptavidin-conjugated reporter antibody was amplified by qPCR for detection and quantification. (B) The same antigen-coupled beads were used to simultaneously capture IgM and IgG host-generated antibodies, which were detected in a multiplex fashion using isotype-specific secondary antibodies coupled to unique reporter oligonucleotides (T1 and T2) similarly amplified by qPCR for detection and quantification.
The PCR plate set-ups for all experiments included, in duplicate, a PCR-negative template control consisting of water and an iPCR bead processing negative control that contained the TBST stock used for processing to determine the sample-to-sample contamination. Additionally, each PCR run included calibrator plasmids carrying the cloned template for the IgM or IgG reporter oligonucleotides that were used to account for run-to-run variation in the threshold calculation between the PCR plates. Briefly, the baseline was manually adjusted such that the Cq values for the calibrator plasmids were set at a constant value for each plate to account for minor variability in the threshold setting.
Positive threshold value and data analysis.
The results of the Lyme disease iPCR assay were reported as ΔCq values. The ΔCq value was calculated as the difference between the antigen-/isotype-specific background threshold Cq value and the Cq value of the sample. The antigen-/isotype-specific background threshold Cq values were calculated as the mean Cq value of each antigen-isotype combination for a group of 16 healthy individuals minus a specific multiple of the standard deviation (SD) of the mean. The antigen-specific multiplier was set at a minimal value (1.9 to 6.6 for IgM and 3.1 to 5 for IgG), such that the samples from all individuals without Lyme disease in CDC research panel I resulted in a Lyme disease iPCR ΔCq value of <0. Using these antigen-/isotype-specific thresholds, any sample that resulted in a Lyme disease iPCR ΔCq of ≥0 was called iPCR positive. The coefficient of variation (CV) was calculated as the ratio of the SD to the mean. Assay sensitivity and specificity and the associated 95% confidence intervals were calculated using GraphPad Prism 5.0 for Windows (GraphPad Software).
RESULTS
Lyme disease iPCR demonstrates strong within-assay precision and reproducible background across a sample population of healthy individuals.
We previously demonstrated proof-of-principle for iPCR detection of human host-generated B. burgdorferi antibodies using VlsE C6 peptide-coated magnetic beads and a panel of serum samples (n = 36) from Lyme disease-positive and Lyme disease-negative patients and healthy controls (10). This feasibility study was accomplished using a small number of samples from healthy controls (n = 5) to determine test efficiency and background threshold levels. In an effort to establish a better understanding of the performance of the Lyme disease iPCR assay, including the repeatability and the variability of the background of the assay across a healthy population, the number of replicates and overall sample size of healthy individuals were expanded. Prospective blood samples were collected from consenting individuals without a history of Lyme disease under the approval of the University of Central Florida's institutional review board. To assess assay repeatability, a serum sample from a single healthy individual was tested 18 times using the same reagent preparation lots, including DbpA antigen-coated beads and oligonucleotide-labeled secondary antibodies. The DbpA protein was selected as a representative in vivo-expressed B. burgdorferi antigen. The results of this analysis demonstrated low within-assay variability for both the IgM- and IgG-specific detection reagents, as indicated by standard deviation values for each data set of 0.39 and 0.73, respectively, and coefficient of variation values for each data set of 1.34% and 2.30%, respectively (Fig. 2).
FIG 2.
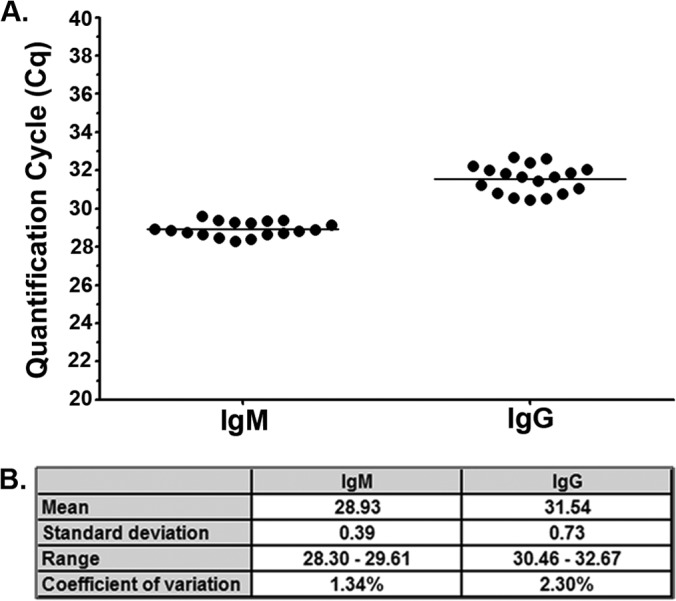
Lyme disease immuno-PCR magnetic bead protocol demonstrates strong within-assay precision. (A) Serum collected from a single healthy individual was assayed 18 times by IgM/IgG multiplex iPCR using recombinant DbpA antigen coupled to magnetic beads. Each dot represents a single replicate, and the horizontal line represents the mean Cq value for all replicates for each isotype. The y axis represents the quantification cycle (Cq) determined by real-time quantitative PCR. (B) The mean, standard deviation (SD), range, and coefficient of variation (CV) (calculated as the ratio of the SD to the mean) were calculated for both the IgM and IgG Cq values.
To determine the variability in the background of the Lyme disease iPCR assay across a healthy human population, the serum samples from 36 healthy individuals were tested in duplicate using magnetic beads coated with the DbpA antigen and the oligonucleotide-labeled IgM and IgG secondary antibodies used for the repeatability analysis. Similar to the within-sample repeatability analysis, the results of the between-sample variability analysis demonstrated a standard deviation across the population of 0.79 for the background detection of IgM antibodies and 0.84 for IgG antibodies; the coefficients of variation were 2.66% and 2.63%, respectively (Fig. 3).
FIG 3.
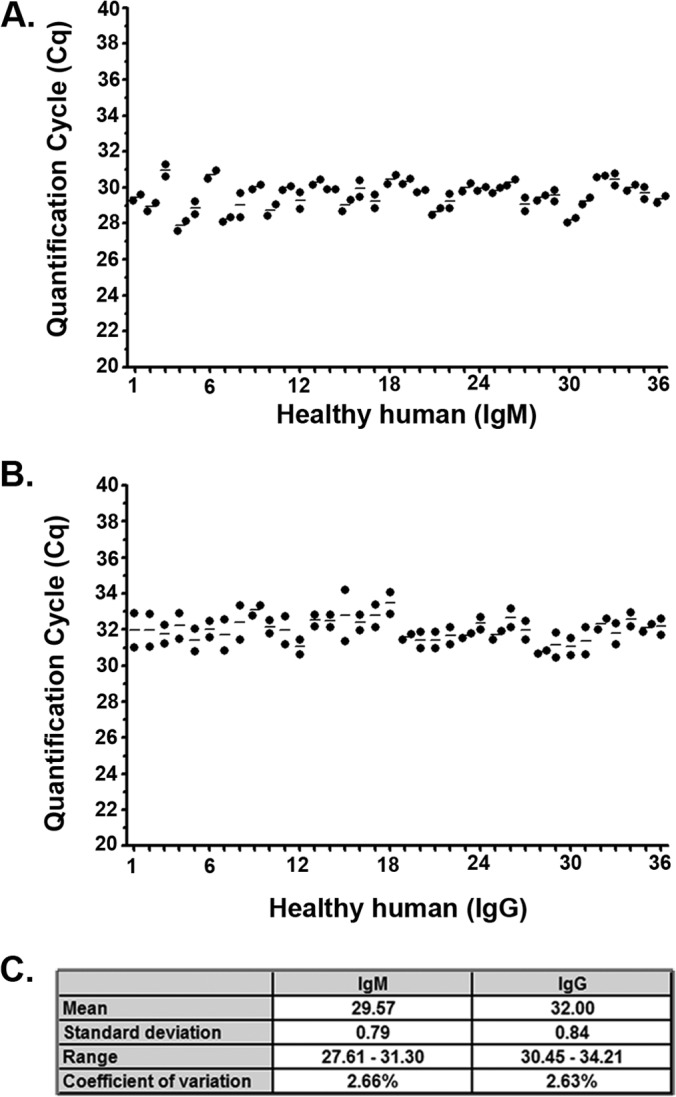
Lyme disease immuno-PCR demonstrates reproducible background across a healthy human population for both IgM and IgG isotypes using the DbpA antigen. Serum samples from 36 healthy individuals were assayed in duplicate by multiplex iPCR using both IgM (A) and IgG (B) secondary antibodies and recombinant DbpA antigen-coupled magnetic beads. Each dot represents a single replicate per individual, with the horizontal lines representing the mean value for duplicate serum samples from each individual. The y axis represents the quantification cycle (Cq) determined by real-time quantitative PCR. (C) The mean, standard deviation (SD), range, and coefficient of variation (CV) (calculated as the ratio of the SD to the mean) is listed for each isotype.
Mean and standard deviation background values across a population of healthy individuals are unique for each Lyme disease iPCR assay antigen-isotype combination.
The analysis of the Lyme disease iPCR assay repeatability and population variability using DbpA-coupled magnetic beads demonstrated that the mean background value for the detection of IgM versus IgG antibodies differed by as much as ∼2.5 Cq values (Fig. 2 and 3). Based on this observation, we predicted that depending on the different antigen used, each Lyme disease iPCR assay would result in a distinct mean background Cq value. If true, this finding would impact the determination of the background threshold setting for the assay, making it necessary to assign a distinct background threshold for each antigen-isotype combination. To test this hypothesis, we compiled a list from the literature of B. burgdorferi proteins that are known or hypothesized to be seroreactive in humans (13–27). From this list, a subset of 8 B. burgdorferi antigens was selected for further analysis in our assay due to their ability to be produced in large quantities as recombinant in-frame N-terminal glutathione S-transferase (GST) fusion proteins in E. coli. To eliminate any possibility of antibody cross-reactivity to the GST tag, this sequence was proteolytically removed. The purity and antigenicity of each recombinant antigen were demonstrated by SDS-PAGE, followed by Coomassie brilliant blue staining and immunoblot analysis using pooled sera collected from B. burgdorferi-infected mice (see Fig. S1 in the supplemental material).
Each antigen was coupled to magnetic beads and examined by Lyme disease iPCR for both IgM and IgG background reactivities across 16 serum samples collected from healthy individuals. As predicted, all antigen-isotype combinations demonstrated unique background values that ranged from a mean Cq of 26.09 to 32.46 for IgM and 25.30 to 36.62 for IgG and a standard deviation of 0.40 to 1.53 for IgM and 0.37 to 1.47 for IgG (Fig. 4).
FIG 4.
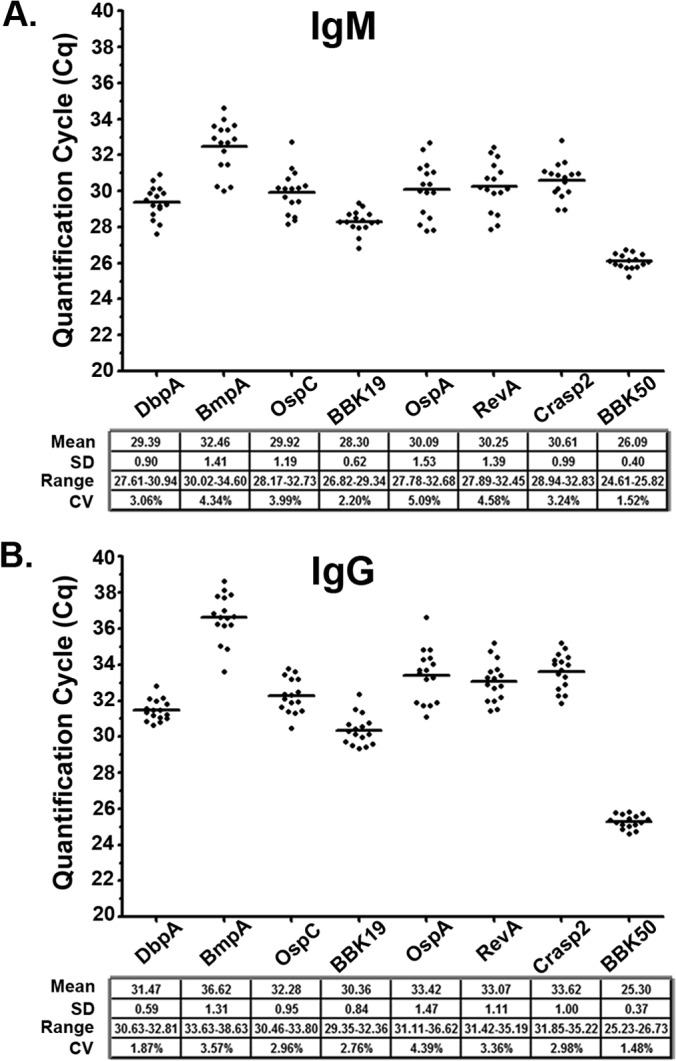
Immuno-PCR demonstrates low intra-antigen background variability for an antigen panel across a healthy human population. Serum samples from 16 healthy individuals were assayed by multiplex iPCR for both IgM (A) and IgG (B) host-generated antibodies against recombinant DbpA, BmpA, OspC, BBK19, OspA, RevA, Crasp2, and BBK50 antigen-coupled magnetic beads. Each point represents a single individual replicate, and the horizontal lines represent the mean Cq values for all individuals for each antigen/isotype combination. Each antigen mean and standard deviation (SD) value is listed. The y axis represents the quantification cycle (Cq) determined by real-time PCR. The across-population mean, standard deviation, range, and coefficient of variation (CV) values are shown for each antigen/isotype combination.
Multiplex iPCR detection of IgM and/or IgG host response antibodies against B. burgdorferi using a panel of antigens has the potential for improved sensitivity compared to 2-tier testing.
Most existing protocols for Lyme disease diagnostics require the use of multiple antigens to diagnose the disease. In an effort to further explore the application of iPCR as a Lyme disease diagnostic, we sought to develop a similar methodology that utilizes a combination of results for different antigens to facilitate diagnosis. The panel of eight B. burgdorferi antigens was tested against the CDC research panel I collection of sera using multiplex iPCR for the simultaneous detection of IgM and IgG host-generated antibodies. The same human serum panel was previously tested according to CDC guidelines by a commercial enzyme-linked immunosorbent assay (ELISA), followed by IgM and IgG immunoblot (IB), and classified for 2-tier testing status (see Table S1 in the supplemental material). Samples were considered positive by iPCR if they resulted in a ΔCq value that was ≥0 for IgM or IgG for one or more of the eight antigens tested. The ΔCq value was calculated as the difference between the antigen-/isotype-specific background threshold Cq value and the Cq value of the sample. The antigen-/isotype-specific background threshold Cq values were calculated as the mean Cq value of each antigen-isotype combination for a group of 16 healthy individuals minus a specific multiple of the standard deviation (SD) of the mean (Fig. 4). Each antigen-specific multiplier was set at a minimum value (1.3 to 6.6 for IgM and 2.8 to 5 for IgG; see Table S2 in the supplemental material), such that the samples from all individuals without Lyme disease in CDC research panel I resulted in a Lyme disease iPCR ΔCq value of <0. Using these criteria, iPCR testing provided similar results to those of 2-tier testing for the Lyme disease patient samples, with one exception (see Table S1 in the supplemental material). A single early Lyme disease patient sample that was deemed negative by 2-tier testing was positive by iPCR (see Table S1, sample A4). It should also be noted that no single antigen provided iPCR-positive results across all Lyme disease patient samples, which comprised different stages and clinical presentations of disease.
Simplified single hybrid antigen iPCR detection of host-generated IgG antibodies alone confirms 2-tier results for a panel of human serum samples.
iPCR testing with the panel of eight B. burgdorferi antigens showed strong potential as a Lyme disease diagnostic method by reproducing the 2-tier test results for CDC research panel I Lyme disease patient samples. Although successful, the use of multiple antigens tested against IgM and IgG increases test complexity by requiring the testing of a single sample with multiple antigens. In an effort to further simplify the Lyme disease iPCR approach, we theorized that a single hybrid antigen composed of the immunogenic epitopes of multiple B. burgdorferi antigens would provide results similar to those of testing with a panel of whole individual antigens. To examine the applicability of a single hybrid antigen for iPCR detection of host-generated antibodies against B. burgdorferi infection, we synthetically constructed a novel hybrid antigen composed of full-length DbpA, the PEPC10 peptide (OspC) (12), and the C6 peptide (VlsE) (11), referred to as the DOC antigen (Fig. 5A). Similar to the previous eight recombinant antigens, we determined the protein purity and seroreactivity toward B. burgdorferi-infected mouse sera of the hybrid protein (see Fig. S1 in the supplemental material). The range of the background reactivity of the DOC antigen in the iPCR assay was determined using the serum from a group of 16 healthy individuals (Fig. 5B). The results of the between-sample variability analysis demonstrated a standard deviation across the population of 0.57 for the background detection of IgM antibodies and 0.51 for the background detection of IgG antibodies; the coefficients of variation were 2.31% and 1.94%, respectively. Using iPCR, we then tested the hybrid antigen in duplicate against the CDC research panel I for IgM and IgG reactivity, utilizing the results to establish the positive call threshold as described above. The DOC antigen IgG results confirmed all 2-tier-positive results (Fig. 6A). Interestingly, the Lyme disease iPCR assay using the DOC antigen tested negative for the detection of host-generated IgM antibodies for all human samples analyzed (Fig. 6B).
FIG 5.
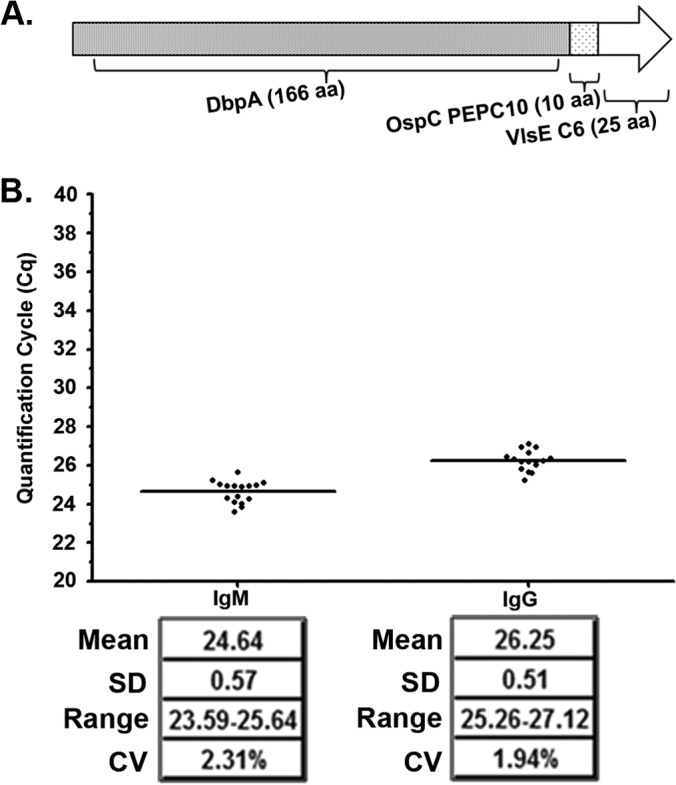
Development of a hybrid antigen for simple detection of Lyme disease. The DOC antigen was assembled using full-length DbpA protein fused to the PEPC10 (OspC) and the C6 (VlsE) peptides (A) and was tested by iPCR using DOC-coated magnetic beads against 16 healthy individuals for IgM and IgG for the range of the background reactivity (B). (B) Each dot represents a single individual replicate, and the horizontal lines represent the mean Cq values for all individuals for IgM and IgG. The mean, standard deviation (SD), range, and CV values are also listed. The y axis represents the quantification cycle (Cq) determined by real-time quantitative PCR.
FIG 6.
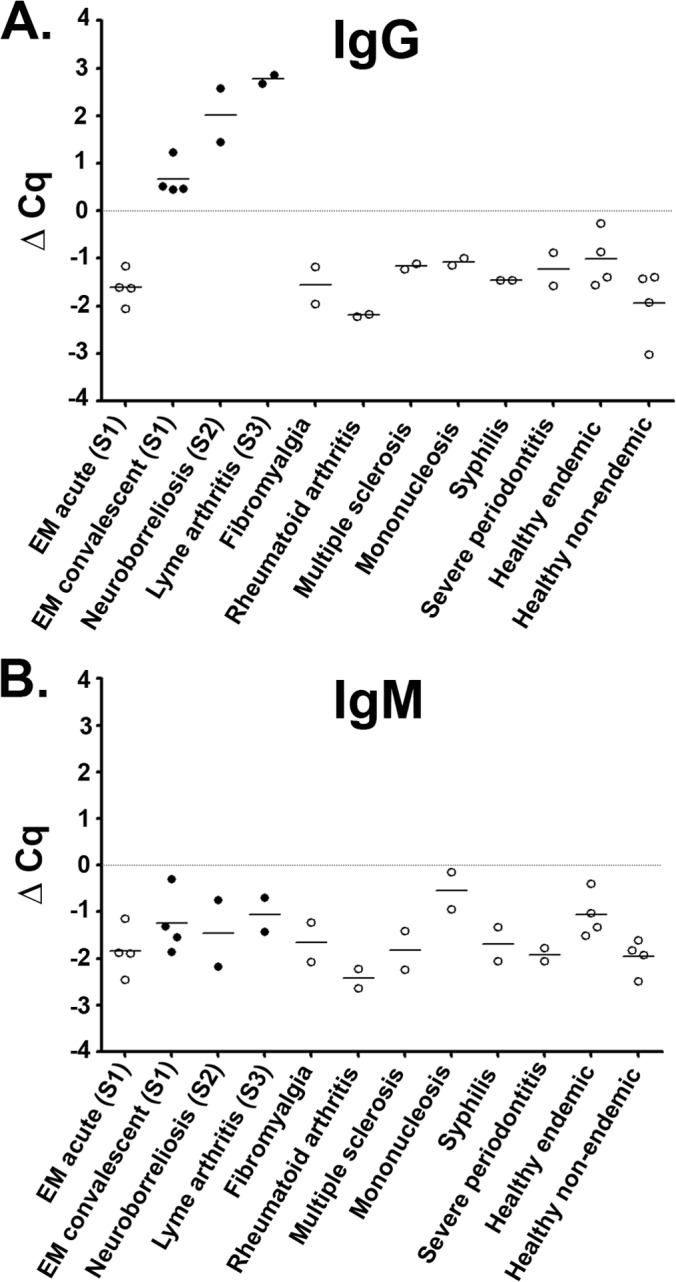
The iPCR assay using the DOC hybrid antigen provides robust detection of Lyme disease. A serum panel composed of 32 samples and consisting of Lyme-infected individuals both early (acute and convalescent) and late (neurologic and arthritis) stage, as well as look-alike diseases and healthy individuals from areas of endemicity and nonendemicity were tested in duplicate using DOC iPCR for both IgG (A) and IgM (B) reactivity. Each dot represents a single individual replicate, and the black horizontal lines represent the mean Cq values for all individuals within each category. The filled circles represent samples that were positive with 2-tier testing, and the open circles signify a 2-tier-negative status. A positive threshold value was established using a multiplier of the standard deviation (SD) above the mean value with the ΔCq threshold (gray horizontal line) representing a value of zero. S1, stage 1; S2, stage 2; S3, stage 3.
Although early, specific diagnosis is the primary goal for any Lyme disease diagnostic, determining the stage of disease progression would provide additional information to aide in the treatment of the disease. It is logical to assume that the amount of host-generated B. burgdorferi antibody will increase with further disease progression. Due to the quantifiable nature of iPCR testing, we hypothesized that the amount of anti-DOC host-generated IgG antibody correlates with disease stage. The mean ± SD iPCR value was −1.61 ± 0.36 for stage 1 acute early Lyme disease patients, 0.67 ± 0.38 for stage 1 convalescent early Lyme disease patients, and 2.39 ± 0.64 for stage 2/stage 3 Lyme disease patients, for a total of n = 4 samples per group. These data may suggest a correlation of increasing antibody capture with disease progression; however, further evaluation with an increased number of clinically defined samples is required to support this finding. It should also be noted that the number of EM rashes documented for each patient showed no correlation with the iPCR value for B. burgdorferi antibody detection (data not shown).
DOC hybrid antigen iPCR demonstrates robust sensitivity and specificity for a blinded panel of human serum samples.
The initial success of DOC IgG iPCR with replicating 2-tier results for a panel of 32 human serum samples provided strong evidence for the application of our approach as a simplified Lyme disease diagnostic. We next sought to perform a larger-scale blinded validation analysis of our assay. The CDC research panel II, composed of 92 samples, including sera collected from patients with early Lyme disease and EM (stage 1), early Lyme disease with neurological or cardiac evidence of dissemination (stage 2), and patients with Lyme arthritis (stage 3), as well as look-alike diseases and healthy donors, was tested by iPCR for host-generated IgG antibodies to the DOC hybrid antigen, and the results were compared to those of the 2-tier test (Table 2). Using the background threshold Cq value for DOC/IgG established above, overall, iPCR provided levels of sensitivity and specificity comparable to those of 2-tier testing (Fig. 7). iPCR replicated all 2-tier-positive results. Moreover, iPCR provided detection of an additional three early Lyme disease samples deemed negative by 2-tier testing, leading to an overall sensitivity for iPCR of 69% (95% confidence interval [CI], 50% to 84%) compared to the sensitivity of 2-tier testing of 59% (95% CI, 41% to 76%). The difference in sensitivity was entirely for detecting stage 1 early Lyme disease samples, with sensitivity for iPCR of 55% (95% CI, 32% to 77%) and of 40% for 2-tier testing (95% CI, 19% to 64%) for this category of samples. iPCR and 2-tier testing showed equivalent sensitivity for stage 2/stage 3 Lyme disease samples of 92% (95% CI, 62% to 100%). iPCR detected only a single false positive for a sample from a healthy control from an area of endemicity (healthy endemic sample), resulting in a specificity of 98% (95% CI, 91% to 100%) compared to 2-tier testing, which detected two false positives for look-alike diseases, providing a specificity of 97% (95% CI, 88% to 100%). For comparison, the sensitivity and specificity for the ELISA first-tier portion of the 2-tier test were calculated to be 75% (95% CI, 57% to 89%) and 77% (95% CI, 64% to 87%), respectively.
TABLE 2.
iPCR using DOC/IgG demonstrates results equivalent to those of 2-tier testing for a panel of Lyme disease patient serum samples
| Sample group | Sample IDa | DOC IgGb | Interpretation forc: |
Tier 2 bands detected for: |
|||
|---|---|---|---|---|---|---|---|
| iPCR | 2-Tier | Tier 1 ELISA | IgM | IgG | |||
| Lyme disease stage 2 | |||||||
| Early Lyme-EMd | B1 | 2.24e | Pos | Pos | Pos | 41, 39, 23 | 58, 41, 39, 23, 18 |
| B2 | 2.20 | Pos | Pos | Pos | 23 | 66, 45, 41, 39, 23, 18 | |
| B3 | 2.07 | Pos | Pos | Pos | 41, 39, 23 | 41, 23 | |
| B4 | 2.05 | Pos | Pos | Pos | 41 | 58, 45, 41, 39, 23, 18 | |
| B5 | 1.59 | Pos | Pos | Pos | 41, 23 | 41, 23 | |
| B6 | 1.45 | Pos | Pos | Pos | 41, 39, 23 | 66, 45, 41, 39, 23, 18 | |
| B7 | 1.08 | Pos | Pos | Pos | 41, 39, 23 | 41, 23 | |
| B8 | 0.80 | Pos | Pos | Pos | 41, 23 | 41 | |
| B9 | 0.52 | Pos | Neg | Pos | 23 | 66, 41, 23 | |
| B10 | 0.08 | Pos | Neg | Equ | |||
| B11 | (0.08) | Neg | Neg | Pos | 23 | 66, 41, 23 | |
| B12 | (0.27) | Neg | Neg | Neg | 66 | ||
| B13 | (0.58) | Neg | Neg | Pos | 23 | ||
| B14 | (0.91) | Neg | Neg | Pos | 23 | 41, 23 | |
| B15 | (1.00) | Neg | Neg | Neg | 67 | ||
| B16 | (1.01) | Neg | Neg | Neg | 39, 23 | 23 | |
| B17 | (1.22) | Neg | Neg | Neg | 23 | ||
| B18 | (1.48) | Neg | Neg | Equ | 23 | 41 | |
| B19 | (1.50) | Neg | Neg | Neg | 23 | ||
| B20 | 1.14 | Pos | Neg | Pos | 41 | 41, 23, 18 | |
| Lyme disease stage 2 | |||||||
| Neuroborreliosis | B21 | 2.64 | Pos | Pos | Pos | 41, 23 | 45, 41, 23 |
| B22 | 2.01 | Pos | Pos | Pos | 41, 39, 23 | 41, 39, 23 | |
| B23 | 0.00 | Pos | Pos | Pos | 41, 39, 23 | 41, 23 | |
| B24 | (0.26) | Neg | Neg | Neg | 41, 23 | 41, 23 | |
| Lyme carditis | B25 | 2.83 | Pos | Pos | Pos | 41, 39, 23 | 66, 45, 41, 23, 18 |
| B26 | 1.37 | Pos | Pos | Pos | 41, 39, 23 | 66, 45, 41, 23, 18 | |
| Lyme disease stage 3 | |||||||
| Lyme arthritis | B27 | 3.44 | Pos | Pos | Pos | 23 | 93, 66, 58, 45, 41, 39, 30, 28, 23, 18 |
| B28 | 2.96 | Pos | Pos | Pos | 41 | 93, 66, 58, 41, 39, 30, 28, 23, 18 | |
| B29 | 2.67 | Pos | Pos | Pos | 41, 23 | 93, 66, 58, 45, 41, 39, 30, 28, 23, 18 | |
| B30 | 2.62 | Pos | Pos | Pos | 66, 58, 45, 41, 39, 28, 23, 18 | ||
| B31 | 2.09 | Pos | Pos | Pos | 23 | 58, 41, 39, 23, 18 | |
| B32 | 1.84 | Pos | Pos | Pos | 93, 66, 58, 41, 39, 30, 23, 18 | ||
| Non-Lyme | |||||||
| Fibromyalgia | B33 | (0.28) | Neg | Neg | Neg | 23 | |
| B34 | (0.81) | Neg | Neg | Neg | 39 | 58, 41 | |
| B35 | (1.70) | Neg | Neg | Neg | 41 | ||
| B36 | (1.89) | Neg | Neg | Neg | 41 | ||
| B37 | (1.93) | Neg | Neg | Neg | |||
| B38 | (2.30) | Neg | Neg | Neg | |||
| Rheumatoid arthritis | B39 | (0.90) | Neg | Neg | Pos | 41 | |
| B40 | (1.17) | Neg | Neg | Neg | 41 | ||
| B41 | (1.56) | Neg | Neg | Neg | |||
| B42 | (1.73) | Neg | Pos | Pos | 41, 23 | ||
| B43 | (1.77) | Neg | Neg | Neg | |||
| B44 | (2.05) | Neg | Neg | Neg | |||
| Multiple sclerosis | B45 | (0.55) | Neg | Neg | Neg | 39, 23 | 41 |
| B46 | (0.78) | Neg | Neg | Pos | 41, 23 | ||
| B47 | (1.09) | Neg | Neg | Neg | |||
| B48 | (1.11) | Neg | Neg | Neg | 39 | ||
| B49 | (1.75) | Neg | Neg | Neg | |||
| B50 | (2.05) | Neg | Neg | Neg | 66 | ||
| Mononucleosis | B51 | (0.09) | Neg | Neg | Neg | 39 | |
| B52 | (0.28) | Neg | Neg | Pos | 41, 39 | ||
| B53 | (0.58) | Neg | Neg | Pos | |||
| B54 | (0.77) | Neg | Neg | Equ | 41 | ||
| B55 | (0.78) | Neg | Neg | Neg | |||
| B56 | (1.25) | Neg | Neg | Neg | 41, 23 | 66, 58, 41 | |
| Syphilis | B57 | (0.56) | Neg | Neg | Pos | ||
| B58 | (0.75) | Neg | Neg | Pos | 41 | ||
| B59 | (0.96) | Neg | Neg | Pos | 41 | ||
| B60 | (1.01) | Neg | Pos | Pos | 39, 23 | ||
| B61 | (1.38) | Neg | Neg | Pos | 41 | ||
| B62 | (1.47) | Neg | Neg | Neg | |||
| Severe periodontitis | B63 | (0.22) | Neg | Neg | Neg | ||
| B64 | (0.29) | Neg | Neg | Neg | |||
| B65 | (0.56) | Neg | Neg | Neg | |||
| B66 | (0.90) | Neg | Neg | Neg | 45, 41 | ||
| B67 | (1.03) | Neg | Neg | Neg | 66 | ||
| B68 | (3.04) | Neg | Neg | Neg | |||
| Healthy controls | |||||||
| From areas of endemicity | B69 | 0.23 | Pos | Neg | Neg | 23 | |
| B70 | (0.04) | Neg | Neg | Pos | 41 | 66 | |
| B71 | (0.53) | Neg | Neg | Pos | 41, 23 | ||
| B72 | (0.87) | Neg | Neg | Neg | 23 | 41 | |
| B73 | (0.87) | Neg | Neg | Equ | 23 | ||
| B74 | (1.11) | Neg | Neg | Neg | 45, 41 | ||
| B75 | (1.16) | Neg | Neg | Neg | |||
| B76 | (1.37) | Neg | Neg | Neg | |||
| B77 | (1.42) | Neg | Neg | Neg | |||
| B78 | (1.49) | Neg | Neg | Neg | 66, 41 | ||
| B79 | (1.95) | Neg | Neg | Neg | 23 | ||
| B80 | (2.47) | Neg | Neg | Pos | 23 | 58, 41, 39, 18 | |
| From areas of nonendemicity | B81 | (0.53) | Neg | Neg | Neg | 41 | |
| B82 | (0.60) | Neg | Neg | Neg | 41, 23 | 41 | |
| B83 | (0.78) | Neg | Neg | Equ | |||
| B84 | (0.80) | Neg | Neg | Pos | |||
| B85 | (0.86) | Neg | Neg | Neg | |||
| B86 | (0.90) | Neg | Neg | Neg | 58, 45 | ||
| B87 | (1.09) | Neg | Neg | Neg | 66, 58, 45, 41 | ||
| B88 | (1.15) | Neg | Neg | Neg | 41 | ||
| B89 | (1.17) | Neg | Neg | Neg | 41 | ||
| B90 | (1.77) | Neg | Neg | Neg | 23 | ||
| B91 | (2.06) | Neg | Neg | Neg | 23 | ||
| B92 | (2.09) | Neg | Neg | Neg | |||
ID, identification.
Values shown represent a ΔCq in reference to the antigen/isotype background threshold Cq value determined using an antigen-specific multiplier of the standard deviation (SD) above the mean value for a set of healthy individuals for each antigen/isotype combination, as described in Materials and Methods. The values in parentheses represent negative iPCR ΔCq values.
Two-tier results were established by standard ELISA and IgG/IgM immunoblot (IB) protocols. Pos, positive; Neg, negative; Equ, equivocal.
EM, erythema migrans.
Bold type indicates positive assay results/interpretations.
FIG 7.
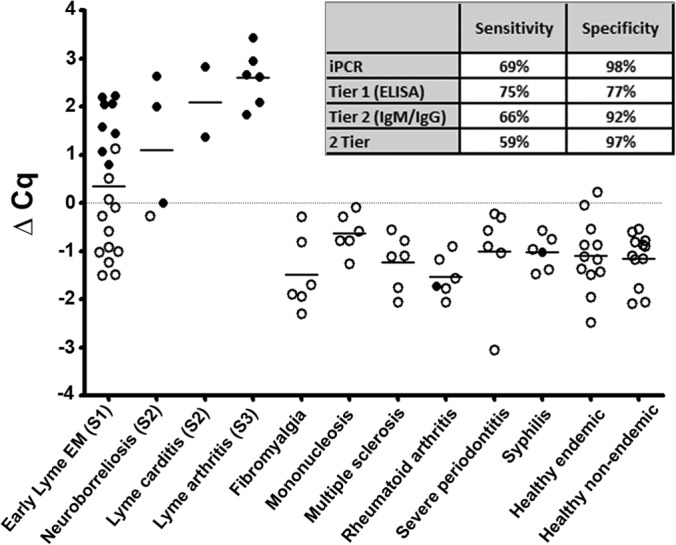
DOC hybrid antigen IgG iPCR demonstrated sensitive and specific detection of Lyme disease for a blinded serum panel. CDC research panel II was tested in a blinded fashion using DOC iPCR for IgG reactivity. Each dot represents a single individual replicate, and the black horizontal lines represent the mean Cq values for all individuals within each category. The filled circles represent samples that were positive with 2-tier testing, and the open circles signify a 2-tier-negative status. A positive threshold value was established using a multiplier of the standard deviation (SD) above the mean value, with the ΔCq threshold (gray horizontal line) representing a value of zero. The sensitivity and specificity values for iPCR, each tier, and combined 2-tier testing are listed.
DISCUSSION
There is an urgent need to develop new tools for improved diagnosis of Lyme disease. This study describes an objective Lyme disease diagnostic method using iPCR detection of host IgG antibody binding to a single recombinant hybrid antigen.
Repeatability is a key parameter of any newly developed diagnostic test that provides confidence the test will identify individuals as disease positive or negative in a reproducible manner across the inherent variability of a human population. iPCR has been shown to be a reproducible approach for detecting other targets (28, 29), although this method generates a background signal in the absence of the analyte being detected (30). The background signal has been attributed to nonspecific binding of the oligonucleotide-labeled secondary antibody, similar to the results observed for other immunodiagnostics (31). Although a number of approaches have been proposed to minimize the level of background amplification (32–34), no approach to date has proven successful at completely eliminating the background signal. For diagnosing Lyme disease, we propose that the iPCR background signal provides an intrinsic advantage due to the fact that a positive result is a relative measure above the established background threshold, thereby limiting the potential contribution of contamination, whereas a positive result for standard PCR is an absolute measure that can be highly sensitive to low-level laboratory contamination (7). The baseline level of amplification using iPCR for a negative sample far surpasses any low-level laboratory contamination that commonly results in false-positive detection for PCR-based clinical diagnostic tests. As a result, the level of PCR contamination required to produce a false positive above background for iPCR is orders of magnitude above that for standard PCR. In addition, critical to the success of this approach is a constant background that remains consistent between sample replicates and is standardized across a healthy human population.
In an effort to determine the consistency of the background amplification for the technique, we tested the serum from a single healthy individual over 18 replicates using iPCR and found the standard deviations of the mean Cq values to be 0.39 and 0.73 for IgM and IgG, respectively, with corresponding coefficients of variation of 1.34% and 2.30%, respectively. The accepted value for PCR sampling error is ∼1 Cq (35), and the coefficient of variation for an ELISA-based test is considered good at <15% (36). These data indicate that our iPCR protocol can provide highly consistent and repeatable results across multiple replicates of a single sample. We proceeded to test serum samples collected from 36 healthy individuals in duplicate for IgM and IgG reactivity using the same antigen to determine the variability of the background across a healthy population. As expected, compared to the within-sample repeatability analysis, we observed a slightly higher standard deviation of the mean Cq values of 0.79 and 0.84 for IgM and IgG, respectively, as well as slightly increased corresponding coefficients of variation of 2.66% and 2.63%, respectively. These data demonstrate that the assay maintains strong repeatability even when compounded with normal human population serum variability. Taken together, these results indicate that the background variability for iPCR detection of host-generated antibodies within and across a healthy human population is well within acceptable levels for the technique.
Previous studies using recombinant antigens have indicated that no single antigen tested to date has the capability to diagnose Lyme disease across its multiple stages and disease manifestations (7). A panel of eight antigens was generated for use in the iPCR assay. These proteins were selected based on previous studies that identified B. burgdorferi immunoreactive antigens (13–27). We first examined the level of variability of the background amplification of each antigen across serum samples collected from healthy individuals for both the IgM and IgG isotypes. Each antigen resulted in a unique background amplification mean and standard deviation value for each antigen-isotype combination. This indicated that each antigen-isotype combination performed uniquely using the current iPCR protocol. These data provided the necessary parameters, including the mean background Cq value and the standard deviation of that mean for determining an individual call threshold for each antigen-isotype combination. The call thresholds were established as the mean background Cq value minus a multiple of the standard deviation. The multiplier of standard deviation was unique for each antigen-isotype combination and established based on the minimum multiplier that resulted in no false-positive calls for the CDC research panel I, which served as the training set for optimizing our assay. The ΔCq was calculated as the established threshold call Cq minus the Cq value of the sample. A sample with a ΔCq value of ≥0 was deemed positive by iPCR. Using the panel of eight antigens, this approach duplicated 2-tier testing results with a single early Lyme patient sample (culture positive) testing positive by iPCR that was negative by 2-tier testing. Samples from individuals in the later stages of disease (neurologic and arthritis) tended to test positive for multiple antigens.
In addition to detecting the presence of host antibodies as laboratory support of an exposure to B. burgdorferi, it would be desirable to link an antibody profile with the clinical stage (i.e., early localized, early disseminated with neurological or cardiac involvement, or Lyme arthritis) of illness to better understand disease progression. The results from the human serum panel iPCR testing classified both late Lyme arthritis samples as strongly positive for IgG using the RevA and Crasp2 proteins, with all other categories of samples testing negative for the same two proteins. These results suggest that these two proteins may specifically illicit an immune response in a Lyme arthritis patient as opposed to those in other stages of Lyme disease. However, analysis of a greater number of clinically defined samples is required to further support these observations.
Limited studies have shown promising results using antigens composed of multiple antigenic portions of various seroreactive proteins to detect B. burgdorferi antibodies in human patient sera (24, 37, 38). The demonstration of iPCR equivalency to 2-tier testing using a panel of antigens led us to surmise that a more simplified version of the protocol using a single hybrid antigen was likely to be successful. Three antigens known to be seroreactive at different stages of the disease (DbpA, OspC, and VlsE) were synthetically joined by combining the seroreactive peptide portions of OspC (39) and VlsE (40) with the full-length DbpA protein into a single recombinant hybrid antigen we termed DOC. The mean background was established for 16 healthy individuals using DOC and showed little variation (standard deviation, 0.57 and 0.51 for anti-B. burgdorferi IgM and IgG antibodies, respectively), similar to the results for the full-length antigens tested. The DOC antigen was then used to test CDC research panel I for anti-B. burgdorferi IgM and IgG antibodies to establish a positive call threshold. Using the positive call threshold, the DOC iPCR IgG assay demonstrated results equivalent to those for 2-tier testing, with all 2-tier positives identified as positive by iPCR. The quantification of the ΔCq for Lyme disease patients showed a trend of increasing average values from early Lyme acute (−1.61) to convalescent early Lyme (0.67) to late-stage Lyme (2.39), suggesting a correlation in the amount of detectable B. burgdorferi antibody with disease stage. Interestingly, DOC iPCR IgM was negative for all samples tested, including Lyme disease patient samples. The full-length DbpA antigen alone resulted in a low-positive IgM iPCR value (0.69) for only a single Lyme disease patient sample. iPCR testing using the full-length OspC antigen resulted in a number of IgM iPCR-positive samples, suggesting that the antibodies detected in these samples may have resulted from OspC epitopes other than the PEPC10 sequence. It is also possible that in the context of the DOC hybrid antigen, the PEPC10 sequence lacks the conformational epitope(s) required for IgM recognition. It is well documented that the VlsE antigen primarily generates IgG rather than IgM antibodies early in infection (41). Therefore, it may not be surprising that the DOC antigen detects IgG antibodies only. These results indicate that testing only the IgG fraction using the DOC hybrid antigen was necessary to achieve a level of sensitivity equivalent to that of 2-tier testing, which required IgM for positive detection in some samples. Given the small sample size, these findings do not rule out the possibility that IgM antibodies might be detected with the DOC iPCR assay in some Lyme disease patient serum samples. Moreover, the additional optimization of the hybrid antigen to include the specific detection of IgM antibodies may contribute to further improved sensitivity for detecting disease in patients with early Lyme disease. Nonetheless, IgM detection has been problematic and controversial due to its contribution to false-positive results and the requirement that IgM testing be used only within the first 4 weeks of infection (7), suggesting that an assay that does not use IgM may represent an improvement over the current methods of testing for Lyme disease. In addition, our data suggest that there exists the potential to determine the stage of disease based on the ΔCq value of the DOC iPCR assay, which represents another possible improvement over current Lyme disease diagnostics.
iPCR testing of the anti-B. burgdorferi IgG antibody fraction using the DOC hybrid antigen was successful at duplicating the 2-tier testing results for a small panel of samples. We then proceeded to test a larger blinded panel of 92 samples composed of serum samples from Lyme patients (early, early disseminated with cardiac or neurological involvement, and Lyme arthritis), those with look-alike diseases (fibromyalgia, mononucleosis, multiple sclerosis, rheumatoid arthritis, severe periodontitis, and syphilis), and healthy (from areas of endemicity and nonendemicity) individuals (CDC research panel II). iPCR demonstrated 69% sensitivity and 98% specificity compared to 59% and 97%, respectively, for 2-tier testing. A single neurologic Lyme patient tested negative by both iPCR and 2-tier testing. This result is most likely due to the fact that the serum sample was taken 7 days post-erythema migrans (EM), which was likely too early in the infection process to produce an adequate immune response.
Currently, the DOC hybrid antigen is composed of B. burgdorferi B31 sequences. Amino acid sequences can vary between strains and species of Lyme disease borreliae by as much as 24% for VlsE C6 (11), 10% for OspC PEPC10C (12), and 44% for DbpA (42). This may be limiting if an individual is infected with other strains or species. It is likely that the incorporation of additional protein/peptide sequences from other species, such as Borrelia afzelii or Borrelia garinii, or other strains might further increase the sensitivity of the assay, especially when samples from patients with Lyme disease from Europe and other diverse locations are analyzed.
The recommended protocol for Lyme disease diagnosis requires a first-tier ELISA, followed by a second-tier IgM and/or IgG immunoblot (7). Here, we demonstrated that the simplified single-tier DOC iPCR assay was sufficient to objectively identify all 2-tier-positive samples across two panels of well-characterized samples from Lyme disease patients. The objective positive/negative call threshold of this sensitive and specific method represents an important improvement over the currently accepted method. Moreover, it is likely that future automation of this protocol will provide additional advantages to the iPCR method. Emerging immunoassay technologies, such as the Erenna system from Singulex and the single-molecule array by Quanterix, provide intriguing options for higher sensitivity and precision. Currently, these systems are considered research and development instruments for biomarker discovery and validation. Although these platforms present new possibilities for assay development and have the potential to provide increased sensitivity, they have yet to be accepted for routine clinical diagnostics. The current iPCR protocol uses real-time quantitative PCR (qPCR) as its method of signal amplification and detection. Real-time qPCR has garnered acceptance for routine use in clinical laboratories as a detection method for a number of assays. Therefore, the use of a qPCR detection system, which is a standard piece of equipment in many clinical laboratories, provides a more direct route for clinical acceptance of an iPCR-based Lyme disease diagnostic assay.
In summary, DOC iPCR shows potential as a novel diagnostic tool for identifying host-generated antibodies against B. burgdorferi. It will be of interest to determine whether this test is useful for monitoring antibody titer changes over time in samples from patients after antibiotic therapy for Lyme disease and for exploring specialty testing using this approach to determine the stage and the type of disease manifestations.
Supplementary Material
ACKNOWLEDGMENTS
We thank Travis Jewett for phlebotomy expertise, helpful discussions, and manuscript review. We also thank Tisha Choudhury Ellis for providing BBK50 purified protein and the students in BSC 6407c for technical support. We thank Dorilyn Hitchcock and the healthy blood donors and the UCF NAF animal care staff.
The research reported in this publication was supported, in part, by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award R01AI099094 (to M.W.J.) and a 2012-2013 UCF College of Medicine competitive research grant (to M.W.J.).
Footnotes
Published ahead of print 4 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00245-14.
REFERENCES
- 1.Bacon RM, Kugeler KJ, Mead PS, Centers for Disease Control and Prevention (CDC) 2008. Surveillance for Lyme disease–United States, 1992–2006. MMWR Surveill. Summ. 57:1–10 [PubMed] [Google Scholar]
- 2.Kuehn BM. 2013. CDC estimates 300,000 US cases of Lyme disease annually. JAMA 310:1110. 10.1001/jama.2013.278331 [DOI] [PubMed] [Google Scholar]
- 3.Campbell GL, Fritz CL, Fish D, Nowakowski J, Nadelman RB, Wormser GP. 1998. Estimation of the incidence of Lyme disease. Am. J. Epidemiol. 148:1018–1026. 10.1093/oxfordjournals.aje.a009568 [DOI] [PubMed] [Google Scholar]
- 4.Young JD. 1998. Underreporting of Lyme disease. N. Engl. J. Med. 338:1629. 10.1056/NEJM199805283382216 [DOI] [PubMed] [Google Scholar]
- 5.Aucott J, Morrison C, Munoz B, Rowe PC, Schwarzwalder A, West SK. 2009. Diagnostic challenges of early Lyme disease: lessons from a community case series. BMC Infect. Dis. 9:79. 10.1186/1471-2334-9-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention (CDC). 1995. Recommendations for test performance and interpretation from the Second National Conference on Serologic Diagnosis of Lyme Disease. MMWR Morb. Mortal. Wkly. Rep. 44:590–591 [PubMed] [Google Scholar]
- 7.Aguero-Rosenfeld ME, Wang G, Schwartz I, Wormser GP. 2005. Diagnosis of lyme borreliosis. Clin. Microbiol. Rev. 18:484–509. 10.1128/CMR.18.3.484-509.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wormser GP, Liveris D, Hanincová K, Brisson D, Ludin S, Stracuzzi VJ, Embers ME, Philipp MT, Levin A, Aguero-Rosenfeld M, Schwartz I. 2008. Effect of Borrelia burgdorferi genotype on the sensitivity of C6 and 2-tier testing in North American patients with culture-confirmed Lyme disease. Clin. Infect. Dis. 47:910–914. 10.1086/591529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binnicker MJ, Jespersen DJ, Harring JA, Rollins LO, Bryant SC, Beito EM. 2008. Evaluation of two commercial systems for automated processing, reading, and interpretation of Lyme borreliosis Western blots. J. Clin. Microbiol. 46:2216–2221. 10.1128/JCM.00200-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halpern MD, Jain S, Jewett MW. 2013. Enhanced detection of host response antibodies to Borrelia burgdorferi using immuno-PCR. Clin. Vaccine Immunol. 20:350–357. 10.1128/CVI.00630-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sillanpää H, Lahdenne P, Sarvas H, Arnez M, Steere A, Peltomaa M, Seppälä I. 2007. Immune responses to borrelial VlsE IR6 peptide variants. Int. J. Med. Microbiol. 297:45–52. 10.1016/j.ijmm.2006.09.001 [DOI] [PubMed] [Google Scholar]
- 12.Arnaboldi PM, Seedarnee R, Sambir M, Callister SM, Imparato JA, Dattwyler RJ. 2013. Outer surface protein C peptide derived from Borrelia burgdorferi sensu stricto as a target for serodiagnosis of early Lyme disease. Clin. Vaccine Immunol. 20:474–481. 10.1128/CVI.00608-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coburn J, Cugini C. 2003. Targeted mutation of the outer membrane protein P66 disrupts attachment of the Lyme disease agent, Borrelia burgdorferi, to integrin alphavbeta3. Proc. Natl. Acad. Sci. U. S. A. 100:7301–7306. 10.1073/pnas.1131117100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Höök M, Skare JT. 2006. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Mol. Microbiol. 59:1591–1601. 10.1111/j.1365-2958.2005.05042.x [DOI] [PubMed] [Google Scholar]
- 15.Brissette CA, Bykowski T, Cooley AE, Bowman A, Stevenson B. 2009. Borrelia burgdorferi RevA antigen binds host fibronectin. Infect. Immun. 77:2802–2812. 10.1128/IAI.00227-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo BP, Norris SJ, Rosenberg LC, Höök M. 1995. Adherence of Borrelia burgdorferi to the proteoglycan decorin. Infect. Immun. 63:3467–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verma A, Brissette CA, Bowman A, Stevenson B. 2009. Borrelia burgdorferi BmpA is a laminin-binding protein. Infect. Immun. 77:4940–4946. 10.1128/IAI.01420-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraiczy P, Stevenson B. 2013. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: structure, function and regulation of gene expression. Ticks Tick Borne Dis. 4:26–34. 10.1016/j.ttbdis.2012.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Padula SJ, Dias F, Sampieri A, Craven RB, Ryan RW. 1994. Use of recombinant OspC from Borrelia burgdorferi for serodiagnosis of early Lyme disease. J. Clin. Microbiol. 32:1733–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simpson WJ, Schrumpf ME, Schwan TG. 1990. Reactivity of human Lyme borreliosis sera with a 39-kilodalton antigen specific to Borrelia burgdorferi. J. Clin. Microbiol. 28:1329–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbour AG, Jasinskas A, Kayala MA, Davies DH, Steere AC, Baldi P, Felgner PL. 2008. A genome-wide proteome array reveals a limited set of immunogens in natural infections of humans and white-footed mice with Borrelia burgdorferi. Infect. Immun. 76:3374–3389. 10.1128/IAI.00048-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panelius J, Lahdenne P, Saxén H, Carlsson SA, Heikkilä T, Peltomaa M, Lauhio A, Seppälä I. 2003. Diagnosis of Lyme neuroborreliosis with antibodies to recombinant proteins DbpA, BBK32, and OspC, and VlsE IR6 peptide. J. Neurol. 250:1318–1327. 10.1007/s00415-003-0205-2 [DOI] [PubMed] [Google Scholar]
- 23.Krause A, Burmester GR, Rensing A, Schoerner C, Schaible UE, Simon MM, Herzer P, Kramer MD, Wallich R. 1992. Cellular immune reactivity to recombinant OspA and flagellin from Borrelia burgdorferi in patients with Lyme borreliosis. Complexity of humoral and cellular immune responses. J. Clin. Invest. 90:1077–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burbelo PD, Issa AT, Ching KH, Cohen JI, Iadarola MJ, Marques A. 2010. Rapid, simple, quantitative, and highly sensitive antibody detection for Lyme disease. Clin. Vaccine Immunol. 17:904–909. 10.1128/CVI.00476-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fikrig E, Feng W, Barthold SW, Telford SR, III, Flavell RA. 2000. Arthropod- and host-specific Borrelia burgdorferi bbk32 expression and the inhibition of spirochete transmission. J. Immunol. 164:5344–5351. 10.4049/jimmunol.164.10.5344 [DOI] [PubMed] [Google Scholar]
- 26.Brissette CA, Rossmann E, Bowman A, Cooley AE, Riley SP, Hunfeld KP, Bechtel M, Kraiczy P, Stevenson B. 2010. The borrelial fibronectin-binding protein RevA is an early antigen of human Lyme disease. Clin. Vaccine Immunol. 17:274–280. 10.1128/CVI.00437-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kraiczy P, Skerka C, Brade V, Zipfel PF. 2001. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect. Immun. 69:7800–7809. 10.1128/IAI.69.12.7800-7809.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer A, von Eiff C, Kuczius T, Omoe K, Peters G, Becker K. 2007. A quantitative real-time immuno-PCR approach for detection of staphylococcal enterotoxins. J. Mol. Med. (Berl.) 85:461–469. 10.1007/s00109-006-0142-5 [DOI] [PubMed] [Google Scholar]
- 29.Shan J, Toye P. 2009. A novel immuno-polymerase chain reaction protocol incorporating a highly purified streptavidin-DNA conjugate. J. Immunoassay Immunochem. 30:322–337. 10.1080/15321810903084764 [DOI] [PubMed] [Google Scholar]
- 30.Banin S, Wilson SM, Stanley CJ. 2004. Demonstration of an alternative approach to immuno-PCR. Clin. Chem. 50:1932–1934. 10.1373/clinchem.2004.037143 [DOI] [PubMed] [Google Scholar]
- 31.McKie A, Samuel D, Cohen B, Saunders NA. 2002. Development of a quantitative immuno-PCR assay and its use to detect mumps-specific IgG in serum. J. Immunol. Methods 261:167–175. 10.1016/S0022-1759(02)00003-0 [DOI] [PubMed] [Google Scholar]
- 32.Zhou H, Fisher RJ, Papas TS. 1993. Universal immuno-PCR for ultra-sensitive target protein detection. Nucleic Acids Res. 21:6038–6039. 10.1093/nar/21.25.6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barletta J, Bartolome A, Constantine NT. 2009. Immunomagnetic quantitative immuno-PCR for detection of less than one HIV-1 virion. J. Virol. Methods 157:122–132. 10.1016/j.jviromet.2008.12.013 [DOI] [PubMed] [Google Scholar]
- 34.Niemeyer CM, Adler M, Wacker R. 2007. Detecting antigens by quantitative immuno-PCR. Nat. Protoc. 2:1918–1930. 10.1038/nprot.2007.267 [DOI] [PubMed] [Google Scholar]
- 35.Karlen Y, McNair A, Perseguers S, Mazza C, Mermod N. 2007. Statistical significance of quantitative PCR. BMC Bioinformatics 8:131. 10.1186/1471-2105-8-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.FDA. 2001. Guidance for industry: bioanalytical method validation. U.S. Food and Drug Administration, Rockville, MD: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf [Google Scholar]
- 37.Gomes-Solecki MJ, Wormser GP, Persing DH, Berger BW, Glass JD, Yang X, Dattwyler RJ. 2001. A first-tier rapid assay for the serodiagnosis of Borrelia burgdorferi infection. Arch. Intern. Med. 161:2015–2020. 10.1001/archinte.161.16.2015 [DOI] [PubMed] [Google Scholar]
- 38.Gomes-Solecki MJ, Dunn JJ, Luft BJ, Castillo J, Dykhuizen DE, Yang X, Glass JD, Dattwyler RJ. 2000. Recombinant chimeric Borrelia proteins for diagnosis of Lyme disease. J. Clin. Microbiol. 38:2530–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathiesen MJ, Christiansen M, Hansen K, Holm A, Asbrink E, Theisen M. 1998. Peptide-based OspC enzyme-linked immunosorbent assay for serodiagnosis of Lyme borreliosis. J. Clin. Microbiol. 36:3474–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang FT, Alvarez AL, Gu Y, Nowling JM, Ramamoorthy R, Philipp MT. 1999. An immunodominant conserved region within the variable domain of VlsE, the variable surface antigen of Borrelia burgdorferi. J. Immunol. 163:5566–5573 [PubMed] [Google Scholar]
- 41.Glatz M, Fingerle V, Wilske B, Ambros-Rudolph C, Kerl H, Müllegger RR. 2008. Immunoblot analysis of the seroreactivity to recombinant Borrelia burgdorferi sensu lato antigens, including VlsE, in the long-term course of treated patients with erythema migrans. Dermatology 216:93–103. 10.1159/000111505 [DOI] [PubMed] [Google Scholar]
- 42.Schulte-Spechtel U, Fingerle V, Goettner G, Rogge S, Wilske B. 2006. Molecular analysis of decorin-binding protein A (DbpA) reveals five major groups among European Borrelia burgdorferi sensu lato strains with impact for the development of serological assays and indicates lateral gene transfer of the dbpA gene. Int. J. Med. Microbiol. 296(Suppl 40):S250–S266. 10.1016/j.ijmm.2006.01.006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.