

ABSTRACT
Apoptosis is a tightly regulated process that plays a crucial role in the removal of virus-infected cells, a process controlled by both pro- and antiapoptotic members of the Bcl-2 family. The proapoptotic proteins Bak and Bax are regulated by antiapoptotic Bcl-2 proteins and are also activated by a subset of proteins known as BH3-only proteins that perform dual functions by directly activating Bak and Bax or by sequestering and neutralizing antiapoptotic family members. Numerous viruses express proteins that prevent premature host cell apoptosis. Vaccinia virus encodes F1L, an antiapoptotic protein essential for survival of infected cells that bears no discernible sequence homology to mammalian cell death inhibitors. Despite the limited sequence similarities, F1L has been shown to adopt a novel dimeric Bcl-2-like fold that enables hetero-oligomeric binding to both Bak and the proapoptotic BH3-only protein Bim that ultimately prevents Bak and Bax homo-oligomerization. However, no structural data on the mode of engagement of F1L and its Bcl-2 counterparts are available. Here we solved the crystal structures of F1L in complex with two ligands, Bim and Bak. Our structures indicate that F1L can engage two BH3 ligands simultaneously via the canonical Bcl-2 ligand binding grooves. Furthermore, by structure-guided mutagenesis, we generated point mutations within the binding pocket of F1L in order to elucidate the residues responsible for both Bim and Bak binding and prevention of apoptosis. We propose that the sequestration of Bim by F1L is primarily responsible for preventing apoptosis during vaccinia virus infection.
IMPORTANCE Numerous viruses have adapted strategies to counteract apoptosis by encoding proteins responsible for sequestering proapoptotic components. Vaccinia virus, the prototypical member of the family Orthopoxviridae, encodes a protein known as F1L that functions to prevent apoptosis by interacting with Bak and the BH3-only protein Bim. Despite recent structural advances, little is known regarding the mechanics of binding between F1L and the proapoptotic Bcl-2 family members. Utilizing three-dimensional structures of F1L bound to host proapoptotic proteins, we generated variants of F1L that neutralize Bim and/or Bak. We demonstrate that during vaccinia virus infection, engagement of Bim and Bak by F1L is crucial for subversion of host cell apoptosis.
INTRODUCTION
Apoptosis, also known as programmed cell death, is a key process in the removal of virus-infected cells in higher order organisms (1–3). The Bcl-2 family plays a central role in maintaining cell survival and promoting cell death, thereby removing infected, damaged, or unwanted cells (4, 5). Members of the Bcl-2 family share one or more Bcl-2 homology (BH) domains and include both prosurvival and prodeath proteins. Prosurvival members of the family include Bcl-2, Bcl-xL, Bcl-w, Mcl-1, A1, and Bcl-b. The proapoptotic proteins Bak and Bax are essential for eliciting cell death and facilitating the release of cytochrome c from the outer mitochondrial membrane (OMM) by forming higher-order homo-oligomers (6–8). Bak exists constitutively at the OMM through its C-terminal transmembrane anchor, whereas Bax exists in the cytosol. Upon the presence of an apoptotic stimulus, Bax undergoes a conformational rearrangement that facilitates its localization to the OMM. This process is tightly governed by the presence of the BH3-only proteins (9). The BH3-only proteins include Puma, Noxa, Bid, Bmf, Bik, Bad, Hrk, and Bim; they function by directly activating Bak and Bax or by sequestering and neutralizing the antiapoptotic family members (10). In contrast to the prosurvival Bcl-2 proteins, which contain multiple BH domains, BH3-only proteins harbor the α-helical BH3 domain, which engages a conserved ligand-binding groove on the prosurvival proteins (11). The BH3-only proteins are upregulated in response to cellular damage signals such as growth factor deprivation or exposure to cytotoxic drugs, thus activating cell death mechanisms (12). The BH3-only protein Bim, in which three primary isoforms are responsible for eliciting cell death, is capable of both directly and indirectly activating Bak and Bax through direct interactions, as well as binding and sequestering the antiapoptotic Bcl-2 family members (13).
Many viruses have evolved strategies to counteract cell death (3). For example, adenoviruses and Epstein-Barr virus (EBV) encode viral Bcl-2-like proteins (11, 14) that are required for successful viral propagation and/or persistence (15). However, other viruses express antiapoptotic proteins that are unrelated by sequence. Included among these viruses are members of the Poxviridae family, including vaccinia virus (VACV) F1L, N1L, and E3L; myxoma virus M11L (16–19); and the more recently identified fowlpox FPV039, orf virus ORF125, deerpox virus DPV022, and sheeppox virus SPPV14 (20–23).
VACV-encoded F1L, which is found exclusively in the Orthopoxviridae family, was originally identified as a potent inhibitor of the mitochondrial apoptotic pathway that localizes to the mitochondria via its C-terminal membrane anchor (19). Our data indicate that F1L interacts with Bak and prevents Bak activation (24). Despite no observable interaction with Bax, F1L was found to be fully capable of preventing Bax activation through an upstream interaction with the BH3-only protein Bim (25, 26). The importance of F1L is highlighted by the F1L-deficient virus VACVΔF1L, which potently causes Bak and Bax activation, and subsequently cell death, in the presence of virus infection alone (26). Recently, we identified divergent BH domains that are responsible for the ability of F1L to interact with Bak and prevent apoptosis (24). Furthermore, biochemical studies revealed interactions with the BH3 domains of Bim, Bak, and Bax (17, 27). The structure of VACV F1L was solved, and despite the lack of sequence similarity to mammalian Bcl-2 family members, F1L adopts a Bcl-2 fold that displays a novel, domain-swapped dimer configuration (27). The structural basis for F1L engagement of prodeath Bcl-2 proteins and the molecular mechanisms underlying F1L-mediated inhibition of apoptosis remain unclear.
Here we report the molecular basis for F1L-mediated inhibition of apoptosis. We determined the crystal structures of modified VACV Ankara strain (MVA)-encoded F1L in complex with the BH3 peptides of Bim and Bak. Using structure-guided mutagenesis we generated mutations within the binding pocket of F1L that, in many instances, abolished the ability to prevent cytochrome c release, as well as an inability to prevent downstream PARP (poly-ADP ribose polymerase) cleavage. Furthermore, immunoprecipitation data reveal several hydrophobic residues within the binding cleft that play a substantial role in facilitating proper binding to the proapoptotic proteins Bak and Bim. The data support the idea that F1L ultimately prevents cell death primarily by sequestering Bim.
MATERIALS AND METHODS
Cells and viruses.
HeLa, HEK 293T, and baby green monkey kidney (BGMK) cells were obtained from the American Type Culture Collection and maintained as previously described (26). VACV strain Copenhagen expressing the enhanced green fluorescence protein (EGFP) and VACV65 (28) expressing β-galactosidase in the thymidine kinase locus were obtained from G. McFadden (University of Florida, Gainesville). cDNAs encoding F1L point mutations were subcloned into pSC66 under the control of a poxviral promoter. Recombinant VACVΔF1L-FLAG-F1L was generated by homologous recombination of pSC66-FLAG-F1L into the thymidine kinase locus of VACVΔF1L as described previously (29). Briefly, 1 × 106 BGMK cells were transfected with 5 μg of pSC66-Flag-F1L and infected with VACVΔF1L at a multiplicity of infection (MOI) of 10. Recombinant viruses were selected by growth on HuTK-143B cells in the presence of 5-bromodeoxyuridine (Sigma-Aldrich), and plaque purification was carried out with 5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside (Rose Scientific Ltd.) to visualize β-galactosidase-positive viruses.
Recombinant protein production and purification.
cDNA of F1L MVA (residues 18 to 186) was amplified by PCR with a forward primer with a BamHI restriction site and a reverse primer containing an EcoRI restriction site (27). The F1L I125F mutant was generated by overlap extension PCR with wild-type (WT) MVA F1L, and the PCR product was cloned into the pET Duet vector (Invitrogen) with BamHI and EcoRI and expressed in Escherichia coli BL21(DE3)pLysS cells. E. coli cells containing the pET Duet vector were grown to an optical density at 600 nm of 1.0 to 1.2. At that point, protein expression was induced with 500 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h. Cells were then pelleted by centrifugation, and cell pellets were homogenized with an Avestin EmulsiFlex homogenizer in a lysis buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 10 mM 2-mercaptoethanol. These cell lysates containing hexahistidine-tagged F1L were centrifuged and filtered prior to loading onto a 1-ml Hi-Trap chelating column (GE Healthcare) that was charged with nickel. Protein elution was accomplished with an elution buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, 10 mM 2-mercaptoethanol, and 250 mM imidazole. Eluted protein was then subjected to gel filtration chromatography with a Superdex 200 column equilibrated with buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, and 10 mM dithiothreitol, where it eluted as a single peak (27).
Crystallization and structure determination.
MVA F1L:Bak or MVA F1L:Bim BH3 complexes were obtained by mixing MVA F1L with human Bak or Bim 26-mer in a 1:1.25 molar ratio and concentrated with a Centricon (Millipore) to 5 mg/ml. The peptides used were described previously (30). MVA F1L:Bak crystals were grown by the sitting-drop method at room temperature in 0.18 M ammonium sulfate–2.25 M LiCl. The crystals belong to space group P6422 with a = b = 116.691 Å, c = 103.639 Å, α = β = 90°, and γ = 120°. The asymmetric unit contains one MVA F1L chain and one Bak BH3 peptide and has 70% solvent content. Diffraction data were collected from crystals flash cooled in 2 M sodium malonate (pH 7.5) at 100K with beamline MX2 at the Australian Synchrotron. MVA F1L:Bim crystals were grown by the sitting-drop method at room temperature in a mixture of 0.2 M KCl, 15% polyethylene glycol 400 (PEG400), 1.44% 2-methyl-2,4-pentanediol, and 0.1 M ammonium citrate (pH 6.2). The crystals belong to space group P41212 with a = b = 56.503 Å, c = 232.685 Å, and α = β = γ = 90°. The asymmetric unit contains two MVA F1L chains and two Bim BH3 peptides and has 35% solvent content. Diffraction data were collected from crystals flash cooled in 25% PEG400–0.1 M sodium acetate (pH 6.2) at 100K with beamline MX2 at the Australian Synchrotron. Diffraction data were processed with HKL2000 (31) or XDS (32) and programs in the CCP4 suite. All complex structures were solved by molecular replacement with PHASER (33) by using a previously determined F1L structure (Protein Data Bank code 2VTY) (27) as a search model. The final models were built with Coot (34) and refined with Refmac5 (35) or PHENIX (36). All of the data collection and refinement statistics are summarized in Table 1. All of the software used, except HKL200, was accessed via SBGrid (37). Figures were prepared with PyMol (38).
TABLE 1.
Crystallographic data collection and refinement statistics
| Parameter | Value(s) for crystal: |
|
|---|---|---|
| F1L:Bak BH3 | F1L:Bim BH3 | |
| Data collection and phasing | ||
| Space group | P6122 | P4122 |
| Resolution range (Å) | 50–2.9 | 46.54–2.1 |
| No. of unique reflections | 9,630 | 23,134 |
| Multiplicitya | 10.0 (10.1) | 11.7 (12.0) |
| Completeness (%)a | 99.4 (99.9) | 99.9 (100.0) |
| Rmergea,b | 0.054 (0.778) | 0.065 (0.482) |
| I/σI | 39.6 (4.2) | 23.2 (5.3) |
| Refinement | ||
| Resolution range (Å) | 38.74–2.9 | 37.79–2.1 |
| No. of reflections (working set/test set) | 9,151/454 | 23,012/1,182 |
| No. of protein atoms | 1,309 | 2,613 |
| No. of solvent (H2O) atoms | 0 | 163 |
| Rcryst/Rfreec | 0.201/0.228 | 0.215/0.255 |
| RMSD, bonds (Å) | 0.007 | 0.006 |
| RMSD, angles (°) | 1.1 | 0.8 |
| Ramachandran plot (%)d | 89.3/10.7/0.0/0.0 | 97.3/2.7/0.0/0.0 |
Values in parentheses are for the highest-resolution shells.
Rmerge = ΣhΣi | Ii(h) − <I(h)> |/ΣhΣiIi(h), where Ii(h) is the ith measurement of reflection h and <I(h)> is the weighted mean of all measurements of h.
R = Σh|Fobs − Fcalc|/ΣhFobs, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively. Rcryst and Rfree were calculated by using the working and test sets, respectively.
Residues in most favored, additionally allowed, generously allowed, and disallowed regions.
Immunoprecipitation and immunoblotting.
HEK 293T cells (1 × 106) were transfected with Lipofectamine 2000 (Invitrogen) with 0.5 μg of pEGFP, 2 μg of pEGFP-F1L, or pEGFP-F1L point mutations and cotransfected with 1 μg of human Flag-BimL (39) for 12 h. Furthermore, HEK 293T cells (1 × 106) were infected with VACVΔF1L, VACV-Flag-F1L, or the VACVΔF1L-Flag-F1L mutants at an MOI of 10 for 12 h. Cells were lysed in 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS; Sigma-Aldrich) lysis buffer supplemented with EDTA-free proteinase inhibitor (Roche Diagnostics), followed by immunoprecipitation with either goat anti-GFP antibody (Luc Berthiaume, University of Alberta, Edmonton, Alberta, Canada) or rabbit anti-FlagM2 antibody (Sigma-Aldrich). Cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes. Detection of protein was accomplished with mouse anti-GFP (1:5,000; Cedarlane Laboratories Ltd.), mouse anti-Flag (1:5,000; Sigma-Aldrich), rabbit anti-BakNT (1:2,000; Upstate), and rabbit anti-Bim (1:1,000; Enzo Life Sciences) antibodies.
Flow cytometry assay for Bak activation.
Bak activation was assessed by infecting 1 × 106 Jurkat cells or Bak- and Bax-deficient Jurkat cells at an MOI of 10 with VACVF1L or VACVmtF1L carrying point mutations. Six hours postinfection, cells were fixed in 0.25% paraformaldehyde, permeabilized with 500 μg/ml digitonin (Sigma-Aldrich), and stained with a conformation-specific anti-Bak Ab-1 antibody (Oncogene Research Products) (40, 41) or an isotype control antibody specific for NK1.1 (PK136) (42). Phycoerythrin-conjugated anti-mouse antibody was used to counterstain cells (Jackson ImmunoResearch) before analysis by flow cytometry (FACScan; Becton Dickinson) with the FL-2 channel equipped with a 585-nm filter (42-nm band pass). Data were analyzed with CellQuest software.
Cytochrome c release.
Jurkat cells (1 × 106) were mock infected, or infected with VVΔF1L, VV65, VACVΔF1L-FLAG-F1L(Y104E), VACVΔF1L-FLAG-F1L(M108W), VACVΔF1L-FLAG-F1L(M111W), VACVΔF1L-FLAG-F1L(A115W), VACVΔF1L-FLAG-F1L(I132F), VACVΔF1L-FLAG-F1L(V141F), VACVΔF1L-FLAG-F1L(L143F), VACVΔF1L-FLAG-F1L(M114R), VACVΔF1L-FLAG-F1L(I125F), VACVΔF1L-FLAG-F1L(N136F), and VACVΔF1L-FLAG-F1L(F148A) at an MOI of 10. After 6 h of infection, cells were treated with or without 2 μM staurosporine (STS) for up to 6 h. Cells were harvested at 6, 8, 10, and 12 h postinfection and lysed in buffer containing 75 mM NaCl, 1 mM Na1H2PO4, 8 mM Na2HPO4, 250 mM sucrose, and 190 μg/ml of digitonin. Lysates were then incubated on ice for 10 min as previously described (43). Mitochondrial and cytoplasmic fractions were separated via ultracentrifugation for 5 min at 10,000 × g. Mitochondrial fractions were resuspended in buffer containing 25 mM Tris (pH 8.0) and 0.1% Triton X-100. Samples were subjected to SDS-PAGE and immunoblotted with mouse anti-cytochrome c (BD PharMingen).
PARP cleavage assay.
To detect the cleavage of PARP, Jurkat cells (1 × 106) were mock infected or infected with VACV65, VACVΔF1L-FLAG-F1L(Y104E), VACVΔF1L-FLAG-F1L(M108W), VACVΔF1L-FLAG-F1L(M111W), VACVΔF1L-FLAG-F1L(A115W), VACVΔF1L-FLAG-F1L(I132F), VACVΔF1L-FLAG-F1L(V141F), VACVΔF1L-FLAG-F1L(L143F), VACVΔF1L-FLAG-F1L(M114R), VACVΔF1L-FLAG-F1L(I125F), VACVΔF1L-FLAG-F1L(N136F), and VACVΔF1L-FLAG-F1L(F148A) at an MOI of 10. After 6 h of infection, cells were treated with 2 μM STS. Cells were harvested 2, 4, and 6 h after STS treatment and lysed in SDS-PAGE sample buffer containing 8 M urea. Samples were subjected to SDS-PAGE and immunoblotted with anti-PARP (BD PharMingen), anti-β-tubulin (EMC Bioscience), and anti-I3L antibodies to detect virus infection (44).
Protein structure accession numbers.
The coordinates determined in this study have been deposited in the Protein Data Bank and assigned accession codes 4d2m (F1L:Bim complex) and 4d21 (F1L:Bak BH3 complex).
RESULTS
Crystal structures of MVA F1L in complex with the Bim and Bak BH3 domains.
Virus-encoded antiapoptotic proteins have been shown to be critical for successful infection by engaging their proapoptotic Bcl-2 counterparts (45, 46). In VACV, the antiapoptotic F1L protein has been shown to inhibit apoptosis during infection (19). The previously determined crystal structure revealed that F1L adopts a Bcl-2 like fold that forms a domain-swapped dimer with α1 helices swapped between two monomeric Bcl-2 domains (27). However, because of extensive crystal contacts involving the putative ligand binding groove and a previously not observed additional helix, α0, the section of the groove comprising helix α3 was disordered in the structure (27). Consequently, only half of the putative ligand binding groove was visible. To investigate how F1L engages the proapoptotic BH3 domains, we determined the crystal structures of a I125F F1L mutant because of an increase in binding efficiency, in complex with the two biochemically identified BH3 domain ligands, Bim and Bak. Both F1L complexes adopt the previously observed domain-swapped dimeric configuration, which results in the formation of two independent ligand binding grooves (Fig. 1A to D). Crystals of F1L bound to the Bak BH3 peptide (F1L:Bak BH3) contain only a monomer in the asymmetric unit, with the full dimer being formed via a crystallographic 2-fold axis (Fig. 1C). In contrast, crystals of F1L bound to the Bim BH3 peptide (F1L:Bim BH3) contain two F1L and two Bim chains in the asymmetric unit which form the expected dimeric F1L configuration (Fig. 1B). Two Bim molecules are simultaneously bound by two binding grooves on dimeric F1L (Fig. 1A). The novel α0 helix observed in ligand-free F1L is noticeably shorter in both F1L complex structures. In the ligand-free structure, the α0 helix spanned 22 residues (27), whereas in the ligand-bound structures, only 7 to 9 residues of α0 are observed (Fig. 1B to D), with the remainder of the N-terminal residues absent and presumed disordered. Overall, our structures indicate that despite the unusual Bcl-2 fold topology, F1L utilizes the canonical ligand binding groove to engage proapoptotic Bcl-2 proteins.
FIG 1.

Crystal structures of F1L in complex with BH3 peptides of both Bak and Bim. (A) Cartoon representation of F1L bound to the Bim BH3 domain. The F1L dimer is shown as a cartoon (one monomer is green, and the other is orange) with helices α1 to α7 labeled. Two BimL BH3 molecules are shown in magenta. The view is down the 2-fold symmetry axis between the domain-swapped α1 helices. (B) Cartoon representation of F1L bound to the BimL BH3 domain. This view is into the hydrophobic binding grove formed by helices α2 to α5. F1L helices α2 to α7 from monomer 1 (green) are labeled, as is helix α1′ from monomer 2 (orange). BimL BH3 is shown in magenta. (C) Cartoon representation of F1L bound to the Bak BH3 complex. F1L (green and orange) is in complex with Bak BH3 (cyan). The view is the same as in panel B. (D) BH3 domain binding to F1L. The BimL (magenta) and Bak (cyan) BH3 domains are shown as traces bound to the hydrophobic binding groove on F1L, which is shown as a molecular surface (gray). (E) Superimposition of the F1L main chains from the two complexes formed with the BimL (green) and Bak (blue) BH3 domains.
The binding interface of F1L with the Bak and Bim BH3 peptides.
Helices α2, α3, α4, and α5 of F1L form an extended binding groove for the BH3 domain and are engaged in nearly identical fashions (Fig. 1E). Superimposition of the Cα backbone of F1L in complex with both BH3 domain ligands yields a root mean square deviation (RMSD) of 0.7 Å (over 103 Cα atoms), highlighting the similarity of F1L in both complexes. In F1L bound to the Bim BH3 peptide (F1L:Bim BH3) (Fig. 2A), residues I58, L62, I65, and F69 protrude into four pockets in the F1L binding groove, similar to the equivalent hydrophobic residues in Bak (V74, L78, I81, and I85) (Fig. 2B). The mutated I125 residue lies in close proximity to Bim I58 and Bak V74, respectively, and the presence of the larger phenylalanine in place of the WT isoleucine may have subtly altered the binding groove. As predicted previously, D67 of Bim is not engaged in a salt bridge with an Arg residue from F1L (27), in contrast to the binding of BimL to mammalian prosurvival Bcl-2, where a D67-mediated salt bridge to an Arg is fully conserved across all mammalian prosurvival Bcl-2–BH3 domain interactions (47). F1L binding to Bim is mediated largely by hydrophobic and van der Waals contacts, whereas no hydrogen bonds between F1L and Bim residues are observed (Fig. 2A). In contrast, F1L forms a salt bridge between D103 and Bak R88, as well as a hydrogen bond between the main chain amino group of G140 and the asparagine side chain of Bak N86 (Fig. 2B).
FIG 2.

Stereo diagrams of F1L in complex with the BH3 peptides of both Bak and Bim. (A) Stereo diagram of the binding interface formed by the F1L (green):Bim (magenta) complex. The F1L surface is gray, except for the orange shading indicating the bottom of the peptide binding groove. F1L residues are labeled in black, and BimL BH3 residues are labeled in magenta. (B) Stereo diagram of the binding interface formed by the F1L (green):Bak (cyan) complex. The view and labeling are the same as in panel A, except for the labeling of Bak residues in cyan.
Superimposition of M11L bound to Bak (48) and F1L bound to Bak yields an RMSD of 3.3 Å over 101 Cα atoms. Of the 16 residues that are conserved between F1L and M11L, 5 are located in the binding groove (Y104, Y112, D113, V141, and A144), with a further six conservative substitutions, highlighting the powerful conservation of features in the binding groove despite the low overall sequence identity (10%). Despite the overall low level of sequence identity between F1L and M11L and their fundamentally different Bcl-2 fold topology, the two viral proteins utilize similar key residues to engage BH3 domain ligands without relying on the hallmark ionic interaction between an arginine from prosurvival Bcl-2 and a BH3 domain aspartic acid observed in mammalian prosurvival Bcl-2:BH3 domain complexes (11).
F1L binding pocket residues are responsible for Bak interaction and preventing Bak activation.
We previously identified VACV F1L as a potent antiapoptotic protein that localizes to the OMM, inhibits cytochrome c release (19, 49), and interacts with Bcl-2 family members Bak and BimL (25, 26). The structure of F1L was previously described and, despite limited sequence similarity, was found to fold in a fashion similar to that of members of the Bcl-2 family (27). The crystal structure led to the elucidation of putative BH domains that are responsible for the observed heteromeric interactions (24). To further characterize the putative BH1 and BH3 domains of F1L, we performed extensive structure-guided mutagenesis to determine the residues responsible for interaction with Bim and Bak. We targeted a number of hydrophobic residues in F1L that were in close contact with the four key hydrophobic residues in the BH3 domain of Bim and Bak that protrude into the F1L binding groove (Fig. 3). We employed immunoprecipitation to determine any observable interaction between the panel of Flag-tagged F1L mutants with endogenous Bak during HeLa cell infection. Upon immunoprecipitation with Flag and immunoblotting with BakNT antibody (40, 41), VACV-Flag-F1L, VACVΔF1L-Flag-F1L(M108W), VACVΔF1L-Flag-F1L(A115W), VACVΔF1L-Flag-F1L(M114R), and VACVΔF1L-Flag-F1L(I125F) interacted with endogenous Bak (Fig. 4A). In contrast, VACVΔF1L, VACVΔF1L-Flag-F1L(Y104E), VACVΔF1L-Flag-F1L(M111W), VACVΔF1L-Flag-F1L(I132F), VACVΔF1L-Flag-F1L(V141F), VACVΔF1L-Flag-F1L(L143F), VACVΔF1L-Flag-F1L(N136F), and VACVΔF1L-Flag-F1L(F148A) had a substantially reduced ability to interact with Bak (Fig. 4A).
FIG 3.
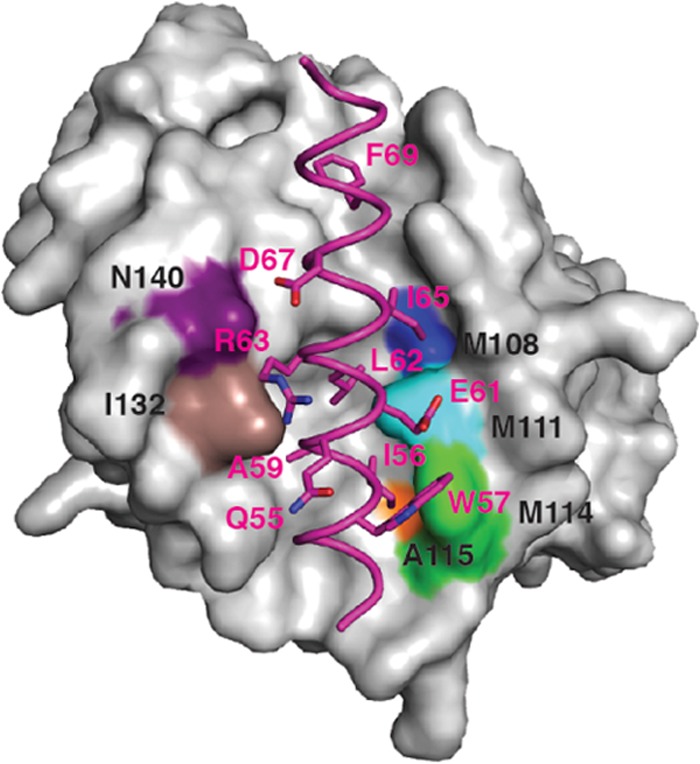
Structure of F1L in complex with the Bim BH3 peptide highlighting the F1L binding pocket hydrophobic residues. Shown is the molecular surface of F1L in complex with the BH3 peptide of BimL (in magenta). The FIL binding pocket residues that facilitate hydrophobic contact with specific amino acids of BimL are highlighted in colors. Included are F1L(N140), which is purple; F1L(I132), which is brown; F1L(A119W), which is orange; F1L(M114), which is green; F1L(M111), which is aqua; and F1L(M108), which is blue.
FIG 4.

F1L binding pocket residues are responsible for Bak interaction and prevention of Bak activation. (A) HeLa cells were mock infected or infected with VACVΔF1L, VACV-FLAG-F1L, or recombinant virus expressing the F1L binding pocket mutations. Infected-cell lysates were then immunoprecipitated (IP) with a monoclonal antibody recognizing FLAG, and Bak was detected by blotting with an anti-Bak monoclonal antibody (40, 41). (B) Jurkat cells were infected with WT VACV expressing EGFP, VACVΔF1L, or a panel of recombinant VACVs carrying F1L point mutations (VACVΔF1L-F1L) for 4 h at an MOI of 10 before treatment with 250 nM STS for 1.5 h to induce apoptosis. Bak N-terminal exposure was monitored by staining cells with the conformation-specific anti-Bak AB-1 antibody (40, 41) or an anti-NK1.1 antibody (42) as an isotype control. Shaded histograms, untreated cells; open histograms, STS-treated cells. WB, Western blotting.
The initial immunoprecipitation experiments demonstrated that numerous hydrophobic residues in the binding cleft of F1L are important for facilitating interactions with Bak. To further understand the effects of abrogating Bak interaction, we examined the ability of the F1L binding cleft mutants to prevent Bak activation with the conformationally specific anti-Bak AB-1 antibody (26). The Bak AB-1 antibody binds Bak upon activation, signifying oligomerization (41). Because of the variability in Bak binding, we predict that the binding pocket mutants will also show heterogeneity in the ability to prevent Bak activation as monitored by flow cytometry. Jurkat cells were infected with WT VACV (VACV-EGFP), VACV devoid of F1L (VACVΔF1L), or VACV expressing Flag-tagged F1L binding pocket mutations. To further promote Bak activation, mock-infected and infected cells were treated in the presence or absence of STS, a potent inducer of intrinsic apoptosis (50). Numerous viruses were able to completely prevent Bak activation, including VACV-EGFP (Fig. 4B, b), which was used as a positive control, VACVΔF1L-Flag-F1L(M108W) (Fig. 4B, e), and VACVΔF1L-Flag-F1L(M111W) (Fig. 4B, f), whereas VACVΔF1L-Flag-F1L(M114R) (Fig. 4B, k), VACVΔF1L-Flag-F1L(V141F) (Fig. 4B, i), VACVΔFlag-F1L(I125F) (Fig. 4B, l), VACVΔFlag-F1L(Y104E) (Fig. 4B, d), VACVΔFlag-F1L(L143F) (Fig. 4B, j), and VACVΔFlag-F1L(F148A) (Fig. 4B, n) caused minimal Bak activation. A subset of viruses including VACVΔF1L (Fig. 4B, c), VACVΔF1L-Flag-F1L(A115W) (Fig. 4B, g), VACVΔF1L-Flag-F1L(I132F) (Fig. 4B, h), and VACVΔF1L-Flag-F1L(N136F) (Fig. 4B, m) were unable to prevent Bak activation. This finding implies not only the importance of the hydrophobic residues in the binding cleft of F1L for Bak interaction but also the possibility of F1L sequestration of an upstream activator of apoptosis, including BimL.
Residues within the F1L binding pocket contribute to BimL binding.
The BH3-only protein Bim is a potent activator of apoptosis that is able to directly activate Bak and Bax, as well as engage and sequester endogenous prosurvival Bcl-2 proteins (9, 10, 51). In mammalian cells, three isoforms of Bim exist that play a role in apoptosis; these include BimS, BimL, and BimEL (52). We have previously shown that F1L can interact with BimL and, through this interaction, prevent Bax activation (25). Since Bim can both directly and indirectly activate Bak, we examined the ability of the F1L binding pocket mutants to immunoprecipitate BimL. To examine the importance of the hydrophobic residues within the binding pocket of F1L that interact with BimL, we cotransfected HEK 293T cells with the EGFP-F1L mutants and Flag-BimL. We observed that upon immunoprecipitation with EGFP and immunoblotting with BimL, EGFP-F1L, EGFP-F1L(M108W), EGFP-F1L(M111W), EGFP-F1L(L143F), and EGFP-F1L(I125F) were capable of interacting with BimL (Fig. 5). Notably, EGFP-F1L(I125F) appeared to bind more BimL than did WT F1L, similar to earlier findings that this mutation increased the affinity of recombinant F1L for Bim BH3 peptides 7-fold (KD = 34 nM) (27). Overall, EGFP-F1L(Y104E), EGFP-F1L(A115W), EGFP-F1L(I132F), EGFP-F1L(V141F), EGFP-F1L(M114R), and EGFP-F1L(N136F) had a diminished ability to interact with BimL. In contrast, the ability of EGFP-F1L(F148A) to interact with BimL was completely abrogated (Fig. 5). The relatively small number of F1L mutants that exhibited WT-like binding to BimL and Bak suggests that the F1L binding groove is highly sensitive to mutations.
FIG 5.
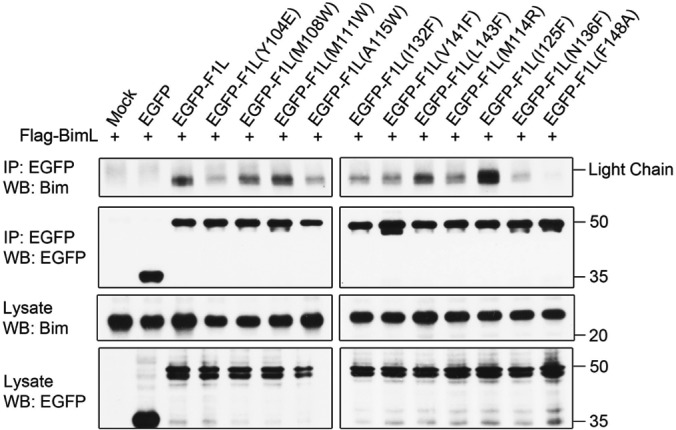
F1L binding pocket residues facilitate interactions with the BH3-only protein BimL. HEK 293T cells were cotransfected with EGFP, EGFP-F1L, or EGFP-tagged F1L containing the binding cleft mutation with FLAG-BimL. Cells were lysed and immunoprecipitated (IP) with goat anti-GFP antibody. Samples were separated by SDS-PAGE, and BimL was detected with anti-Bim antibody (Enzo Life Sciences). WB, Western blotting.
Hydrophobic residues within the binding cleft of F1L are responsible for preventing cell death.
Previous results have elucidated the importance of the hydrophobic residues for Bak and BimL interaction. To prevent mitochondrial dysfunction, we assayed individual mutants for the ability to prevent cytochrome c release in the presence of STS. As controls, cells were mock infected (Fig. 6a) or infected with VACVΔF1L (Fig. 6b) or VACV65 (Fig. 6c). VACVΔF1L-Flag-F1L(Y104E) (Fig. 6d), VACVΔF1L-Flag-F1L(A115W) (Fig. 6g), VACVΔF1L-Flag-F1L(I132F) (Fig. 6h), and VACVΔF1L-Flag-F1L(N136F) (Fig. 6m) were unable to prevent cytochrome c release. The viruses fully capable of maintaining mitochondrial potential included WT VACV65 (Fig. 6c), VACVΔF1L-Flag-F1L(M108W) (Fig. 6e), and VACVΔF1L-Flag-F1L(I125F) (Fig. 6l). Finally, VACVΔF1L-Flag-F1L(M111W) (Fig. 6f), VACVΔF1L-Flag-F1L(V141F) (Fig. 6i), VACVΔF1L-Flag-F1L(L143F) (Fig. 6j), VACVΔF1L-Flag-F1L(M114R) (Fig. 6k), and VACVΔF1L-Flag-F1L(F148A) (Fig. 6n) were only partially protective in the ability to prevent cytochrome c release. This finding exemplifies the importance of the binding cleft residues in preventing the release of proapoptotic molecules from the mitochondria in the presence of apoptotic stimuli.
FIG 6.

Prevention of cytochrome c release by infection with VACV encoding F1L binding pocket mutants. Jurkat cells (1 × 106) were mock infected (a) or infected with VACVΔF1L (b), VACV65 (c), or a panel of recombinant VACVs carrying F1L point mutations (d to n) at an MOI of 10 for 6, 8, 10, or 12 h. At 6 h postinfection, cells were treated with 2 μM STS. Mitochondrial and cytoplasmic fractions were separated in lysis buffer containing digitonin. Mitochondrial fractions were subsequently resuspended in 0.1% Triton X-100 lysis buffer. Twenty percent of the mitochondrial fractions and 50% of the cytoplasmic fractions were Western blotted (WB) with anti-cytochrome c antibody to determine the presence of cytochrome c in the separated fractions or with anti-BakNT antibody to ensure the separation of the two fractions.
To further investigate the ability of F1L mutants to prevent apoptosis, we determined the extent of PARP cleavage, an indicator of cell death (53). VACVΔF1L, VACVΔF1L-Flag-F1L(Y104E) (Fig. 7d), VACVΔF1L-Flag-F1L(A115W) (Fig. 7g), VACVΔF1L-Flag-F1L(I132F) (Fig. 7h), and VACVΔF1L-Flag-F1L(N136F) (Fig. 7m) were again compromised in the ability to prevent apoptosis, while WT VACV65 (Fig. 7c) and VACVΔF1L-Flag-F1L(M111W) (Fig. 7f) prevented PARP cleavage. Additionally, VACVΔF1L-Flag-F1L(M108W) (Fig. 7e), VACVΔF1L-Flag-F1L(M114R) (Fig. 7k), VACVΔF1L-Flag-F1L(V141F) (Fig. 7i), VACVΔF1L-Flag-F1L(L143F) (Fig. 7j), VACVΔF1L-Flag-F1L(I125F) (Fig. 7l), and VACVΔF1L-Flag-F1L(F148A) (Fig. 7n) were only slightly impaired in the ability to prevent PARP cleavage. These data, in combination with the immunoprecipitation and Bak activation data, provide significant evidence of the importance of the hydrophobic residues within the binding cleft of F1L in regard to both binding capabilities and prevention of cell death. Furthermore, these experiments provide support for the critical importance of BimL interaction and sequestration by F1L in the prevention of mitochondrion-mediated cell death.
FIG 7.

Inhibition of PARP cleavage by infection with VACV encoding F1L binding pocket mutations. Jurkat cells (1 × 106) were mock infected (a) or infected with VACV65 (b), VACVΔF1L (c), or a panel of recombinant VACVs carrying the F1L binding pocket point mutations (d to n) at an MOI of 10. After 6 h of infection, cells were treated with 2 μM STS for 2, 4, or 6 h. Whole-cell lysates were then collected in SDS lysis buffer containing 8 M urea. Samples were subjected to SDS-PAGE and Western blotted (WB) for PARP to determine apoptosis, β-tubulin as a loading control, and I3L as a sign of infection (44).
DISCUSSION
Apoptosis is an important innate defense mechanism utilized by higher organisms to fight viral infections (2). Consequently, viruses have evolved an array of proteins to ensure survival and proliferation. Numerous viruses, including the families Adenoviridae (14), Herpesviridae (15, 54, 55), Asfarviridae (56), and Poxviridae (18, 20, 22, 23, 57–59), encode proteins that are crucial for subverting host apoptotic defenses. The ability to prevent host cell death hinges on a group of viral Bcl-2-like proteins that, under certain instances and despite limited sequence identity, share structural homology with members of the Bcl-2 family. Ultimately, loss of the viral Bcl-2 proteins can lead to widespread apoptosis upon viral infection, resulting in reduced viral dissemination (19, 46), underscoring the importance of neutralizing host apoptotic responses.
Despite significant advances in our understanding of how viruses neutralize apoptosis by using Bcl-2-like proteins, redundancy in mammalian apoptotic signaling has hampered the identification of key proapoptotic effector Bcl-2 proteins that have to be targeted at a minimum for successful viral infection and proliferation. Myxoma virus M11L has been shown to act primarily by sequestering Bax and Bak (48); however, these experiments were not performed with live myxoma virus. Other viral Bcl-2 proteins have been shown to utilize targeting strategies with respect to proapoptotic Bcl-2 proteins. For example, ORF virus ORFV125 has been shown to engage Bim, Bik, Hrk, Noxa, Puma, and Bax (60), whereas VACV N1L binds peptides from Bid, Bim, and Bak (57) and immunoprecipitates Bid, Bad, and Bax (58). Sheeppox SPPV14 binds Bim, Bid, Bmf, Hrk, and Puma, as well as Bax and Bak (59), and fowlpox FPV039 engages Bik and Bim, as well as Bak and Bax (20). However, of all of the poxviral Bcl-2-like inhibitors characterized to date, VACV F1L harbors the most restricted proapoptotic Bcl-2 ligand binding profile, making it a suitable candidate for careful dissection of the relative importance of each proapoptotic ligand during VACV infection. F1L has previously been shown to engage a very limited number of proapoptotic Bcl-2 proteins and displays higher affinity for the Bim BH3 domain, whereas the Bak and Bax BH3 domains are bound with modest affinity in biosensor assays (17, 27). Notably, in vitro interaction with Bax is not observed in cells (25). Mechanistically, F1L operates by replacing the antiapoptotic activity of Mcl-1 (24) and sequesters Bak (26, 61) and Bim (25), although other lines of evidence suggest that Noxa plays a role in F1L-mediated inhibition of apoptosis despite the lack of a direct interaction (62).
Here we report the identification of BimL, which was recently shown to be the sole BH3-only protein that is targeted by all mammalian prosurvival Bcl-2 proteins (63), as a critical target of F1L-mediated subversion of apoptotic signaling during VACV infection. F1L point mutants that selectively bind either BimL or Bak showed antiapoptotic activity in a cell-based model of VACV infection that correlated primarily with F1L engagement of BimL, with a limited association with Bak, in immunoprecipitation assays. The binding and sequestration of BimL by F1L can potentially prevent direct activation of Bak and Bax, as well as enable the release of prosurvival proteins in the process. Examination of the binding phenotypes and presence or absence of cell death inhibition in F1L binding cleft mutants provides a clearer scenario of apoptosis mechanisms during VACV infection.
On the basis of immunoprecipitation data, we distinguished four subgroups of F1L binding cleft mutants, (i) mutants that behaved like WT F1L by binding both BimL and Bak, (ii) mutants that lost binding to both BimL and Bak, (iii) mutants that were able to bind Bak but unable to interact with BimL, and (iv) mutants that bound only BimL but not Bak. F1L binding pocket mutants with binding phenotypes similar to those of WT F1L were fully capable of preventing Bak activation. Furthermore, these mutants were able to prevent cytochrome c release from the mitochondria and were capable of preventing the downstream cleavage of the apoptotic indicator PARP (2, 50). Binding cleft mutants whose ability to bind both BimL and Bak was abrogated were dramatically impaired in these assays, including both F1L(N136F) and F1L(F148A) (Table 2). However, F1L(Y104E) appears to contain a unique binding profile that is completely abrogated in both Bak and BimL binding yet retains the ability to prevent Bak activation (Table 2). This supports the idea that F1L may sequester another upstream BH3-only protein. Of particular interest, the F1L(A115W) mutant, which binds only Bak but not BimL, was unable to prevent Bak activation, cytochrome c release, or PARP cleavage, indicating complete abrogation of antiapoptotic activity. In contrast, F1L (L143F) and F1L(M111W) were able to bind BimL yet lost the ability to pull down Bak (Table 2). Interestingly, this activity was sufficient to prevent Bak activation, cytochrome c release, and PARP cleavage, providing evidence of the importance of Bim sequestration. Overall, our data indicate that the ability to prevent Bak activation, cytochrome c release, and PARP cleavage coincided with BimL interactions and less so for F1L binding to Bak (Fig. 4B). This suggests that BimL sequestration by F1L is the predominant mechanism that enables F1L to protect against both virus- and STS-induced Bak activation and subsequent apoptosis.
TABLE 2.
Comprehensive list of F1L binding pocket mutants and their abilities to interact with both Bak and Bim and prevent Bak activationa
| Strain | Bak interaction | Prevention of Bak activation | Bim interaction | Prevention of cytochrome c release | Prevention of PARP cleavage |
|---|---|---|---|---|---|
| F1L (WT) | +++ | +++ | ++ | +++ | +++ |
| F1L(Y104E) | − | ++ | − | − | − |
| F1L(M108W) | ++ | +++ | ++ | ++ | ++ |
| F1L(M111W) | − | +++ | ++ | ++ | + |
| F1L(A115W) | +++ | − | + | + | − |
| F1L(I132F) | + | − | + | + | − |
| F1L(V141F) | + | ++ | + | + | + |
| F1L(L143F) | − | ++ | ++ | + | + |
| F1L(M114R) | ++ | ++ | + | + | + |
| F1L(I125F) | ++ | ++ | +++ | ++ | ++ |
| F1L(N136F) | − | − | + | − | − |
| F1L(F148A) | − | − | − | + | + |
Symbols: +++, full ability to interact with Bak or Bim or to prevent Bak activation; ++, moderately reduced ability to interact with Bak or Bim or to prevent Bak activation; +, severely reduced ability to interact with Bak or Bim or to prevent Bak activation; −, abrogation of interaction or prevention of Bak activation capability.
BimL has previously been shown to play a role during measles infection because of the induction of endoplasmic reticulum stress (64). However, in that study, measles infection led to the rapid onset of apoptosis, with the virus evidently not initiating suppression of host cell apoptosis. In the case of EBV, BHRF1 has been shown to target BimL (65), although additional lines of evidence also suggest a role for Bak inhibition (66). Using a combination of structural, biochemical, and cell biological approaches, we now show that F1L-mediated sequestration of BimL is an important step in preventing host cell apoptosis during VACV infection. Loss of Bak binding appears to have a minor impact on the ability of F1L to counter apoptosis, whereas loss of Bim binding correlates with a loss of antiapoptotic activity even if significant binding to Bak is maintained. By sequestering and preventing normal BimL functions, F1L can prevent direct activation of both Bak and Bax. Furthermore, this binding and occlusion can ultimately promote the release of prosurvival Bcl-2 members, thus indirectly preventing the oligomerization of Bak and Bax.
ACKNOWLEDGMENTS
We thank A. Wardak for technical assistance, the staff at beamline MX2 at the Australian Synchrotron for help with X-ray data collection, and the CSIRO C3 Collaborative Crystallization Centre for assistance with crystallization. We thank Emeka Enwere, Ryan Noyce, Jacquie Gulbis, Silke Fischer, Brian Smith, David Huang, Hamsa Puthalakath, and Sofia Caria for discussions and thank H. Rabinowich and G. Hacker for reagents.
This work was supported by the National Health and Medical Research Council Australia (project grant APP1007918 and fellowship 637372 to M.K. and a fellowship and program grant to P.M.C.), the Australian Research Council (fellowship FT130101349 to M.K.), and Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS. Our work is supported by the Canadian Institute of Health Research. Michele Barry is a Tier 1 Canada Research Chair.
Footnotes
Published ahead of print 21 May 2014
This article is dedicated to John Thibault (20 July 1981 to 10 May 2014), esteemed colleague, brilliant scientist, and outstanding friend.
REFERENCES
- 1.Cuconati A, White E. 2002. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev. 16:2465–2478. 10.1101/gad.1012702 [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog. 4:e1000018. 10.1371/journal.ppat.1000018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor JM, Barry M. 2006. Near death experiences: poxvirus regulation of apoptotic death. Virology 344:139–150. 10.1016/j.virol.2005.09.032 [DOI] [PubMed] [Google Scholar]
- 4.Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9:47–59. 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- 5.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. 2010. The BCL-2 family reunion. Mol. Cell 37:299–310. 10.1016/j.molcel.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM. 2013. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152:519–531. 10.1016/j.cell.2012.12.031 [DOI] [PubMed] [Google Scholar]
- 7.Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, Kluck RM. 2012. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 19:661–670. 10.1038/cdd.2011.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green DR, Kroemer G. 2004. The pathophysiology of mitochondrial cell death. Science 305:626–629. 10.1126/science.1099320 [DOI] [PubMed] [Google Scholar]
- 9.Happo L, Strasser A, Cory S. 2012. BH3-only proteins in apoptosis at a glance. J. Cell Sci. 125:1081–1087. 10.1242/jcs.090514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. 2011. BH3-only proteins: orchestrators of apoptosis. Biochim. Biophys. Acta 1813:508–520. 10.1016/j.bbamcr.2010.11.024 [DOI] [PubMed] [Google Scholar]
- 11.Kvansakul M, Hinds MG. 2013. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 4:e909. 10.1038/cddis.2013.436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strasser A. 2005. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5:189–200. 10.1038/nri1568 [DOI] [PubMed] [Google Scholar]
- 13.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. 2010. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 330:1390–1393. 10.1126/science.1190217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White E, Sabbatini P, Debbas M, Wold WS, Kusher DI, Gooding LR. 1992. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor alpha. Mol. Cell. Biol. 12:2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. 1993. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. U. S. A. 90:8479–8483. 10.1073/pnas.90.18.8479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartlett N, Symons JA, Tscharke DC, Smith GL. 2002. The vaccinia virus N1L protein is an intracellular homodimer that promotes virulence. J. Gen. Virol. 83:1965–1976 http://vir.sgmjournals.org/content/83/8/1965.long [DOI] [PubMed] [Google Scholar]
- 17.Fischer SF, Ludwig H, Holzapfel J, Kvansakul M, Chen L, Huang DC, Sutter G, Knese M, Hacker G. 2006. Modified vaccinia virus Ankara protein F1L is a novel BH3-domain-binding protein and acts together with the early viral protein E3L to block virus-associated apoptosis. Cell Death Differ. 13:109–118. 10.1038/sj.cdd.4401718 [DOI] [PubMed] [Google Scholar]
- 18.Graham KA, Opgenorth A, Upton C, McFadden G. 1992. Myxoma virus M11L ORF encodes a protein for which cell surface localization is critical in manifestation of viral virulence. Virology 191:112–124. 10.1016/0042-6822(92)90172-L [DOI] [PubMed] [Google Scholar]
- 19.Wasilenko ST, Stewart TL, Meyers AF, Barry M. 2003. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100:14345–14350. 10.1073/pnas.2235583100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banadyga L, Gerig J, Stewart T, Barry M. 2007. Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein Bak. J. Virol. 81:11032–11045. 10.1128/JVI.00734-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Best SM. 2008. Viral subversion of apoptotic enzymes: escape from death row. Annu. Rev. Microbiol. 62:171–192. 10.1146/annurev.micro.62.081307.163009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westphal D, Ledgerwood EC, Hibma MH, Fleming SB, Whelan EM, Mercer AA. 2007. A novel Bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J. Virol. 81:7178–7188. 10.1128/JVI.00404-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banadyga L, Lam SC, Okamoto T, Kvansakul M, Huang DC, Barry M. 2011. Deerpox virus encodes an inhibitor of apoptosis that regulates Bak and Bax. J. Virol. 85:1922–1934. 10.1128/JVI.01959-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell S, Hazes B, Kvansakul M, Colman P, Barry M. 2010. Vaccinia virus F1L interacts with Bak using highly divergent Bcl-2 homology domains and replaces the function of Mcl-1. J. Biol. Chem. 285:4695–4708. 10.1074/jbc.M109.053769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor JM, Quilty D, Banadyga L, Barry M. 2006. The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J. Biol. Chem. 281:39728–39739. 10.1074/jbc.M607465200 [DOI] [PubMed] [Google Scholar]
- 26.Wasilenko ST, Banadyga L, Bond D, Barry M. 2005. The vaccinia virus F1L protein interacts with the proapoptotic protein Bak and inhibits Bak activation. J. Virol. 79:14031–14043. 10.1128/JVI.79.22.14031-14043.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kvansakul M, Yang H, Fairlie WD, Czabotar PE, Fischer SF, Perugini MA, Huang DC, Colman PM. 2008. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 15:1564–1571. 10.1038/cdd.2008.83 [DOI] [PubMed] [Google Scholar]
- 28.Perkus ME, Goebel SJ, Davis SW, Johnson GP, Norton EK, Paoletti E. 1991. Deletion of 55 open reading frames from the termini of vaccinia virus. Virology 180:406–410. 10.1016/0042-6822(91)90047-F [DOI] [PubMed] [Google Scholar]
- 29.Earl PL, Moss B, Wyatt LS, Carroll MW. 1998. Generation of recombinant vaccinia viruses, p 16.17.11–16.17.19 In Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl KK. (ed), Current protocols in molecular biology. Wiley Interscience, New York, NY [Google Scholar]
- 30.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17:393–403. 10.1016/j.molcel.2004.12.030 [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-x [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W. 2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storoni LC, McCoy AJ, Read RJ. 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. Biol. Crystallogr. 60:432–438. 10.1107/S0907444903028956 [DOI] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 36.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P. 2013. Collaboration gets the most out of software. eLife 2:e01456. 10.7554/eLife.01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeLano WL. 2002. The PyMOL molecular graphics system. http://www.pymol.org
- 39.Lei K, Davis RJ. 2003. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100:2432–2437. 10.1073/pnas.0438011100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Griffiths GJ, Corfe BM, Savory P, Leech S, Esposti MD, Hickman JA, Dive C. 2001. Cellular damage signals promote sequential changes at the N-terminus and BH-1 domain of the pro-apoptotic protein Bak. Oncogene 20:7668–7676. 10.1038/sj.onc.1204995 [DOI] [PubMed] [Google Scholar]
- 41.Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, Dive C, Hickman JA. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903–914. 10.1083/jcb.144.5.903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koo GC, Peppard JR. 1984. Establishment of monoclonal anti-Nk-1.1 antibody. Hybridoma 3:301–303. 10.1089/hyb.1984.3.301 [DOI] [PubMed] [Google Scholar]
- 43.Wasilenko ST, Meyers AF, Vander Helm K, Barry M. 2001. Vaccinia virus infection disarms the mitochondrion-mediated pathway of the apoptotic cascade by modulating the permeability transition pore. J. Virol. 75:11437–11448. 10.1128/JVI.75.23.11437-11448.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Welsch S, Doglio L, Schleich S, Krijnse Locker J. 2003. The vaccinia virus I3L gene product is localized to a complex endoplasmic reticulum-associated structure that contains the viral parental DNA. J. Virol. 77:6014–6028. 10.1128/JVI.77.10.6014-6028.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altmann M, Hammerschmidt W. 2005. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 3:e404. 10.1371/journal.pbio.0030404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang G, Barrett JW, Nazarian SH, Everett H, Gao X, Bleackley C, Colwill K, Moran MF, McFadden G. 2004. Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J. Virol. 78:7097–7111. 10.1128/JVI.78.13.7097-7111.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. 2003. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity 19:341–352. 10.1016/S1074-7613(03)00234-6 [DOI] [PubMed] [Google Scholar]
- 48.Kvansakul M, van Delft MF, Lee EF, Gulbis JM, Fairlie WD, Huang DC, Colman PM. 2007. A structural viral mimic of prosurvival Bcl-2: a pivotal role for sequestering proapoptotic Bax and Bak. Mol. Cell 25:933–942. 10.1016/j.molcel.2007.02.004 [DOI] [PubMed] [Google Scholar]
- 49.Stewart TL, Wasilenko ST, Barry M. 2005. Vaccinia virus F1L protein is a tail-anchored protein that functions at the mitochondria to inhibit apoptosis. J. Virol. 79:1084–1098. 10.1128/JVI.79.2.1084-1098.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chae HJ, Kang JS, Byun JO, Han KS, Kim DU, Oh SM, Kim HM, Chae SW, Kim HR. 2000. Molecular mechanism of staurosporine-induced apoptosis in osteoblasts. Pharmacol. Res. 42:373–381. 10.1006/phrs.2000.0700 [DOI] [PubMed] [Google Scholar]
- 51.Walensky LD, Gavathiotis E. 2011. BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem. Sci. 36:642–652. 10.1016/j.tibs.2011.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marani M, Tenev T, Hancock D, Downward J, Lemoine NR. 2002. Identification of novel isoforms of the BH3 domain protein Bim which directly activate Bax to trigger apoptosis. Mol. Cell. Biol. 22:3577–3589. 10.1128/MCB.22.11.3577-3589.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gobeil S, Boucher CC, Nadeau D, Poirier GG. 2001. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 8:588–594. 10.1038/sj.cdd.4400851 [DOI] [PubMed] [Google Scholar]
- 54.Cheng EH, Nicholas J, Bellows DS, Hayward GS, Guo HG, Reitz MS, Hardwick JM. 1997. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. U. S. A. 94:690–694. 10.1073/pnas.94.2.690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nava VE, Cheng EH, Veliuona M, Zou S, Clem RJ, Mayer ML, Hardwick JM. 1997. Herpesvirus saimiri encodes a functional homolog of the human bcl-2 oncogene. J. Virol. 71:4118–4122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brun A, Rodríguez F, Escribano JM, Alonso C. 1998. Functionality and cell anchorage dependence of the African swine fever virus gene A179L, a viral bcl-2 homolog, in insect cells. J. Virol. 72:10227–10233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aoyagi M, Zhai D, Jin C, Aleshin AE, Stec B, Reed JC, Liddington RC. 2007. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 16:118–124. 10.1110/ps.062454707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cooray S, Bahar MW, Abrescia NG, McVey CE, Bartlett NW, Chen RA, Stuart DI, Grimes JM, Smith GL. 2007. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 88:1656–1666. 10.1099/vir.0.82772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okamoto T, Campbell S, Mehta N, Thibault J, Colman PM, Barry M, Huang DC, Kvansakul M. 2012. Sheeppox virus SPPV14 encodes a Bcl-2-like cell death inhibitor that counters a distinct set of mammalian proapoptotic proteins. J. Virol. 86:11501–11511. 10.1128/JVI.01115-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Westphal D, Ledgerwood EC, Tyndall JD, Hibma MH, Ueda N, Fleming SB, Mercer AA. 2009. The orf virus inhibitor of apoptosis functions in a Bcl-2-like manner, binding and neutralizing a set of BH3-only proteins and active Bax. Apoptosis 14:1317–1330. 10.1007/s10495-009-0403-1 [DOI] [PubMed] [Google Scholar]
- 61.Postigo A, Cross JR, Downward J, Way M. 2006. Interaction of F1L with the BH3 domain of Bak is responsible for inhibiting vaccinia-induced apoptosis. Cell Death Differ. 13:1651–1662. 10.1038/sj.cdd.4401853 [DOI] [PubMed] [Google Scholar]
- 62.Eitz Ferrer P, Potthoff S, Kirschnek S, Gasteiger G, Kastenmuller W, Ludwig H, Paschen SA, Villunger A, Sutter G, Drexler I, Hacker G. 2011. Induction of Noxa-mediated apoptosis by modified vaccinia virus Ankara depends on viral recognition by cytosolic helicases, leading to IRF-3/IFN-beta-dependent induction of pro-apoptotic Noxa. PLoS Pathog. 7:e1002083. 10.1371/journal.ppat.1002083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rautureau GJ, Yabal M, Yang H, Huang DC, Kvansakul M, Hinds MG. 2012. The restricted binding repertoire of Bcl-B leaves Bim as the universal BH3-only prosurvival Bcl-2 protein antagonist. Cell Death Dis. 3:e443. 10.1038/cddis.2012.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129:1337–1349. 10.1016/j.cell.2007.04.027 [DOI] [PubMed] [Google Scholar]
- 65.Desbien AL, Kappler JW, Marrack P. 2009. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. U. S. A. 106:5663–5668. 10.1073/pnas.0901036106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kvansakul M, Wei AH, Fletcher JI, Willis SN, Chen L, Roberts AW, Huang DC, Colman PM. 2010. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 6:e1001236. 10.1371/journal.ppat.1001236 [DOI] [PMC free article] [PubMed] [Google Scholar]